

Abstract
Chronic hypoxia caused by migration of native sea-level dwellers to high altitude or chronic lung disease leads to the development of increased pulmonary vascular resistance and pulmonary hypertension. This altitude-induced hypertension offers no obvious benefit and may indeed be maladaptive. A major mechanism thought to contribute to the development of pulmonary hypertension is hypoxia-induced loss of small blood vessels, sometimes termed rarefaction or pruning. More recent evidence caused us to question this widely accepted concept including the potent angiogenic effect of chronic hypoxia in all other vascular beds and the demonstration that new vessels can form in the pulmonary circulation when stimulated by chronic infection and lung resection. We tested the hypothesis that chronic environmental hypoxia causes angiogenesis in the adult pulmonary circulation by using stereological techniques combined with confocal microscopy to examine the resultant changes in pulmonary vascular structure in rats. We found that chronic hypoxia resulted in increased total pulmonary vessel length, volume, endothelial surface area and number of endothelial cells in vivo. This is the first reported demonstration of hypoxia-induced angiogenesis in the mature pulmonary circulation, a structural adaptation that may have important beneficial consequences for gas exchange. These findings imply that we must revise the widely accepted paradigm that hypoxia-induced loss of small vessels is a key structural change contributing to the development of pulmonary hypertension in high altitude adaptation and chronic lung disease.
Chronic hypoxia caused by migration of native sea-level dwellers to high altitude or chronic lung disease leads to the development of increased pulmonary vascular resistance and pulmonary hypertension (Heath et al. 1973; Rabinovitch et al. 1979; Grover et al. 1983; Meyrick & Reid, 1983; Fishman, 1985; Jones & Reid, 1995; Hopkins & McLoughlin, 2003). This altitude-induced hypertension offers no obvious benefit and may indeed be maladaptive. When sustained pulmonary arterial hypertension complicates the course of chronic lung diseases, it increases morbidity and reduces survival (Skwarski et al. 1991; MacNee, 1994a,b; Incalzi et al. 1999). During the early period of hypoxic exposure, pulmonary vascular resistance is elevated largely due to hypoxic vasoconstriction. However, following sustained exposure to hypoxia, it is thought that structural changes in the pulmonary vascular bed become the major determinant of elevated vascular resistance (Sime et al. 1971; Lockhart et al. 1976; Fried et al. 1983; Grover et al. 1983; Fishman, 1985).
The structural changes that are thought to underlie the increased vascular resistance can be broadly classified into two processes: firstly, remodelling of the walls of the pulmonary resistance vessels and, secondly, a reduction in the total number of blood vessels in the lung (Ryland & Reid, 1975; Hislop & Reid, 1976; Meyrick & Reid, 1978; Rabinovitch et al. 1979). Remodelling results in thickening of the arterial wall and is said to increase resistance by causing the vessel walls to encroach into the lumen and reduce its diameter. These structural changes include muscularisation of previously non-muscular arterioles, increased medial thickness of previously partially and completely muscular arterioles, adventitial hypertrophy and deposition of additional matrix components, including collagen and elastin, in the vascular walls (Rabinovitch et al. 1979; Grover et al. 1983; Fishman, 1985; Stenmark & Mecham, 1997; Rabinovitch, 1999, 2001). The second major structural alteration caused by chronic hypoxia is loss of small blood vessels, sometimes termed rarefaction or pruning, which is said to increase vascular resistance by reducing the extent of parallel vascular pathways (Hislop & Reid, 1976, 1977; Rabinovitch et al. 1979; Meyrick& Reid, 1983; Jones & Reid 1995; Partovian et al. 2000). Thus, it is generally thought that rarefaction is an important component of the structural basis for chronic hypoxic pulmonary hypertension.
More recent evidence caused us to question this widely accepted paradigm. The reported hypoxia-induced loss of blood vessels in the pulmonary circulation is in puzzling contrast to the marked angiogenesis observed in the systemic circulation in hypoxia, a clearly beneficial adaptive response (LaManna et al. 1992; Marti & Risau, 1999; Smith & Marshall, 1999; Griffioen & Molema, 2000). The specific vascular endothelial mitogen, vascular endothelial growth factor (VEGF), and its receptors are extensively expressed in the lung (Monacci et al. 1993; Tuder et al. 1995; Christou et al. 1998; Marti & Risau, 1998) and VEGF has an essential homeostatic role in maintaining the normal pulmonary vasculature, demonstrating that the endothelium of the pulmonary circulation responds to this cell-specific mitogen (Kasahara et al. 2000; Taraseviciene-Stewart et al. 2001). Moreover, chronic hypoxia leads to increased VEGF expression in the lung (Tuder et al. 1995; Christou et al. 1998; Marti & Risau, 1998). We have recently reported that chronic airway infection stimulates angiogenesis in the pulmonary circulation (Hopkins et al. 2001) and others have shown that partial pneumonectomy stimulates pulmonary angiogenesis (Hsia et al. 1994; Thet & Law, 1984). Taken together, these data demonstrate that the pulmonary circulation is capable of angiogenesis when appropriately stimulated.
Because of the enormously vascular nature of the lung when compared with any other organ, it is extremely difficult to detect newly formed blood vessels. Many previous investigations of hypoxia-induced remodelling have used casting techniques that specifically identify the pulmonary arterial vessels and concluded that chronic hypoxia leads to loss of such vessels (Hislop & Reid, 1976; Rabinovitch et al. 1979; Jones & Reid, 1995), although not all are in agreement (Meyrick & Reid, 1979b; Finlay et al. 1986). The reason for focusing on these vessels was to determine the role of arterial remodelling in causing pulmonary hypertension. Such techniques by design exclude capillary and venous vessels and cannot therefore detect angiogenesis that takes place at those sites. Yet, in the systemic circulation the predominant sites of angiogenesis are the capillaries and venules (Ausprunk & Folkman, 1977; Diaz-Flores et al. 1994; Patan et al. 2001). If those were also the sites of hypoxia-induced new vessel formation in the pulmonary circulation, previous studies would not have detected those new vessels.
Taken together, these considerations lead us to hypothesise that chronic hypoxia causes angiogenesis in the adult pulmonary circulation. To test this hypothesis, we used, for the first time, stereological techniques combined with confocal microscopy to examine the changes in pulmonary vascular structure induced by chronic normobaric hypoxia in adult rats. We examined the structural changes induced by hypoxia in arterial, capillary and venous vessels. These techniques allow representative, unbiased, quantitative analysis of the three-dimensional structure of the lung vasculature based on the two-dimensional information provided by histological images. We report the first demonstration that chronic hypoxia causes new vessel formation in the adult pulmonary circulation. These findings imply that we must revise the widely accepted paradigm that hypoxia-induced loss of small vessels is a key structural change contributing to the development of pulmonary hypertension in chronic lung disease.
METHODS
All study protocols were approved by the University Ethics Committee and conducted under licence from the Department of Health. Adult male specific pathogen-free Sprague-Dawley rats (Harlan, Bicester, UK) were maintained for a 2 week period in a normobaric environmental chamber (volume 325 l), as previously described (Ooi et al. 2000). In brief, animals were maintained in normal oxygen conditions (inspired O2 fraction, Fi,O2 0.21) or hypoxia (Fi,O2 0.10). Chamber gases were monitored continuously using an O2 analyser (model 1175, Servomex, Crowborough, UK) and a CO2 analyser (model 1505, Servomex). Excess CO2 was removed by continuously recirculating gases from the chamber through soda lime. Excess humidity and ammonia were removed using a cooled condensing unit and an activated charcoal filter (Carbon-Cap 150, Whatman, Maidstone, UK) respectively. Food and water were available ad libitum, and exposure to the chamber environment was continuous apart from brief periods each day when the chamber was opened to clean cages and replenish food and water.
Lung isolation and fixation
At the end of 2 weeks of exposure to experimental conditions, rats were anaesthetised (70 mg kg−1 sodium pentobarbital intraperitoneally), anti-coagulated (1000 units kg−1 heparin intravenously) and killed by exsanguination. Cannulae were inserted into the trachea and the pulmonary artery and an incision was then made in the left atrium. The heart and lung were removed en bloc from the thorax and the pulmonary circulation was perfused with normal saline (37 °C) until the effluent was clear of blood. Calcium-free physiological saline solution (PSS with EGTA) was introduced into the pulmonary circulation to induce complete relaxation of the pulmonary vessels. The lungs were fully inflated at a pressure of 25 cmH2O with fixative (4 % w/v paraformaldehyde), followed by simultaneous infusion of this solution through the pulmonary artery (62.5 cmH2O). A ligature was placed to obstruct outflow from the pulmonary veins, so that the pulmonary vasculature was fixed under ‘no flow’ conditions. This manoeuvre produced a constant, transmural distending pressure (37.5 cmH2O) at all locations along the vascular bed. After one hour, the main-stem bronchi and pulmonary artery were tied off at the level of the hilum, and the lung volumes determined by water displacement (Scherle, 1970).
