

Abstract
PSD-95 is one of the most abundant proteins found in the postsynaptic density of excitatory synapses. However, the precise functional role played by PSD-95 in regulating synaptic transmission and plasticity remains undefined. To address this issue, we have overexpressed PSD-95 in cortical pyramidal neurons in organotypic brain slices using particle-mediated gene transfer and assessed the consequences on synaptic transmission and plasticity. The AMPA receptor/NMDA receptor (AMPAR/NMDAR) ratio of evoked EPSCs recorded at +40 mV was greater in PSD-95-transfected pyramidal neurons than in controls. This difference could not be accounted for by a change in rectification of AMPAR-mediated synaptic currents since the current-voltage curves obtained in controls and in PSD-95-transfected neurons were indistinguishable. However, the amplitude of AMPAR-mediated evoked EPSCs was larger in PSD-95-transfected neurons compared to matched controls. Paired-pulse ratio analysis suggested that overexpression of PSD-95 did not alter presynaptic release probability. Transfection of PSD-95 was further accompanied by an increase in the frequency, but not amplitude, of AMPAR-mediated mEPSCs. Together, these results indicate that transfection of PSD-95 increased AMPAR-mediated synaptic transmission. Furthermore, they suggest that this phenomenon reflects an increased number of synapses expressing AMPARs rather than an increased number or function of these receptors at individual synapses. We tested the consequences of these changes on synaptic plasticity and found that PSD-95 transfection greatly enhanced the probability of observing long-term depression. These results thus identify a physiological role for PSD-95 and demonstrate that this protein can play a decisive role in controlling synaptic strength and activity-dependent synaptic plasticity.
Release of glutamate from synaptic terminals in the central nervous system elicits fast excitatory postsynaptic potentials by activating AMPA and NMDA receptors (AMPARs and NMDARs). Both these receptors are embedded within a dense protein matrix forming a distinct specialization known as the postsynaptic density (PSD; Sheng, 2001). PSD-95, a member of the MAGUK (membrane-associated guanylate kinase) superfamily of proteins, is one of the most abundant proteins in the PSD (Hunt et al. 1996). This protein is preferentially, although not exclusively, targeted to dendritic spines where it is thought to interact directly or indirectly with both NMDARs and AMPARs (Kornau et al. 1995, 1997; Tomita et al. 2001; Sheng, 2001).
Previous studies in cultured neurons have shown that PSD-95 is involved in synapse maturation (Okabe et al. 1999, 2001; El Husseini et al. 2000, 2002; Tomita et al. 2001; Prange & Murphy, 2001) while work on a PSD-95 transgenic mouse (Migaud et al. 1998) has suggested PSD-95 is involved in regulating synaptic plasticity at central synapses. Unfortunately, technical limitations in these experimental preparations have made it difficult to sort developmental effects of PSD-95 from more acute effects on synaptic function (El Husseini et al. 2000). To address this issue, we have taken advantage of particle-mediated gene transfer techniques to introduce PSD-95 into postnatal cultured brain slices. We find that short-term overexpression of PSD-95 dramatically changes the properties of cortico-cortical synapses by selectively facilitating AMPAR-mediated synaptic transmission and markedly increasing the ability of these synapses to express long-term depression (LTD).
Methods
Slice preparation and culturing
The procedures used for slice preparation were approved by the animal investigation committee of Wayne State University School of Medicine. Briefly, male Sprague-Dawley rats (P8-P12) were anaesthetized with halothane (by inhalation) and killed by decapitation. Slices containing prefrontal cortex were prepared as previously described (Haj-Dahmane & Andrade, 1996; Walker et al. 2000). Coronal slices (300-µm-thick) were cut in Ringer solution of standard composition (mm): 119 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 1 NaH2PO4, 26.2 NaHCO3 and 11 glucose, bubbled to saturation with 95 % O2-5 % CO2) and cultured essentially as described by Stoppini et al. (1991) except that glial cell line-derived neurotrophic factor (GDNF; 10 ng ml−1) was added to the culture media.
Preparation of cDNA and transfection
Four plasmids were used in the present study: PSD-95 subcloned in pcDNA3.1-, pEGFP-N1 (Clontech, Palo Alto, CA, USA), pDsRed2-N1 (Clontech) and a PSD-95-GFP fusion protein (PSD-95 subcloned in pEGFP-N1). Neuronal transfection was achieved using particle-mediated gene transfer methods using a Bio-Rad Helios gene gun. The procedure used was essentially as recommended by the manufacturer except for the use of a modified ‘gene gun barrel’ (O'Brien et al. 2001) which allowed for lower pressures and less gold particles per ‘bullet’ (half to a quarter of the manufacturer's recommendation). Cortical slices were transfected (50–100 p.s.i. at 3–5 mm distance) with the appropriate set of ‘bullets’ within 2 h after cutting.
