

Abstract
Inhibition of dephosphorylation of the 20 kDa myosin light chain (MLC20) is an important mechanism for the Ca2+-induced sensitization of vascular smooth muscle contraction. We investigated whether this mechanism operates in prostaglandin F2α (PGF2α)-induced contraction of rabbit aortic smooth muscle and, if so, whether protein kinase C (PKC) or rho-associated kinase (rho kinase) contribute to the inhibition of dephosphorylation. In normal medium, PGF2α (10 μm) increased the phosphorylation of MLC20 and developed tension. The rho-kinase inhibitors fasudil and hydroxyfasudil inhibited these changes, despite having no effect on a phorbol-ester-induced MLC20 phosphorylation. After treatment with verapamil or chelation of external Ca2+ with EGTA, PGF2α increased the MLC20 phosphorylation and tension without an increase in[Ca2+]i, all of which were sensitive to fasudil and hydroxyfasudil. ML-9, a MLC kinase inhibitor, quickly reversed the KCl-induced MLC20 phosphorylation and contraction to the resting level. However, fractions of PGF2α-induced contraction and MLC20 phosphorylation were resistant to ML-9 but were sensitive to fasudil. Ro31-8220 (10 μm), a PKC inhibitor, did not affect the phosphorylation of MLC20 and the tension caused by PGF2α, thus excluding the possibility of the involvement of PKC in the PGF2α-induced MLC20 phosphorylation. PGF2α increased phosphorylation at Thr654 of the myosin binding subunit (MBS) of myosin phosphatase, which is a target of rho kinase, and fasudil decreased the phosphorylation. These data suggest that the PGF2α-induced contraction is accompanied by the inhibition of MLC20 dephosphorylation through rho kinase-induced MBS phosphorylation, leading to Ca2+ sensitization of contraction. An actin-associated mechanism may also be involved in the PGF2α-induced sensitization.
Over the past decade, in studies of vascular smooth muscle physiology and pathophysiology much interest has been paid to the ‘Ca2+-sensitization’ of contraction (Somlyo & Somlyo, 1994). Initially, this term was simply an expression that described the enhancement of contraction at a given cytoplasmic Ca2+ concentration ([Ca2+]i), but recently a molecular basis for this phenomenon has been provided. Ca2+ sensitization refers to when phosphorylation of the 20 kDa myosin light chain (MLC20), which is catalysed by Ca2+/calmodulin-dependent myosin light chain (MLC) kinase and is a primary determinant of contraction, is increased over the level expected from[Ca2+]i (Horowitz et al. 1996). Alternatively, this term is also used when the developed tension is relatively high at a given level of MLC20 phosphorylation. The latter enhancement may be related to alterations in regulatory proteins on thin filaments (Katsuyama et al. 1992; Itoh et al. 1995; Je et al. 2001).
Ca2+ sensitization resulting from an increase in MLC20 phosphorylation can occur either when smooth muscle myosin phosphatase (SMPP-1M), which is responsible for the dephosphorylation of MLC20, is inhibited (Somlyo et al. 1989; Kitazawa et al. 1991) or when MLC20 is phosphorylated in a Ca2+/calmodulin-independent manner (Kureishi et al. 1997; Weber et al. 1999). It has been reported that SMPP-1M is negatively regulated by several factors including small GTPase rho-associated kinase (rho kinase, Noda et al. 1995; Kimura et al. 1996), CPI-17 (Li et al. 1998) or arachidonic acid (Gong et al. 1992). Since the inhibition of phosphatase by arachidonic acid or CPI-17 is associated with the activation of protein kinase C (PKC; Gong et al. 1992; Gailly et al. 1997; Hartshorne et al. 1998; Li et al. 1998) and Ca2+-independent phosphorylation of MLC20 can also be caused by CPI-17 or rho kinase (Kureishi et al. 1997; Li et al. 1998), it is believed that PKC and rho kinase are the two major determinants for the Ca2+ sensitization observed in vascular smooth muscles.
When a receptor coupled to a heterotrimeric GTP binding protein is activated, Ca2+ sensitization as well as Ca2+ mobilization occurs. If diacylglycerol, a product of phosphatidylinositol hydrolysis, increases to a level sufficient to activate PKC, PKC-dependent Ca2+ sensitization should play a role in the enhancement of contraction. However, the role of PKC in receptor-mediated Ca2+-sensitization remains controversial, as some authors are in favour of the idea (Collins et al. 1992; Khalil & Morgan, 1992; Shimamoto et al. 1992; Parsons et al. 1996; Buus et al. 1998; Eto et al. 2001), while others are not (Singer et al. 1989; Hori et al. 1993; Jensen et al. 1996). On the other hand, the presence of GTP is required for mediation of Ca2+ sensitization by some types of receptor (Nishimura et al. 1988; Kitazawa et al. 1991), and this requirement was explained by the involvement of rho, which activates rho kinase. The involvement of rho kinase in receptor-mediated contractions has been claimed in some studies in which Botulinum C3 exoenzyme, which ADP-ribosylates and inactivates rho (Hirata et al. 1992; Fujita et al. 1995), or rho kinase inhibitors were used (Uehata et al. 1997; Nagumo et al. 2000).
Stimulation of rho induces the inhibition of myosin phosphatase activity by stimulating rho kinase-mediated phosphorylation of the 130 kDa myosin binding subunit (MBS) of SMPP-1M in smooth muscle (Noda et al. 1995; Kimura et al. 1996; Nagumo et al. 2000). Some heterotrimeric GTP-binding proteins coupled to a receptor such as Gα12 or Gα13 can activate rho through the action on the guanine nucleotide exchange factor (GEF) for rho (Hart et al. 1998; Kozasa et al. 1998; Sakurada et al. 2001). Such a link indicates that rho can be involved in receptor-mediated Ca2+ sensitization. However, it is still unclear whether PKC or rho kinase is more important for the Ca2+ sensitization caused by the inhibition of MLC20 phosphatase following receptor activation, or whether both kinases synergistically play a role to bring about the inhibition of the phosphatase. Moreover, the involvement of rho kinase has been proved in some but not all types of receptor-mediated contraction. Prostaglandin (PG)F2α is a unique Ca2+ sensitizer, because it causes diphosphorylation of MLC20 in rabbit aortic smooth muscles (Seto et al. 1990b). Since PGF2α is supposed to be involved in a delayed spastic contraction of the cerebral arteries after subarachnoid haemorrhage (Hagen et al. 1977; Chehrazi et al. 1989), it could be an important mediator of pathological vasospasm. In order to clarify the mechanism underlying PGF2α-induced Ca2+ sensitization, we investigated the following issues: (1) whether MLC20 dephosphorylation is inhibited during PGF2α receptor-mediated contraction, (2) whether MBS is phosphorylated during the contraction and (3) whether it is PKC or rho kinase that is involved in the inhibition of dephosphorylation.
Methods
Force measurement
The following experimentation was approved by the Animal Care and Use Committee at Miyazaki University Faculty of Agriculture. Adult male rabbits were anaesthetized with sodium pentobarbitone (45 mg (kg body weight)−1) and killed by cervical dislocation. The thoracic aorta was isolated, the endothelium was removed by rubbing the intimal surface with a cotton swab, and a strip 4–5 mm wide and 7–8 mm long was made. The removal of endothelium was confirmed by abolition of the relaxant response to acetylcholine (1 µm) in each strip. The strip was mounted in an organ bath containing 5 ml physiological saline solution (PSS) with the following composition (mm): NaCl 136.8, KCl 5.4, MgCl2 1.0, CaCl2 2.5, NaHCO3 11.9 and glucose 5.5 (when saturated with 95 % O2 and 5 % CO2, the pH was 7.3–7.4). A resting tension of 9.8 mN was applied throughout the experiment. Ca2+-free EGTA solution was made by omitting CaCl2 from the PSS and adding 1 mm EGTA. A high KCl (65.4 mm) solution was made by substituting 60 mmNaCl in PSS with iso-osmolar KCl.
[Ca2+]i measurement
For measurement of[Ca2+]i, the aortic strip was loaded with the Ca2+ indicator Fura-PE3/AM (5 µm, TEF LABS, Austin, TX, USA) that had been sonicated with 0.02 % cremophore EL (polyethoxylated castor oil) in PSS. The strip was then mounted in a bath constructed in a fluorimeter (CAF-100, JASCO, Tokyo, Japan). Fluorescence at 500 nm following alternate excitation at 340 nm (F340) and 380 nm (F380) was monitored together with tension. The ratio of F340/F380 was taken as an index of[Ca2+]i.
Protein extraction for Western blotting
At an appropriate time during the tension experiments, a strip was quickly removed from the organ bath, while taking care not to stretch it, and immersed in dry-ice/acetone containing 10 % (v/v) trichloroacetic acid (TCA) and 5 mm dithiothreitol (DTT). It took about 3 s for the preparation to freeze. This demounting did not affect the MLC20 phosphorylation level, since the phosphorylation level following this procedure in 65.4 mm KCl-contracted muscles was not different from when a mounted muscle was snap-frozen by dry-ice/acetone in the organ bath. Frozen tissues were rinsed with acetone containing 10 mm DTT to remove TCA and then dried. The strip was cut into tiny pieces and proteins were extracted with 8 m urea and 10 mm DTT.
MLC20 phosphorylation
MLC20 phosphorylation was measured as described previously (Miura et al. 1997). Briefly, the proteins were subjected to glycerol-polyacrylamide gel electrophoresis (glycerol-PAGE). The proteins were electro-transferred from the gylcerol-PAGE gel onto a nitrocellulose membrane. The membrane was soaked in skimmed milk to prevent non-specific binding of the antibody. The membrane was then incubated overnight with 10 µg ml−1 of polyclonal anti-MLC20 antibody, prepared as described elsewhere (Seto et al. 1990b) and thereafter exposed to a peroxidase-labelled secondary antibody (anti-rabbit IgG, Amersham NA934). A band was visualized using electrochemiluminescence (ECL; kit from Amersham). The ratio of the sum of monophosphorylated and diphosphorylated forms to total MLC20 was calculated.
