

Abstract
Glucocorticoids have been used for 30 years to accelerate fetal lung maturation in human pregnancy at risk of preterm delivery. Exposure to inappropriate levels of steroid, however, leads to altered maturation of the cardiovascular, metabolic and central nervous systems. The effects of betamethasone on neuronal development and function were determined in the fetal baboon brain by examination of cytoskeletal microtubule associated proteins (MAPs) and the presynaptic marker protein synaptophysin. At 0.73 gestation, commencing 28 weeks of gestation, pregnant baboons received four doses of saline (n = 8) or 87.5 µg (kg body weight)−1 betamethasone i.m. (n = 7) 12 h apart. This dose is equivalent to 12 mg betamethasone administered daily over two consecutive days to a 70 kg woman. Baboons underwent Caesarean section 12 h after the last injection. Paraffin sections of the fetal neocortex and the underlying white matter were labelled immunohistochemically against MAP1B, MAP2abc, MAP2ab and synaptophysin and stained histochemically with hematoxylin-eosin and silver. Tissue staining was quantified morphometrically. Betamethasone exposure resulted in decreased immunoreactivity (IR) of MAP1B by 34.3 % and MAP2abc by 34.1 % (P < 0.05). Loss of MAP2 IR was due to loss of IR of the juvenile isoform MAP2c (P < 0.05). MAP1B and MAP2c are involved in neuritogenesis and neuronal plasticity. Synaptophysin IR was reduced by 51.8 % (P < 0.01). These changes might reflect functional neuronal disturbances because they were not accompanied by an alteration of the density of neurofibrils or neuronal necrosis. These results are in agreement with earlier findings of alterations of cytoskeletal proteins and presynaptic terminals in the fetal sheep brain after betamethasone infusion directly to the fetus and support a common effect of inappropriate fetal exposure to glucocorticoids on neuronal cytoskeleton and synapses in mammalian species.
It is well established that the prepartum increase in fetal cortisol concentrations is essential for the normal maturation of the brain (for reviews see Meyer, 1985; De Kloet et al. 1988) as it is for other organs such as the lungs, adrenals, pituitary, liver and gut (for review see Fowden, 1995). Antenatal administration of synthetic glucocorticoids to pregnant women who threaten to deliver prematurely lessens the incidence of respiratory distress syndrome in their offspring (Liggins & Howie, 1972). Exposure of the fetus to levels of cortisol that are inappropriately high for the current stage of fetal development has been proposed to be a major link between adverse intrauterine conditions and the altered development of fetal tissues and organs that may lead to an increased prevalence of diseases in later life such as hypertension, cardiovascular disease and type II diabetes mellitus (Barker, 1998).
Overexposure to glucocorticoids in late gestation (Welberg et al. 2001) as well as prenatal stress, maternal deprivation and neonatal handling (Vallee et al. 1997; Takahashi, 1998; Weinstock, 2001) affect brain development in rats and have long-term effects on adult brain function. Significant behavioural changes have been shown to occur in the human fetus following maternal administration of synthetic glucocorticoids (Mulder et al. 1997; Senat et al. 1998). These behavioural changes are in agreement with our observation of acutely altered complex neuronal activity in the fetal sheep brain during intravenous infusion of betamethasone over 48 h to the fetus at 0.87 of gestation (Schwab et al. 2001b). Glucocorticoid-induced functional changes of electrocortical fetal brain activity were accompanied by an acute loss of microtubule associated proteins (MAPs) (Schwab et al. 2001a) and synaptophysin as a marker protein of presynaptic terminals (Antonow-Schlorke et al. 2001).