Electrophysiology and imaging
Pyramidal neurons of the medial prefrontal cortex were targeted for whole-cell patch-clamp recording using differential interference contrast imaging on a fixed-stage upright microscope (Olympus, BX50WI). Targeted pyramidal neurons were restricted to layers III and V, which in this region are adjacent to each other. A small number of recordings were also obtained from comparable cells in the middle layers of somatosensory cortex. Since no obvious differences were observed between cells in these two areas, results were pooled for analysis. All recordings were obtained 2–3 days after transfection.
EGFP (enhanced green fluorescent protein)-tagged PSD-95 was used in a small number of imaging experiments aimed at ascertaining the locus of expression of PSD-95 (Fig. 1A). Since PSD-95 is a multifunctional protein containing multiple protein-protein interaction domains, we carried out the electrophysiological recordings in neurons cotransfected with untagged PSD-95 and EGFP (PSD-95/EGFP) in order to avoid any possible impact of the GFP tag on PSD-95 function. Control experiments using β-galactosidase and EGFP as independent reporters indicated that the vast majority of transfected cells (> 95 %) identified by the expression of GFP also express the cotransfected gene; see supplementary information at:
Figure 1. Expression of PSD-95 in pyramidal neurons of the prefrontal cortex increases the AMPAR/NMDAR ratio.
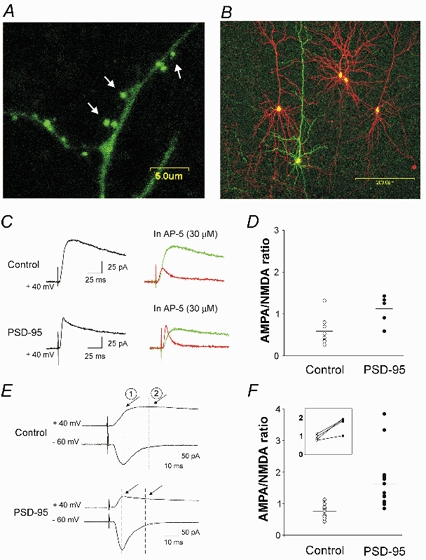
A, confocal image taken after 2 days in vitro illustrating the preferential targeting of PSD-95-GFP fusion protein to dendritic spines (see arrows). B, illustration of the experimental preparation used for these experiments. Prefrontal cortex pyramidal neurons in this slice were transfected with PSD-95/EGFP (green) and (in some cases) with pcDNA3.1/DsRed (red) to serve as within-slice transfected controls. C, current traces recorded at +40 mV from neurons transfected with pcDNA3.1/EGFP (control) or PSD95/EGFP, before (black trace) and after (red trace) administration of 30 µm (d)-AP-5. The green traces depict the NMDAR (AP-5-sensitive) component of the evoked current obtained by subtraction. D, computed AMPAR/NMDAR ratios obtained from PSD-95-transfected neurons (n = 6) and controls (5 cells transfected with pcDNA3.1/EGFP and 3 untransfected neurons; P < 0.02). E, current traces recorded at −60 and +40 mV from a control (pcDNA3.1/EGFP) and from a PSD-95-transfected neuron. By superimposing the traces, the AMPAR (1) and NMDAR (2) components of the traces at +40 mV were estimated. F, computed AMPAR/NMDAR ratios obtained using this method for PSD-95-transfected (n = 14) and control neurons (6 untransfected cells and 7 cells transfected with empty plasmid (i.e. pcDNA3.1/EGFP; sister slice comparisons, n = 5; or pcDNA3.1/DsRed; same slice comparisons, n = 2). A subset of these experiments involving pairwise sequential recordings from neighbouring neurons is re-plotted in the inset. PSD-95 transfection increased the AMPAR/NMDAR ratio (P < 0.01 for pooled data and P < 0.03 for pairwise recordings).
http://www.jphysiol.org/cgi/content/full/546/3/859
PSD-95/EGFP-transfected neurons were compared to three different types of control groups. The first involved untransfected neurons. The second involved neurons in sister slices transfected with pcDNA3.1 (empty vector)/EGFP (to control for the transfection procedure). The third control group involved neurons in the same slice transfected with pcDNA3.1 (empty vector)/DsRed (red fluorescent protein) (the emission spectra difference between EGFP and DsRed allowed discrimination between the two populations of transfected neurons in the same slice). Unfortunately, the yield from the latter approach was low and thus impractical to use for all experiments. Since no electrophysiological differences between these control groups were detected, results were pooled. While in the text we collectively refer to these groups as ‘controls’, the number of cells from each type of control is specified in the figure legends. Some of the untransfected control neurons were filled with the fluorescent dye Alexa Fluor 568 hydrazide (Molecular Probes, Eugene, OR, USA; 250 µm in the tip of the electrode) to assure comparable dendritic arborization. The maximal distance between two neurons in the experiments carried out in close, neighbouring neurons (referred to as pairwise or triad recordings) was of ≈300 µm.