Preparation of antibodies against MBS phosphorylated at Thr654 and MBS
A polyclonal antibody against MBS phosphorylated at Thr654 (antibody pMBS-654) was raised in New Zealand White rabbits, using a synthetic peptide corresponding to residues 648-660 (RQSRRSTQGVTLTC) of 130 kDa MBS containing phosphorylated Thr654 (chicken sequence) as the antigen (see Fig. 7), as described previously (Hartshorne et al. 1998). Antiserum was purified through sequential affinity columns of immobilized phospho-MBS and non-phospho-MBS peptides. A polyclonal antibody against MBS (antibody N-MBS) was raised using a synthetic peptide corresponding to the amino-terminal peptide (MKMADAKQKRNEC) of chicken MBS.
Figure 7. Characterization of the antibody pMBS-T654.
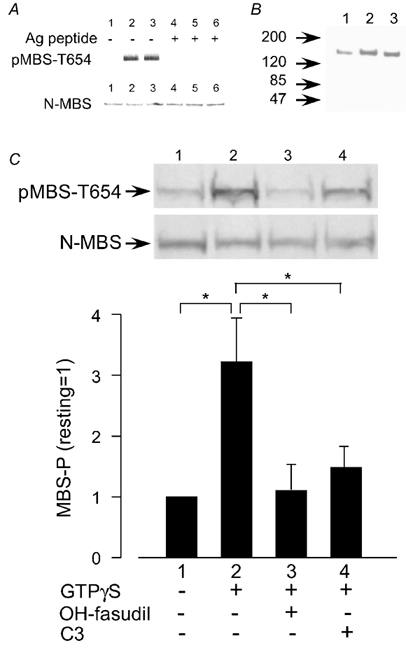
A, detection of rho kinase-induced phosphorylation of the myosin binding subunit (MBS) by pMBS-T654 in vitro. Recombinant MBS was phosphorylated by rho kinase for 0 min (lanes 1 and 4), 10 min (lanes 2 and 5) or 20 min (lanes 3 and 6) and subjected to SDS-PAGE, followed by Western blotting with pMBS-T654 (upper panel) or with the antibody N-MBS, which recognizes the N-terminus of MBS (lower panel), in the absence (lanes 1–3) or presence (lanes 4–6) of phosphorylated antigen peptide (T654-phosphopeptide). B, PGF2α-induced phosphorylation at Thr654 of MBS in the rabbit carotid artery. Extracts of arteries before stimulation (lane 1) or 1 (lane 2) and 2 min (lane 3) after the stimulation with PGF2α (30 µm) were subjected to SDS-PAGE followed by Western blotting with pMBS-T654. C, GTPγS-induced phosphorylation at Thr654 of MBS (MBS-P) and its inhibition by hydroxyfasudil or C3 toxin in permeabilized pig aortic cells. Cells were treated with GTPγS for 10 min. When used, hydroxyfasudil (OH-fasudil, 10 µm) or C3 toxin (1 µg ml−1) was used to pretreat cells for 15 min before application of GTPγS. Photographs: band reacted with pMBS-T654 (upper panel) or N-MBS (lower panel). MBS phosphorylation is expressed as a percentage of the level in the resting state. Data are means ± s.e.m. (n = 4). *P < 0.05.
Preparation of recombinant MBS
The 3.8 kilobase cDNA encoding the entire coding region of MBS was cloned by hybridization screening of a pig aortic smooth muscle cDNA library, using the 480 base pairs rat MBS cDNA as a probe, and ligated into pAcHTL-C vector (PharMingen, San Diego, CA, USA) at an Eco RI site downstream of the polyhedrin promoter to produce pAcHTL-MBS. Sf9 cells were co-transfected with pAcHTL-MBS and Baculogold baculovirus DNA (PharMingen, San Diego, CA, USA) by the lipofection method, and the recombinant baculovirus encoding the MBS cDNA was recovered.
MBS phosphorylation
Characterization of the antibody against phosphorylated MBS (pMBS-654) was determined in cultured pig aortic smooth muscle cells between the 5th and the 15th passages and in the intact rabbit mesenteric artery. Aortic cells were permeabilized with β-escin (20 µm) as described previously (Noda et al. 1995; Nagumo et al. 2000).
Equal volumes of extracts from arteries or cells were subjected to two sets of sodium dodecylsulphate (SDS)-PAGE. The proteins were transferred from the SDS-PAGE gel onto two nitrocellulose membranes in a buffer containing 20 mm Tris base (pH 7.5). One membrane was probed with the N-MBS antibody and the other with the pMBS-654 antibody using the ECL system. The extent of phosphorylation of MBS on Thr654 was normalized for the total expression levels of MBS.
Phosphorylation at Ser19 of MLC20
An antibody against MLC20 phosphorylated at Ser19 was prepared as described previously (Sakurada et al. 1994). The proteins were electrophoresed as described in the above section and were transferred onto two nitrocellulose membranes. One membrane was probed with the monoclonal anti-phosphoSer19 antibody and the other with the anti-MLC20 antibody. Details of the method have been described previously (Miura et al. 1997).
Assay of protein kinase activity
Rho kinase was purified from bovine brain according to the method of Matsui et al. (1996). The Ki value of fasudil and hydroxyfasudil for rho kinase was determined, as described previously (Nagumo et al. 2000). For the immunoblot or autoradiography assay, recombinant MBS (35 µg ml−1) was incubated with rho kinase in a reaction mixture composed of 50 mm Tris/HCl (pH 7.5), 5 mm MgCl2, 100 µm ATP in the absence or presence of [γ-32P]ATP at 37 °C.
MLC kinase and MLC20 were prepared from chicken gizzard as described previously (Walsh et al. 1983; Yoshida & Yagi, 1988). Calmodulin was prepared from porcine brain, by the method of Yazawa et al. (1980). Myosin light chain kinase (MLCK) activity was determined by the method of Sakurada et al. (1994). Protein kinase C (PKC) was prepared from porcine brain by the method of Manenti et al. (1992). Cyclic AMP-dependent protein kinase (PKA) was prepared from bovine heart by the method of Beavo et al. (1974). PKC and PKA activities were determined by using the NRPK assay kit (MBL, Japan).
Drugs
Drugs used were fasudil (HA1077), hydroxyfasudil (HA1100, Asahi Chemical, Tokyo, Japan), Y-27632 (Welfide, Tokyo, Japan), ONO-RS-082 (Ono Pharmaceutical, Osaka, Japan), Ro31-8220 (bisindoylmaleimide IX, Sigma-RBI, St Louis, MO, USA) and verapamil (Eisai, Tokyo, Japan).
Statistics
Data are expressed as means ± s.e.m. Significance was tested by Student's t test for a single comparison. For a multiple comparison, one-way analysis of variance followed by the Dunnet (comparison with a single control group) or Student-Newman-Keuls (comparison between all pairs) test was performed. The level of statistical significance was set at the level of P < 0.05.
Results
Effects of fasudil and hydroxyfasudil on the KCl-, 12-deoxyphorbol 13-isobutyrate (DPB)- and PGF2α-induced contraction and MLC20 phosphorylation of rabbit aortae
In this study, we used mainly fasudil and hydroxyfasudil as rho kinase inhibitors (Uehata et al. 1997; Nagumo et al. 2000; Swärd et al. 2000). Table 1 shows the Ki values of both compounds against rho kinase, PKC, PKA and MLC kinase. Hydroxyfasudil is an active metabolite of fasudil that is produced in the liver and is equally potent against rho kinase but less potent than fasudil against MLC kinase and PKC. Therefore, if rho kinase is involved in the Ca2+ sensitization caused by a constrictor, fasudil and hydroxyfasudil should inhibit the sensitization to a similar degree, or alternatively, if PKC is involved, fasudil should exhibit more potent inhibition than hydroxyfasudil. Based on these predictions, we compared the inhibitory action of fasudil and hydroxyfasudil on contractions mainly dependent on MLC kinase (KCl), PKC (DPB, a PKC activator) or receptor activation (PGF2α). Each compound was added cumulatively at intervals of 30 min during the sustained phase of 65.4 mm KCl-, 1 µm DPB- or 10 µm PGF2α-induced contraction. Both compounds dose-dependently decreased the tension elevated by each constrictor. The IC50 of both compounds is summarized in Table 2. Fasudil and hydroxyfasudil inhibited the PGF2α-induced contraction equally, whereas the IC50 of hydroxyfasudil against the KCl- or the DPB-induced contraction was greater than that of fasudil (P < 0.05, n = 7–8). Although the IC50 of fasudil against the PGF2α-induced contraction was not significantly different from that against the DPB-induced contraction calculated from the cumulative dose study, the fasudil-induced inhibition of the DPB-induced contraction was significantly less than that of the PGF2α-induced contraction when a single dose of fasudil (10 µm) was applied (see Fig. 1). The order of potency of another rho kinase inhibitor, Y-27632, was similar to that of fasudil (Table 2).
Table 1.
Ki value of fasudil and hydroxyfasudil against rho kinase, protein kinase C (PKC), cyclic AMP-dependent protein kinase (PKA) and myosin light chain kinase (MLCK)
| Rho kinase | PKC | PKA | MLCK | |
|---|---|---|---|---|
| Fasudil (μm) | 0.35 | 3.3 | 1.6 | 36 |
| Hydroxyfasudil (μm) | 0.56 | 18 | 2.5 | 140 |
Ki values of hydroxyfasudil are from the data by Seto et al. (1991).
Table 2.