Measurement of right ventricular weights
The right ventricular free wall (RV) was dissected from the left ventricle and septum (LV + S), each ventricle weighed separately and the ratio of RV to LV + S weight calculated.
Sectioning strategy
Vertical uniform random (VUR) sections and isotropic uniform random (IUR) sections were prepared from each left lung, as we have previously described (Hopkins et al. 2001). In brief, the vertical axis of each left lung was identified, and 4 mm thick slices cut perpendicular to the axis starting from a randomly chosen position within the first 4 mm. The slices were cut into bars at 2 mm intervals, and every fourth bar selected from a randomly chosen start point between one and four. For the VUR sections, the selected bars were embedded in resin, and then marked into 2 mm blocks at random angles. Every fourth block was selected and cut from the resin, again from a random start point between one and four. Sections (1–2 µm) were taken from random positions within each tissue block and stained with Toluidine Blue; the vertical axis of the section could be identified throughout. IUR sections were then obtained from the resin-embedded blocks of tissue as previously described (Hopkins et al. 2001). Random fields of view from each section were examined by light microscopy (Leica, Laboratory Instruments), captured using video camera (JVC KY-F55B Eurotek, Dublin, Ireland), digitised (Adobe Photoshop) and imported into Stereology Toolbox (Morphometrix, CA, USA) for analysis.
Estimation of the volumes of pulmonary tissue compartments
To determine the volumes of the vascular compartments of interest, a cascade approach was used (Bolender et al. 1993; Hopkins et al. 2001). Volume densities of specific tissues in the lung (volume per unit volume of lung) were estimated by point counting, and the absolute volumes were calculated using the previously measured lung volumes. The volume densities of the conducting airways and their associated structures (extra-acinar tissues) and the tissues and air spaces in the gas exchange regions (intra-acinar tissues) were estimated by point counting. Extra-acinar tissue was defined as large vessels and airways (down to and including terminal bronchioles) and their associated connective tissue, vasa vasorum and nerves, while intra-acinar tissue included respiratory bronchioles, alveolar tissue and the associated vessels within the gas exchange region of the lung. Random fields of view were digitised, imported into Stereology Toolbox and a point counting grid superimposed. The intra-acinar gas exchange region of the lung was next considered as three sub-compartments: the intra-acinar blood vessels meaning all blood vessels other than capillaries, the intra-acinar airspaces and, finally, the alveolar walls including capillaries. Volume densities of these compartments were obtained by point counting on random fields of view obtained at a higher magnification. To estimate the volume densities of the vessel lumen and the layers of the vessel wall tunica intima, tunica media and tunica adventitia respectively, images of randomly selected intra-acinar vessels were captured and placed randomly within point counting grids. The internal elastic lamina and the cells internal to it were considered as a single layer since, in most places, where the cytoplasm of the endothelial cells is attenuated, it was not possible to separately resolve these two structures. The media was defined as the layer of vessel wall between the adventitia and the internal elastic lamina (including the external elastic lamina). The adventitia was identified as the loose connective tissue external to the external elastic lamina or the single elastic lamina. Capillary lumen volume density was estimated by point counting on random fields of view obtained at high magnification.
Estimation of vessel length and radius
To estimate the length of intra-acinar vessels per unit lung volume (length density), the number of intra-acinar vessels that transected counting frames randomly superimposed over images taken from IUR sections was counted (Bolender et al. 1993; Hopkins et al. 2001). Since the vessels were in a relaxed state during fixation and distension, they were considered to be cylindrical in shape; thus, the average radius of the vessels was calculated from the measured length and volume densities using standard formulae. Capillary length density and average radius were calculated in the same way.
Estimation of alveolar epithelial and capillary endothelial surface areas
To estimate the alveolar epithelial and capillary endothelial surface areas, horizontal cycloids were superimposed on randomly chosen fields of view taken from VUR sections (Bolender et al. 1993). The intersections of the alveolar epithelium and capillary endothelium with the cycloid lines were counted to give estimates of alveolar and capillary surface areas per unit area of lung (surface density).
Estimation of gas exchange membrane thickness
We calculated the arithmetic mean thickness of the tissue layer between the alveolar airspace and the plasma as the alveolar wall volume less the capillary lumen volume, all divided by the alveolar epithelial surface area (Weibel & Knight, 1964; Vock & Weibel, 1993). To provide an estimate of the total barrier (tissue plus plasma) between the airspace and the centre of the capillary, the harmonic mean total barrier thickness was determined from the distribution of random intercept lengths across the alveolar wall from epithelial surface to the middle of the capillary. This length was taken as half the length of the intercepts that ran through capillaries and across the whole of the alveolar wall (from epithelial surface to epithelial surface), measured using linear probes, as previously described (Weibel & Knight, 1964). The harmonic mean thickness of the barrier was used to calculate membrane-diffusing capacity according to standard formulae, taking the permeability coefficient for oxygen in both tissue and plasma as 5.5 × 10−10 cm−2 s −1 mmHg−1 (Weibel et al. 1993).
Confocal estimation of endothelial cell number
The estimation of endothelial cell numerical density was obtained by visualising immunofluorescently stained endothelial cells with a laser-scanning confocal microscope, and quantified using the disector method (Bolender et al. 1993; Hopkins et al. 2001). To permit immunostaining, wax-embedded tissue was prepared from a second series of control and hypoxic rats and sectioned using a VUR sampling strategy, as previously described. Endothelial cells were labelled using an anti-vascular endothelial growth factor receptor-2 (VEGFR-2) antibody (Alpha Diagnostics, USA) and then visualised using a biotin-conjugated secondary antibody and streptavidin conjugated to fluorescein isothiocyanate (FITC). Nuclei were stained using propidium iodide. Images of the fluorescently stained sections were acquired using a confocal laser-scanning microscope (Biorad MRC 1024, ×63 oil immersion objective; NA 1.4, confocal aperture 2.6). Randomly chosen pairs of vertically aligned images separated by 5 µm in the z-axis were obtained from each tissue section by optical sectioning. These dissector pairs were imported into Stereology Toolbox for estimation of numerical density.
Data analysis
Values are expressed as means ± s.e.m. Total volumes of specific tissue compartments, vessel and capillary lengths, alveolar epithelial and capillary endothelial surface areas and endothelial cell number are reported per left lung. Statistical comparisons of means were made using Student's unpaired t test. A value of P < 0.05 was accepted as statistically significant.
RESULTS
Mean body weight (306 ± 4.9 g) in the hypoxic group was significantly (P < 0.01) lower following exposure to chronic hypoxia than that (376 ± 6.2 g) of the control group. Chronic hypoxia produced an increase (P < 0.01) in mean haematocrit (64.4 ± 1.09 %) when compared with control values (45.5 ± 0.55 %) and also caused right ventricular hypertrophy (Table 1). Although total left ventricular plus septal weight was significantly reduced by hypoxia (Table 1), this effect was due to the reduced size of the hypoxic animals, as the mean left ventricular weight corrected for body weight was not significantly different in the two groups (Table 1). Mean lung volume was increased by chronic hypoxic exposure (Table 1), predominantly due to a significant (P < 0.05, t test) increase in tissue volume in the gas exchange (intra-acinar) regions of hypoxic (3.07 ± 0.22 cm3 per left lung (LL−1)) when compared with control animals (1.66 ± 0.15 cm3 LL−1). In contrast, mean extra-acinar tissue volume in hypoxic rats (0.45 ± 0.06 cm3 LL−1) was not significantly different from that of control rats (0.37 ± 0.06 cm3 LL−1). While the hypoxia-induced increase in intra-acinar tissue volume was due to a significant increase in all sub-compartment volumes, namely the airspaces, intra-acinar vessels and alveolar walls, the increase in the vascular compartment was proportionately greater than that in the other compartments (Fig. 1).
Table 1.