Whole-cell recordings were carried out with an Axopatch 200 amplifier (Axon Instruments). All recordings were filtered at 2 kHz, digitized at 10 kHz and acquired with Clampex (Axon Instruments). The recording pipettes were pulled from borosilicate glass (1.2 mm outer diameter) using a Flaming/ Brown horizontal puller (Model P97, Sutter Instrument Co., Novato, CA, USA). Liquid junction potentials were measured to be 10–11 mV and voltages were left uncompensated except for the determination of the AMPAR current-voltage (I-V) curves. A bipolar stimulation electrode was placed 50–150 µm away from the apical dendrite of the recorded neuron and 100–200 µm towards the pial surface to activate excitatory afferents. The intracellular solution for all experiments (except those depicted in Fig. 6) contained (mm): 77 caesium gluconate, 10 tetracaesium BAPTA, 5 TEA-Cl, 3 CaCl2, 10 Hepes, 4 ATP, 0.5 GTP, 5 QX-314 and 10 sodium phoshocreatine. The pH was adjusted to 7.3 and sucrose was added to reach 280 mosmol l−1. The Ringer solution was supplemented with 30 µm (-)bicuculline and 12 mm each of MgSO4 and CaCl2 (Sah & Nicoll, 1991) to block GABAA synaptic potentials, block epileptiform activity and minimize polysynaptic responses.
Figure 6. PSD-95 overexpression favours the emergence of LTD.
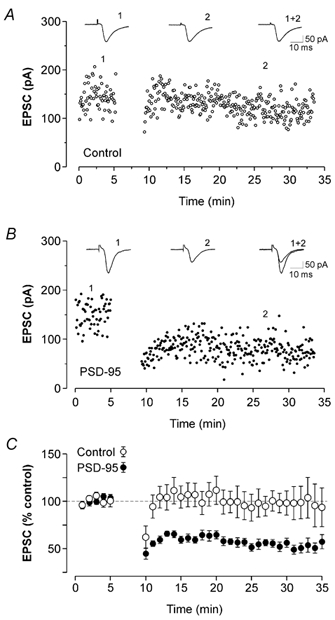
A, in this untransfected control neuron, pairing presynaptic stimulation and postsynaptic depolarization did not induce any significant change in synaptic strength. Each data point represents the amplitude of EPSCs recorded at −70 mV. B, in a PSD-95-transfected cell, the same pairing protocol resulted in the appearance of robust LTD. C, ensemble average plot for the experiments illustrated in panels A and B. The pairing protocol did not induce any significant changes in EPSC amplitudes in control cells (○; n = 10; 5 untransfected cells recorded in a sequential pairwise fashion with a PSD-95-transfected neurons, 3 cells transfected with pcDNA3.1/EGFP and 2 untransfected cells). In contrast, the same pairing protocol induced a significant and lasting reduction of EPSCs amplitude in PSD-95-transfected neurons (•; n = 12; P < 0.01).
For the experiments designed to examine synaptic plasticity, we used control Ringer solution and an intracellular solution of the following composition (mm): 129 potassium MeSO4, 5 KCl, 1 NaCl, 1 MgCl2, 0.02 EGTA, 10 Hepes, 4 ATP, 0.5 GTP and 10 sodium phosphocreatine. This solution brings the reversal potential for chloride to the holding potential for these experiments and was designed to avoid GABAA contamination of the evoked EPSCs. Excitatory inputs were stimulated at 0.2 Hz while holding the cell at −70 mV. The pairing protocol was started 6–8 min after break-in and involved holding the cell at 0 mV for 50 pulses without changing the frequency of stimulation. Stimulus intensity was adjusted to produce robust, presumably monosynaptic, EPSCs of similar amplitudes between control and transfected neurons (baseline EPSCs: 66 ± 10 and 71 ± 10 pA in control and PSD-95-transfected neurons, respectively). Access resistance was continuously monitored during the experiments by delivering a 5 mV hyperpolarizing voltage step (100 ms duration; 100 ms after the delivery of the stimulus pulse).
Miniature EPSCs (mEPSCs) were acquired from every neuron for 5 min at −70 mV in control Ringer solution containing 30 µm (-)bicuculline and 30 µm (d)-AP-5 ((d-)-2-amino-5-phosphonovaleric acid). Analysis was carried out using Mini Analysis Program (version 5.2; Synaptosoft, Leonia, NJ, USA). The average amplitude and frequency of mEPSCs was first determined for each recording and then averaged together according to the experimental group. Likewise, for the ensemble cumulative distribution plots for both the amplitude and frequency (i.e. interevent-intervals) of mEPSCs, distribution plots were first determined for each recording and then averaged together according to the experimental group. Only events greater than 7.5 pA were included in the analysis, which corresponds to the event detection limit used for the noisiest recording analysed. Statistical significance was assessed using the Student's unpaired t test.