IC50 values of rho kinase inhibitors for PGF2α-, KCl- or DPB-induced contractions in rabbit aortae
| PGF2α (3 μm) | Constrictor KCl (65.4 mM) | DPB(1 μm) | |
|---|---|---|---|
| Fasudil (μm) | 3.7 ± 1.4* | 22.1 ± 5.0 | 4.6 ± 1.4* |
| Hydroxyfasudil (μm) | 4.0 ± 0.8*§ | 48.8 ± 11.4† | 13.0 ± 3.0*† |
| Y−27632(μm) | 0.85 ± 0.29* | 2.8 ± 0.1 | 1.23 ± 0.26* |
Each inhibitor was cumulatively added at an interval of 30 min on the sustained contraction due to PGF2α, KCl or DPB. Data are means ± s.e.m. of 7−8 preparations.
P < 0.05 (vs. IC50 against the KCl-induced contraction).
P < 0.05 (vs. IC50 against the DPB-induced contraction).
P < 0.05 (vs. the IC50 of fasudil). Comparison between IC50 of Y−27632 and that of fasudil or hydroxyfasudil is not shown.
Figure 1. Effects of fasudil and hydroxyfasudil on the prostaglandin F2α (PGF2α)- or 12-deoxyphorbol 13-isobutyrate (DPB)-induced contraction and 20 kDa myosin light chain (MLC20) phosphorylation in rabbit aortae.
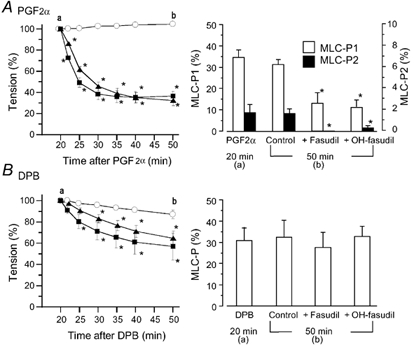
Fasudil (▪) or hydroxyfasudil (OH-fasudil, (Δ) at 10 µm was added 20 min after the application of 10 µm PGF2α (A) or 1 µm DPB (B). Left panels, a change in tension after the addition of fasudil or hydroxyfasudil (○, agonist alone). Tension is expressed as a percentage of contraction just before the addition of fasudil or hydroxyfasudil. In the right panels, MLC20 phosphorylation (MLC-P) was observed 20 min after the addition of PGF2α or DPB (just before the addition of fasudil or hydroxyfasudil (a) or 50 min after the addition of PGF2α or DPB (b), and is expressed as a percentage of phosphorylated MLC20 of total MLC20. MLC20 phosphorylation in the resting state was 8–9 % in each case (data not shown). Since PGF2α caused diphosphorylation of MLC20, monophosphorylation (MLC-P1, open columns) and diphosphorylation (MLC-P2, black columns) are shown (A, right panel). Data are presented as the mean ± s.e.m. (n = 5–6). * Significantly different from the time-matched control (P < 0.05).
Next, we examined whether the inhibition of PGF2α- or DPB-induced contraction by fasudil and hydroxyfasudil was accompanied by inhibition of MLC20 phosphorylation (Fig. 1). After PGF2α (10 µm) or DPB (1 µm) induced a sustained contraction, fasudil or hydroxyfasudil was added. PGF2α increased the monophosphorylation level from 7.0 ± 0.6 % in the resting state to 34.7 ± 3.5 % and caused diphosphorylation of MLC20 (1.8 ± 0.8 %, n = 6) at 20 min. Diphosphorylation of MLC20 due to PGF2α was greater at earlier times, but then the phosphorylation declined (Seto et al. 1990b, 1991), so in the later experiments we did not quantify the density of the band corresponding to diphosphorylation when the MLC20 phosphorylation was measured 15 min after the application of PGF2α. Fasudil or hydroxyfasudil (10 µm) added during the sustained phase of PGF2α-induced contraction lowered the tension by 63.1 ± 6.1 and 67.4 ± 5.0 %, respectively (n = 6). The decrease in tension effected by fasudil or hydroxyfasudil was accompanied by inhibition of MLC20 phosphorylation. Diphosphorylation due to PGF2α was almost completely abolished by both compounds (Fig. 1A).
DPB (1 µm) also increased MLC20 phosphorylation, which attained a sustained phase at 5 min, whereas the tension gradually developed and attained a peak at 20–30 min (Miura et al. 1997). Fasudil or hydroxyfasudil (10 µm) did not affect the DPB-induced MLC20 phosphorylation measured at 30 min after DPB, although these compounds decreased the tension by 43.2 ± 12.6 and 35.4 ± 6.9 %, respectively (n = 6, Fig. 1B).
Dependence of the PGF2α-induced contraction and MLC20 phosphorylation on Ca2+
We examined how PGF2α mobilized Ca2+ and how the PGF2α-induced contraction depended on[Ca2+]i in fura-PE3-loaded aortae. In normal PSS, the pattern of[Ca2+]i increase induced by PGF2α (10 µm) was variable.[Ca2+]i rapidly increased following application of PGF2α and the level was sustained at a slightly depressed level (n = 10, Fig. 2) or[Ca2+]i slowly increased and was then sustained (n = 12). In rare cases,[Ca2+]i rapidly increased to a peak within 1 min and then decreased to a sustained level of less than 50 % of the peak (n = 5). Irrespective of the pattern in[Ca2+]i, the developed tension persisted during the observation period (20–40 min), although the tension development was faster when the upstroke of[Ca2+]i was rapid. On average, the maximal increase in[Ca2+]i and the contraction induced by PGF2α were 63.7 ± 4.6 and 117.2 ± 6.9 %, respectively (n = 12), of those induced by 65.4 mm KCl. Verapamil (10 µm) slightly decreased[Ca2+]i in the resting state and the subsequent addition of PGF2α only slightly increased[Ca2+]i (6.6 ± 2.8 % of the response obtained in the presence of external Ca2+, n = 8), but increased the tension to 73.6 ± 6.1 % (n = 8) of the contraction obtained in normal PSS (Fig. 2). In a separate experiment, chelation of external Ca2+ with 4 mm EGTA lowered[Ca2+]i to a greater degree than verapamil. In this situation, PGF2α did not alter[Ca2+]i but induced a contraction that was 22.0 ± 3.0 % (n = 6) of that induced in normal PSS. Thus, it is likely that the PGF2α-induced Ca2+ mobilization depended on Ca2+ entry through L-type Ca2+ channels in this artery. From these results it is evident that PGF2α can cause a sizeable contraction when an increase in[Ca2+]i is absent or is very small.
Figure 2. Changes in[Ca2+]i, tension and MLC20 phosphorylation due to PGF2α under normal conditions or conditions where an increase in [Ca2+]i was inhibited.
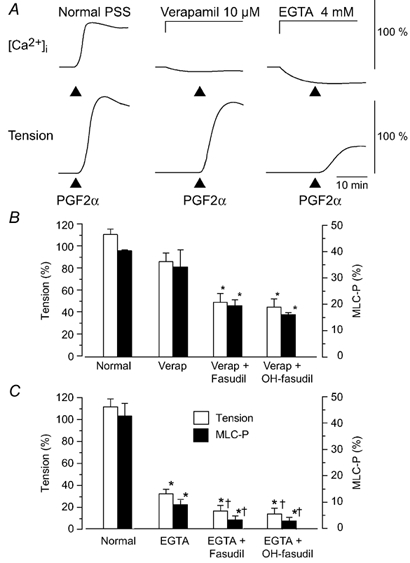
A, examples of changes in[Ca2+]i in normal physiological saline solution (PSS), in the presence of verapamil (10 µm) or EGTA (4 mm). For[Ca2+]i and tension, 100 % represents the change induced by 65.4 mm KCl, which was obtained before the application of PGF2α. Verapamil or EGTA was applied 10 min before the addition of PGF2α (10 µm). B, summarized data of the PGF2α-induced contraction and MLC20 phosphorylation in the normal medium or in the presence of verapamil and fasudil or hydroxyfasudil (OH-fasudil). Developed tension is expressed as a percentage of the maximum contraction to 65.4 mm KCl. C, summarized data of the PGF2α-induced contraction and MLC20 phosphorylation in the normal medium or in the presence of EGTA and fasudil or OH-fasudil. In B and C, MLC20 phosphorylation (MLC-P) was measured at 5 min after the addition of PGF2α (10 µm). When used, fasudil (10 µm) or hydroxyfasudil (OH-fasudil, 10 µm) was added 30 min before PGF2α. The protocol in B and C was the same as that shown in A, but the experiments were performed in a different series. MLC20 phosphorylation under the resting condition in normal PSS was 8.9 ± 2.4 % (data not shown). Verap: verapamil.
The contraction induced by PGF2α under the conditions where an increase in[Ca2+]i was inhibited was accompanied by an increase in MLC20 phosphorylation (Fig. 2). In the presence of verapamil, PGF2α increased the MLC20 monophosphorylation (33.9 ± 6.1 % at 5 min after application of PGF2α) together with a slight diphosphorylation of MLC20 (0.4 ± 0.1 %, data not shown in Fig. 2B). Pretreatment with fasudil or hydroxyfasudil (10 µm) for 30 min before the application of PGF2α abolished the diphosphorylation of MLC20 and inhibited both the monophosphorylation and the contraction. In EGTA solution, PGF2α increased the MLC20 phosphorylation from 1.5 ± 0.3 to 9.1 ± 1.5 % (n = 12) without any detectable diphosphorylation (Fig. 2C). The MLC20 phosphorylation caused by PGF2α in the presence of EGTA was similar to that measured in normal PSS with no stimulation (8.9 ± 2.4 %). Fasudil and hydroxyfasudil decreased the phosphorylation to 3.4 ± 1.0 and 3.0 ± 0.8 % (n = 6), respectively, and inhibited the contraction.
