

Abstract
In CA3 pyramidal neurons from organotypic slice cultures, activation of Gq-coupled group I metabotropic glutamate receptors (mGluRs) induces a non-selective cationic conductance that enhances excitability. We have found that this response shares several properties with conductances that are mediated by the transient receptor potential (TRP) family of ion channels, including inhibition by La3+, 2-aminoethoxydiphenylborane (2APB), cis-N-(2-phenylcyclopentyl)azacyclotridec-1-en-2-amine (MDL 12,330A) and a doubly rectifying current-voltage relationship. Stimulation of mGluR1 and mGluR5 converged to activate the TRP-like conductance in a synergistic manner, and activation of either subtype alone produced only a fraction of the normal response. Activation of the cationic current required elevated intracellular Ca2+. Chelating intracellular Ca2+ or blocking Ca2+ entry through voltage-gated Ca2+ channels attenuated responses to the activation of mGluRs. Conversely, raising intracellular Ca2+ potentiated mGluR activation of the TRP-like conductance. Under control conditions, blocking G protein activation using intracellular GDPβS with or without N-(2, 6-dimethylphenylcarbamoylmethyl) triethylammonium chloride (QX-314) prevented mGluR-mediated activation of the TRP-like conductance. Following G protein blockade, however, the coupling between mGluRs 1 and/or 5 and the TRP-like conductance was rescued by increasing intracellular Ca2+. This suggests that a G protein-independent signalling pathway is also activated by group I mGluRs. Such a pathway may represent an alternative transduction mechanism to maintain metabotropic responses under conditions where G proteins are functionally uncoupled from their cognate receptors.
Metabotropic glutamate receptors (mGluRs) regulate excitability in the nervous system by gating diverse ionic conductances and by modulating synaptic transmission (Conn & Pin, 1997). The group I mGluRs, comprising mGluR1 and mGluR5, couple mainly to G proteins of the Gq family to activate phospholipase C and to target ionic channels (reviewed in Hermans & Challiss, 2001), thereby generally enhancing excitability by depolarising neurons. The mechanism responsible for neuronal depolarisation is best understood in hippocampal pyramidal cells, where both G protein-dependent inhibition of K+ conductances and activation of an unidentified non-selective cationic conductance induce inward currents (see Anwyl, 1999 for review). The transient receptor potential (TRP) family of ion channels are good candidates for mediating the cationic conductance as these can be activated by Gq-dependent signalling cascades (Boulay et al. 1997; Okada et al. 1999; Delmas et al. 2002), and are widely expressed in the brain (Mizuno et al. 1999) including in hippocampal pyramidal cells (Philipp et al. 1998; Mezey et al. 2000; Strübing et al. 2001).
To date, few studies have sought to ascribe a particular role to currents mediated by TRP channels in native systems or to examine aspects of the coupling to native G protein-coupled receptors. Furthermore, conflicting results have been reported on the role of G protein signalling cascades in transducing the mGluR-mediated cationic current, with both G protein-dependent (Crepel et al. 1994; Pozzo Miller et al. 1995; Congar et al. 1997) and G protein-independent (Guérineau et al. 1995; Heuss et al. 1999) pathways being implicated. Here, we examined the cationic current evoked by activating group I mGluRs in hippocampal CA3 pyramidal cells and found that the current exhibits several properties consistent with mediation by channels of the TRP family.
Methods
All experiments were carried out according to the guidelines laid down by the Swiss Department for Veterinary Affairs.
Hippocampal organotypic slice cultures were prepared from 6-day-old Wistar rats using the roller-tube technique, as described previously (Gähwiler et al. 1998). Rats were killed by decapitation. After 3–4 weeks in vitro, slice cultures were transferred to a 1 ml recording chamber, maintained at 28 °C and continuously superfused (1.5 ml min−1) with (mm): 137 NaCl, 2.7 KCl, 11.6 NaHCO3, 0.4 NaH2PO4, 2 MgCl2, 3 CaCl2, 5.6 d-glucose, 0.001 % phenol red, pH 7.4, 305 mosmol l−1. Somatic whole-cell voltage-clamp recordings were made from visualised CA3 pyramidal cells using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA, USA). Patch pipettes had resistances of 2–3 MΩ. The standard pipette solution contained (mm): 130 CsCH3SO3, 10 CsCl, 10 Hepes, 1 EGTA, 1 MgCl2, 0.4 NaGTP, 2 MgATP, pH 7.25, 280–290 mosmol l−1. When indicated, K+ replaced Cs+ ions, Cl− replaced CH3SO3− or 1 mm guanosine 5′-(β-thio)diphosphate trilithium salt (GDPβS) replaced GTP. For experiments with high BAPTA, EGTA was replaced with 40 mm Cs4-BAPTA and CsCH3SO3 was reduced to keep osmolality constant (280–290 mosmol l−1). When LaCl3 was used, NaH2PO4 was replaced with NaCl for all responses. Liquid junction potentials were corrected. Immediately after obtaining whole-cell access, cells were discarded if the membrane potential was less negative than −55 mV, if input resistance was less than 120 MΩ or if series resistance was more than 14 MΩ. Series resistance was measured using the amplifier and was 4–14 MΩ except when GDPβS was used, in which case series resistance was less than 10 MΩ. Hyperpolarising steps (-5 mV) were applied to monitor input resistance. With intracellular Cs+, the current often shifted in the outward direction in response to the steps due to the closure of persistent Ca2+ conductances present in these cells (Avery & Johnston, 1996; C. E. Gee & P. Benquet, unpublished observations), therefore no values for input resistance are reported here. All cells were dialysed for 18–25 min before applying the group I mGluR agonist (S)-3,5-dihydroxyphenylglycine (DHPG). Responses to DHPG at intervals longer than 10 min were then stable, with second, third and fourth responses being 102.8 ± 7.4 % (n = 22, P = 0.31), 95.7 ± 12.2 % (n = 16, P = 0.16) and 82.0 ± 9.8 % (n = 4, P = 0.13), respectively, of the first responses obtained in control cells periodically throughout the study. To prevent network activity, all experiments were performed with 0.5–1 µm TTX, 200 µm picrotoxin, 10 µm 6-cyano-7-nitroquinoxaline-2,3-dione disodium (CNQX) and 20 µm 3-((R)-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (D-CPP) in the superfusate. Substances were applied via the superfusate.
Data were filtered at 1–2 kHz, digitised and stored on a PC at 500 Hz using pCLAMP7 software and analysed off-line. Data were compared using Student's paired or unpaired t tests as appropriate. For presentation purposes, traces were re-filtered with a low-pass Gaussian filter with a cut-off of −3 db at 50–200 Hz.