Results
The particle-mediated gene transfer technique was used to overexpress PSD-95 in cortical pyramidal neurons maintained in organotypic slices. As illustrated in Fig. 1A, and consistent with previous work by others (Marrs et al. 2001), transfection of PSD-95-GFP results in robust expression and preferential targeting of the protein to dendritic spines. We took advantage of these observations to examine the functional role played by PSD-95 in regulating cortical synaptic function by overexpressing the native form of the protein in pyramidal neurons of prefrontal cortex (Fig. 1B).
PSD-95 increases the AMPAR/NMDAR ratio of excitatory postsynaptic currents
Both AMPARs and NMDARs mediate the postsynaptic effects of glutamate at central synapses (Nicoll et al. 1990). Therefore, we examined the effect of overexpressing PSD-95 on the AMPAR/NMDAR ratio of excitatory postsynaptic currents (EPSCs) evoked by stimulation of cortical afferents. Pyramidal neurons were held at +40 mV under voltage-clamp, and bath administration of (d)-AP-5 (30 µm; Fig. 1C) was used to identify, by subtraction, the NMDAR component (Hsia et al. 1998; Ungless et al. 2001). The (d)-AP-5-insensitive component of the EPSC was blocked by 30 µm CNQX (not shown) and was thus taken as a measure of the AMPAR component. As illustrated in Fig. 1C and D, the AMPAR/NMDAR ratio was significantly increased in PSD-95-transfected neurons (n = 6) compared to controls (untransfected cells or cells transfected with empty vector/EGFP, P < 0.02, Student's unpaired t test; n = 8).
A potential limitation of this experiment is that it relies on cross slice comparisons. Since the NMDAR component of the EPSC is much slower than the AMPAR component (Edmonds et al. 1995) this property can be used to estimate the AMPAR/NMDAR ratio (Fig. 1E) from neighbouring cells recorded sequentially. Therefore we next conducted pairwise comparisons of PSD-95-transfected cells and control cells within single slices. This approach again showed that the AMPAR/NMDAR ratio was greater in PSD-95-transfected neurons than in controls (n = 4 paired sequential recordings; inset of Fig. 1F; P < 0.03). Similar results were obtained using this method in a larger sample of unpaired cells (Fig. 1E and F; 13 PSD-95-transfected neurons and 14 controls; P < 0.01, Student's unpaired t test). Combined, these results indicate that overexpression of PSD-95 leads to a marked increase in the AMPAR/NMDAR ratio at cortical synapses.
Since AMPARs lacking GluR2 subunits show inward rectification (Verdoorn et al. 1991), a change in subunit composition could, in principle, have been responsible for the increase in AMPAR/NMDAR ratio observed at +40 mV. However, as illustrated in Fig. 2A and B, the current-voltage (I-V) relationship of the AMPAR-EPSC for both PSD-95-transfected (n = 5) and control (n = 7) neurons did not differ. Thus, the observed change in the AMPAR/NMDAR ratio could not be explained simply by a change in channel rectification. Rather, it pointed to a change in the AMPAR and/or NMDAR component of the EPSC.
Figure 2. PSD-95 does not change the rectification properties of AMPAR-mediated synaptic currents.
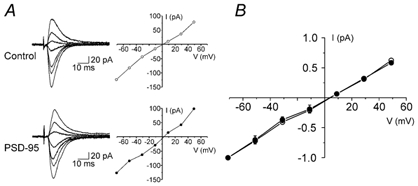
A, current traces and I-V relationships for AMPAR-mediated EPSCs in a control (non-transfected) and a PSD-95-transfected cell. B, average I-V relationships for AMPAR-mediated EPSCs in PSD-95-transfected (n = 5) and control neurons (n = 7; 6 untransfected neurons and 1 neuron transfected with pcDNA3.1/EGFP). The experiments were carried out in the presence of 30 µm (d)-AP-5. Data were normalized for every cell to the amplitude of the synaptic current obtained at −71 mV.
PSD-95 increases the AMPAR/NMDAR ratio by increasing the AMPAR-mediated component of the evoked EPSCs
PSD-95 could increase the AMPAR/NMDAR ratio by increasing the AMPAR component of the synaptic response, by reducing the NMDAR component, or a combination of both of these effects. To distinguish between these possibilities, we directly assessed AMPAR-mediated synaptic currents in neighbouring PSD-95-transfected and control neurons using sequential recordings while holding stimulus intensity constant (Fig. 3A). To control for spatial relationship with respect to the stimulation electrode, we recorded from PSD-95 transfected neurons that were spatially flanked by one (paired sequential recordings) or two (triad sequential recordings) control cells (Fig. 3A and B). Some control cells were transfected with DsRed (n = 2) or filled with Alexa 568 (n = 7) dye to ascertain comparable dendritic arborizations. As illustrated in Fig. 3B and C, transfection of PDS-95 induced a significant increase in the amplitude of AMPAR-mediated EPSCs when compared to matched controls in the same slice (P < 0.02, Student's unpaired t test).