It is possible that during the contraction induced by PGF2α in muscles exposed to EGTA, PGF2α caused the release of Ca2+ from the sarcoplasmic reticulum (SR), but the increase in Ca2+ was restricted to a region around the contractile machinery so that it could not be detected as an averaged Fura-PE3-Ca2+ signal. To completely exclude the possible participation of Ca2+, the SR was depleted of Ca2+ by application of ryanodine (3 µm) and phenylephrine (10 µm) during incubation in Ca2+-free EGTA (1 mm) solution for 30 min. In Ca2+-depleted muscles, PGF2α (10 µm) increased both the MLC20 phosphorylation and the tension. The MLC20 phosphorylation and the contraction caused by PGF2α (9.0 ± 2.5 and 29.3 ± 7.8 %, respectively, n = 6) were similar to those obtained under the condition where EGTA (4 mm) was added to the medium (Fig. 2). The similar degree of contraction and MLC20 phosphorylation observed under conditions where the SR had been depleted of Ca2+ or only external Ca2+ was chelated by EGTA suggests that PGF2α did not induce Ca2+ release from the SR. Pretreatment with fasudil (10 µm) 60 min before the addition of PGF2α inhibited the increase in MLC20 phosphorylation and the tension caused by PGF2α (data not shown).
Inhibition of MLC20 dephosphorylation during the PGF2α-induced contraction
Next, we examined how the relaxation and the MLC20 dephosphorylation took place when the MLC20 phosphorylation by MLC kinase was inhibited by a reduction in[Ca2+]i during the PGF2α-induced contraction (Fig. 3). After a muscle was maximally contracted with 30 µm PGF2α in normal PSS, external Ca2+ was removed by switching the medium to Ca2+-free EGTA (1 mm) solution (5 min after PGF2α, Fig. 3). The tension decreased to about 30 % of the maximal contraction in 20 min and was sustained at that level. The MLC20 phosphorylation was maintained between 20 and 25 % during the sustained phase. When fasudil or hydroxyfasudil (10 µm) was present in Ca2+-free EGTA solution, the MLC20 phosphorylation and the tension rapidly decreased (Fig. 3, left).
Figure 3. Effects of fasudil, hydroxyfasudil or ONO-RS-082 on MLC20 phosphorylation and tension maintained in Ca2+-free, EGTA solution in the presence of PGF2α.
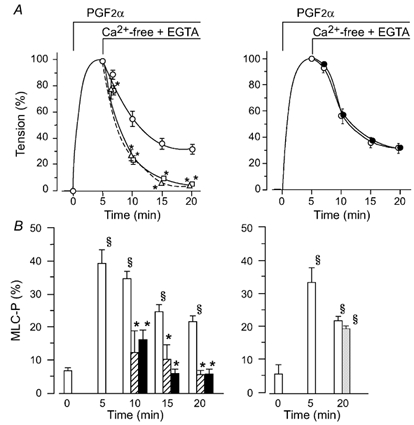
A, change in tension. After PGF2α (10 µm)-induced contraction had reached a peak, the external medium was changed from normal PSS to Ca2+ free, 1 mm EGTA solution (5 min in the figure; ○, PGF2α alone). When used, fasudil (δ, 10 µm) or hydroxyfasudil (□, 10 µm) was added at the time when the external Ca2+ was omitted (the left panel). ONO-RS-082 (•, 5 µm, the right panel) was applied 15 min before the addition of PGF2α. Data are expressed as a percentage of the maximal contraction to PGF2α in normal medium (n = 6-7). B, MLC20 phosphorylation (MLC-P) measured in the resting state (0 min), just before switching the medium to Ca2+ free, EGTA solution (5 min), 5, 10 and 15 min after Ca2+ removal (10, 15 and 20 min after the addition of PGF2α). Open columns: controls; hatched columns: fasudil-treated muscles; filled columns: hydroxyfasudil-treated muscles; grey column: ONO-RS-082-treated muscles. * Significantly different from the level in the absence of fasudil or hydroxyfasudil (P < 0.05); § significantly different from the resting level (P < 0.05).
In a previous paper we reported that DPB inhibited the dephosphorylation of MLC20, and that the phospholipase A2 inhibitor ONO-RS-082 antagonized that inhibition, suggesting that arachidonic acid is involved in the DPB-induced inhibition of dephosphorylation (Miura et al. 1997). Therefore, if arachidonic acid is involved in the inhibition of MLC20 dephosphorylation by PGF2α, ONO-RS-082 should antagonize the effect of PGF2α on the MLC20 phosphorylation. To test this possibility, ONO-RS-082 (5 µm) was applied 15 min before PGF2α. Pretreatment with ONO-RS-082 did not affect either the MLC20 phosphorylation or the contraction induced by PGF2α in the same protocol, as shown in Fig. 3 (right).
To examine further the involvement of PKC, we compared the effects of the PKC inhibitor Ro31-8220 on the PGF2α- or DPB-induced inhibition of MLC20 dephosphorylation and relaxation. Since phorbol ester-induced MLC20 phosphorylation involves phosphorylation at sites directly phosphorylated by PKC (Ser1, Ser2) and at sites dependent on MLC kinase (Ser19, Thr18; Ikebe & Hartshorne, 1985; Ikebe et al. 1987; Miura et al. 1997) and if rho kinase is involved in the DPB-induced contraction, the phosphorylation should increase at MLC kinase-dependent sites. We observed the Ser19 phosphorylation in this comparison. The protocol used to examine the inhibition of dephosphorylation by PGF2α was the same as in Fig. 3. The protocol for DPB was that initially MLC20 was MLC kinase-dependently phosphorylated by KCl (65.4 mm) in normal PSS, then the muscle was rinsed with Ca2+-free, EGTA (1 mm) PSS (5.4 mm K+) to dephosphorylate MLC20. DPB (1 µm), which was applied when the KCl-contracted muscle was rinsed with Ca2+-free, EGTA PSS inhibited the dephosphorylation and maintained the tension at about 30–50 % of the maximum level (Miura et al. 1997). Ro31-8220 (10 µm), which was applied when the external Ca2+ was chelated with EGTA, did not affect the Ser19 phosphorylation of MLC20 or the tension caused by PGF2α, whereas it significantly inhibited these parameters caused by 1 µm DPB (Fig. 4).
Figure 4. Effects of the protein kinase C (PKC) inhibitor Ro31-8220 on phosphorylation of MLC20 at Ser19 and contraction induced by PGF2α and DPB.
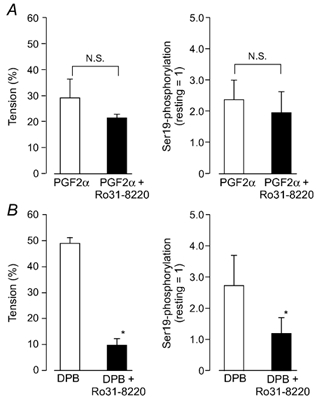
A, the effects of Ro31-8220 on the PGF2α-induced inhibition of Ser19 dephosphorylation and relaxation. The protocol was the same as in Fig. 3. When the external medium was changed from normal PSS to Ca2+-free, 1 mm EGTA solution, Ro31-8220 (5 µm) was simultaneously added to one group (filled columns) but not to the control group (open columns). B, the effects on the DPB-induced inhibition of Ser19 dephosphorylation and relaxation. After 65.4 mm KCl caused a maximal contraction (5 min), the external medium was switched to Ca2+ free, 1 mm EGTA solution (5.4 mm K+) containing 1 µm DPB. When present, Ro31-8220 (5 µm) was added simultaneously with DPB. Data were taken 15 min after the omission of external Ca2+. The left panels represent a relative value of the 65.4 mm KCl-induced contraction, which was obtained before PGF2α. The right panels are Ser19 phosphorylation expressed as a value relative to the phosphorylation in the resting state. Data are mean ± s.e.m. (n = 4). * Significantly different from DPB alone (P < 0.05, N.S. not significant).
Sensitivity to MLC kinase inhibitor of MLC20 phosphorylation and the contraction induced by KCl, PGF2α or calyculin A
The MLC kinase inhibitor ML-9 (100 µm), which was added during the sustained phase of a 65.4 mm KCl-induced contraction, rapidly decreased the MLC20 phosphorylation and the tension generated (Fig. 5). Fasudil (10 µm), which was added 15 min after ML-9, slightly accelerated the relaxation but did not accelerate the MLC20 dephosphorylation under this condition. When 100 µm ML-9 was applied during the PGF2α-induced contraction, it scarcely affected the MLC20 phosphorylation or the tension, so we used 200 µm ML-9 instead of 100 µm for the PGF2α-induced contraction. ML-9 (200 µm) partially inhibited the PGF2α-induced MLC20 phosphorylation and contraction. Subsequent addition of fasudil (10 µm) 15 min after ML-9 further decreased the MLC20 phosphorylation and the tension (Fig. 5). The addition of the PKC inhibitor calphostin C (10 µm) instead of fasudil had no effect on the time course of the change in tension (data not shown).
Figure 5. Sensitivity of KCl- and PGF2α-induced contraction and MLC20 phosphorylation to ML-9 and fasudil.
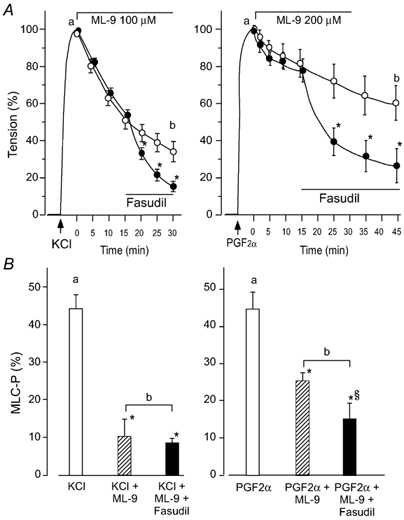
After KCl (65.4 mm, left)- or PGF2α (10 µm, right)-induced contraction reached a respective peak (5 min), ML-9 (100 or 200 µm, respectively) was applied. Fasudil (10 µm) was added 15 min later. A, a change in tension in fasudil-treated (•) or untreated muscles (○). The ordinate represents the relative tension of maximum contraction to KCl or PGF2α, respectively. B, MLC20 phosphorylation (MLC-P) measured in the protocol shown in A. Data were sampled just before the addition of ML-9 (open columns, corresponding to a in A) and 30 min after the addition of ML-9 for KCl-induced contraction or 45 min after addition of ML-9 for PGF2α-induced contraction (corresponding to b in A). Hatched columns: in the absence of fasudil, filled columns: in the presence of fasudil. * Significantly different from the level before the addition of ML-9 (P < 0.05), § significantly different from the level in the absence of fasudil (P < 0.05n = 5–8).