MAPs and synaptophysin are key proteins in brain morphogenesis and function. MAP1B and MAP2 are predominantly neuronal cytoskeletal tubulin binding proteins that determine stability and arrangement of the neuronal microtubules (Tucker, 1990). The age-related expression of MAP1B and MAP2 indicates a significant role in neuronal differentiation and neuritogenesis (Matus, 1994). MAP1B (350 kDa) is the major fetal MAP present in the embryonic brain (Tucker et al. 1988). The developmentally controlled decline of MAP1B parallels the process of synaptogenesis (Riederer et al. 1990). In the immature brain, MAP2 gene expression reveals two MAP2 isoforms, MAP2c (70 kDa), which is only present during brain development (Tucker, 1990), and MAP2b (350 kDa), which is continuously expressed during life (Riederer & Matus, 1985). In rats, down-regulation of MAP2c after birth parallels the up-regulation of another MAP2 isoform, MAP2a (350 kDa) (Riederer & Matus, 1985). Synaptophysin is a specific Ca2+ binding protein located in the synaptic vesicle membrane of presynaptic nerve endings in vertebrates (Wiedenmann & Franke, 1985). These vesicles are abundant in at least 95 % of all neocortical synapses (Navone et al. 1986). Thus, synaptophysin is a widely accepted cytochemical standard marker for nerve terminals (Jahn & De Camilli, 1991). It has been suggested that synaptophysin performs important functions in the generation and maintenance of small synaptic vesicles and their interaction with cytoskeletal elements (Johnston et al. 1989).
In the present study we wished to evaluate whether changes occurred in cytoskeletal proteins and presynaptic terminals in non-human primates that are similar to the pronounced changes observed in fetal sheep following exposure to inappropriately high levels of glucocorticoid (Schwab et al. 2001a; Antonow-Schlorke et al. 2001). Demonstration of similarities in brain morphogenesis of non-human primates and other mammals will enable more confident extrapolation of observations in non-human primates to the human fetal brain. The age of gestation at which we performed the present study is in the middle of the period of gestation at which human fetuses are exposed therapeutically to exogenous glucocorticoids. Additionally, we investigated glucocorticoid-induced changes in the density of neurofibrils to examine whether a loss of MAPs is accompanied by a loss of neuronal processes. Loss of MAP2 immunostaining has been shown to be associated with dendritic injury in neuronal trauma (Folkerts et al. 1998) and cerebral ischaemia (Matesic & Lin, 1994; Malinak & Silverstein, 1996).
In our previous sheep studies we infused betamethasone directly to the fetus to avoid differences in the placental transfer of betamethasone between the ovine and the human placenta. In the present study, we administered betamethasone intramuscularly to the mother at a daily dose equivalent on a weight adjusted basis to that given to pregnant women in threatened preterm labour. The non-human primate and human placenta show similar permeability to glucocorticoids as shown by the maternal to fetal concentration gradients (Walsh et al. 1984; Gitau et al. 1998). Effects of glucocorticoid exposure on organ maturation may differ between maternal admimistration and direct glucocorticoid administration to the fetus (Newnham et al. 1999).
METHODS
Experimental protocol
All procedures were approved by the Cornell University Animal Use and Care Committee. Pregnant baboons (Papio cynocephalus) of 12.1 ± 0.4 kg body weight (mean ± s.e.m.) were maintained for least 6 weeks in individual cages in sight of at least one other animal in rooms with controlled light-dark cycles (14 h light-10 h dark) and had free access to water and food (Teklad 25 % protein primate diet 2055, Harlan, Madison, WI, USA). Animals interacted at least twice a day with the same care-giver and were provided with fresh fruits and vegetables. Four injections of 87.5 µg (kg body weight)−1 betamethasone (Celestone phosphate, Schering, Kenilworth, NJ, USA; n = 7) or of the equivalent volume of saline (n = 8) were administered intramuscularly (i.m.) 12 h apart beginning at 134 ± 1 dGA (mean ± s.e.m.; 0.73 of gestation equivalent to 28 weeks of gestation in humans). This dose of betamethasone (175 µg per day) is equivalent to the clinically used dose regimen (12 mg per day) over two consecutive days when weight adjusted to a 70 kg pregnant woman. Twelve hours after the fourth injection animals were premedicated with 0.0075 mg kg−1 buprenorphine (Reckitt & Coleman, Richmond, VA, USA) and 0.0125 mg kg−1 glycopyrrolate (Robinul, Baxter, Deerfield, IL, USA) i.m. Ketamine at 10–15 mg kg−1 (KetaFlo, Abbott, North Chicago, IL, USA) was administered i.m. and general anaesthesia was initiated with halothane and maintained with 1.5 % halothane (Halothane, Halocarbon, River Edge, NJ, USA) in 1–2 l min−1 O2. Fetuses were delivered by Caesarean section and killed by exsanguination while still under halothane general anaesthesia. Mothers recovered from surgery in individual cages and were treated with 0.015 mg kg−1 per day buprenorphine in two equal i.m. injections for 3 days, and given antibiotic treatment with clavamox orally, 30 mg kg−1 day−1, in equally divided doses, twice a day in peanut butter for five days. After recovery in individual cages the mothers were returned to the social context of gang cages.