Figure 3. PSD-95 increases the amplitude of AMPAR-mediated EPSCs.
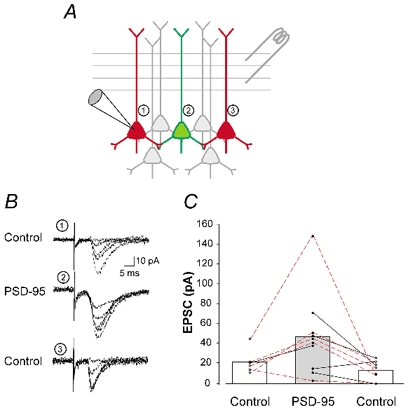
A, illustration of the experimental approach used for the experiments depicted in B and C. Evoked AMPAR-mediated EPSCs recorded from a PSD-95-transfected neuron (green) were compared to those recorded from 2 control neurons (red), spatially bracketing the transfected cell. B, superimposed individual current traces from 3 neurons in one of the triads illustrated in C. C, amplitude of EPSCs recorded from control (n = 13 untransfected and n = 2 pcDNA3.1/DsRed-transfected) and PSD-95-transfected neurons (n = 10). The data are plotted as a graph (control-PSD-95-control) so as to illustrate the spatial organization in which the recordings were obtained (as depicted in A). Triad recordings (n = 5) are outlined by the red connecting lines. In some cases, only paired recordings could be obtained (n = 5; black lines). PSD-95-transfection resulted in a significant increase in the amplitude of AMPAR-mediated evoked EPSCs (P < 0.05 for triads and P < 0.02 for the pooled data (triad and pairwise recordings).
Overall, transfection of PSD-95 resulted in a 2.7-fold increase in the AMPAR-mediated synaptic current. This change alone quantitatively accounts for the ≈2-fold increase in the AMPAR/NMDAR ratio. As such, these results suggest that PSD-95 induced little or no change in the NMDAR component of the synaptic response. Consistent with this idea, we failed to detect any change in evoked NMDAR currents recorded at +40 mV using sequential pairwise recordings (23 ± 9 pA in controls vs. 26 ± 14 pA in PSD-95-transfected neurons, n = 5; P = 0.87, Student's unpaired t test). These results indicated that the principal effect of PSD-95 is to increase the AMPAR component of the evoked EPSC.
PSD-95 increases AMPAR-mediated synaptic responses by increasing the fraction of synapses expressing AMPARs
Excitatory glutamate synapses are thought to exist in two forms; one expresses both AMPARs and NMDARs and the second, termed ‘silent’ synapses, expresses only NMDARs (Isaac et al. 1995; Liao et al. 1995). Previous studies in dissociated cell culture have suggested that PSD-95 can transynaptically facilitate neurotransmitter release (El Husseini et al. 2000). Therefore the results obtained thus far could be explained by a selective increase in release probabilities at synapses containing both AMPARs and NMDARs. We thus examined whether PSD-95 overexpression altered the probability of release at AMPAR-expressing synapses. To that effect, we carried out paired-pulse ratio analysis, a sensitive index of presynaptic release probability (Regehr & Stevens, 2001). As illustrated in Fig. 4, the paired-pulse ratio obtained in PSD-95-transfected neurons (n = 4) did not differ from that observed in controls (n = 4) at any of the intervals tested (Fig. 4; P > 0.5, Student's unpaired t test). These results indicated that a change in release probability is unlikely to account for the effects of PSD-95 on AMPAR-mediated synaptic transmission.
Figure 4. PSD-95 does not alter presynaptic release probability.
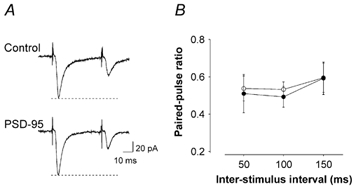
A, current traces from a control (pcDNA3.1/EGFP) and from a PSD-95-transfected neuron illustrating the response to two successive stimulations (50 ms interval). B, paired-pulse ratios calculated at three distinct inter-stimulus intervals (ISI) were not different between controls (n = 4; 3 cells transfected with pcDNA3.1/EGFP and 1 untransfected neuron) and PSD-95-transfected neurons (n = 4; P > 0.5 for all intervals).
PSD-95 could alternatively increase the amplitude of evoked EPSCs by increasing the number or function of AMPARs at individual synapses. To test this possibility we examined quantal AMPAR-mediated synaptic currents recorded in TTX. As illustrated in Fig. 5A-C, we did not observe any significant differences in the amplitude of AMPAR-mediated mEPSCs between control (n = 8) and PSD-95-transfected (n = 6) neurons (P = 0.81, Student's unpaired t test). These results indicate that, within the limits of detection of this approach, PSD-95 did not increase the number or function of AMPARs at individual synapses.
Figure 5. PSD-95 increases the frequency, but not the amplitude of AMPAR mEPSCs.