Body weight, haematocrit, cardiac ventricular weights and lung volumes in control and hypoxic animals
| Control(n = 11) | Hypoxic(n = 11) | |
|---|---|---|
| RV weight (g) | 0.17 ± 0.02 | 0.27 ± 0.01* |
| Normalised RV weight (g (100 g body wt)−1) | 0.05 ± 0.003 | 0.08 ± 0.004* |
| LV + S weight (g) | 0.77 ± 0.04 | 0.65 ± 0.02† |
| Normalised LV + S weight (g (100 g body wt)−1) | 0.21 ± 0.007 | 0.22 ± 0.005 |
| RV/(LV + S) ratio | 0.24 ± 0.02 | 0.38 ± 0.02* |
| Left lung volume (ml) | 2.50 ± 0.012 | 3.61 ± 0.015* |
| Right lung volume (ml) | 4.43 ± 0.13 | 6.88 ± 0.45* |
Values are means (± S.E.M.). RV weight, right ventricular weight. Normalised RV weight, right ventricular weight corrected for body weight. LV + S weight, left ventricular plus septum weight. Normalised LV + S weight, left ventricular plus septum weight corrected for body weight. RV/(LV + S) ratio, ratio of weight of right ventricular free wall to that of left ventricle plus septum
and
indicate significant differences from control values (P < 0.05 and P < 0.01, respectively).
Figure 1. Mean (± s.e.m.) volume of intra-acinar sub-compartments for control (n = 7) and hypoxic (n = 6) animals.
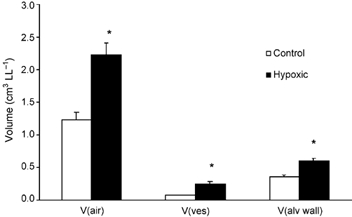
V(air), volume of intra-acinar airspaces; V(ves), volume of intra-acinar pulmonary blood vessels excluding capillaries; V(alv wall), volume of intra-acinar alveolar wall including capillaries. *Significant difference from controls (P < 0.05, t test).
Intra-acinar vascular dimensions
In the control lungs the majority of intra-acinar vessels were typically thin-walled with a single elastic lamina and no discernable tunica media (Fig. 2A). However in the hypoxic lungs, typical features of hypoxic pulmonary vascular wall thickening were observed, with the development of separate internal and external elastic laminae and hypertrophied media and adventitial layers (Fig. 2B). Stereological analysis showed that the mean total volumes of the tunica intima plus internal elastic lamina together, the tunica media, and the tunica adventitia of the intra-acinar pulmonary blood vessels were significantly augmented following chronic exposure to hypoxia (Fig. 3). The mean total luminal volume of the intra-acinar pulmonary vessels was also significantly increased in hypoxic compared with control animals (Fig. 3).
Figure 2. Photomicrographs of intra-acinar blood vessels in control and hypoxic lungs.
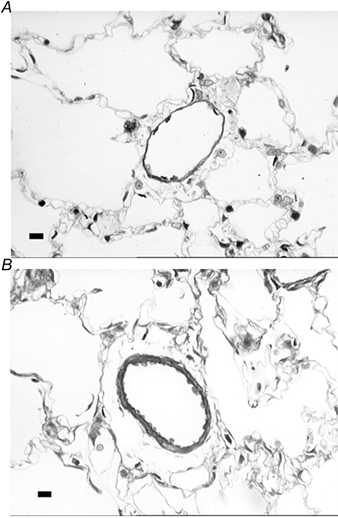
Both images were taken from semi-thin resin sections stained with Toluidine Blue. A, photomicrograph of intra-acinar vessel from control lungs. Vessel is typically thin walled with no medial layer and a single elastic lamina. B, photomicrograph of a typically remodelled intra-acinar vessel from chronically hypoxic lungs. Vessel demonstrates medial thickening, with both an internal and an external elastic lamina present. Scale bars indicate 20 µm.
Figure 3. Mean (± s.e.m.) volume of intra-acinar vessel lumen, tunica intima plus internal elastic lamina together, tunica media and tunica adventitia in intra-acinar pulmonary blood vessels in the left lungs of control (n = 7) and hypoxic (n = 6) animals.
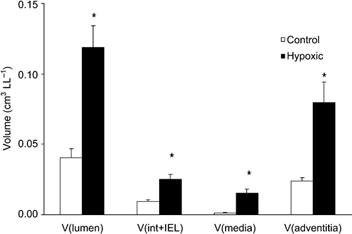
V(lumen), volume of intra-acinar blood vessel lumen; V(int + IEL), volume of tunica intima plus internal elastic lamina considered together (see Methods for details); V(media), volume of tunica media; V(adventitia), volume of tunica adventitia. *Significant difference from control animals (P < 0.001, t test).
The mean total length of the intra-acinar pulmonary blood vessels was significantly greater in the chronically hypoxic lungs than in the control group (Fig. 4). There was a significant increase in both the volume of the tunica media and the adventitia per unit vessel length, indicating that these layers had become thickened as a result of chronic hypoxia (Table 2). The total vessel wall thickness (intima, media and adventitia together) was significantly (P < 0.01) greater in the hypoxic group (9.9 ± 0.7 µm) than in controls (7.0 ± 0.5 µm). However, the mean diameter of the vascular lumen in the hypoxic lungs (43.8 ± 4.1 µm) was not significantly different from that of the control group (37.5 ± 2.4 µm). To facilitate comparison with previous literature (see Discussion), the mean thickness of the internal elastic lamina and the intima internal to it were calculated in both groups; the mean value in the chronically hypoxic lungs was 2.4 (± 0.14) µm, a value not significantly different from that of the control group (2.5 ± 0.14 µm).
Figure 4. Mean (± s.e.m.) intra-acinar vessel length per left lung in control and hypoxic animals.
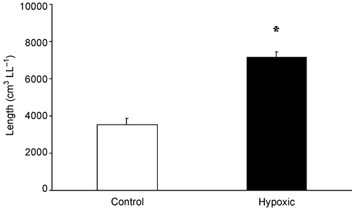
*Significant difference from controls (P < 0.01, t test).
Table 2.
Volumes per unit vessel length of tunica intima plus internal elastic lamina, tunica media and tunica adventitia in control and hypoxic intra-acinar pulmonary blood vessels
| Control (n = 7) | Hypoxic (n = 6) | |
|---|---|---|
| Volume of intima + IEL per unit length (× 10−6 cm3 cm−1) | 2.66 ± 0.18 | 3.50 ± 0.50 |
| Volume of media per unit length (× 10−6 cm3 cm−1) | 0.35 ± 0.03 | 2.06 ± 0.47* |
| Volume of adventitia per unit length (× 10−6 cm3 cm−1) | 6.82 ± 0.56 | 11.50 ± 1.73* |
Values are means (± S.E.M.). Volume of intima + IEL, volume of intima and internal elastic lamina considered together (see Methods for details.
Significant difference from controls (P < 0.05, t test).
Capillary dimensions
In control lungs, a typically highly vascularised alveolar wall was observed with an extensive capillary network (Fig. 5A). Hypoxic lungs also demonstrated an extensive capillary network, but capillaries were observed that appeared to protrude into the alveolar spaces suggesting new vessel formation (Fig. 5B). The mean capillary volume density, total capillary volume, capillary length density and total capillary length were significantly increased in the hypoxic lungs compared with control lungs (Table 3). Mean capillary diameter did not differ significantly between control (5.5 ± 0.14 µm) and hypoxic (5.6 ± 0.14 µm) lungs. The total alveolar epithelial and capillary endothelial surface areas were significantly augmented in the chronically hypoxic lungs (Table 4). However, the increase in capillary endothelial surface area was disproportionately greater, as indicated by the observation that the ratio of capillary endothelium to alveolar epithelium was significantly augmented in the hypoxic lungs compared with controls (Table 4). The mean ratio of capillary length to alveolar epithelial surface area was also significantly (P < 0.01) augmented in the chronically hypoxic (767 ± 27.1 cm cm−2) compared with control (588 ± 45.7 cm cm−2) lungs. Similarly, the ratio of capillary volume to alveolar surface area was significantly (P < 0.01) increased in hypoxic lungs (0.00019 ± 0.00001 cm3 cm−2) when compared with control lungs (0.00014 ± 0.00001 cm3 cm−2). The arithmetic mean tissue thickness was significantly reduced in hypoxic lungs (Table 5); however, the harmonic mean barrier thickness was not significantly increased in the hypoxic lungs (Table 5). The combined effect of the increased surface area and harmonic mean barrier thickness led to a mean membrane diffusing capacity in the hypoxic group (0.0071 ± 0.0009 ml O2 s−1 mmHg−1 LL−1) that was greater than that of the control group (0.0050 ± 0.0006 ml O2 s−1 mmHg−1 LL−1), although this difference was not statistically significant (P = 0.07). However, when corrected for body weight the hypoxic value (0.0023 ± 0.0002 ml O2 s−1 mmHg−1 LL−1 (100 g body weight)−1) was significantly (P < 0.01) larger than that of the control group (0.0014 ± 0.0001 ml O2 s−1 mmHg−1 LL−1 (100 g body weight)−1).