Calyculin A (300 nm), which inhibits protein phosphatase types 1 and 2A (Hartshorne et al. 1989), gradually increased the MLC20 phosphorylation and the tension. At 30 min after addition of calyculin A, MLC20 phosphorylation and the tension attained a steady-state level. Addition of hydroxyfasudil (1-300 µm) at that time did not affect the MLC20 phosphorylation or contraction. On the other hand, ML-9 at 100 µm significantly inhibited the contraction, and at 300 µm it completely inhibited both the MLC20 phosphorylation and the contraction (Fig. 6).
Figure 6. Differential effects of hydroxyfasudil and ML-9 on the calyculin A-induced contraction and MLC20 phosphorylation.
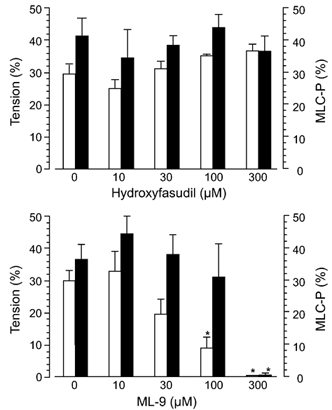
Open columns: tension expressed as a percentage of the maximum contraction due to 65.4 mm KCl, which was obtained before the addition of calyculin A, filled columns: MLC-P. At 30 min after the application of calyculin A (300 nm), hydroxyfasudil or ML-9 at each concentration was added. * Significantly different from the level in the absence of ML-9 (P < 0.05).
Phosphorylation by PGF2α of the MBS of phosphatase and the effects of fasudil and hydroxyfasudil
Rho kinase phosphorylates the MBS of SMPP-1M at Thr654, thereby inhibiting phosphatase activity (Feng et al. 1999a). To determine whether PGF2α inhibits the MLC20 dephosphorylation through rho kinase, the phosphorylation of MBS at Thr654 was measured using the antibody pMBS-T654, which recognizes MBS phosphorylated at T654.
At first, we characterized the specificity of the antibody pMBS-T654. The antibody recognized rho kinase-phosphorylated recombinant 130 kDa MBS, but not the non-phosphorylated MBS (compare lane 1 and lanes 2 and 3 of Fig. 7A). The pMBS-T654-reactive band was completely quenched with excess T654-phosphopeptide (lanes 4-6 in Fig. 7A). In contrast, two other phosphopeptides, EKRRS(PO3H2)TGVSF and EKRRST(PO3H2)GVSF, which correspond to the amino acid 804-813 of chicken MBS and are similar in their core sequence to the T654-phosphopeptide, could not quench the pMBS-T654-reactive band (data not shown). A homology search of MBS revealed that with the exception of the amino acid sequence 804-813 in MBS, there were no other sequences homologous to the amino acid sequence 648–660. These results indicate that the antibody pMBS-T654 specifically recognizes MBS phosphorylated at Thr654.
Figure 7B shows the MBS phosphorylation probed with pMBS-T654 in intact rabbit mesenteric arteries. Only a single band corresponding to 130 kDa was detected between 47 and 200 kDa, indicating that the antibody does not react with any protein other than MBS. PGF2α (30 µm) increased the phosphorylation of MBS at 1 min (lane 2) and slightly less at 2 min (lane 3) of application. Similar results were obtained with the carotid arteries (data not shown). In another preliminary study with β-escin-permeabilized pig aortic cells, GTPγS (30 µm, 10 min) increased the MBS phosphorylation (Fig. 7C). Pretreatment with hydroxyfasudil (10 µm, 10 min) or C3 toxin (1 µg ml−1, 15 min), which inactivates rho, significantly decreased the GTPγS-induced MBS phosphorylation.
Using the pMBS-T654 antibody we tested whether PGF2α increased the phosphorylation of MBS in rabbit aortae. PGF2α significantly increased the phosphorylation at Thr654 of MBS 1 min after application. At 15 min post-application, the level of MBS phosphorylation was less than at 1 min post-application. Pretreatment with fasudil (10 µm) decreased the phosphorylation of MBS at 1 and 15 min (Fig. 8).
Figure 8. PGF2α-induced phosphorylation of the MBS of phosphatase, and its inhibition by fasudil.
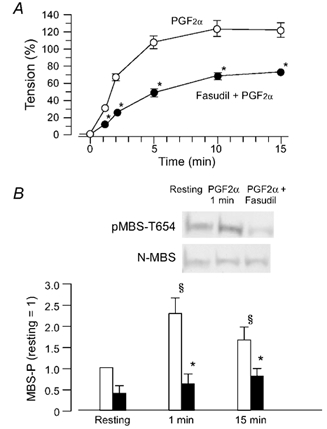
A, time-course of the PGF2α (10 µm)-induced contraction in the absence (○) or presence (•) of fasudil (10 µm). When present, fasudil was applied 15 min before addition of PGF2α. B, (photographs) example of Western blots with the pMBS-T654 antibody or the N-MBS antibody. Samples were taken before the addition of PGF2α (resting) and 1 min after PGF2α. Bar graph, quantified phosphorylation of MBS (MBS-P) in the resting state, 1 or 15 min after the addition of PGF2α in the absence (open columns) or presence (filled columns) of fasudil. The ratio of the density of a band with the pMBS-T654 antibody to that with the N-MBS antibody in the resting state is referred as 1. Data are presented as the means ± s.e.m. of four experiments. * Significantly different from the resting level (P < 0.05); § significantly different from PGF2α alone (P < 0.05).
Discussion
PGF2α-induced Ca2+ sensitization
The Ca2+ senstization of vascular smooth muscle contraction induced by PGF2α has been observed by several groups (e.g. Bradley & Morgan, 1987; Balwierczak, 1991; Kurata et al. 1993) and this sensitization has been attributed to the enhancement of MLC20 phosphorylation (Seto et al. 1990a; Suematsu et al. 1991; Hori et al. 1992; Shin et al. 2002). The kinetics of MLC20 phosphorylation by PGF2α in rabbit aortae was analysed by Seto et al. (1990b). They reported that PGF2α increased MLC20 monophosphorylation, which was above the resting level during a 30 min observation period, and caused transient diphosphorylation at the initial stage, and suggested that the diphosphorylation contributes to acceleration of the rate of force generation. The diphosphorylation is specific for PGF2α since neither KCl nor histamine caused diphosphorylation (Seto et al. 1990b). Since the kinetic data of PGF2α-induced MLC20 phosphorylation in rabbit aortae are available from the paper by Seto et al. (1990b), we did not measure the entire time course of MLC20 phosphorylation and did not quantify the diphosphorylation caused by PGF2α except for a few experiments. Here, we focussed our attention on the mechanism underlying the PGF2α-induced increase in MLC20 phosphorylation.
Verapamil or removal of extracellular Ca2+ inhibited the PGF2α-induced rise in[Ca2+]i. In the absence of external Ca2+, the MLC20 phosphorylation and the tension development induced by PGF2α in muscles that had been depleted of Ca2+ in the SR were similar to those in muscles that had been treated with EGTA alone. This suggests that PGF2α does not employ Ca2+ released from the SR for MLC20 phosphorylation and contraction. Thus, it is likely that the PGF2α-induced increase in[Ca2+]i depends exclusively upon Ca2+ influx through L-type Ca2+ channels in rabbit aortae. This observation is consistent with the data from ferret aortae showing that PGF2α does not stimulate Ca2+ release from the intracellular Ca2+ stores (Bradley & Morgan, 1987). This is another unique property of PGF2α-induced contraction, since other receptor agonists usually utilize the Ca2+ released from intracellular Ca2+ stores as well as the Ca2+ that enters from the extracellular space. However, this feature may not be shared by other types of vascular smooth muscle, because PGF2α induces not only Ca2+ influx but also Ca2+ release from the SR in rat aortae (Kurata et al. 1993). PGF2α increased both MLC20 phosphorylation and tension even when an increase in[Ca2+]i was inhibited by verapamil or Ca2+-free solution, although the extent of MLC20 phosphorylation under these conditions was smaller than that observed under the normal condition. Hence, PGF2α can enhance the MLC20 phosphorylation without a rise in[Ca2+]i or with a very small increase in[Ca2+]i. The MLC20 phosphorylation and the tension due to PGF2α were higher in the presence of verapamil than after treatment with EGTA. This may be related to higher[Ca2+]i in verapamil-treated muscles than in EGTA-treated muscles, thereby causing MLC kinase activity to be higher in the former. Alternatively, we cannot exclude the possibility that in the presence of verapamil, PGF2α still increased Ca2+ at a restricted region, and this increase was not detected by the fluorescent dye. It is also possible that this increase partly contributes to an increase in MLC20 phosphorylation.