To image intracellular Ca2+, 20 µm Oregon Green 488 BAPTA-2 was added to the intracellular solution (Kd ≈ 580 nm). Excitation illumination was applied at 488 nm using a TILL Photonics Polychrome I monochromator (Planegg, Germany) and emitted images were collected with a cooled CCD camera (Princeton Instruments, Trenton, NJ, USA) after passing them through a TILL FITC filter set, stored and then analysed using Axon Imaging Workbench (Axon Instruments). Images were collected for 1–2 s at 8 s intervals. Average fluorescence was determined for regions of interest over the soma (avoiding the nucleus) and the background fluorescence of a region away from the filled cell was subtracted. ΔF/F was then calculated for the region of interest in each image (ΔF/F = (fluorescence - average baseline fluorescence)/ average baseline fluorescence). For each condition in a given cell, the ΔF/F value is the average of 3–7 successive images.
GDPβS (Sigma) was dissolved in water at 20 × the final concentration and kept frozen for up to 1 week before adding to the intracellular solution on the day of the experiment. Internal solutions were kept on ice during experiments. Other compounds were dissolved in water, dimethylsulphoxide or fresh dilute NaOH, as appropriate, at × 1000 or higher final concentrations and kept frozen in small aliquots at −20 °C until just before use. DHPG was used within 2 weeks. TTX was from Latoxan (Valence, France). cis-N-(2-phenylcyclopentyl)azacyclotridec-1-en-2-amine (MDL 12,330A) was from Calbiochem (Juro; Lucerne, Switzerland). DHPG, (+)-2-methyl-4 carboxyphenylglycine (LY367385), 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP), 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB), 2-aminoethoxydiphenylborane (2APB), and CNQX were purchased from Tocris-Cookson (Bristol, UK). N-(2,6-dimethylphenylcarbamoylmethyl) triethylammonium chloride (QX-314) was from Alomone Labs (Jerusalem, Israel). Cs4-BAPTA and Oregon Green 488 BAPTA-2 were from Molecular Probes (Leiden, The Netherlands). ω-Agatoxin IVA was from The Peptide Institute (Osaka, Japan), ω-conotoxins MVIIC and GVIA were from Bachem (Dubendorf, Switzerland). Baclofen and D-CPP were gifts from Novartis (Basel, Switzerland). All other chemicals were purchased from Sigma.
Results
TRP-like channels mediate the inward current induced by group I mGluRs in CA3 pyramidal cells
With a Cs+-based intracellular solution, CA3 pyramidal cells voltage clamped at −50 mV responded to the group I mGluR-specific agonist DHPG with an inward current of −45 ± 4 pA (n = 111, Fig. 1A). Note that when n values are displayed in figures they are not repeated in the text. The inward current was often followed by an outward current, which we did not study further. The current-voltage (I-V) relationship was assessed using a ramp protocol, applied prior to the application of DHPG and near the peak inward current, or by holding the cell at different potentials (Fig. 1B). The average reversal potential from the ramps was −17 ± 10 mV, indicative of a mixed cationic conductance, which has been demonstrated previously in CA3 pyramidal cells and is carried mainly by monovalent cations (Caeser et al. 1993; Guérineau et al. 1995; Pozzo Miller et al. 1995; Chuang et al. 2000). The I-V relationship exhibited a negative slope between −40 mV and −120 mV but no second reversal potential, suggesting that with a Cs+-based intracellular solution the block of K+ channels by activating group I mGluRs contributes little to the inward current, as is the case following muscarinic activation in cortical neurons (Haj-Dahmane & Andrade, 1996). We also monitored Ca2+ levels with Oregon Green BAPTA-2 fluorescence in four CA3 pyramidal cells while the voltage-clamp command was increased from −90 to 20 or 40 mV in 10 mV steps (Fig. 1B). We found that Ca2+ levels began to rise as the voltage reached −60 mV and reached a plateau at around −40 mV, which corresponds to the peak of the inward current. This suggests that the negative slope reflects the Ca2+ dependence of the conductance (see below). The inward current was inhibited by NPPB (P = 0.001; Fig. 1C), which antagonises some non-selective cation channels (Popp et al. 1993). NPPB is more widely known to inhibit Cl− channels and anion exchangers. However, shifting the Cl− reversal potential from −65 to −1 mV with CsCl-filled patch electrodes failed to change the peak amplitude of the DHPG-induced inward currents (P = 0.83; Fig. 1D), indicating that the mGluR-induced inward current is not mediated by Cl− channels or an anion exchanger. Replacing extracellular Na+ with Li+ also did not affect the peak current, suggesting that transporter currents are not involved (-40.5 ± 9.6 pA, n = 5, P = 0.88; data not shown).
Figure 1. Transient receptor potential (TRP)-like channels conduct the current induced by (S)-3, 5-dihydroxyphenylglycine (DHPG) in CA3 pyramidal cells.

A, sample response to a 30 s application of 10 µm DHPG in a cell voltage clamped at −50 mV. B, the average subtracted I-V relationship of the response to DHPG, calculated from 3 s voltage ramps (+40 to −120 mV, n = 5), indicates that the response is associated with an increase in a mixed cationic conductance. Inset is the peak current in response to DHPG vs. different holding potentials in a typical cell. Superimposed on the I-V relationship is the change in Ca2+ levels (ΔF/F) with increasing holding potential from cells loaded with Oregon Green BAPTA-2 (circles, n = 4). Intracellular Ca2+ rose with increasing voltage beginning from −60 mV and began to plateau at −40 mV, close to the peak of the inward current. C, 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB), an inhibitor of Cl− channels and anion exchangers that also inhibits some cationic conductances, inhibited the DHPG-induced current. D, exchanging intracellularCH3SO3− for Cl− to reverse the direction of current flow through Cl− channels and anion exchangers had no effect on the response to DHPG. E, La3+, an antagonist of both TRP channels and Ca2+ channels, inhibited responses to DHPG. F, the TRP channel and adenylyl cyclase antagonist cis-N-(2-phenylcyclopentyl)azacyclotridec-1-en-2-amine (MDL 12,330A) inhibited the response to DHPG. G, 2-aminoethoxydiphenylborane (2APB), which inhibits TRP channels and inositol trisphosphate receptor (IP3R)- mediated events, inhibited the DHPG response. In this and subsequent figures, traces of control data (cntl) and traces obtained after drug application are from the same cell voltage clamped at −50 mV. The line below traces indicates time of DHPG entering the bath. The n values are given on the figures; n =x indicates that the same x cells are included in each condition. Numbers in parentheses indicate either that a subpopulation is included, especially after washes, or that different cells were used in each condition. Pooled data are shown as means ±s.e.m. *P≤ 0.05.