Histological processing
Fetal brains were perfusion-fixed via the carotid artery with a neutrally buffered solution containing 2.5 % Acrolein (Sigma, St Louis, MO, USA) and 4 % paraformaldehyde (Sigma). After about 2 h brains were removed from the skull and stored in 1 % buffered paraformaldehyde until they were embedded in paraffin. Serial brain slices of 6 µm thickness were cut from the frontal cortex at 10 mm from the anterior pole and stained immunohistochemically with monoclonal antibodies against MAP1B (mouse anti-MAP1B, 1:500, Sigma), MAP2 (mouse anti-MAP2abc, 1:500, mouse anti-MAP2ab, 1:500, Sigma) and synaptophysin (mouse anti-synaptophysin, 1:2000, Sigma) using the ABC-technique (Vectastain Elite ABC staining kit, Vector Labs, Burlingame, CA, USA). After dewaxing, tissue slices were treated with 0.3 % hydrogen peroxide and incubated with 1.5 % normal horse serum (Vector) to minimise background activity. The primary antibody was added overnight (4 °C), followed by a secondary biotinylated antibody for 2 h (1:200, 37 °C, Vector). After incubating the tissue with a preformed avidin-horseradish peroxidase complex (Vector) immunostaining was developed by diaminobenzidine (Sigma) resulting in a brown precipitate. Slices were counterstained with hematoxylin to visualise cell bodies. Additional, conventional histochemical staining with haematoxylin- eosin was used to discriminate irreversible neuronal damage by typical features such as triangular shape and presence of a dense eosinophilic nucleus. Histochemical staining of neurofibrils was performed by silver staining according to Servier & Munger (1965).
Quantitative image analysis
Quantification of tissue staining was performed morphometrically by an individual (I. A.-S.) blinded to the experimental protocol. Immunoreactivity (IR) of synaptophysin and MAPs was estimated within the cortical layers III and IV, and the amount of neurofibrils was estimated in the cortical white matter. Light microscopic micrographs at × 150 magnification were digitised using a 3CCD colour video camera (Sony, MC3215) and areas of tissue staining were analysed using an image analysis program (Scion Image 6.21, NIH, USA). For each animal and histochemical staining, three fields of 0.2 mm2 per section were measured and the results were averaged. Background immunoreactivity was estimated by negative-staining controls. Measurements were standardised using white calibration. A blue filter was used to prevent signals resulting from counterstaining with haematoxylin. The amount of IR of the MAP2c isoform was assessed by subtraction of the IR values of MAP2abc and MAP2ab of serial tissue slices (Schwab et al. 2001a). Statistical significance between the values of the vehicle- and betamethasone-treated groups was tested by the Mann-Whitney U test with the significance level of P < 0.05. Results are presented as means ± s.e.m..
RESULTS
Fetal weights were 532.9 ± 48.5 g in the vehicle- and 436.1 ± 36.7 g in the betamethasone-treated group and did not differ significantly between both experimental groups.