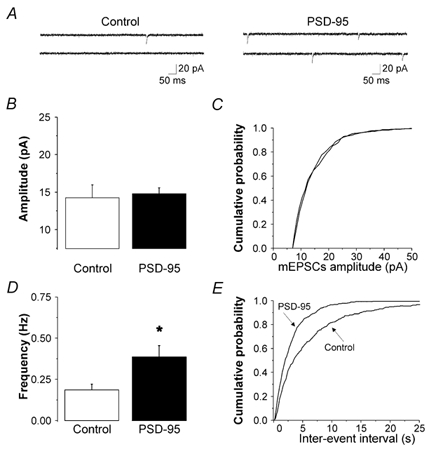
A, current traces (recorded in the presence of TTX, bicuculline and (d)-AP-5) from a control (untransfected) cell and a PSD-95-transfected cell. B, PSD-95 (n = 6) did not induce any significant change in the amplitude of mEPSC (n = 8 untransfected control). C, average cumulative probability plot for mEPSC amplitudes in all PSD-95-transfected and control neurons. D, PSD-95 resulted in a significant increase in the frequency of mEPSCs (P < 0.02). E, average cumulative probability plots for the mEPSC inter-event intervals in all PSD-95-transfected and control neurons.
Finally, PSD-95 could increase AMPAR-mediated synaptic responses by increasing the number of AMPAR-expressing synapses. Consistent with this idea, we observed a marked increase in the frequency of AMPAR-mediated mEPSCs in PSD-95-transfected neurons compared to controls (Fig. 5A, D and E: P < 0.02, Student's unpaired t test). Overall we detected a 2.1-fold increase in AMPAR-mediated mEPSC frequency in PSD-95-transfected neurons. This effect, in principle, could quantitatively account for the increase in the AMPAR-mediated synaptic current (Fig. 3) as well as for the increase in the AMPAR/NMDAR ratio (Fig. 1). Thus, these results suggest that most of the effect of PSD-95 on AMPAR synaptic transmission can be accounted for by an increase in the number of AMPAR-expressing synapses. These results, as an ensemble, can thus be interpreted most parsimoniously as indicating that transfection of PSD-95 facilitated AMPAR synaptic transmission by increasing the fraction of synapses expressing AMPARs.
PSD-95 favours the emergence of LTD
Previous studies have assigned a key role to silent synapses in synaptic plasticity. Therefore we next examined whether overexpression of PSD-95 would alter the ability of these synapses to express synaptic plasticity. To test this idea, we used a standard pairing protocol that yielded little or no change in the amplitude of EPSCs in control cells (Fig. 6A and C; 97 ± 2 % of baseline; calculated from a 5 min period 20 min after the onset of pairing, n = 10 cells). Surprisingly, when this normally innocuous protocol was applied to PSD-95-transfected cells, the amplitude of the EPSCs was markedly depressed following the pairing (Fig. 6B and C; 52 ± 2 % of baseline; n = 12; P < 0.01). The ability of the pairing protocol to induce LTD in PSD-95-transfected cells was abolished by (d)-AP-5 (30 µm; n = 6; not shown) indicating that it was dependent on NMDAR activation. These results indicate that transfection of PSD-95 facilitated the appearance of NMDAR-dependent LTD at cortical synapses.
Discussion
In the present study, we used an overexpression strategy in organotypic brain slice cultures using the particle-mediated gene transfer technique to examine the role of PSD-95 in regulating synaptic transmission and synaptic plasticity in cortical neurons. We found that PSD-95 transfection greatly facilitates AMPAR-mediated synaptic transmission while favouring the emergence of LTD.
PSD-95 overexpression in cortical pyramidal cells induced an increase in the AMPAR/NMDAR ratio of EPSCs recorded at +40 mV. This effect of PSD-95 could not be accounted for by altered rectification of AMPAR channels or by a selective change in release probabilities at AMPAR-containing synapses. Rather, this increase in AMPAR/ NMDAR ratio reflected a selective increase in the AMPAR component of the synaptic response. Furthermore, the frequency, but not the amplitude, of AMPAR-mediated mEPSCs was enhanced in neurons transfected with PSD-95. As such, these observations suggest that the increase in AMPAR-mediated synaptic function induced by transfection with PSD-95 did not involve an increase in AMPAR number or function at individual synapses but, instead, reflected an increase in the number of AMPAR-expressing synapses. A likely scenario that would account for these observations is that overexpressing PSD-95 led to an increase of the fraction of synapses expressing AMPARs, at the expense of the ‘silent’ synapse population. Such a role is consistent with previous morphological studies showing that PSD-95 is associated with the conversion of transient filipodia-like spines, the probable morphological correlates of silent synapses (Takumi et al. 1999; Matsuzaki et al. 2001), into stable spines (Okabe et al. 1999; Marrs et al. 2001; Okabe et al. 2001; Prange & Murphy, 2001).