In this study, ML-9, which was added during the sustained contraction observed in response to PGF2α, only partially decreased MLC20 phosphorylation and the contraction. Therefore, it seems that MLC kinase-dependent MLC20 phosphorylation does not play an exclusive role in the maintenance of a sustained contraction. This is compatible with the data showing that PGF2α increased MLC20 phosphorylation in the situation where a rise in[Ca2+]i was inhibited so that the MLC kinase activity was low (Fig. 2). If[Ca2+]i in a contracted muscle is lowered, the MLC kinase activity would be decreased and the relative activity of phosphatase would increase, causing dephosphorylation of MLC20 and relaxation. When the PGF2α-contracted muscle was rinsed with Ca2+-free EGTA solution, the MLC20 phosphorylation and the tension did not easily return to the resting level (Fig. 3). This suggests that the MLC20 dephosphorylation induced by the phosphatase was partially inhibited in the presence of PGF2α. The data by Shin et al. (2002) showing that PGF2α caused dissociation of the catalytic subunit (PP1c) of myosin phosphatase in ferret portal vein cells supports the idea that PGF2α inhibits dephosphorylation. It is thus likely that the inhibition of MLC20 dephosphorylation is responsible for the maintenance of MLC20 phosphorylation and tension during the sustained contraction to PGF2α.
Fasudil and hydroxyfasudil as protein kinase inhibitors
Fasudil and hydroxyfasudil are equally potent against rho kinase, while fasudil is more potent than hydroxyfasudil against MLC kinase and PKC (Table 1, Sakurada et al. 1994; Nagumo et al. 1998, 2000). In the tension experiment, fasudil and hydroxyfasudil inhibited the PGF2α-induced contractions equally. Fasudil at 10 µm was less potent on the KCl- or the DPB-induced contraction than on the PGF2α-induced contraction, and hydroxyfasudil was an even weaker inhibitor of these contractions. The similar sensitivity to fasudil and hydroxyfasudil of the PGF2α-induced contraction indirectly suggests the involvement of a rho kinase-dependent mechanism. At concentrations of up to 300 µm, hydroxyfasudil did not decrease the phosphatase inhibitor calyculin A-induced MLC20 phosphorylation and contraction, whereas ML-9 decreased these parameters. This indicates that the calyculin A-induced MLC20 phosphorylation was dependent on MLC kinase, which was partially active in the resting state. Although fasudil is about four times more potent than hydroxyfasudil on MLC kinase, the fact that 300 µm hydroxyfasudil did not affect the MLC20 phosphorylation induced by calyculin A suggests that fasudil is unlikely to exert an inhibitory effect on MLC kinase at the concentration used in this study (10 µm). Therefore, the target site of this compound, which is responsible for inhibition of the PGF2α-induced MLC20 phosphorylation, should be a kinase other than MLC kinase.
Rho kinase is responsible for the inhibition of phosphatase
If PGF2α produces diacylglycerol in an amount sufficient to activate PKC, PKC could participate in the inhibition of myosin phosphatase through mechanisms including that involving CPI-17 (Li et al. 1998) or arachidonic acid (Gong et al. 1992, 1995; Hartshorne et al. 1998; Feng et al. 1999b). For example, 5-hydroxytryptamine-induced Ca2+ sensitization of rabbit mesenteric artery involved a PKC- and arachidonic acid-dependent mechanism because it was sensitive to Ro31-8220, a non-selective PKC inhibitor, and quinacrine, a phospholipase A2 inhibitor (Parsons et al. 1996). Likewise, histamine increased the phosphorylation of CPI-17, thereby causing Ca2+ sensitization (Kitazawa et al. 2000; Eto et al. 2001). The report by Eto et al. (2001) showed that Ro31-8220 inhibits the phosphorylation of CPI-17, suggesting that the PKC isozyme responsible for the activation of CPI-17 is sensitive to Ro31-8220. We have shown previously that activation of PKC by the phorbol ester DPB causes the inhibition of MLC20 dephosphorylation in rabbit aortae, which is partially dependent upon arachidonic acid, since the phospholipase A2 inhibitor ONO-RS-082 decreased the DPB-induced MLC20 phosphorylation and contraction (Miura et al. 1997). An increase in MLC20 phosphorylation at Ser19 by DPB was also inhibited by Ro31-8220 (Fig. 4). Therefore, if PKC plays a role in the PGF2α-induced Ca2+ sensitization, the MLC20 phosphorylation should be sensitive to Ro31-8220 or ONO-RS-082 in rabbit aortae. In the present study, neither ONO-RS-082 nor Ro31-8220 affected the PGF2α-induced MLC20 phosphorylation and contraction (Fig. 3 and Fig. 4). These data suggest that PKC is not responsible for the PGF2α-enhanced MLC20 phosphorylation. In contrast, the paper by Katsuyama & Morgan (1993) showed that a PKC pseudosubstrate inhibitor effectively inhibited the 100 µm PGF2α-induced contraction of permeabilized ferret aortic cells clamped at pCa 7. It is possible that in the study of Katsuyama & Morgan (1993), in which a 10 times higher concentration of PGF2α was used compared with the present study, PKC might have participated in the contraction.
The rho kinase inhibitors fasudil, hydroxyfasudil and Y-27632 inhibited the MLC20 phosphorylation caused by PGF2α. Since it is unlikely that fasudil or hydroxyfasudil inhibited contractions dependent on PKC or MLC kinase in this study, as discussed above, the most probable kinase that was inhibited by these inhibitors is rho kinase. Feng et al. (1999a) reported that rho kinase inhibited SMPP-1M by phosphorylating Thr695 of 133 kDa MBS, which corresponds to Thr654 of the 130 kDa isoform (Hartshorne et al. 1998). To detect whether MBS is phosphorylated by PGF2α, we raised the antibody pMBS-T654 against recombinant phospho-MBS, which recognizes the phosphorylation at Thr654 of 130 kDa MBS. The identity of the site that is phosphorylated by rho kinase in MBS has been controversial. It was suggested that rho kinase purified from the brain phosphorylated the same site (Thr654) as the site for endogenous kinase in gizzard myosin phosphatase (Hartshorne et al. 1998). In contrast, Kimura et al. (1996) reported that rho kinase phosphorylated the C-terminal part of gizzard MBS (residues 753-1004) but not the N-terminal segment (residue 1–721) in an in vitro study. In this well-controlled immunoblotting experiment using the antibody pMBS-T654, we showed that rho kinase phosphorylated Thr654 of full-length recombinant MBS (Fig. 7A). Furthermore, using permeabilized aortic smooth muscle cells, pMBS-T654 specifically detected a GTPγS-induced increase in phosphorylation of MBS, which was abolished by C3 toxin and the rho kinase inhibitor (Fig. 7C). We conclude, therefore, that the rho kinase-phosphorylated site includes Thr654 of MBS.
An endogenous inhibitor against MLC phosphatase other than rho kinase is CPI-17 (Kimura et al. 1996; Li et al. 1998; Kitazawa et al. 2000). Since the activation of CPI-17 by PKC is unlikely to be involved in the PGF2α-induced increase in MLC20 phosphorylation, a candidate for the phosphorylation of MBS could be narrowed down to rho kinase. The data showing that Thr654 of MBS was phosphorylated by PGF2α and fasudil inhibited the phosphorylation confirm this idea. Consistent with this, Shin et al. (2002) found that PGF2α transiently increased the phosphorylation of MBS at Thr695 in ferret portal veins. Based on these discussions, we conclude that the PGF2α-induced enhancement of MLC20 phosphorylation is caused by the inhibition of phosphatase by rho kinase. However, we cannot strictly exclude the possible involvement of CPI-17, because rho kinase or protein kinase N, a downstream effector of Rho, can phosphorylate CPI-17, leading to the inhibition of the phosphatase (Hamaguchi et al. 2000; Kitazawa et al. 2000; Koyama et al. 2000).
The PGF2α-induced contraction involves an MLC20 phosphorylation-independent mechanism
In Ca2+-free medium, PGF2α increased the MLC20 phosphorylation and the tension without an increase in[Ca2+]i. Rho kinase can directly phosphorylate MLC20 and cause contraction, independently of Ca2+ (Kureishi et al. 1997). Therefore, the MLC20 phosphorylation and the contraction induced by PGF2α in Ca2+-depleted muscles could be due to rho kinase. Alternatively, it is possible that the inhibition of MLC20 dephosphorylation by rho kinase increased the phosphorylation level despite the very low MLC kinase activity. Regardless, the MLC20 phosphorylation in these muscles (about 9 %) was similar to the level recorded under resting conditions in Ca2+-containing medium (8.9 ± 2.4 %), and thus it was not high enough to contribute to the tension development. Therefore, it is highly likely that the PGF2α-induced contraction includes a mechanism independent of MLC20 phosphorylation.
A determinant other than MLC20 phosphorylation for smooth muscle contraction is the regulation of thin filaments by calponin or caldesmon, which bind to actin and inhibit actin-activated myosin Mg2+-ATPase (Miki et al. 1992; Malmqvist et al. 1997; Earley et al. 1998; Je et al. 2001). When calponin or caldesmon are phosphorylated, these proteins lose the ability to inhibit the Mg2+-ATPase (Winder & Walsh, 1990; Itoh et al. 1995; Horowitz et al. 1996). It has been reported that PGF2α increases calponin phosphorylation in porcine coronary arteries during the initial stage and that fasudil or hydroxyfasudil inhibit this phosphorylation with an ED50 value of 26 µm (Nagumo et al. 1998). Therefore, it is possible that PGF2α phosphorylated calponin and elevated the tension independently of MLC20 phosphorylation in Ca2+-depleted muscles. PKC is known to phosphorylate calponin and caldesmon. Caldesmon can also be phosphorylated by p21-activated protein kinase, another rho family GTPase (Van Eyk et al. 1998). However, it has not been determined whether rho kinase can phosphorylate calponin or caldesmon. The sensitivity of PGF2α-induced contraction in Ca2+-depleted muscles to fasudil indicates the possible involvement of PKC or rho kinase. It should be clarified in a future study whether or not PGF2α phosphorylates calponin or caldesmon in Ca2+-depleted muscles.