The region of negative slope conductance is reminiscent of the I-V relationship reported for TRPC1 + TRPC4 or TRPC5 (Strübing et al. 2001), or TRPV1 (Gunthorpe et al. 2002) members of the TRP family of cationic channels, when transfected in human embryonic kidney 293 cells. We therefore examined whether the DHPG-induced current exhibits additional TRP-like properties. As no TRP-selective pharmacological antagonists are yet available, we used the unrelated compounds La3+, MDL12,330A and 2APB, which, while having other actions, also block several TRP-mediated currents (Van Rossum et al. 2000; Clapham et al. 2001). The inward current induced by DHPG was inhibited by 10–15 min applications of La3+ (Fig. 1E; P = 0.017), MDL 12,330A (Fig. 1F; P = 0.015) and 2APB (Fig. 1G; P = 0.0097).
La3+ also blocks some voltage-gated Ca2+ channels (VGCCs), but unlike Cd2+ (see below) did not cause an outward shift in the holding current (P = 0.44; data not shown). MDL 12,330A is also known to inhibit adenylyl cyclase (Siegel & Wiech, 1976). However, the cAMP analogue 8-bromoadenosine-3′,5′-cyclic monophosphate had no effect on the DHPG-induced inward current (500 µm, P = 0.51; n = 5; data not shown), suggesting that cAMP production by adenylyl cyclase was not required for the inward current. Staurosporine, a general protein kinase inhibitor, did not affect the DHPG-induced current (1–2 µm, control −39.5 ± 15.4 pA, staurosporine −35.2 ± 18 pA, n = 4, P = 0.35), suggesting that the activation of neither protein kinase A nor C was required and that MDL 12,330A was acting via an adenylyl cyclase→cAMP→protein kinase A-independent mechanism. 2APB is also an inhibitor of inositol trisphosphate (IP3)-receptor-mediated Ca2+ release from intracellular stores. If 2APB acted solely by preventing intracellular Ca2+ release, then raising intracellular Ca2+ by increasing the extracellular K+ concentration to 8 mm should have ‘rescued’ the block by 2APB (explanation below). This was not the case, however, as 2APB still significantly inhibited the response to DHPG in 8 mm K+ (vs. 2APB in 2.7 mm K+P = 0.82, n = 4; data not shown). Taken together, the results suggest strongly that the inward current induced by group I mGluR activation in CA3 pyramidal cells is mediated by non-selective cationic TRP-like channels.
Both mGluR1 and mGluR5 contribute to the inward current in CA3 pyramidal cells
We examined the mGluR subtype specificity for the induction of inward current using the mGluR1 specific antagonist LY367385 (IC50 8–12 µm) and the mGluR5 specific antagonist MPEP (IC50 0.03 µm), both of which are reported not to affect the other subtype at the concentrations used here (Clark et al. 1997; Bruno et al. 1999; Gasparini et al. 1999). The mGluR1-specific agonist LY367385 reversibly reduced the DHPG-induced inward current to 10.9 ± 9.9 % of the control response, whereas the mGluR5-specific antagonist MPEP irreversibly reduced the DHPG response to 36.6 ± 7.3 % of the control response (Fig. 2). Therefore, activation of mGluR5 alone causes virtually no inward current and activating mGluR1 alone induces only about 37 % of the response to DHPG in our normal conditions, suggesting that group I mGluRs operate synergistically to induce the inward current in CA3 pyramidal cells.
Figure 2. Metabotropic glutamate receptors (mGluRs) 1 (mGluR1) and 5 (mGluR5) contribute synergistically to activate a cationic conductance in CA3 pyramidal cells.
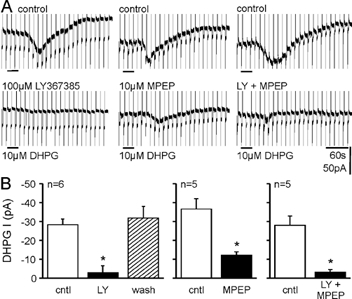
A, top traces show responses to DHPG in control conditions and bottom traces show responses to DHPG after application of either the mGluR1 antagonist LY367385 (LY), the mGluR5 antagonist 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP), or both. B, pooled data show that blocking either mGluR1 or mGluR5 reduced the response to DHPG by more than 50 % in CA3 pyramidal neurons. Only the block by the competitive antagonist LY367385 was reversible.
G protein dependence
We then determined whether induction of the cationic current by DHPG depends on the activation of G proteins. G protein function was blocked by dialysing cells with the non-hydrolysable GDP analogue GDPβS (Eckstein et al. 1979) at 1 mm, a concentration that effectively blocks the G protein-dependent potentiation of NMDA receptors by mGluR5 in our preparation (Benquet et al. 2002). For these experiments, a K+-based intracellular solution was used, allowing us to assess the effectiveness of G protein blockade by monitoring the G protein-dependent K+ currents induced by periodic application of the GABAB receptor agonist baclofen. The baclofen responses induced during the first 3 min of whole-cell recording (208.5 ± 14.0 pA) were completely blocked after 12–60 min (Fig. 3A1; 4.0 ± 2.3 pA, P = 2 × 10−10, n = 15), whereas in control cells with GTP, baclofen responses were 104.0 ± 33.2 pA after 35–78 min (n = 5, P = 2 × 10−6vs. GDPβS cells). Following G protein blockade, responses to application of the group I mGluR agonist DHPG were blocked (Fig. 3A2 and B; P = 0.04vs. K+/GTP-containing cells). The effect was not due to the presence of Li+ because with 3 mm intracellular Li+, responses to DHPG were not different from control cells (P = 0.42, n = 3).
Figure 3. Intracellular GDPβS (1 mm) prevents activation of the cationic current to DHPG.
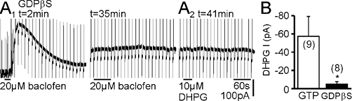
A1, with intracellular Cs+ exchanged for K+, the GABAB receptor agonist baclofen, applied within 2 min after obtaining whole-cell access, induced a G protein-dependent K+ current. This response was completely blocked after 12–60 min dialysis with GDPβS. A2, subsequent application of DHPG induced no response in CA3 pyramidal cells. B, pooled responses to DHPG from cells with intracellular K+ and either GTP or GDPβS.