MAP1B and MAP2 were demonstrated immunohistochemically in the frontal neocortex of the fetal baboon brain. Both MAP1B and MAP2 IR stained neuronal perikarya and processes. MAP2c contributed 39.9 ± 2.1 % of total MAP2 isoforms. The immunohistochemical distribution of synaptophysin showed a widespread granular and dense staining pattern within the grey matter (Fig. 1).
Figure 1. Representative photomicrographs of the cerebral neocortex of the fetal baboon.
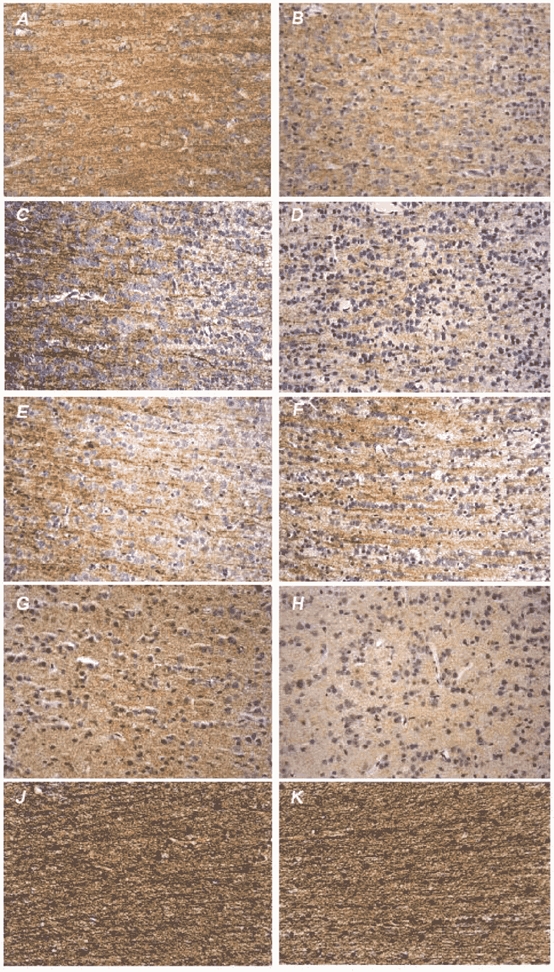
Left: fetus of vehicle-treated pregnant baboon; right: fetus of betamethasone-treated pregnant baboon. Immunohistochemical staining (brown precipitate, hematoxylin counterstaining to visualise cell nuclei) of microtubule-associated proteins MAP1B (A and B), MAP2abc (C and D), MAP2ab (E and F) and presynaptic protein synaptophysin (G and H). J and K, histochemical staining (black precipitate) of argyrophil neurofibrils. All photomicrographs were taken at × 320 magnification. Note the loss of MAP1B, MAP2abc and synaptophysin IR after antenatal betamethasone exposure in the absence of neuronal necrosis. The amount of the high molecular weight isoform MAP2ab remained unchanged. Histochemical staining of neurofibrils of the cortical white matter did not demonstrate effects of betamethasone.
At 12 h after the last betamethasone injection, antenatal exposure to betamethasone resulted in an acute loss of MAP1B IR by 34.3 % and of MAP2 IR by 34.1 % in the frontal neocortex of fetal baboon brain (P < 0.05, Fig. 1 and Fig. 2). Loss of MAP2 IR was due to a loss of MAP2c IR (P < 0.05, Fig. 1 and Fig. 2). MAP2ab IR remained unchanged (Fig. 1 and Fig. 2). Antenatal betamethasone exposure also reduced synaptophysin IR by 51.8 % (P < 0.01, Fig. 1 and Fig. 2).
Figure 2. Effect of antenatal betamethasone (βM) treatment on the cerebral neocortex of the fetal baboon.