This study provided evidence that PGF2α activates rho kinase and possibly also an actin-associated sensitizing mechanism, both of which cause Ca2+ sensitization. The latter mechanism also seems to be sensitive to fasudil. It should be determined in future studies which kinase is responsible for the mechanism.
References
- Balwierczak JL. The relationship of KCl- and prostaglandin F2α-mediated increases in tension of the porcine coronary artery with changes in intracellular Ca2+ measured with fura-2. Br J Pharmacol. 1991;104:373–378. doi: 10.1111/j.1476-5381.1991.tb12438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beavo JA, Bechtel PJ, Krebs EG. Preparation of homogeneous cyclic AMP-dependent protein kinase(s) and its subunits from rabbit skeletal muscle. Methods Enzymol. 1974;38:299–308. doi: 10.1016/0076-6879(74)38046-9. [DOI] [PubMed] [Google Scholar]
- Bradley AL, Morgan KG. Alteration in cytoplasmic calcium sensitivity during porcine coronary artery contraction as detected by aequorin. J Physiol. 1987;385:437–448. doi: 10.1113/jphysiol.1987.sp016500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buus CL, Aalkjar C, Nilsson H, Juul B, Moller JV, Mulvany MJ. Mechanisms of Ca2+ sensitization of force production by noradrenaline in rat mesenteric small arteries. J Physiol. 1998;510:577–590. doi: 10.1111/j.1469-7793.1998.577bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chehrazi BB, Giri S, Joy RM. Prostaglandins and vasoactive amines in cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Stroke. 1989;20:217–224. doi: 10.1161/01.str.20.2.217. [DOI] [PubMed] [Google Scholar]
- Collins EM, Walsh MP, Morgan KG. Contraction of single vascular smooth muscle cells by phenylephrine at constant[Ca2+]i. Am J Physiol. 1992;262:H754–762. doi: 10.1152/ajpheart.1992.262.3.H754. [DOI] [PubMed] [Google Scholar]
- Earley JJ, Su X, Moreland RS. Caldesmon inhibits active crossbridges in unstimulated vascular smooth muscle. An antisense oligodeoxynucleotide approach. Circ Res. 1998;83:661–667. doi: 10.1161/01.res.83.6.661. [DOI] [PubMed] [Google Scholar]
- Eto M, Kitazawa T, Yazawa M, Mukai H, Ono Y, Brautigan DL. Histamine-induced vasoconstriction involves phosphorylation of a specific inhibitor protein for myosin phosphatase by protein kinase C α and δ isoforms. J Biol Chem. 2001;276:29072–29078. doi: 10.1074/jbc.M103206200. [DOI] [PubMed] [Google Scholar]
- Feng J, Ito M, Ichikawa K, Isaka N, Nishikawa M, Hartshorne DJ, Nakano T. Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J Biol Chem. 1999a;274:37385–37390. doi: 10.1074/jbc.274.52.37385. [DOI] [PubMed] [Google Scholar]
- Feng J, Ito M, Kureishi Y, Ichikawa K, Amano M, Isaka N, Okawa K, Iwamatsu A, Kaibuchi K, Hartshorne DJ, Nakano T. Rho-associated kinase of chicken gizzard smooth muscle. J Biol Chem. 1999b;274:3744–3752. doi: 10.1074/jbc.274.6.3744. [DOI] [PubMed] [Google Scholar]
- Fujita A, Takeuchi T, Nakajima H, Nishio H, Hata F. Involvement of heterotrimeric GTP-binding protein and rho protein, but not protein kinase C, in agonist-induced Ca2+ sensitization of skinned muscle of guinea pig vas deferens. J Pharmacol Exp Ther. 1995;274:555–561. [PubMed] [Google Scholar]
- Gailly P, Gong MC, Somlyo AV, Somlyo AP. Possible role of atypical protein kinase C activated by arachidonic acid in Ca2+ sensitization of rabbit smooth muscle. J Physiol. 1997;500:95–100. doi: 10.1113/jphysiol.1997.sp022002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong MC, Fuglsang A, Alessi D, Kobayashi S, Cohen P, Somlyo AV, Somlyo AP. Arachidonic acid inhibits myosin light chain phosphatase and sensitizes smooth muscle to calcium. J Biol Chem. 1992;267:21492–21498. [PubMed] [Google Scholar]
- Gong MC, Kinter MT, Somlyo AV, Somlyo AP. Arachidonic acid and diacylglycerol release associated with inhibition of myosin light chain dephosphorylation in rabbit smooth muscle. J Physiol. 1995;486:113–122. doi: 10.1113/jphysiol.1995.sp020795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen AA, Gerber JN, SweeleY CC, White RP, Robertson JT. Pleocytosis and elevation of prostaglandin F2α and E2 in cerebrospinal fluid following intracisternal injection of thrombin. Stroke. 1977;8:236–238. doi: 10.1161/01.str.8.2.236. [DOI] [PubMed] [Google Scholar]
- Hamaguchi T, Ito M, Feng J, Seko T, Koyama M, Machida H, Takase K, Amano M, Kaibuchi K, Hartshorne DJ, Nakano T. Phosphorylation of CPI-17, an inhibitor of myosin phosphatase, by protein kinase N. Biochem Biophys Res Commun. 2000;274:825–830. doi: 10.1006/bbrc.2000.3225. [DOI] [PubMed] [Google Scholar]
- Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G. Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Gα13. Science. 1998;280:2112–2114. doi: 10.1126/science.280.5372.2112. [DOI] [PubMed] [Google Scholar]
- Hartshorne DJ, Ishihara H, Karaki H, Ozaki H, Sato K, Hori M, Watabe S. Okadaic acid and calyculin A: effects on smooth muscle systems. Adv Protein Phosphatases. 1989;5:219–231. [Google Scholar]
- Hartshorne DJ, Ito M, Erdodi F. Myosin light chain phosphatase: subunit composition, interactions and regulation. J Muscle Res Cell Motil. 1998;19:325–341. doi: 10.1023/a:1005385302064. [DOI] [PubMed] [Google Scholar]
- Hirata K, Kikuchi A, Sasaki T, Kuroda S, Kaibuchi K, Matsuda Y, Seki H, Saida K, Takai A. Involvement of rho p21 in the GTP-enhanced calcium ion sensitivity of smooth muscle contraction. J Biol Chem. 1992;267:8719–8722. [PubMed] [Google Scholar]
- Hori M, Sato K, Miyamoto S, Ozaki H, Karaki H. Different pathways of calcium sensitization activated by receptor agonists and phorbol esters in vascular smooth muscle. Br J Pharmacol. 1993;110:1527–1531. doi: 10.1111/j.1476-5381.1993.tb13996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori M, Sato K, Sakata K, Ozaki H, Takano-Ohmuro H, Tsuchiya TS, Sugi H, Kato I, Karaki H. Receptor agonists induce myosin phosphorylation-dependent and phosphorylation-independent contractions in vascular smooth muscle. J Pharmacol Exp Ther. 1992;261:506–512. [PubMed] [Google Scholar]
- Horowitz A, Menice CB, Laporte R, Morgan KG. Mechanisms of smooth muscle contraction. Physiol Rev. 1996;76:967–1003. doi: 10.1152/physrev.1996.76.4.967. [DOI] [PubMed] [Google Scholar]
- Ikebe M, Hartshorne DJ. Phosphorylation of smooth muscle myosin at two distinct sites by myosin light chain kinase. J Biol Chem. 1985;260:10027–10031. [PubMed] [Google Scholar]
- Ikebe M, Hartshorne DJ, Ellzinga M. Phosphorylation of the 20 000-dalton light chain of smooth muscle myosin by the calcium-activated, phospholipid-dependent protein kinase. Phosphorylation sites and effects of phosphorylation. J Biol Chem. 1987;262:9569–9573. [PubMed] [Google Scholar]
- Itoh T, Suzuki A, Watanabe Y, Mino T, Naka M, Tanaka T. A calponin peptide enhances Ca2+ sensitivity of smooth muscle contraction without affecting myosin light chain phosphorylation. J Biol Chem. 1995;270:20400–20403. doi: 10.1074/jbc.270.35.20400. [DOI] [PubMed] [Google Scholar]
- Je H-D, Gangopadhyay SS, Ashworth TD, Morgan KG. Calponin is required for agonist-induced signal transduction-evidence from an antisense approach in ferret smooth muscle. J Physiol. 2001;537:567–577. doi: 10.1111/j.1469-7793.2001.00567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen PE, Gong MC, Somlyo AV, Somlyo AP. Separate upstream and convergent downstream pathways of G-protein- and phorbol ester-mediated Ca2+ sensitization of myosin light chain phosphorylation in smooth muscle. Biochem J. 1996;318:469–475. doi: 10.1042/bj3180469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuyama H, Morgan KG. Mechanisms of Ca2+-independent contraction in single permeabilized ferret aorta cells. Circ Res. 1993;72:651–657. doi: 10.1161/01.res.72.3.651. [DOI] [PubMed] [Google Scholar]
- Katsuyama H, Wang C-LA, Morgan KG. Regulation of vascular smooth muscle tone by caldesmon. J Biol Chem. 1992;267:14555–14558. [PubMed] [Google Scholar]
- Khalil RA, Morgan KG. Phenylephrine-induced translocation of protein kinase C and shortening of two types of vascular cells of the ferret. J Physiol. 1992;455:585–599. doi: 10.1113/jphysiol.1992.sp019317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Eto M, Woodsome TP, Brautigan DL. Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J Biol Chem. 2000;275:9897–9900. doi: 10.1074/jbc.275.14.9897. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Gaylinn BD, Denney GH, Somlyo AP. G-protein-mediated Ca2+ sensitization of smooth muscle contraction through myosin light chain phosphorylation. J Biol Chem. 1991;266:1708–1715. [PubMed] [Google Scholar]