Calcium dependence
Including 40 mm BAPTA in the pipette solution significantly reduced the inward current induced by DHPG compared with interleaved controls (Fig. 4A; P = 0.0007). With this concentration of intracellular BAPTA, the increase in intracellular Ca2+ levels seen when the holding potential was increased from −70 to −50 mV was still significant (0.05 ± 0.01 ΔF/F, n = 4, P = 0.02) but was significantly reduced when compared to control cells (0.35 ± 0.11 ΔF/F, n = 4, P = 0.03), as detected with Oregon Green BAPTA-2 fluorescence. Bathing the slice with 50 µm Cd2+ also reduced responses to DHPG (Fig. 4B; P = 0.017), suggesting that entry through VGCCs contributes to the rise in intracellular Ca2+ necessary for the inward current to be activated. Cd2+ also induced an outward shift in the holding current (62.8 ± 14.8 pA), suggesting that it blocked a tonically active Ca2+ conductance. Exchanging extracellular Ca2+ for Ba2+ also prevented activation of the inward current, suggesting that Ba2+ is not able to substitute for Ca2+ in supporting activation of the underlying channels (Fig. 4C; P = 0.001). We next raised the extracellular K+ from 2.7 to 8 mm, which caused an inward current (-136.5 ± 22.5 pA, n = 8) and significantly increased the current induced by DHPG (Fig. 4D; P = 0.0027). The application of 100 µm Cd2+ + 50 µm Ni2+ (Cd2+/Ni2+) reduced the K+-induced inward current in four out of four cells (-134.3 ± 14.7 pA vs. −93.0 ± 21.2 pA, P = 0.06), indicating that 8 mm K+ depolarised unclamped dendrites, resulting in Ca2+ influx through VGCCs. Cd2+/Ni2+ also blocked the enhancement of the DHPG-induced inward current by increasing extracellular K+ (Fig. 4E; P = 0.133), suggesting that Ca2+ entry through VGCCs potentiates the group I mGluR-induced inward current. To rule out a non-specific action by Cd2+/Ni2+, we used a cocktail of 200 nm AgaIVA, 1 µm MVIIC, 2 µm GVIA and 10 µm nifedipine to block P/Q-, N-, and L-type VGCCs. The DHPG-induced inward current in 2.7 mm K+ was attenuated by this cocktail (Fig. 4F), and blocking VGCCs also prevented the enhancement of the DHPG-induced inward current by elevating extracellular K+ (Fig. 4F; P = 0.95). Thus, the current induced by activation of group I mGluRs in CA3 pyramidal cells is dependent on Ca2+ entry through VGCCs.
Figure 4. The response to DHPG is Ca2+ dependent.
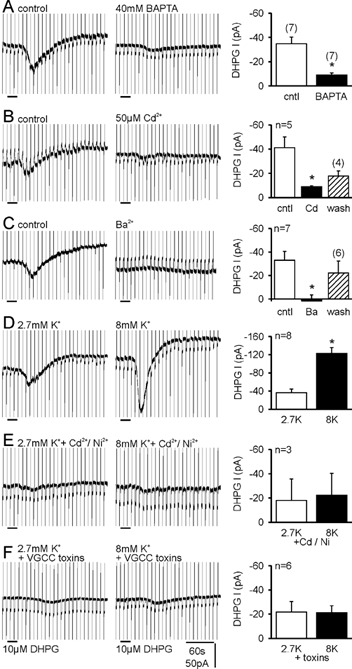
A, including 40 mm BAPTA in the recording pipette suppressed the inward current induced by DHPG. B, blocking voltage-gated Ca2+ channels (VGCCs) with Cd2+ strongly reduced the inward current. C, substituting Ba2+ for extracellular Ca2+ prevented activation of the inward current by DHPG. D, increasing extracellular K+ from 2.7 mm (2.7K)to 8 mm (8K) increased the response to DHPG. E, in the continuous presence of 100 µm Cd2++ 50 µm Ni2+, increasing the extracellular K+ concentration no longer enhanced responses to DHPG. F, the increase in inward current by raising extracellular K+ was also prevented by a cocktail of toxins that block VGCCs (see text).
Increasing intracellular Ca2+ rescues the inward current following G protein blockade
The role of G proteins in the activation of cationic current by group I mGluRs in hippocampal pyramidal cells is presently unclear (see Introduction). As the inward current is Ca2+ sensitive, it is possible that GDPβS acts by preventing G protein-mediated release of Ca2+ from IP3-sensitive stores. We checked whether this mechanism is involved by first dialysing cells with GDPβS until responses to baclofen and DHPG were fully blocked (Fig. 5A, B1 and C). When the extracellular K+ concentration was then raised from 2.7 to 8 mm to increase intracellular Ca2+, we observed a recovery of the cationic current in response to DHPG (Fig. 5A3vs. A5 and B1; P = 0.013vs. 2.7 mm K+ GDPβS, P = 0.54vs. 8 mm K+ GTP). Therefore, by increasing intracellular Ca2+, a G protein-independent pathway coupling mGluRs to activation of the inward current was revealed. That G protein blockade was unaffected by 8 mm K+ was verified by the continued lack of response to baclofen (Fig. 5C). Several G proteins including Gq and Go are directly blocked by QX-314 (Hollmann et al. 2001), which was added to the intracellular solution containing GDPβS in five cells. Responses to baclofen were now abolished within 10–36 min (Fig. 5C). Raising extracellular K+ from 2.7 to 8 mm again rescued responses to DHPG (Fig. 5B1, P = 0.0007) without affecting the block of responses to baclofen (Fig. 5C; P = 0.31). We also tested subtype-specific mGluR antagonists and found that elevated K+ was able to rescue responses to both group I mGluR subtypes (Fig. 5A4, A6 and D). We confirmed the ‘rescue’ of the inward current by 8 mm K+ in a separate series using our standard Cs+-based solution (Fig. 5B2; data shown in Fig. 3B are reproduced here for ease of comparison) in which GDPβS effectively suppressed DHPG responses in 2.7 mm K+ in five out of six cells (P = 0.02vs. GTP, n = 5; Fig. 5B2). Upon increasing extracellular K+ to 8 mm, the response to DHPG was again ‘rescued’ and a G protein-independent mode of activation was confirmed. We also tested whether raising intracellular Ca2+ rescues responses (after presumable blockade of G proteins) by facilitating the ability of residual unblocked G proteins to activate phospholipase C. In five cells, two with GDPβS and three with GDPβS plus QX-314, the response to DHPG in 8 mm K+ was not significantly affected by 20–40 min applications of the phospholipase C inhibitor U73122 (Fig. 5E, P = 0.25).
Figure 5. Raising intracellular Ca2+ rescues the response to DHPG following G protein blockade.