Figure 5. Images of alveolar walls taken from semi-thin (1–2 µm) sections stained with Toluidine Blue.
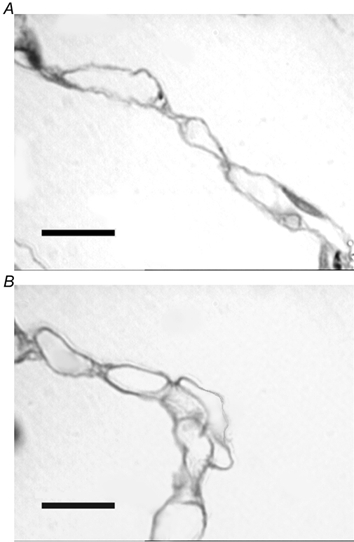
A, alveolar wall taken from control lung with numerous capillaries discernible within the alveolar wall. B, alveolar wall taken from chronically hypoxic lung tissue. Some capillaries appear to protrude from alveolar wall into alveolar lumen, a pattern not seen in control lungs. Scale bars indicate 10 µm.
Table 3.
Capillary volume and length in control and hypoxic animals
| Control (n = 7) | Hypoxic (n = 6) | |
|---|---|---|
| Capillary volume density (cm3 cm−3) | 0.09 ± 0.005 | 0.13 ± 0.01* |
| Total capillary volume (cm3 LL−1) | 0.19 ± 0.019 | 0.43 ± 0.03* |
| Capillary length density (× 105 cm cm−3) | 4.02 ± 0.19 | 5.24 ± 0.29* |
| Total capillary length (× 105 cm LL−1) | 8.07 ± 0.60 | 17.56 ± 0.56* |
Values are means (± S.E.M.). Capillary volume density, capillary volume per unit volume of lung. Total capillary volume, capillary volume per left lung. Capillary length density, capillary length per unit volume of lung. Total capillary length, capillary length per left lung.
Significant difference from controls (P < 0.01, t test).
Table 4.
Alveolar epithelial and capillary endothelial surface densities, areas and ratio of capillary endothelial to epithelial surface areas
| Control (n = 7) | Hypoxic (n = 6) | |
|---|---|---|
| Epithelial surface density (cm2 cm−3) | 711.9 ± 73.9 | 686.9 ± 55.7 |
| Total epithelial area (cm2 LL−1) | 1436 ± 169 | 2302 ± 119* |
| Endothelial surface density (cm2 cm−3) | 806.8 ± 79.6 | 959.4 ± 61.6 |
| Total endothelial area (cm2 LL−1) | 1642 ± 186 | 3261 ± 230* |
| Endothelial/epithelial ratio | 1.14 ± 0.02 | 1.41 ± 0.07* |
Values are means (± S.E.M.). Epithelial surface density, alveolar epithelial area per unit volume of left lung. Total epithelial area, alveolar epithelial surface area per left lung. Endothelial surface density, capillary endothelial surface area per unit volume of left lung. Total endothelial area, capillary endothelial surface area per left lung.
Significant difference from controls (P < 0.005, t test).
Table 5.
Arithmetic and harmonic mean barrier thicknesses
| Control (n = 7) | Hypoxic (n = 6) | |
|---|---|---|
| Arithmetic mean tissue thickness (μm) | 1.2 ± 0.12 | 0.8 ± 0.13* |
| Harmonic mean total barrier thickness (μm) | 1.6 ± 0.09 | 1.9 ± 0.17 |
Values are means (± S.E.M.). Tissue barrier thickness is the thickness of the tissue barrier to gas exchange, i.e. the barrier formed by the epithelial cell, interstitium, basement membrane(s) and endothelial cells. Total barrier thickness refers to the thickness of the tissue barrier together with the plasma lying between the endothelium and the centre of the capillary (see text for detailed discussion).
Significant difference from controls (P < 0.05, t test).
Endothelial cell number
Pulmonary vascular endothelial cells were identified by anti-VEGFR-2 labelling (Fig. 6). Mean total endothelial cell number in the left lung was significantly greater in the hypoxic (4.84 ± 0.62 × 108 LL−1) than in the control (1.60 ± 0.09 × 108 LL−1) group (Fig. 7).
Figure 6. Photomicrograph showing a confocally acquired image of control lung tissue.
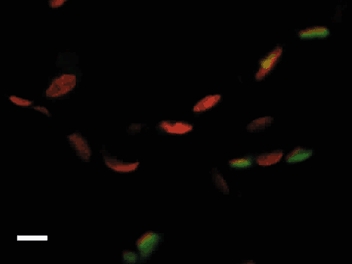
Nuclei were stained with Propidium Iodide (red) while endothelial cells were stained using anti-VEGFR-2 antibody visualised using FITC (green). Scale bar represents 10 µm.
Figure 7. Photomicrographs showing confocally acquired images of endothelial cells stained immunofluorescently with anti-VEGFR-2 antibody labelled with FITC.
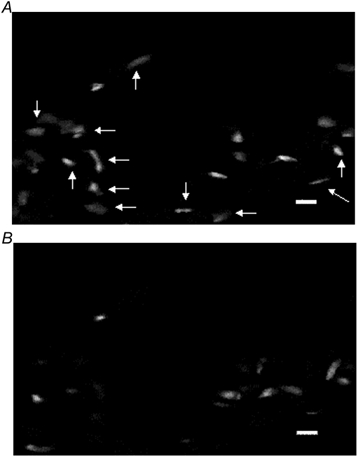
The number of endothelial cells seen in the control lung section (A) is less than that observed in the hypoxic lung section (B). Scale bars represent 10 µm.
DISCUSSION
We have shown that chronic hypoxia led to increased blood vessel volume and length in the pulmonary circulation, increased capillary surface area and increased vascular endothelial cells when compared with control animals maintained in normal oxygen conditions. These results demonstrate that new vessel formation had occurred in response to hypoxia and is the first reported demonstration of hypoxia-induced angiogenesis in the adult pulmonary circulation.
General effects of hypoxia
In agreement with previous findings, hypoxic rats demonstrated elevated haematocrits and right ventricular hypertrophy (Table 1), indicative of pulmonary arterial hypertension (Abraham et al. 1971; Heath et al. 1973; Hunter et al. 1974; Hislop & Reid, 1976; Meyrick & Reid, 1978; Rabinovitch et al. 1979; Jones et al. 1984; Ooi et al. 2000). Quantification of intra-acinar remodelling revealed typical medial and adventitial thickening, which was evident both by histological examination (Figs 5A and B) and stereological analysis (Fig. 2) (Hislop & Reid, 1976; Meyrick & Reid, 1978; Rabinovitch et al. 1979). Hypoxic lung volumes (Table 1) were significantly greater than those of controls, in agreement with the findings of Cunningham et al. (1974), who attributed the increase in lung volume in adult rats to an increase in mean alveolar size.
Validation of the stereological approach
In order to validate our use of quantitative stereology, we compared the vascular dimensions that we obtained in control lungs with those previously obtained using conventional microscopic approaches. The structure of the pulmonary vasculature of the rat has been extensively studied, most notably by Reid and colleagues (Hislop & Reid, 1976, 1978; Meyrick et al. 1978; Meyrick & Reid, 1978, 1979a). They reported that rat intra-acinar arterial vessels range in external diameter from 15 to 150 µm. Since mean luminal diameter, as determined by stereological techniques, depends upon the relative numbers of vessels of different diameters and their lengths, and since smaller vessels are more numerous than the larger ones, the value of 37.5 µm for mean luminal diameter determined in our control lungs seems to be in good agreement with those previous reports. Haworth et al. (1991), using casting techniques, determined the slope of the power relationship between the numbers of intra-pulmonary vessels and their diameters. Based on their model, we calculated that the expected volume-derived mean diameter of the intra-acinar vessels in normal lungs is 35 µm, a value in close agreement with our estimate. Our stereologically determined value for the thickness of tunica intima including the internal elastic lamina in control animals (2.5 ± 0.24 µm) is in good agreement with the data of Meyrick & Reid (1979a). They found, using electron microscopy, that the internal elastic lamina was up to 1.4 µm thick and the mean thickness of the cells of the tunica intima internal to this varies from 0.2 to 4.0 µm. The ratio of capillary endothelial to alveolar epithelial surface area that we observed in control lungs (Table 4) is also in close agreement with those previously reported (Crapo et al. 1978, 1983; Haies et al. 1981). Furthermore, the arithmetic mean thickness of the tissue barrier of the gas exchange membrane is similar to the values previously reported (Crapo et al. 1983; Vock & Weibel, 1993), as is the ratio of the capillary volume to the alveolar surface area (Vock & Weibel, 1993). Taken together, these comparisons demonstrate that our stereological approach produced estimates of pulmonary vascular dimensions in normal lungs that are in close agreement with those obtained previously.