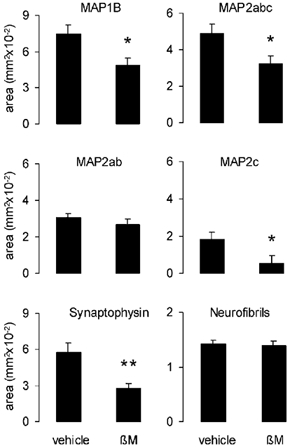
Area of immunostaining of microtubule-associated proteins MAP1B, MAP2abc and presynaptic vesicle membrane protein synaptophysin was reduced significantly by betamethasone. Three fields of 0.2 mm2 per section were measured for each animal and immunohistochemical marker protein. Loss of MAP2 IR was due to loss of the isoform MAP2c. Histochemical staining of neurofibrils did not show any alteration. Vehicle-exposed fetuses n = 8; betamethasone-exposed fetuses n = 7; means ±s.e.m.; *P < 0.05, **P < 0.01.
No indication of neuronal necrosis was observed in the fetal baboon neocortex after betamethasone administration (Fig. 2). Antenatal betamethasone exposure did not alter the density of neurofibrils in the cortical white matter (Fig. 1 and Fig. 2).
DISCUSSION
To our knowledge, this is the first demonstration of the cytoskeletal proteins MAP1B and the isoforms of MAP2 in the fetal brain of non-human primates. Fetal exposure to betamethasone over 48 h at the weight adjusted dose used clinically to enhance fetal lung maturation acutely affects neuronal cytoskeletal proteins MAP1B and MAP2 and presynaptic terminals in the fetal brain of the non-human primate at 0.73 of gestation. These changes were not accompanied by an acute loss of neuronal processes or irreversible neuronal damage.
The juvenile MAP1B that is supposed to be expressed primarily in the fetal period (Tucker et al. 1988) appeared as the predominant MAP. The considerable levels of juvenile MAP1B and MAP2c, cytoskeletal proteins that are essential to establish synapses (Tucker, 1990), observed in our study indicate that cytoskeletal maturation and synaptogenesis have not been completed by 0.73 gestation in the fetal baboon brain. This finding is in agreement with another study showing a continuous increase of MAP2 in the primary visual cortex of macaque monkeys from 75 dGA to birth (Mehra & Hendrickson, 1993). In that study, however, the isoforms of MAP2 were not differentiated and the development of MAP1B was not examined. Maturation of the human neuronal cytoskeleton has been reported to continue until the second postnatal year (Arnold & Trojanowski, 1996). In our study, the dense immunohistochemical distribution of synaptophysin demonstrates the existence of a considerable number of synapses in the fetal baboon brain at 0.73 of gestation. The developmental rise of synaptophysin is closely linked with the time course of synaptogenesis (Grunnet, 1995). The granular pattern of synaptophysin IR is associated with clusters of synaptic vesicles in the synaptic terminals (Wiedenmann & Franke, 1985; Navone et al. 1986; Leclerc et al. 1989). In macaque monkeys, the number of cortical synapses increases rapidly during the last 2 months of gestation (Rakic et al. 1986).
The exact mechanisms responsible for the loss of MAPs and synaptophysin IR are not known. Cerebral blood flow decreases also following betamethasone exposure in fetal sheep (Schwab et al. 2000) but it did not correlate well with the loss of immunostaining for MAP1B and MAP2 (Schwab et al. 2001a). Thus, we favour the view that the glucocorticoid effects described here reflect a direct action on the neurons rather than effects secondary to a decrease in cerebral blood flow.
The selective binding of the synthetic glucocorticoid betamethasone to glucocorticoid receptors (GRs) and its higher biological potency in comparison to cortisol (Yang et al. 1990) will shift the ratio of activated mineralocorticoid receptors and GRs in the direction of predominant activation of GRs. Thus, betamethasone may cause overactivation of GRs with potentially harmful effects. GRs have been demonstrated in most brain regions including the cerebral cortex (Cintra et al. 1994). Excessive GR activation contributes to neurotoxicity in rats (Packan & Sapolsky, 1990; Hassan et al. 1996) and non-human primates in which it induces dendrite degeneration and depletion of pyramidal and dentate granular cells in the hippocampus (Uno et al. 1990).