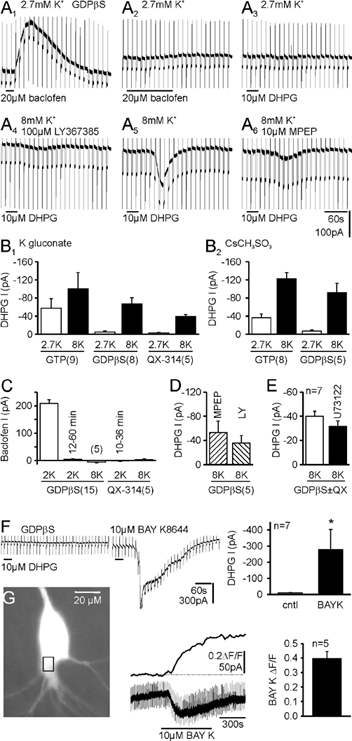
A, sample traces from one cell recorded with intracellular K+ and 1 mm GDPβS are shown in chronological order. A1, 2, the loss of response to baclofen indicates G protein blockade. A3, following G protein blockade, responses to DHPG were blocked. A4, raising intracellular Ca2+ by increasing extracellular K+ rescued the response to selective activation of mGluR5 (DHPG + LY367385). A5, rescue is also observed by activation of both group I mGluRs after washing LY367385 from the slice. A6, response in 8 mm K+ to selective activation of mGluR1 (DHPG + MPEP). B1, pooled data with GTP, GDPβS or GDPβS plus N-(2,6-dimethylphenylcarbamoylmethyl) triethylammonium chloride (QX-314; 5 mm) showing the block of responses to DHPG in 2.7 mm K+, potentiation of the response in GTP cells with 8 mm K+ and rescue of the response after G protein blockade. B2, with intracellular Cs+, GDPβS still blocked the response to DHPG in 2.7 mm K+ and responses were rescued with 8 mm K+. C, with intracellular GDPβS, responses to baclofen recorded within 3 min of gaining whole-cell access were abolished within 12–60 min. Raising extracellular K+ to 8 mm did not affect G protein blockade by GDPβS. With QX-314 added to the GDPβS-containing solution, baclofen responses were blocked within 10–36 min, and again were not affected by the rise in intracellular Ca2+ induced by 8 mm K+. D, pooled data from five cells in which responses to DHPG with the mGluR1- and mGluR5-selective antagonists were obtained in 8 mm K+ following G protein blockade. E, rescue of responses to DHPG by 8 mm K+ was not affected by the phospholipase C antagonist U73122 (10 µm). In two cells, G proteins were blocked by GDPβS, and in five cells they were blocked by GDPβS plus QX-314. F, following G protein blockade, the L-type Ca2+ channel agonist BAY K8644 also rescued the response to DHPG. Traces are from one cell, with baclofen responses abolished by GDPβS. G, BAY K8644 increased intracellular Ca2+ levels (holding potential −50 mV). A typical CA3 pyramidal cell showing the region of interest along with the simultaneous Ca2+ and electrophysiological recordings.
Finally, instead of activating VGCCs indirectly by raising K+, we modulated Ca2+ influx directly through L-type Ca2+ channels with the agonist BAY K8644. BAY K8644 induced an inward current (-71 ± 20 pA, n = 7) and ‘rescued’ responses to DHPG (P = 0.05, Fig. 5F). BAY K8644 significantly increased intracellular Ca2+ in our conditions, as detected by Oregon Green BAPTA-2 imaging (Fig. 5G, P = 0.001), providing further evidence that G protein-independent activation of the inward current is unmasked by raising intracellular Ca2+.
Discussion
Our results demonstrate that both subtypes of group I mGluRs participate in activating native TRP-like channels in CA3 pyramidal cells in a Ca2+-dependent manner. The cationic conductance evoked by group I mGluRs is dependent on G proteins in ‘control’ conditions; however, the apparent G protein dependence can be overcome by raising intracellular Ca2+.
Identity of channels mediating the inward current
We found that the current activated by mGluR activation in CA3 pyramidal cells has many properties consistent with channels formed by members of the TRP family of cation channels, and several members of the TRP family have been localised to CA3 pyramidal cells (Philipp et al. 1998; Okada et al. 1999; Mezey et al. 2000; Strübing et al. 2001). We determined a I-V relationship similar to that published for TRPC1 + TRPC4 or TRPC5 (Strübing et al. 2001), or TRPV1 (Gunthorpe et al. 2002). Moreover, activation of many TRP channels occurs following agonist activation of seven transmembrane receptors and the G protein→phospholipase C→diacylglycerol/IP3 pathway (for review see Minke & Cook, 2002). Ca2+ is required for opening of several TRPs including TRPC4 and TRPC5 (Okada et al. 1998; Philipp et al. 1998; Schaefer et al. 2000). Ca2+ initiates or enhances currents through some TRPs (Strübing et al. 2001; Zhang et al. 2001), whereas co-expression of TRPC1 and TRPC3 induces the formation of channels that are inactivated by Ca2+ (Lintschinger et al. 2000). We found that La3+ blocked the inward current. This finding suggests that TRPC1 + TRPC4 or TRPC5, which are facilitated by La3+ (Strübing et al. 2001), do not mediate the cationic current in CA3 pyramidal cells, or that in CA3 cells these channel subunits form heteromultimers in combination with other TRPs to yield channels that are blocked by La3+. As the biophysical properties of TRP channels are different in combination than when expressed alone, a combined molecular- biochemical-physiological approach will be required to identify the TRPs mediating the mGluR-induced cationic current in CA3 pyramidal cells.
A further class of channels mediating cationic currents, which are expressed in principal cells of the hippocampus, are the ‘olfactory’-type cyclic nucleotide gated (CNG) channels (Bradley et al. 1997). It has been suggested that CNG channels underlie a similar membrane depolarisation that occurs in CA1 pyramidal neurons following activation of muscarinic receptors (Kuzmiski & MacVicar, 2001). To date we are not aware of any reports in which it has been demonstrated that MDL 12,330A, 2APB or NPPB are able to block responses that are mediated by cGMP-activated CNG channels.
A curious observation is that the outward current seen in many of the recordings was blocked in parallel with the inward current. At least part of this current is due to the mGluR-dependent inhibition of (tonically active) VGCCs (Anwyl, 1999), which explains its disappearance in many of our manipulations.
Synergism between mGluR1 and mGluR5 activation
That both mGluR1 and mGluR5 receptors contributed to activation of the inward current is novel, as in several neuronal types these mGluR subtypes are found to have largely divergent roles in regulating synaptic transmission and general excitability (for review see Valenti et al. 2002). We found, however, that while both receptor subtypes were involved in mediating the response, the involvement of the two was not additive but synergistic, pointing to divergence in the role of each. A similar synergism is present in interneurons (Mori & Gerber, 2002). It is known that the mGluRs are part of a multiprotein complex that is associated with cytoskeletal elements and interacting proteins, and if the receptors were linked by such a scaffolding network one can imagine that blocking one could infringe on the ability of the other to be activated. An alternative possibility is that a mediating second messenger has multiple binding sites on either an intermediate molecule or directly on the target channels so that non-linear activation occurs. Thus, the second-messenger levels produced by activation of a single group I mGluR subtype may barely reach threshold for opening the channels, and a small increase in the messenger concentration caused by activation of the second subtype increases exponentially the number of channels with enough binding sites occupied to become active.