To identify endothelial cells we used an anti-VEGFR-2 antibody; VEGFR-2 expression is specific to endothelial cells and it is constitutively expressed by the quiescent endothelial cells of the normal lung (Jakeman et al. 1992; Peters et al. 1993; Tuder et al. 1995; Sandner et al. 1997; Ferrara & Davis-Smyth, 1997; Marti & Risau, 1998). This endothelial cell specificity is conferred by an endothelial cell-specific promoter in the VEGFR-2 gene (Patterson et al. 1995). Thet & Law (1984) estimated, using a combined electron microscopy and stereological approach, that the number of endothelial cells per right lung in a group of normal adult rats was 2.01 (± 0.11) × 108. To allow direct comparison, we estimated the mean endothelial cell number in control right lungs (2.9 ± 0.38 × 108 cells per lung) and found it to be in close agreement with the previous report (Thet & Law, 1984).
The effects of chronic hypoxia on pulmonary vascular structure
The walls of the intra-acinar vessels were remodelled as would be expected following chronic hypoxic exposure. Thus we observed increased wall volume per unit length and increased wall thickness in the hypoxic animals (Fig. 2 and Table 2). However, the mean diameter of the maximally distended vascular lumen was not reduced in the hypoxic lungs, despite the fact that these animals had developed pulmonary hypertension, as evidenced by the presence of right ventricular hypertrophy (Table 1). Additionally, the total volume of the lumen of these vessels was increased (Fig. 3). This at first seems surprising since it is generally thought that wall thickening in these vessels encroaches on the lumen, contributing substantially to increased vascular resistance (Hislop & Reid, 1976; Meyrick & Reid, 1978; Rabinovitch et al. 1979; Grover et al. 1983; Fishman, 1985; Jones & Reid, 1995; Stenmark & Mecham, 1997). A potential explanation is that the structural increase in vessel wall thickness in these hypoxic lungs may have occurred in a predominantly outward direction without encroachment on the lumen. This has previously been reported in chronically infected rat lungs (Hopkins et al. 2001) and in atherosclerotic disease of the systemic arteries (Glagov et al. 1993).
The changes in the capillary bed were broadly similar to those observed in the intra-acinar vessels and included an increase in the mean lumen volume (Table 3), an increase in mean length (Table 3) and an increase in the total endothelial surface area (Table 4), whilst mean lumen diameter remained unchanged. This finding of an increased extent of the pulmonary vascular bed is supported by the work of Emery and colleagues (Emery et al. 1981). They measured the pulmonary vascular volume, not by microscopic examination of vessels, but by measuring the quantity of radiolabelled albumin contained in the pulmonary circulation, and demonstrated an increase in pulmonary vascular volume in chronically hypoxic rats lungs compared with controls of a similar weight (Emery et al. 1981). This observation is in close agreement with our finding of an increase in total pulmonary vascular volume in hypoxia.
Could the structural changes that we observed in the vasculature have resulted from simple dilatation of pre-existing vessels or did they result from angiogenesis? The total length of pulmonary blood vessels was significantly increased in the hypoxic animals by approximately 2.5-fold (Table 3 and Fig. 4). Since volume is related to linear dimensions by the third power, it would have been necessary for the lung volume in hypoxic animals to have increased 15-fold compared with control lungs, if dilatation alone had accounted for this increase in vessel length. However, the observed increase in lung volume following hypoxia was considerably less at approximately 1.5-fold (Table 1). Similarly, the increase in the mean volumes of the lumen of intra-acinar and capillary vessels in chronically hypoxic lungs was approximately 3-fold, disproportionately larger than the increase in lung volume (Fig. 2 and Table 3). The ratio of capillary endothelial surface area to alveolar epithelial surface area was significantly increased (Table 4) following chronic hypoxia, as were the ratios of capillary length and volume to alveolar epithelial area. These observations demonstrate that the changes in vascular dimensions were greater than could be expected on the basis of distension alone, suggesting that angiogenesis rather than hypertrophy was responsible for the observed hypoxia-induced changes in vessel structure. Our observation that chronic hypoxia caused a significant increase in the number of endothelial cells in the intra-acinar vessels and capillaries confirmed that new vessel formation had taken place (Fig. 7).
It is important to note that altered expression of VEGFR-2 in hypoxia cannot account for our findings. The effect of chronic hypoxia on VEGFR-2 receptor expression within endothelial cells is controversial; some groups report increased expression (Tuder et al. 1995; Brogi et al. 1996; Christou et al. 1998) while others do not (Sandner et al. 1997; Marti & Risau, 1998; Olfert et al. 2001). However, the stereological dissector technique is unaffected by increasing staining intensity within individual cells, since cells are counted if they express VEGFR-2 regardless of the intensity of the staining. As discussed earlier, the similarity of total endothelial cell numbers detected in control lungs in the present study and those reported previously using electron microscopy (Thet & Law, 1984) shows that the confocal approach was able to detect normal constitutive VEGFR-2 on the pulmonary endothelial cells. Thus, the increase in endothelial cell numbers detected in our hypoxic lungs cannot be an artefact resulting from the identification of undetected endothelial cells in control lungs.
Chronic hypoxia led to larger lung volumes, greater vascular volumes, length, capillary endothelial surface area and endothelial cell number despite the fact that the hypoxic animals were smaller than the corresponding control animals. Exactly similar conclusions are reached if all these parameters are expressed following normalisation for body weight (data not shown).
Why has hypoxia-induced angiogenesis not been previously detected in the pulmonary circulation? The normal lung is a highly vascularised structure with a vast blood vessel network, which is required to provide adequate surface area for gas exchange. Given the extent of the normal vasculature when compared with other organs, it is extremely difficult to detect newly formed blood vessels in the lung, a problem that has been reviewed and commented upon previously (Mooi & Wagenvoort, 1983; Schraufnagel et al. 1996). Furthermore, it is not possible to detect a change in total vessel length by simply counting the number of vessels viewed per microscopic field of view in a single section of lung taken from a standard location in a fixed orientation (Gundersen et al. 1988; Bolender et al. 1993), the most commonly adopted technique in previous studies. However, the stereological techniques used in the present study circumvent these difficulties and allow unbiased quantitative analysis of the three-dimensional structure of the lung vasculature based on the two-dimensional information provided by histological images. By using strict random sampling from throughout the lung, stereological approaches ensure that the data obtained are representative of the whole organ. Another important feature of stereology is that it carefully defines and quantifies a reference volume within which a particular parameter is to be measured. This step allows absolute quantities to be measured, even in circumstances where the total lung volume changes, as in chronic hypoxia (Gundersen et al. 1988; Bolender et al. 1993). Conventional approaches make no allowance for changes in lung volume, a further potential reason for failing to detect new vessel formation (Gundersen et al. 1988; Bolender et al. 1993). It is interesting to note again that Emery and colleagues observed an increase in total pulmonary vascular volume in response to chronic hypoxia when using radiolabelled albumin, a method that is independent of the difficulties involved in the use of conventional microscopic approaches (Emery et al. 1981).
In this context, a further aspect of our techniques is worthy of specific note. By fixing vessels at high infusion pressures under ‘no-flow’ conditions in the fully relaxed state, we ensured that the transmural pressure was constant at all points along the vessels and independent of any alteration of upstream resistance. This approach meant that all vessels were recruited and maximally dilated at a constant, standard, distending pressure, which was identical in both hypoxic and control lungs, and was independent of any changes in upstream vascular resistance. As a result, we were able to examine the structure of all the vessels of the pulmonary circulation: arterial, venous and capillary. The latter two segments are particularly important since these are the major sites of new vessel formation in the systemic circulation and this may also be the case in the pulmonary circulation (Ausprunk & Folkman, 1977; Diaz-Flores et al. 1994; Patan et al. 2001). In many previous investigations of hypoxia-induced remodelling, gelatine infusion techniques have been used to specifically identify the pulmonary arterial vessels (Hislop & Reid, 1976; Rabinovitch et al. 1979; Jones & Reid, 1995). Such techniques by design exclude capillary and venous vessels and cannot therefore detect angiogenesis that takes place at those sites. Indeed, even within the arterial segment, the extent and diameters of vessels identified by gelatine are critically influenced by increased vascular resistance (Meyrick & Reid, 1979b;Finlay et al. 1986).