Alteration of MAPs and synaptophysin may have functional relevance. MAP1B and MAP2c, whose abundance was diminished by betamethasone, are involved in neuronal plasticity during neuronal outgrowth and synaptogenesis (Gordon-Weeks, 1997). Synaptophysin is potentially involved in vesicle release (Thomas et al. 1988). Therefore, alteration of synaptophysin may result in a disturbance of synaptic transmission. The changes in the abundance of MAPs and synaptophysin may contribute to the altered neuronal activity in the fetal sheep brain (Schwab et al. 2001b) and to behavioural changes shown in human infants (Mulder et al. 1997; Senat et al. 1998) after antenatal glucocorticoid exposure. We do not know whether the alterations of neuronal proteins that are necessary for normal neuronal morphology and function shown here are reversible. It has been reported that loss of MAP2 correlates with neuronal degeneration (Matesic & Lin, 1994). Alteration of the cytoskeleton was found to be a major step in the initiation of apoptosis in neurons (Bonfoco et al. 1995). In the present study, the lack of any effect on the amount of neurofibrils indicates that loss of MAPs is not associated with loss of neuronal processes. Further investigations of persistence or recovery of these changes after different time intervals of observation are necessary to clarify whether the alterations of MAPs and presynaptic terminals we report here have a long-term effect on brain development.
Considering the similar effects we demonstrated in the late gestation fetal sheep after intravenous fetal betamethasone administration (Antonow-Schlorke et al. 2001; Schwab et al. 2001a), the acute alterations of cytoskeletal proteins and presynaptic terminals appear to be independent of the mammalian species and the route of betamethasone administration. An extensive loss of MAP2 IR has also been reported in adult rats in the brain regions investigated (striatum, hippocampus) 24 h after a single i.p. injection of 0.7 mg (kg body weight)−1 dexamethasone (Haynes et al. 2001). Moreover, the glucocorticoid effects on the neuronal cytoskeleton and on the presynaptic terminals seem to occur relatively independently of the gestational age during the last third of gestation. We showed these effects in previous studies in the precocious sheep brain at 0.87 gestation (Antonow-Schlorke et al. 2001; Schwab et al. 2001a) and in the present study in primates at 0.73 gestation. The similar glucocorticoid effects on sheep and baboon brain, the design of our study in a primate species mimicking the clinical situation in all aspects, i.e. regarding gestational age, dose and route of betamethasone administration, and the use of a non-instrumented fetus make it highly likely that similar effects occur in the human fetal brain.
In summary, the present study demonstrates acute structural alterations of the neuronal cytoskeleton and presynaptic terminals in the immature primate brain following exposure to therapeutically used levels of glucocorticoid at 0.73 gestation. The betamethasone dose therapeutically used may represent an inappropriate exposure of the fetal brain to glucocorticoids that clearly has the potential to alter brain structures. The alteration of proteins that are essential for brain development and neuronal function may contribute to the programming of life-time brain function. Programming has been described to occur following many sub-optimal situations that occur during mammalian pregnancy since these have been shown to depend at least in part on increased maternal secretion and transplacental passage of glucocortioids (Barbazanges et al. 1996; Gardner et al. 1997).
Acknowledgments
We are grateful to Dr Xiu-Ying Ding for skilful surgical assistance and Claudia Hiepe and Renate Klupsch for excellent technical assistance in histological processing. This work was supported by HL 55416, NS 34805, BMBF 01ZZ0105 and the IZKF ‘Clinical Neuroscience’ at the Friedrich Schiller University Jena.