Ca2+ dependence
The requirement for Ca2+ entry through VGCCs in mGluR-mediated signalling has been described in several systems. In CA3 pyramidal cells, mGluR-mediated Ca2+ release evoked by mossy-fibre stimulation requires Ca2+ entry through VGCCs (Kapur et al. 2001). In hippocampal interneurons (Woodhall et al. 1999) and lamprey spinal cord neurons (Kettunen et al. 2002), the mGluR-mediated depolarisation and rise in intracellular Ca2+ is attenuated if either Ca2+ entry through VGCCs or Ca2+ release from intracellular stores is blocked. Furthermore, potentiation of the parallel fibre-evoked mGluR response in cerebellar Purkinje cells is seen following Ca2+ spikes (Batchelor & Garthwaite, 1997).
We were able to attenuate responses to DHPG with high concentrations of intracellular BAPTA, suggesting a requirement for a rise in intracellular Ca2+. In addition, Ba2+ was not able to substitute for Ca2+, suggesting a requirement for Ca2+per se. Ca2+ is not, however, solely responsible for activating the inward current in pyramidal cells, which is different from the case of the calcium-activated non-selective cationic current in cardiac cells (Colquhoun et al. 1981). Even in conditions of high intracellular Ca2+, activation of mGluRs was necessary to induce the inward current. In a previous study from this laboratory (Guérineau et al. 1995), it was reported that the cationic current activated by mGluRs was Ca2+ independent; however, this conclusion was based on the lack of inhibition by 20 mm BAPTA with 16 mm extracellular K+. In that condition, when there would be a steady influx of Ca2+ from the effectively infinite extracellular supply, the small amount of BAPTA actually loaded into the cell would have become saturated. Even with 40 mm BAPTA, the rise in intracellular Ca2+ induced by increasing the voltage command potential from −70 to −50 mV was not completely prevented.
G protein-independent activation
Group I mGluRs are known to release Ca2+ from intracellular stores in a Gq/G11→phospholipase C→IP3-dependent manner and our data suggest that this pathway underlies the G protein dependence of the cationic current in CA3 pyramidal cells. Our findings resolve much of the controversy over the G protein dependence of this conductance. G protein independence was reported when the extracellular K+ concentration was increased (Guérineau et al. 1995) or Ca2+ was elevated in the intracellular solution (Heuss et al. 1999). Further support for G protein-independent activation comes from studies with Gq and G11 knockout mice in which, in CA1 pyramidal neurons, the depolarising response to group I mGluR activation persists (Krause et al. 2002). Interestingly we have also noted a block of mGluR-mediated current with intracellular GDPβS in CA1 pyramidal cells, dentate granule cells and interneurons, which then could be recovered by raising extracellular K+ concentration (Gee & Gerber, 2001). With low extracellular K+ (2.7–3.5 mm; Pozzo Miller et al. 1995; Congar et al. 1997; this study) when intracellular Ca2+ levels would be expected to be low, the G protein dependence of the mGluR-mediated cationic conductance is observed. However, a change in the membrane potential to −50 mV in CA3 pyramidal cells is sufficient to induce a persistent Ca2+ current (Avery & Johnston, 1996), which may be adequate to allow G protein-independent signalling.
The ability to ‘rescue’ the cationic current after G protein blockade by elevating Ca2+ entry through VGCCs suggests strongly that a G protein-independent pathway links mGluRs to opening of the channel(s), in addition to the G protein-dependent pathway. It is unlikely that elevated Ca2+ inactivated GDPβS, as responses were also rescued when G proteins were blocked with QX-314 (Hollmann et al. 2001). In addition, in a previous study it was shown that with elevated extracellular K+, GTPγS, which permanently activates G proteins, does not occlude the mGluR-mediated inward current (Guérineau et al. 1995). While we have yet to demonstrate and identify the components of the G protein-independent cascade, a diffusible second messenger should be involved as, with cell-attached recordings, application of mGluR or muscarinic receptor agonists outside the area of the patch can activate cationic channels that lie directly under the recording pipette (Guérineau et al. 1995).
G proteins can become uncoupled from their cognate receptors following receptor phosphorylation (Schaffhauser et al. 2000) and during ischaemia (Tanabe et al. 1998), when intracellular Ca2+ and glutamate increase in tandem (Lipton, 1999). Our data indicate that under these conditions, alternate transduction mechanisms can be activated to maintain metabotropic signalling in response to glutamate. This mechanism could play an important role in mGluR-mediated long-term depression and long-term potentiation or, if unchecked, could contribute to excitotoxic processes. Indeed, antagonists for group I mGluRs confer some measure of neuroprotection in several models of neurological disease (Bruno et al. 1999; Wong et al. 1999; Gasparini et al. 2002; Valenti et al. 2002).
Acknowledgments
We thank Beat Gähwiler for continuous support and for providing slice cultures. We thank H. Blum, S. Giger, H. Kasper, L. Rietschin and M. Studer for excellent technical assistance, and A. Cowan, B. Gähwiler, M. Mori, M. Scanziani and C. Stricker for helpful discussions and critical reading of the manuscript. This work was funded by the Swiss National Science Foundation and the NCCR on Neural Plasticity and Repair.