Role of vascular remodelling in the development of pulmonary hypertension
It is well established that chronic hypoxia leads to the development of pulmonary hypertension (Grover et al. 1983; Meyrick & Reid, 1983; Fishman, 1985; MacNee 1994a,b; Jones and Reid 1995) and the finding of right ventricular hypertrophy in hypoxic rats in the present series of experiments is in good agreement with this. Moreover, in our previous work we have shown that the hypoxic conditions used in the present study lead to an increase in pulmonary vascular resistance (Ooi et al. 2000). Yet the structural changes in the vasculature that we report here may seem, at first sight, to be at variance with the observed increase in pulmonary vascular resistance. It is important to understand that this is not the case and that our results are potentially completely compatible with the development of pulmonary hypertension.
Three mechanisms could account for pulmonary hypertension in these conditions: (i) reduction in lumen narrowing restricted to one critical region of the vasculature; (ii) angiogenesis by elongation; and (iii) differences between vascular dimensions in vivo and in fixed tissues.
(i) Our method of lung preparation meant that we could not reliably distinguish arterial and venous vessels and we therefore considered them together when undertaking stereological measurement. As a result, the mean lumen diameter is a reflection of all the intra-acinar vessels excluding capillaries. It is possible that the lumen of the smaller more distal arterioles might have been preferentially reduced in diameter and become a dominant determinant of increased vascular resistance while that of the venous side increased. In this event the mean vessel diameter, as we determined it, could have remained unchanged in hypoxic lungs despite a reduction in the mean diameter of small arterial vessels. In essence, vascular resistance may increase following chronic hypoxia due to a focal reduction in vessel diameter within small arterioles while all other segments of the intra-acinar vessels increase in diameter, length and volume. In such circumstances, the remodelled pulmonary circulation could behave like the normal systemic circulation so that total pulmonary vascular resistance might be dominated by the contribution of constricted arterioles.
(ii) We have shown an approximately 2.5-fold increase in vessel length (Fig. 4) without significant change in lumen diameter. If this increased length resulted from elongation of existing vessels, as has been shown in other organs (Hansen-Smith et al. 1996; Gambino et al. 2002), and not from formation of new parallel vascular pathways, it would have resulted in a substantial increase in vascular resistance. Thus, the effect of new capillary formation on vascular resistance similarly depends on the exact structural arrangement of these new vessels. New vessels laid down in series would increase vascular resistance whereas new vessels in parallel would reduce it.
(iii) The complete distension of vessels prior to fixation produced by our protocol allowed us to investigate the structural changes in these vessels. However, the lumen diameter of both the non-capillary and capillary vessels determined in this circumstance clearly does not reflect the dimensions that existed in vivo, where in the presence of vascular smooth muscle tone and hypoxic vasoconstriction, the lumen diameter would have been considerably different. Moreover, as in the capillaries of the normal lung, large numbers of these new capillaries may not be perfused at any given moment and thus not have any influence on total resistance.
Thus, while our findings clearly demonstrate for the first time new vessel formation in the pulmonary circulation in response to chronic hypoxia, they are compatible with the development of pulmonary hypertension as a result of vascular remodelling. Nonetheless, since we have shown that the pulmonary arterial tree expands in chronic hypoxia, increased vascular resistance does not simply result from a reduced number of pulmonary vessels or rarefaction.
Effects on gas exchange
The observation that the arithmetic mean tissue barrier thickness was slightly reduced following chronic hypoxia (Table 5) demonstrated that the alveolar wall had not become thickened by oedema or enlargement of the cells or matrix. However, the diffusion barrier to oxygen consists not just of the tissue of the alveolar-capillary membrane but also of the plasma interposed between the endothelial surface and the red blood corpuscles (Weibel et al. 1993). The size of this plasma barrier depends in turn on the size of the capillary lumen and on the haematocrit. The haematocrit is increased in chronic hypoxia and can alter the thickness of the plasma barrier in a manner that is independent of structural changes in the lung vasculature.
To exclude the influence of haematocrit on barrier thickness, we have considered a total barrier that extends from the alveolar surface of the epithelium to a point mid-way across the capillary lumen. As has been previously shown, the tissue of the alveolar wall and the plasma behave so similarly in relation to the diffusion of oxygen, they may be treated as a single barrier whose surface area is reasonably approximated as the surface area of the alveolar epithelium (Weibel et al. 1993). Using this model we found a significant increase in the total membrane diffusing capacity in chronic hypoxia, a potentially beneficial adaptation during acclimatisation to high altitude. It must be emphasised again that the barrier considered here deliberately excludes the effect of red blood corpuscles in order to allow an examination of the effect of structural changes in the vasculature. In vivo, the increase in haematocrit caused by hypoxia would increase the number of red blood corpuscles in the pulmonary capillaries and reduce the thickness of the plasma barrier, thus increasing the actual in vivo membrane diffusing capacity.
Other features of the changes in the capillary structure would tend to increase total oxygen diffusing capacity in vivo independently of the increase in membrane diffusing capacity. The total diffusing capacity of the lung depends not just on the membrane diffusing capacity but also on the reaction of oxygen with haemoglobin. This latter component depends in part on the total volume of blood available within the alveolar wall. Our finding that the capillary volume per unit area of surface epithelium and the total capillary volume were increased in chronically hypoxic lungs suggests that in vivo a greater volume of blood could be made available in the alveolar walls and could lead to an increase in the total pulmonary diffusing capacity.
It must be emphasised that the inferences made about pulmonary diffusing capacity based on morphometric data give an indication of the theoretical maximum diffusing capacity. In vivo these capillaries may not all be perfused and, if perfused, may not be fully distended. Thus, the actual gas exchange surface area and the volume of blood in the lung are unlikely to be as large as those suggested by the morphometric analysis of fixed tissue (Weibel et al. 1993).
Finally, when considering the potential effects of the structural changes reported here on gas exchange, it must be remembered that in normal sea level conditions, both at rest and during exercise, oxygen uptake is not limited by the diffusing capacity of the lung but is instead perfusion-limited (Weibel, 1999), i.e. the alveolar capillary blood has come into equilibrium with alveolar gas before the blood has completely traversed the capillary. However, during exercise by native sea-level dwellers at high altitude, end-capillary PO2 may fall substantially below alveolar values (Weibel, 1999). The increased capillary length caused by chronic hypoxia that we report here, would prolong the time that red blood corpuscles spend in the alveolar capillaries at any given cardiac output, allowing more time for complete equilibration of PO2 between alveolar gas and blood. This is a further mechanism that would enhance oxygen uptake even though the maximal membrane diffusing capacity was unchanged.
Conclusions
We have shown that chronic hypoxia resulted in increased total pulmonary vessel length and volume and augmented capillary endothelial surface area, together with increased endothelial cell number in vivo in the adult pulmonary circulation, a structural adaptation that may have important beneficial consequences for gas exchange. This is the first reported demonstration of hypoxia-induced angiogenesis in the mature pulmonary circulation. These findings imply that we must revise the widely accepted paradigm that hypoxia-induced loss of small vessels is a key structural change contributing to the development of pulmonary hypertension during acclimatisation to high altitude environments, in chronic lung disease and congenital cyanotic heart disease.
Acknowledgments
This work was supported by a grant from the Health Research Board of Ireland.