Transfection of cortical pyramidal neurons with PSD-95 increased AMPAR-mediated synaptic transmission while having no detectable effect on NMDAR function. This is a surprising result in view of the extensive evidence linking PSD-95 and NMDARs (Kornau et al. 1995, 1997; Kennedy, 1997; Craven & Bredt, 1998). These observations, however, are consistent with the failure of a targeted disruption of PSD-95 to affect NMDAR function in transgenic animals (Migaud et al. 1998). They are also consistent with the previous observations that NMDAR synaptic clustering can form independently of PSD-95 (Washbourne et al. 2002) and that overexpression of PSD-95 fails to alter NMDAR clustering in cultured neurons (El Husseini et al. 2000).
A previous study in which PSD-95 was overexpressed in dissociated embryonic hippocampal neurons (El Husseini et al. 2000) reported that PSD-95 had multiple effects on synapses including increases in: presynaptic release probability, the level of expression of AMPAR at individual synapses and the total number of AMPAR containing synapses. As such, these results contrast and converge with the present findings. Differences, however, are perhaps not unexpected given the use of very different experimental preparations. In the earlier work, embryonic neurons were transfected during dissociation and examined 11–21 days later, thus ensuring overexpression of PSD-95 throughout the re-establishment of synaptic connections. This allowed for an examination of the effects of PSD-95 on the development of synapses and led the authors to conclude that PSD-95 promoted the maturation of synapses (El Husseini et al. 2000). In the present study, PSD-95 was transfected into postnatal brain slices (P8-P12) where synaptogenesis is substantially advanced. This made it possible to study the more acute effects of PSD-95 and allowed us to identify the regulation of AMPAR expression at synapses as one of the proximal actions of PSD-95. It will be of interest to understand the mechanisms that underlie the differences between the present observations in slices and those of earlier studies in dissociated cultures. Such understanding may help dissect differences and commonalities in the roles played by PSD-95 during synaptic development (El Husseini et al. 2000) and synaptic transmission at mature synapses.
The use of an organotypic brain slice preparation allowed us to study the role of PSD-95 on activity-dependent synaptic plasticity. Transfection with PSD-95 favoured the emergence of LTD. These results are consistent with the previous observation that LTP was enhanced in transgenic mice expressing a targeted disruption of PSD-95 (Migaud et al. 1998). The exact mechanism by which PSD-95 regulates synaptic plasticity however is unclear. It is possible that the ‘upregulated’ state of the synapse population triggered by transfection with PSD-95 would be more amenable to depression (or depotentiation). This possibility is consistent with recent studies indicating that the propensity to synaptic depression is dependent on the state and history of the synapse (Montgomery & Madison, 2002). Alternatively, it is possible that PSD-95 may favour LTD by directly facilitating its intracellular signalling pathway. Distinguishing between these, and other, possibilities will need to await a molecular level understanding of PSD-95 function.
In conclusion, the results presented here show that transfection of pyramidal neurons with PSD-95, one of the most abundant proteins found in the PSD, increases AMPAR function and favours the emergence of LTD at cortico-cortical synapses. As such, these results demonstrate that PSD-95 can play a decisive role in controlling synaptic strength and activity-dependent synaptic plasticity.
Acknowledgments
We would like to thank Dr Roger A. Nicoll for helpful discussions, Drs G. Kapatos and S. Haj-Dahmane for critically reading earlier versions of the manuscript, P. D. Perring, Lj. Mladenovic and A. Andrade for expert technical assistance and Dr Vivian Budnik for kindly providing the PSD-95 cDNA. The present study was supported by grant MH43985 from the National Institutes of Health (R.A.). J.C.B. is in receipt of a postdoctoral fellowship from the Canadian Institutes for Health Research (CIHR).
Supplementary material
The online version of this paper can be found at:
http://www.jphysiol.org/cgi/content/full/546/3/859
and contains material entitled:
Coexpression of EGFP and β-gal by neurons in cultured cortical slices
‘Bullets’ were coated with EGFP and β-gal in pcDNA3 at a 1:1 ratio. After 2 days in culture, slices were processed for immunocytochemistry using anti-β-gal and Cy3 as a fluorochrome. We examined 110 GFP+ cells and then assessed whether these cells were also β-gal+. We then examined 151 β-gal+ cells and determined whether these cells were GFP+. There was an almost perfect correspondence between β-gal+ and GFP+ cells.