- Koyama M, Ito M, Feng J, Seko T, Shiraki K, Takase K, Hartshorne DJ, Nakano T. Phosphorylation of CPI-17, an inhibitory phosphoprotein of smooth muscle myosin phosphatase, by Rho-kinase. FEBS Lett. 2000;475:197–200. doi: 10.1016/s0014-5793(00)01654-9. [DOI] [PubMed] [Google Scholar]
- Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC. p115 RhoGEF, a GTPase activating protein for Gα12 and Gα13. Science. 1998;280:2109–2111. doi: 10.1126/science.280.5372.2109. [DOI] [PubMed] [Google Scholar]
- Kurata R, Takayanagi I, Hisayama T. Eicosanoid-induced Ca2+ release and sustained contraction in Ca2+-free media are mediated by different signal transduction pathways in rat aorta. Br J Pharmacol. 1993;110:875–881. doi: 10.1111/j.1476-5381.1993.tb13894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kureishi Y, Kobayashi S, Amano M, Kimura K, Kanaide H, Nakano T, Kaibuchi K, Ito M. Rho-associated kinase directly induces smooth muscle contraction through myosin light chain phosphorylation. J Biol Chem. 1997;272:12257–12260. doi: 10.1074/jbc.272.19.12257. [DOI] [PubMed] [Google Scholar]
- Li L, Eto M, Lee MR, Morita F, Yazawa M, Kitazawa T. Possible involvement of the novel CPI-17 protein in protein kinase C signal transduction of rabbit arterial smooth muscle. J Physiol. 1998;508:871–881. doi: 10.1111/j.1469-7793.1998.871bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmqvist U, Trybus KM, Yagi S, Carmichael J, Fay FS. Slow cycling of unphosphorylated myosin is inhibited by calponin, thus keeping smooth muscle relaxed. Proc Natl Acad Sci U S A. 1997;94:7655–7660. doi: 10.1073/pnas.94.14.7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manenti S, Sorokine O, Van Dorsselaer A, Taniguchi H. Affinity purification and characterization of myristoylated alanine-rich protein kinase C substrate (MARCKS) from bovine brain. Comparison of the cytoplasmic and the membrane-bound forms. J Biol Chem. 1992;267:22310–22315. [PubMed] [Google Scholar]
- Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J. 1996;15:2208–2216. [PMC free article] [PubMed] [Google Scholar]
- Miki M, Walsh MP, Hartshorne DJ. The mechanism of inhibition of the actin-activated myosin MgATPase by calponin. Biochem Biophys Res Commun. 1992;187:867–871. doi: 10.1016/0006-291x(92)91277-w. [DOI] [PubMed] [Google Scholar]
- Miura M, Iwanaga T, Ito KM, Seto M, Sasaki Y, Ito K. The role of myosin light chain kinase-dependent phosphorylation of myosin light chain in phorbol ester-induced contraction of rabbit aorta. Pflugers Arch. 1997;434:685–693. doi: 10.1007/s004240050452. [DOI] [PubMed] [Google Scholar]
- Nagumo H, Sasaki Y, Ono Y, Okamoto H, Seto M, Takuwa Y. Rho kinase inhibitor HA-1077 prevents Rho-mediated myosin phosphatase inhibition in smooth muscle cells. Am J Physiol Cell Physiol. 2000;278:C57–65. doi: 10.1152/ajpcell.2000.278.1.C57. [DOI] [PubMed] [Google Scholar]
- Nagumo H, Seto M, Sakurada K, Walsh MP, Sasaki Y. HA1077, a protein kinase inhibitor, inhibits calponin phosphorylation on Ser175 in porcine coronary artery. Eur J Pharmacol. 1998;360:257–264. doi: 10.1016/s0014-2999(98)00676-1. [DOI] [PubMed] [Google Scholar]
- Nishimura J, Kolber M, Van Breemen C. Norepinephrine and GTP-γ-S increase myofilament Ca2+ sensitivity in α-toxin permeabilized arterial smooth muscle. Biochem Biophys Res Commun. 1988;157:677–683. doi: 10.1016/s0006-291x(88)80303-6. [DOI] [PubMed] [Google Scholar]
- Noda M, Yasuda-Fukazawa C, Moriishi K, Kato T, Okuda T, Kurokawa K, Takuwa Y. Involvement of rho in GTPγS-induced enhancement of phosphorylation of 20 kDa myosin light chain in vascular smooth muscle cells: inhibition of phosphatase activity. FEBS Lett. 1995;367:246–250. doi: 10.1016/0014-5793(95)00573-r. [DOI] [PubMed] [Google Scholar]
- Parsons SJW, Sumner MJ, Garland CJ. Phospholipase A2 and protein kinase C contribute to myofilament sensitization to 5-HT in the rabbit mesenteric artery. J Physiol. 1996;491:447–453. doi: 10.1113/jphysiol.1996.sp021228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurada K, Ikuhara T, Seto M, Sasaki Y. An antibody for phosphorylated myosin light chain of smooth muscle: application to a biochemical study. J Biochem. 1994;115:18–21. doi: 10.1093/oxfordjournals.jbchem.a124297. [DOI] [PubMed] [Google Scholar]
- Sakurada S, Okamoto H, Takuwa N, Sugimoto N, Takuwa Y. Rho activation in excitatory agonist-stimulated vascular smooth muscle. Am J Physiol Cell Physiol. 2001;281:C571–578. doi: 10.1152/ajpcell.2001.281.2.C571. [DOI] [PubMed] [Google Scholar]
- Seto M, Sasaki Y, Sasaki Y. Alteration in the myosin phosphorylation pattern of smooth muscle by phorbol ester. Am J Physiol. 1990a;259:C769–774. doi: 10.1152/ajpcell.1990.259.5.C769. [DOI] [PubMed] [Google Scholar]
- Seto M, Sasaki Y, Sasaki Y. Stimulus-specific patterns of myosin light chain phosphorylation in smooth muscle of rabbit thoracic artery. Pflugers Arch. 1990b;415:484–489. doi: 10.1007/BF00373627. [DOI] [PubMed] [Google Scholar]
- Seto M, Sasaki Y, Sasaki Y, Hidaka H. Effects of HA1077, a protein kinase inhibitor, on myosin phosphorylation and tension in smooth muscle. Eur J Pharmacol. 1991;195:267–272. doi: 10.1016/0014-2999(91)90545-2. [DOI] [PubMed] [Google Scholar]
- Shimamoto H, Shimamoto Y, Kwan C-Y, Daniel EE. Participation of protein kinase C in endothelin-1-induced contraction in rat aorta: studies with a new tool, calphostin C. Br J Pharmacol. 1992;107:282–287. doi: 10.1111/j.1476-5381.1992.tb12739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H-M, Je H-D, Gallant C, Tao TC, Hartshorne DJ, Ito M, Morgan K G. Differential association and localization of myosin phosphatase subunits during agonist-induced signal transduction in smooth muscle. Circ Res. 2002;90:546–553. doi: 10.1161/01.res.0000012822.23273.ec. [DOI] [PubMed] [Google Scholar]
- Singer HA, Oren JW, Benscoter HA. Myosin light chain phosphorylation in 32P-labeled rabbit aorta stimulated by phorbol 12, 13-dibutyrate and phenylephrine. J Biol Chem. 1989;264:21215–21222. [PubMed] [Google Scholar]
- Somlyo AP, Kitazawa T, Himpens S, Matthijs G, Horiuti K, Kobayashi S, Goldman YE, Somlyo AV. Modulation of Ca2+-sensitivity and of the time course of contraction in smooth muscle: a major role of protein phosphatase? Adv Protein Phosphatases. 1989;5:181–195. [Google Scholar]
- Suematsu E, Resnick M, Morgan KG. Change of Ca2+ requirement for myosin phosphorylation by prostaglandin F2α. Am J Physiol. 1991;261:C253–258. doi: 10.1152/ajpcell.1991.261.2.C253. [DOI] [PubMed] [Google Scholar]
- Swärd K, Dreja K, Susnjar M, Hellstrand P, Hartshorne DJ, Walsh MP. Inhibition of Rho-associated kinase blocks agonist-induced Ca2+ sensitization of myosin phosphorylation and force in guinea-pig ileum. J Physiol. 2000;522:33–49. doi: 10.1111/j.1469-7793.2000.0033m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- Van Eyk JE, Arrell DK, Foster DB, Strauss JD, Heinonen TyK, Furmaniak-Kazmierczak E, Cote GP, Mak AS. Different molecular mechanisms for Rho family GTPase-dependent, Ca2+-independent contraction of smooth muscle. J Biol Chem. 1998;273:23433–23439. doi: 10.1074/jbc.273.36.23433. [DOI] [PubMed] [Google Scholar]
- Walsh MP, Hinkins S, Dabrowska R, Hartshorne DJ. Smooth muscle myosin light chain kinase. Methods Enzymol. 1983;99:279–288. doi: 10.1016/0076-6879(83)99063-8. [DOI] [PubMed] [Google Scholar]
- Weber LP, Van Lierop JE, Walsh MP. Ca2+-independent phosphorylation of myosin in rat caudal artery and chicken gizzard myofilaments. J Physiol. 1999;516:805–824. doi: 10.1111/j.1469-7793.1999.0805u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder SJ, Walsh MP. Smooth muscle calponin. Inhibition of actomyosin MgATPase and regulation by phosphorylation. J Biol Chem. 1990;265:10148–10155. [PubMed] [Google Scholar]
- Yazawa M, Sakuma M, Yagi K. Calmodulins from muscles of marine invertebrates, scallop and sea anemone. J Biochem. 1980;87:1313–1320. doi: 10.1093/oxfordjournals.jbchem.a132869. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Yagi K. Two kinds of myosin phosphatases with different enzymatic properties from fresh chicken gizzard smooth muscle. Purification and characterization. J Biochem. 1988;103:380–385. doi: 10.1093/oxfordjournals.jbchem.a122278. [DOI] [PubMed] [Google Scholar]