References
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Avery RB, Johnston D. Multiple channel types contribute to the low-voltage-activated calcium current in hippocampal CA3 pyramidal neurons. J Neurosci. 1996;16:5567–5582. doi: 10.1523/JNEUROSCI.16-18-05567.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor AM, Garthwaite J. Frequency detection and temporally dispersed synaptic signal association through a metabotropic glutamate receptor pathway. Nature. 1997;385:74–77. doi: 10.1038/385074a0. [DOI] [PubMed] [Google Scholar]
- Benquet P, Gee CE, Gerber U. Two distinct signaling pathways upregulate NMDA receptor responses via two distinct metabotropic glutamate receptor subtypes. J Neurosci. 2002;22:9679–9686. doi: 10.1523/JNEUROSCI.22-22-09679.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay G, Zhu X, Peyton M, Jiang M, Hurst R, Stefani E, Birnbaumer L. Cloning and expression of a novel mammalian homolog of Drosophila transient receptor potential (Trp) involved in calcium entry secondary to activation of receptors coupled by the Gq class of G protein. J Biol Chem. 1997;272:29672–29680. doi: 10.1074/jbc.272.47.29672. [DOI] [PubMed] [Google Scholar]
- Bradley J, Zhang Y, Bakin R, Lester HA, Ronnett GV, Zin K. Functional expression of the heteromeric ‘olfactory’ cyclic nucleotide-gated channel in the hippocampus: a potential effector of synaptic plasticity in brain neurons. J Neurosci. 1997;17:1993–2005. doi: 10.1523/JNEUROSCI.17-06-01993.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno V, Battaglia G, Kingston A, O'Neill MJ, Catania MV, Di Grezia R, Nicoletti F. Neuroprotective activity of the potent and selective mGlu1a metabotropic glutamate receptor antagonist, (+)-2-methyl-4 carboxyphenylglycine ( LY367385): comparison with LY357366, a broader spectrum antagonist with equal affinity for mGlu1a and mGlu5 receptors. Neuropharmacology. 1999;38:199–207. doi: 10.1016/s0028-3908(98)00159-2. [DOI] [PubMed] [Google Scholar]
- Caeser M, Brown DA, Gähwiler BH, Knöpfel T. Characterization of a calcium-dependent current generating a slow afterdepolarization of CA3 pyramidal cells in rat hippocampal slice cultures. Eur J Neurosci. 1993;5:560–569. doi: 10.1111/j.1460-9568.1993.tb00521.x. [DOI] [PubMed] [Google Scholar]
- Chuang SC, Bianchi R, Wong RK. Group I mGluR activation turns on a voltage-gated inward current in hippocampal pyramidal cells. J Neurophysiol. 2000;83:2844–2853. doi: 10.1152/jn.2000.83.5.2844. [DOI] [PubMed] [Google Scholar]
- Clapham DE, Runnels LW, Strübing C. The TRP ion channel family. Nat Rev Neurosci. 2001;2:387–396. doi: 10.1038/35077544. [DOI] [PubMed] [Google Scholar]
- Clark BP, Baker SR, Goldsworthy J, Harris JR, Kingston AE. (+)-2-Methyl-4-carboxyphenylglycine ( LY367385). selectively antagonises metabotropic glutamate mGluR1 receptors. Bioorg Med Chem Lett. 1997;7:2777–2780. [Google Scholar]
- Colquhoun D, Neher E, Reuter H, Stevens CF. Inward current channels activated by intracellular Ca in cultured cardiac cells. Nature. 1981;294:752–754. doi: 10.1038/294752a0. [DOI] [PubMed] [Google Scholar]
- Congar P, Leinekugel X, Ben-Ari Y, Crepel V. A long-lasting calcium-activated nonselective cationic current is generated by synaptic stimulation or exogenous activation of group I metabotropic glutamate receptors in CA1 pyramidal neurons. J Neurosci. 1997;17:5366–5379. doi: 10.1523/JNEUROSCI.17-14-05366.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Crepel V, Aniksztejn L, Ben-Ari Y, Hammond C. Glutamate metabotropic receptors increase a Ca(2+)-activated nonspecific cationic current in CA1 hippocampal neurons. J Neurophysiol. 1994;72:1561–1569. doi: 10.1152/jn.1994.72.4.1561. [DOI] [PubMed] [Google Scholar]
- Delmas P, Wanaverbecq N, Abogadie FC, Mistry M, Brown DA. Signaling microdomains define the specificity of receptor-mediated InsP3 pathways in neurons. Neuron. 2002;34:209–220. doi: 10.1016/s0896-6273(02)00641-4. [DOI] [PubMed] [Google Scholar]
- Eckstein F, Cassel D, Levkovitz H, Lowe M, Selinger Z. Guanosine 5′-O-(2-thiodiphosphate). An inhibitor of adenylate cyclase stimulation by guanine nucleotides and fluoride ions. J Biol Chem. 1979;254:9829–9834. [PubMed] [Google Scholar]
- Gähwiler BH, Thompson SM, McKinney RA, Debanne D, Robertson RT. Organotypic slice cultures of neural tissue. In: Banker G, Goslin K, editors. Culturing Nerve Cells. Cambridge, MA, USA: MIT Press; 1998. pp. 461–498. [Google Scholar]
- Gasparini F, Kuhn R, Pin JP. Allosteric modulators of group I metabotropic glutamate receptors: novel subtype-selective ligands and therapeutic perspectives. Curr Opin Pharmacol. 2002;2:43–49. doi: 10.1016/s1471-4892(01)00119-9. [DOI] [PubMed] [Google Scholar]
- Gasparini F, Lingenhohl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Velicelebi G, Kuhn R. 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology. 1999;38:1493–1503. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- Gee CE, Gerber U. Comparison of ionic currents induced by activation of mGluRs in hippocampal CA1 and CA3 pyramidal cells, dentate granule cells and interneurons. Soc Neurosci Abstr. 2001;27:493. [Google Scholar]
- Guérineau NC, Bossu JL, Gähwiler BH, Gerber U. Activation of a nonselective cationic conductance by metabotropic glutamatergic and muscarinic agonists in CA3 pyramidal neurons of the rat hippocampus. J Neurosci. 1995;15:4395–4407. doi: 10.1523/JNEUROSCI.15-06-04395.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunthorpe MJ, Benham CD, Randall A, Davis JB. The diversity in the vanilloid (TRPV) receptor family of ion channels. Trends Pharmacol Sci. 2002;23:183–191. doi: 10.1016/s0165-6147(02)01999-5. [DOI] [PubMed] [Google Scholar]
- Haj-Dahmane S, Andrade R. Muscarinic activation of a voltage-dependent cation nonselective current in rat association cortex. J Neurosci. 1996;16:3848–3861. doi: 10.1523/JNEUROSCI.16-12-03848.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans E, Challiss RAJ. Structural, signalling and regulatory properties of the group I metabotropic glutamate receptors: prototypic family C G-protein coupled receptors. Biochem J. 2001;359:465–484. doi: 10.1042/0264-6021:3590465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuss C, Scanziani M, Gähwiler BH, Gerber U. G-protein-independent signaling mediated by metabotropic glutamate receptors. Nat Neurosci. 1999;2:1070–1077. doi: 10.1038/15996. [DOI] [PubMed] [Google Scholar]