References
- Craven SE, Bredt DS. PDZ proteins organize synaptic signaling pathways. Cell. 1998;93:495–498. doi: 10.1016/s0092-8674(00)81179-4. [DOI] [PubMed] [Google Scholar]
- Edmonds B, Gibb AJ, Colquhoun D. Mechanisms of activation of glutamate receptors and the time course of excitatory synaptic currents. Annu Rev Physiol. 1995;57:495–519. doi: 10.1146/annurev.ph.57.030195.002431. [DOI] [PubMed] [Google Scholar]
- El Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- El Husseini AE, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier-Campbell C, Aguilera-Moreno A, Nicoll RA, Bredt DS. Synaptic strength regulated by palmitate cycling on PSD-95. Cell. 2002;108:849–863. doi: 10.1016/s0092-8674(02)00683-9. [DOI] [PubMed] [Google Scholar]
- Haj-Dahmane S, Andrade R. Muscarinic activation of a voltage-dependent cation nonselective current in rat association cortex. J Neurosci. 1996;16:3848–3861. doi: 10.1523/JNEUROSCI.16-12-03848.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia AY, Malenka RC, Nicoll RA. Development of excitatory circuitry in the hippocampus. J Neurophysiol. 1998;79:2013–2024. doi: 10.1152/jn.1998.79.4.2013. [DOI] [PubMed] [Google Scholar]
- Hunt CA, Schenker LJ, Kennedy MB. PSD-95 is associated with the postsynaptic density and not with the presynaptic membrane at forebrain synapses. J Neurosci. 1996;16:1380–1388. doi: 10.1523/JNEUROSCI.16-04-01380.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Kennedy MB. The postsynaptic density at glutamatergic synapses. Trends Neurosci. 1997;20:264–268. doi: 10.1016/s0166-2236(96)01033-8. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Seeburg PH, Kennedy MB. Interaction of ion channels and receptors with PDZ domain proteins. Curr Opin Neurobiol. 1997;7:368–373. doi: 10.1016/s0959-4388(97)80064-5. [DOI] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Marrs GS, Green SH, Dailey ME. Rapid formation and remodeling of postsynaptic densities in developing dendrites. Nat Neurosci. 2001;4:1006–1013. doi: 10.1038/nn717. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Montgomery JM, Madison DV. State-dependent heterogeneity in synaptic depression between pyramidal cell pairs. Neuron. 2002;33:765–777. doi: 10.1016/s0896-6273(02)00606-2. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC, Kauer JA. Functional comparison of neurotransmitter receptor subtypes in mammalian central nervous system. Physiol Rev. 1990;70:513–565. doi: 10.1152/physrev.1990.70.2.513. [DOI] [PubMed] [Google Scholar]
- O'Brien JA, Holt M, Whiteside G, Lummis SC, Hastings MH. Modifications to the hand-held Gene Gun: improvements for in vitro biolistic transfection of organotypic neuronal tissue. J Neurosci Methods. 2001;112:57–64. doi: 10.1016/s0165-0270(01)00457-5. [DOI] [PubMed] [Google Scholar]
- Okabe S, Kim HD, Miwa A, Kuriu T, Okado H. Continual remodeling of postsynaptic density and its regulation by synaptic activity. Nat Neurosci. 1999;2:804–811. doi: 10.1038/12175. [DOI] [PubMed] [Google Scholar]
- Okabe S, Miwa A, Okado H. Spine formation and correlated assembly of presynaptic and postsynaptic molecules. J Neurosci. 2001;21:6105–6114. doi: 10.1523/JNEUROSCI.21-16-06105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prange O, Murphy TH. Modular transport of postsynaptic density-95 clusters and association with stable spine precursors during early development of cortical neurons. J Neurosci. 2001;21:9325–9333. doi: 10.1523/JNEUROSCI.21-23-09325.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Stevens CF. Physiology of synaptic transmission and short-term plasticity. In: Cowan WM, Südhof T, Stevens CF, editors. Synapses. Baltimore: The Johns Hopkins University Press; 2001. pp. 135–175. [Google Scholar]
- Sah P, Nicoll RA. Mechanisms underlying potentiation of synaptic transmission in rat anterior cingulate cortex in vitro. J Physiol. 1991;433:615–630. doi: 10.1113/jphysiol.1991.sp018446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M. Molecular organization of the postsynaptic specialization. Proc Natl Acad Sci U S A. 2001;98:7058–7061. doi: 10.1073/pnas.111146298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Takumi Y, Ramirez-Leon V, Laake P, Rinvik E, Ottersen OP. Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nat Neurosci. 1999;2:618–624. doi: 10.1038/10172. [DOI] [PubMed] [Google Scholar]
- Tomita S, Nicoll RA, Bredt DS. PDZ protein interactions regulating glutamate receptor function and plasticity. J Cell Biol. 2001;153:F19–24. doi: 10.1083/jcb.153.5.f19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Verdoorn TA, Burnashev N, Monyer H, Seeburg PH, Sakmann B. Structural determinants of ion flow through recombinant glutamate receptor channels. Science. 1991;252:1715–1718. doi: 10.1126/science.1710829. [DOI] [PubMed] [Google Scholar]
- Walker PD, Andrade R, Quinn JP, Bannon MJ. Real-time analysis of preprotachykinin promoter activity in single cortical neurons. J Neurochem. 2000;75:882–885. doi: 10.1046/j.1471-4159.2000.0750882.x. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Bennett JE, McAllister AK. Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci. 2002;5:751–759. doi: 10.1038/nn883. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.