REFERENCES
- Abraham AS, Kay JM, Cole RB, Pincock AC. Haemodynamic and pathological study of the effect of chronic hypoxia and subsequent recovery of the heart and pulmonary vasculature of the rat. Cardiovasc Res. 1971;5:95–102. doi: 10.1093/cvr/5.1.95. [DOI] [PubMed] [Google Scholar]
- Ausprunk DH, Folkman J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc Res. 1977;14:53–65. doi: 10.1016/0026-2862(77)90141-8. [DOI] [PubMed] [Google Scholar]
- Bolender RP, Hyde DM, Dehoff RT. Lung morphometry: a new generation of tools and experiments for organ, tissue, cell, and molecular biology. Am J Physiol. 1993;265:L521–548. doi: 10.1152/ajplung.1993.265.6.L521. [DOI] [PubMed] [Google Scholar]
- Brogi E, Schatteman G, Wu T, Kim EA, Varticovski L, Keyt B, Isner JM. Hypoxia-induced paracrine regulation of vascular endothelial growth factor receptor expression. J Clin Invest. 1996;97:469–476. doi: 10.1172/JCI118437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christou H, Yoshida A, Arthur V, Morita T, Kourembanas S. Increased vascular endothelial growth factor production in the lungs of rats with hypoxia-induced pulmonary hypertension. Am J Respir Cell Mol Biol. 1998;18:768–776. doi: 10.1165/ajrcmb.18.6.2980. [DOI] [PubMed] [Google Scholar]
- Crapo JD, Peters-Golden M, Marsh-Salin J, Shelburne JS. Pathologic changes in the lungs of oxygen-adapted rats: a morphometric analysis. Lab Invest. 1978;39:640–653. [PubMed] [Google Scholar]
- Crapo JD, Young SL, Fram EK, Pinkerton KE, Barry BE, Crapo RO. Morphometric characteristics of cells in the alveolar region of mammalian lungs. Am Rev Respir Dis. 1983;128:S42–46. doi: 10.1164/arrd.1983.128.2P2.S42. [DOI] [PubMed] [Google Scholar]
- Cunningham EL, Brody JS, Jain BP. Lung growth induced by hypoxia. J Appl Physiol. 1974;37:362–366. doi: 10.1152/jappl.1974.37.3.362. [DOI] [PubMed] [Google Scholar]
- Diaz-Flores L, Gutierrez R, Varela H. Angiogenesis: an update. Histol Histopathol. 1994;9:807–843. [PubMed] [Google Scholar]
- Emery CJ, Bee D, Barer GR. Mechanical properties and reactivity of vessels in isolated perfused lungs of chronically hypoxic rats. Clin Sci (Lond) 1981;61:569–580. doi: 10.1042/cs0610569. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- Finlay M, Barer GR, Suggett AJ. Quantitative changes in the rat pulmonary vasculature in chronic hypoxia - relation to haemodynamic changes. Q J Exp Physiol. 1986;71:151–163. doi: 10.1113/expphysiol.1986.sp002975. [DOI] [PubMed] [Google Scholar]
- Fishman AP. In: American Handbook of Physiology, The Respiratory System. Fishman AP, Fisher AB, editors. I. Bethesda, MD: American Physiological Society; 1985. pp. 93–165. [Google Scholar]
- Fried R, Meyrick B, Rabinovitch M, Reid L. Polycythemia and the acute hypoxic response in awake rats following chronic hypoxia. J Appl Physiol. 1983;55:1167–1172. doi: 10.1152/jappl.1983.55.4.1167. [DOI] [PubMed] [Google Scholar]
- Gambino LS, Wreford NG, Bertram JF, Dockery P, Lederman F, Rogers PA. Angiogenesis occurs by vessel elongation in proliferative phase human endometrium. Hum Reprod. 2002;17:1199–1206. doi: 10.1093/humrep/17.5.1199. [DOI] [PubMed] [Google Scholar]
- Glagov S, Zarins CK, Masawa N, Xu CP, Bassiouny H, Giddens DP. Mechanical functional role of non-atherosclerotic intimal thickening. Front Med Biol Eng. 1993;5:37–43. [PubMed] [Google Scholar]
- Griffioen AW, Molema G. Angiogenesis: potentials for pharmacologic intervention in the treatment of cancer, cardiovascular diseases, and chronic inflammation. Pharmacol Rev. 2000;52:237–268. [PubMed] [Google Scholar]
- Grover RF, Wagner WW, McMurtry IF, Reeves JT. In: The Cardiovascular System. Shepard JT, Aboud FM, editors. Vol. 3. Bethesda, MD, USA: American Physiological Society; 1983. pp. 103–136. [Google Scholar]
- Gundersen HJ, Bagger P, Bendtsen TF, Evans SM, Korbo L, Marcussen N, Moller A, Nielsen K, Nyengaard JR, Pakkenberg B, et al. The new stereological tools: disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. APMIS. 1988;96:857–881. doi: 10.1111/j.1699-0463.1988.tb00954.x. [DOI] [PubMed] [Google Scholar]
- Haies DM, Gil J, Weibel ER. Morphometric study of rat lung cells. I. Numerical and dimensional characteristics of parenchymal cell population. Am Rev Respir Dis. 1981;123:533–541. doi: 10.1164/arrd.1981.123.5.533. [DOI] [PubMed] [Google Scholar]
- Hansen-Smith FM, Hudlicka O, Egginton S. In vivo angiogenesis in adult rat skeletal muscle: early changes in capillary network architecture and ultrastructure. Cell Tissue Res. 1996;286:123–136. doi: 10.1007/s004410050681. [DOI] [PubMed] [Google Scholar]
- Haworth ST, Linehan JH, Bronikowski TA, Dawson CA. A hemodynamic model representation of the dog lung. J Appl Physiol. 1991;70:15–26. doi: 10.1152/jappl.1991.70.1.15. [DOI] [PubMed] [Google Scholar]
- Heath D, Edwards C, Winson M, Smith P. Effects on the right ventricle, pulmonary vasculature, and carotid bodies of the rat of exposure to, and recovery from, simulated high altitude. Thorax. 1973;28:24–28. doi: 10.1136/thx.28.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hislop A, Reid L. New findings in pulmonary arteries of rats with hypoxia-induced pulmonary hypertension. Br J Exp Pathol. 1976;57:542–554. [PMC free article] [PubMed] [Google Scholar]
- Hislop A, Reid L. Changes in the pulmonary arteries of the rat during recovery from hypoxia-induced pulmonary hypertension. Br J Exp Pathol. 1977;58:653–662. [PMC free article] [PubMed] [Google Scholar]
- Hislop A, Reid L. Normal structure and dimensions of the pulmonary arteries in the rat. J Anat. 1978;125:71–83. [PMC free article] [PubMed] [Google Scholar]
- Hopkins N, Cadogan E, Giles S, McLoughlin P. Chronic airway infection leads to angiogenesis in the pulmonary circulation. J Appl Physiol. 2001;91:919–928. doi: 10.1152/jappl.2001.91.2.919. [DOI] [PubMed] [Google Scholar]
- Hopkins N, McLoughlin P. The structural basis of pulmonary hypertension in chronic lung disease: remodelling, rarefaction or angiogenesis. J Anat. 2003 doi: 10.1046/j.1469-7580.2002.00096.x. (in the Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia CC, Herazo LF, Fryder-Doffey F, Weibel ER. Compensatory lung growth occurs in adult dogs after right pneumonectomy. J Clin Invest. 1994;94:405–412. doi: 10.1172/JCI117337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter C, Barer GR, Shaw JW, Clegg EJ. Growth of the heart and lungs in hypoxic rodents: a model of human hypoxic disease. Clin Sci Mol Med. 1974;46:375–391. doi: 10.1042/cs0460375. [DOI] [PubMed] [Google Scholar]
- Incalzi RA, Fuso L, De Rosa M, Di Napoli A, Basso S, Pagliari G, Pistelli R. Electrocardiographic signs of chronic cor pulmonale: A negative prognostic finding in chronic obstructive pulmonary disease. Circulation. 1999;99:1600–1605. doi: 10.1161/01.cir.99.12.1600. [DOI] [PubMed] [Google Scholar]
- Jakeman LB, Winer J, Bennett GL, Altar CA, Ferrara N. Binding sites for vascular endothelial growth factor are localized on endothelial cells in adult rat tissues. J Clin Invest. 1992;89:244–253. doi: 10.1172/JCI115568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones R, Reid L. In: Pulmonary Vascular Remodelling. Bishop JE, Reeves JT, Laurent GJ, editors. London: Portland Press Ltd; 1995. pp. 47–116. [Google Scholar]
- Jones R, Zapol WM, Reid L. Pulmonary artery remodeling and pulmonary hypertension after exposure to hyperoxia for 7 days. A morphometric and hemodynamic study. Am J Pathol. 1984;117:273–285. [PMC free article] [PubMed] [Google Scholar]
- Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest. 2000;106:1311–1319. doi: 10.1172/JCI10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamanna JC, Vendel LM, Farrell RM. Brain adaptation to chronic hypobaric hypoxia in rats. J Appl Physiol. 1992;72:2238–2243. doi: 10.1152/jappl.1992.72.6.2238. [DOI] [PubMed] [Google Scholar]
- Lockhart A, Zelter M, Mensch-Dechene J, Antezana G, Paz-Zamora M, Vargas E, Coudert J. Pressure-flow-volume relationships in pulmonary circulation of normal highlanders. J Appl Physiol. 1976;41:449–456. doi: 10.1152/jappl.1976.41.4.449. [DOI] [PubMed] [Google Scholar]