- Hollmann MW, Wieczorek KS, Berger A, Durieux ME. Local anesthetic inhibition of G protein-coupled receptor signaling by interference with Gαq protein function. Mol Pharmacol. 2001;59:294–301. doi: 10.1124/mol.59.2.294. [DOI] [PubMed] [Google Scholar]
- Kapur A, Yeckel MF, Johnston D. Hippocampal mossy fiber activity evokes Ca2+ release in CA3 pyramidal neurons via a metabotropic glutamate receptor pathway. Neuroscience. 2001;107:59–69. doi: 10.1016/s0306-4522(01)00293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettunen P, Krieger P, Hess D, El Manira A. Signaling mechanisms of metabotropic glutamate receptor 5 subtype and its endogenous role in a locomotor network. J Neurosci. 2002;22:1868–1873. doi: 10.1523/JNEUROSCI.22-05-01868.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M, Offermanns S, Stocker M, Pedarzani P. Functional specificity of Gαq and G α11 in the cholinergic and glutamatergic modulation of potassium currents and excitability in hippocampal neurons. J Neurosci. 2002;22:666–673. doi: 10.1523/JNEUROSCI.22-03-00666.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmiski JB, MacVicar BA. Cyclic nucleotide-gated channels contribute to the cholinergic plateau potential in hippocampal CA1 pyramidal neurons. J Neurosci. 2001;21:8707–8714. doi: 10.1523/JNEUROSCI.21-22-08707.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lintschinger B, Balzer-Geldsetzer M, Baskaran T, Graier W, Romanin C, Zhu MX, Groschner K. Coassembly of TRP1 and TRP3 proteins generates diacylglycerol-and Ca2+-sensitive cation channels. J Biol Chem. 2000;275:27799–27805. doi: 10.1074/jbc.M002705200. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Mezey E, Toth ZE, Cortright DN, Arzubi MK, Krause JE, Elde R, Guo A, Blumberg PM, Szallasi A. Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc Natl Acad Sci U S A. 2000;97:3655–3660. doi: 10.1073/pnas.060496197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minke B, Cook B. TRP channel proteins and signal transduction. Physiol Rev. 2002;82:429–472. doi: 10.1152/physrev.00001.2002. [DOI] [PubMed] [Google Scholar]
- Mizuno N, Kitayama S, Saishin Y, Shimada S, Morita K, Mitsuhata C, Kurihara H, Dohi T. Molecular cloning and characterization of rat trp homologues from brain. Brain Res Mol Brain Res. 1999;64:41–51. doi: 10.1016/s0169-328x(98)00296-4. [DOI] [PubMed] [Google Scholar]
- Mori M, Gerber U. Slow feedback inhibition in the CA3 area of the rat hippocampus by synergistic synaptic activation of mGluR1 and mGluR5. J Physiol. 2002;544:793–799. doi: 10.1113/jphysiol.2002.030163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Inoue R, Yamazaki K, Maeda A, Kurosaki T, Yamakuni T, Tanaka I, Shimizu S, Ikenaka K, Imoto K, Mori Y. Molecular and functional characterization of a novel transient receptor potential protein homologue TRP7. J Biol Chem. 1999;274:27359–27370. doi: 10.1074/jbc.274.39.27359. [DOI] [PubMed] [Google Scholar]
- Okada T, Shimizu S, Wakamori M, Maeda A, Kurosaki T, Takada N, Imoto K, Mori Y. Molecular cloning and functional characterization of a novel receptor-activated TRP Ca2+ channel from mouse brain. J Biol Chem. 1998;273:10279–10287. doi: 10.1074/jbc.273.17.10279. [DOI] [PubMed] [Google Scholar]
- Philipp S, Hambrecht J, Braslavski L, Schroth G, Freichel M, Murakami M, Cavalié A, Flockerzi V. A novel capacitative calcium entry channel expressed in excitable cells. EMBO J. 1998;17:4274–4282. doi: 10.1093/emboj/17.15.4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp R, Englert HC, Lang HJ, Gögelein H. Inhibitors of nonselective cation channels in cells of the blood-brain barrier. EXS. 1993;66:213–218. doi: 10.1007/978-3-0348-7327-7_16. [DOI] [PubMed] [Google Scholar]
- Pozzo Miller LD, Petrozzino JJ, Connor JA. G protein-coupled receptors mediate a fast excitatory postsynaptic current in CA3 pyramidal neurons in hippocampal slices. J Neurosci. 1995;15:8320–8330. doi: 10.1523/JNEUROSCI.15-12-08320.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer M, Plant T, Obukhov AG, Hofmann T, Gudermann T, Schultz G. Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J Biol Chem. 2000;275:17517–17526. doi: 10.1074/jbc.275.23.17517. [DOI] [PubMed] [Google Scholar]
- Schaffhauser H, Cai Z, Hubalek F, Macek TA, Pohl J, Murphy TJ, Conn PJ. cAMP-dependent protein kinase inhibits mGluR2 coupling to G-proteins by direct receptor phosphorylation. J Neurosci. 2000;20:5663–5670. doi: 10.1523/JNEUROSCI.20-15-05663.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel BW, Wiech NL. RMI 12,330A: an inhibitor of cholera toxin induced intestinal hypersecretion which also inhibits adenylate cyclase activity. Gastroenterology. 1976;70:937–945. [Google Scholar]
- Strübing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron. 2001;29:645–655. doi: 10.1016/s0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- Tanabe M, Gähwiler BH, Gerber U. Effects of transient oxygen-glucose deprivation on G-proteins and G-protein-coupled receptors in rat CA3 pyramidal cells in vitro. Eur J Neurosci. 1998;10:2037–2045. doi: 10.1046/j.1460-9568.1998.00215.x. [DOI] [PubMed] [Google Scholar]
- Valenti O, Conn PJ, Marino MJ. Distinct physiological roles of the Gq-coupled metabotropic glutamate receptors co-expressed in the same neuronal populations. J Cell Physiol. 2002;191:125–137. doi: 10.1002/jcp.10081. [DOI] [PubMed] [Google Scholar]
- Van Rossum DB, Patterson RL, Ma H-T, Gill DL. Ca2+ entry mediated by store depletion, S-nitrosylation, and TRP3 channels. J Biol Chem. 2000;275:28562–28568. doi: 10.1074/jbc.M003147200. [DOI] [PubMed] [Google Scholar]
- Wong RK, Bianchi R, Taylor GW, Merlin LR. Role of metabotropic glutamate receptors in epilepsy. Adv Neurology. 1999;79:685–698. [PubMed] [Google Scholar]
- Woodhall G, Gee CE, Robitaille R, Lacaille JC. Membrane potential and intracellular Ca2+ oscillations activated by mGluRs in hippocampal stratum oriens/alveus interneurons. J Neurophysiol. 1999;81:371–382. doi: 10.1152/jn.1999.81.1.371. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Tang J, Tikunova S, Johnson JD, Chen Z, Qin N, Dietrich A, Stefani E, Birnbaumer L, Zhu MX. Activation of Trp3 by inositol 1, 4, 5-trisphosphate receptors through displacement of inhibitory calmodulin from a common binding domain. Proc Natl Acad Sci U S A. 2001;98:3168–3173. doi: 10.1073/pnas.051632698. [DOI] [PMC free article] [PubMed] [Google Scholar]