- Marti HH, Risau W. Systemic hypoxia changes the organ-specific distribution of vascular endothelial growth factor and its receptors. Proc Natl Acad Sci U S A. 1998;95:15809–15814. doi: 10.1073/pnas.95.26.15809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease. Part One. Am J Respir Crit Care Med. 1994a;150:833–852. doi: 10.1164/ajrccm.150.3.8087359. [DOI] [PubMed] [Google Scholar]
- MacNee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease. Part Two. Am J Respir Crit Care Med. 1994b;150:1158–1168. doi: 10.1164/ajrccm.150.4.7921453. [DOI] [PubMed] [Google Scholar]
- Marti HH, Risau W. Systemic hypoxia changes the organ-specific distribution of vascular endothelial growth factor and its receptors. Proc Natl Acad Sci U S A. 1998;95:15809–15814. doi: 10.1073/pnas.95.26.15809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti HH, Risau W. Angiogenesis in ischemic disease. Thromb Haemost. 1999;82(suppl. 1):44–52. [PubMed] [Google Scholar]
- Meyrick B, Hislop A, Reid L. Pulmonary arteries of the normal rat: the thick walled oblique muscle segment. J Anat. 1978;125:209–221. [PMC free article] [PubMed] [Google Scholar]
- Meyrick B, Reid L. The effect of continued hypoxia on rat pulmonary arterial circulation. An ultrastructural study. Lab Invest. 1978;38:188–200. [PubMed] [Google Scholar]
- Meyrick B, Reid L. Ultrastructural features of the distended pulmonary arteries of the normal rat. Anat Rec. 1979a;193:71–97. doi: 10.1002/ar.1091930106. [DOI] [PubMed] [Google Scholar]
- Meyrick B, Reid L. Hypoxia and incorporation of 3H-thymidine by cells of the rat pulmonary arteries and alveolar wall. Am J Pathol. 1979b;96:51–70. [PMC free article] [PubMed] [Google Scholar]
- Meyrick B, Reid L. Pulmonary hypertension. Anatomic and physiologic correlates. Clin Chest Med. 1983;4:199–217. [PubMed] [Google Scholar]
- Monacci WT, Merrill MJ, Oldfield EH. Expression of vascular permeability factor/vascular endothelial growth factor in normal rat tissues. Am J Physiol. 1993;264:C995–1002. doi: 10.1152/ajpcell.1993.264.4.C995. [DOI] [PubMed] [Google Scholar]
- Mooi W, Wagenvoort CA. Decreased numbers of pulmonary blood vessels: reality or artifact? J Pathol. 1983;141:441–447. doi: 10.1002/path.1711410403. [DOI] [PubMed] [Google Scholar]
- Olfert IM, Breen EC, Mathieu-Costello O, Wagner PD, O'Regan RG, McLoughlin P. Chronic hypoxia attenuates resting and exercise-induced VEGF, flt-1, and flk-1 mRNA levels in skeletal muscle. Am J Physiol Heart Circ Physiol. 2001;90:1532–1538. doi: 10.1152/jappl.2001.90.4.1532. [DOI] [PubMed] [Google Scholar]
- Ooi H, Cadogan E, Sweeney M, Howell K, O'Regan RG, McLoughlin P. Chronic hypercapnia inhibits hypoxic pulmonary vascular remodeling. Am J Physiol Heart Circ Physiol. 2000;278:H331–338. doi: 10.1152/ajpheart.2000.278.2.H331. [DOI] [PubMed] [Google Scholar]
- Patterson C, Perrella MA, Hsieh CM, Yoshizumi M, Lee ME, Haber E. Cloning and functional analysis of the promoter for KDR/flk-1, a receptor for vascular endothelial growth factor. J Biol Chem. 1995;270:23111–23118. doi: 10.1074/jbc.270.39.23111. [DOI] [PubMed] [Google Scholar]
- Partovian C, Adnot S, Raffestin B, Louzier V, Levame M, Mavier IM, Lemarchand P, Eddahibi S. Adenovirus-mediated lung vascular endothelial growth factor overexpression protects against hypoxic pulmonary hypertension in rats. Am J Respir Cell Mol Biol. 2000;23:762–771. doi: 10.1165/ajrcmb.23.6.4106. [DOI] [PubMed] [Google Scholar]
- Patan S, Tanda S, Roberge S, Jones RC, Jain RK, Munn LL. Vascular morphogenesis and remodeling in a human tumor xenograft: blood vessel formation and growth after ovariectomy and tumor implantation. Circ Res. 2001;89:732–739. doi: 10.1161/hh2001.097872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters KG, De Vries C, Williams LT. Vascular endothelial growth factor receptor expression during embryogenesis and tissue repair suggests a role in endothelial differentiation and blood vessel growth. Proc Natl Acad Sci U S A. 1993;90:8915–8919. doi: 10.1073/pnas.90.19.8915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitch M. EVE and beyond, retro and prospective insights. Am J Physiol. 1999;277:L5–12. doi: 10.1152/ajplung.1999.277.1.L5. [DOI] [PubMed] [Google Scholar]
- Rabinovitch M. Pathobiology of pulmonary hypertension. Extracellular matrix. Clin Chest Med. 2001;22:433–449. viii. doi: 10.1016/s0272-5231(05)70282-3. [DOI] [PubMed] [Google Scholar]
- Rabinovitch M, Gamble W, Nadas AS, Miettinen OS, Reid L. Rat pulmonary circulation after chronic hypoxia: hemodynamic and structural features. Am J Physiol. 1979;236:H818–827. doi: 10.1152/ajpheart.1979.236.6.H818. [DOI] [PubMed] [Google Scholar]
- Ryland D, Reid L. The pulmonary circulation in cystic fibrosis. Thorax. 1975;30:285–292. doi: 10.1136/thx.30.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandner P, Wolf K, Bergmaier U, Gess B, Kurtz A. Hypoxia and cobalt stimulate vascular endothelial growth factor receptor gene expression in rats. Pflügers Arch. 1997;433:803–808. doi: 10.1007/s004240050348. [DOI] [PubMed] [Google Scholar]
- Scherle W. A simple method for volumetry of organs in quantitative stereology. Mikroskopie. 1970;26:57–60. [PubMed] [Google Scholar]
- Schraufnagel DE, Sekosan M, McGee T, Thakkar MB. Human alveolar capillaries undergo angiogenesis in pulmonary veno-occlusive disease. Eur Respir J. 1996;9:346–350. doi: 10.1183/09031936.96.09020346. [DOI] [PubMed] [Google Scholar]
- Sime F, Penaloza D, Ruiz L. Bradycardia, increased cardiac output, and reversal of pulmonary hypertension in altitude natives living at sea level. Br Heart J. 1971;33:647–657. doi: 10.1136/hrt.33.5.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skwarski K, MacNee W, Wraith PK, Sliwinski P, Zielinski J. Predictors of survival in patients with chronic obstructive pulmonary disease treated with long-term oxygen therapy. Chest. 1991;100:1522–1527. doi: 10.1378/chest.100.6.1522. [DOI] [PubMed] [Google Scholar]
- Smith K, Marshall JM. Physiological adjustments and arteriolar remodelling within skeletal muscle during acclimation to chronic hypoxia in the rat. J Physiol. 1999;521:261–272. doi: 10.1111/j.1469-7793.1999.00261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark KR, Mecham RP. Cellular and molecular mechanisms of pulmonary vascular remodeling. Annu Rev Physiol. 1997;59:89–144. doi: 10.1146/annurev.physiol.59.1.89. [DOI] [PubMed] [Google Scholar]
- Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, McMahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15:427–438. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- Thet LA, Law DJ. Changes in cell number and lung morphology during early postpneumonectomy lung growth. J Appl Physiol. 1984;56:975–978. doi: 10.1152/jappl.1984.56.4.975. [DOI] [PubMed] [Google Scholar]
- Tuder RM, Flook BE, Voelkel NF. Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia. Modulation of gene expression by nitric oxide. J Clin Invest. 1995;95:1798–1807. doi: 10.1172/JCI117858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vock R, Weibel ER. Massive hemorrhage causes changes in morphometric parameters of lung capillaries and concentration of leukocytes in microvasculature. Exp Lung Res. 1993;19:559–577. doi: 10.3109/01902149309031728. [DOI] [PubMed] [Google Scholar]
- Weibel ER. Understanding the limitation of O2 supply through comparative physiology. Respir Physiol. 1999;118:85–93. doi: 10.1016/s0034-5687(99)00084-5. [DOI] [PubMed] [Google Scholar]
- Weibel ER, Federspiel WJ, Fryder-Doffe F, Hsia CC, Konig M, Stalder-Navarro V, Vock R. Morphometric model for pulmonary diffusing capacity. I. Membrane diffusing capacity. Respir Physiol. 1993;93:125–149. doi: 10.1016/0034-5687(93)90001-q. [DOI] [PubMed] [Google Scholar]
- Weibel ER, Knight BW. A morphometric study on the thickness of the pulmonary air-blood barrier. J Cell Biol. 1964;21:367–384. doi: 10.1083/jcb.21.3.367. [DOI] [PMC free article] [PubMed] [Google Scholar]