REFERENCES
- Antonow-Schlorke I, Kühn B, Müller T, Schubert H, Sliwka U, Nathanielsz PW, Schwab M. Effect of antenatal betamethasone treatment on density of presynaptic terminals in the fetal sheep. Neurosci Lett. 2001;297:147–150. doi: 10.1016/s0304-3940(00)01605-0. [DOI] [PubMed] [Google Scholar]
- Arnold SE, Trojanowski JQ. Human fetal hippocampal development: II The neuronal cytoskeleton. J Comp Neurol. 1996;367:293–307. doi: 10.1002/(SICI)1096-9861(19960401)367:2<293::AID-CNE10>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Barbazanges A, Piazza PV, Le Moal M, Maccari S. Maternal glucocorticoid secretion mediates long-term effects of prenatal stress. J Neurosci. 1996;16:3943–3949. doi: 10.1523/JNEUROSCI.16-12-03943.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJP. Mothers, Babies and Health in Later Life. Edinburgh: Churchill Livingstone; 1998. [Google Scholar]
- Bonfoco E, Ceccatelli S, Manzo L, Nicotera P. Colchicine induces apoptosis in cerebellar granule cells. Exp Cell Res. 1995;218:189–200. doi: 10.1006/excr.1995.1147. [DOI] [PubMed] [Google Scholar]
- Cintra A, Zoli M, Rosén L, Agnati LF, Okret S, Wikström AC, Gustafsson JA, Fuxe K. Mapping and computer assisted morphometry and microdensitometry of glucocorticoid receptor immunoreactive neurons and glial cells in the rat central nervous system. Neuroscience. 1994;62:843–897. doi: 10.1016/0306-4522(94)90481-2. [DOI] [PubMed] [Google Scholar]
- De Kloet ER, Rosenfeld P, Van Eekelen JA, Sutanto W, Levine S. Stress glucocorticoids and development. Prog Brain Res. 1988;73:101–120. doi: 10.1016/S0079-6123(08)60500-2. [DOI] [PubMed] [Google Scholar]
- Folkerts MM, Berman RF, Muizelaar JP, Rafols JA. Disruption of MAP-2 immunostaining in rat hippocampus after traumatic brain injury. J Neurotrauma. 1998;15:349–363. doi: 10.1089/neu.1998.15.349. [DOI] [PubMed] [Google Scholar]
- Fowden AL. Endocrine regulation of fetal growth. Reprod Fertil Dev. 1995;7:351–363. doi: 10.1071/rd9950351. [DOI] [PubMed] [Google Scholar]
- Gardner DS, Jackson AA, Langley-Evans SC. Maintenance of maternal diet-induced hypertension in the rat is dependent on glucocorticoids. Hypertension. 1997;30:1525–1530. doi: 10.1161/01.hyp.30.6.1525. [DOI] [PubMed] [Google Scholar]
- Gitau R, Cameron A, Fisk NM, Glover V. Fetal exposure to maternal cortisol. Lancet. 1998;352:707–708. doi: 10.1016/S0140-6736(05)60824-0. [DOI] [PubMed] [Google Scholar]
- Gordon-Weeks PR. MAPs in growth cones. In: Avila J, Brandt R, Kosik KS, editors. Brain Microtubule Associated Proteins. Modifications in Disease. Sydney: Harwood Academic Publishers; 1997. pp. 53–72. [Google Scholar]
- Grunnet ML. A lectin and synaptophysin study of developing brain. Ped Neurol. 1995;13:157–160. doi: 10.1016/0887-8994(95)00147-8. [DOI] [PubMed] [Google Scholar]
- Hassan AH, Von Rosenstiel P, Patchev VK, Holsboer F, Almeida OF. Exacerbation of apoptosis in the dentate gyrus of the aged rat by dexamethasone and the protective role of corticosterone. Exp Neurol. 1996;140:43–52. doi: 10.1006/exnr.1996.0113. [DOI] [PubMed] [Google Scholar]
- Haynes LE, Griffiths MR, Hyde RE, Barber DJ, Mitchell IJ. Dexamethasone induces limited apoptosis and extensive sublethal damage to specific subregions of the striatum and hippocampus: implications for mood disorders. Neuroscience. 2001;104:57–69. doi: 10.1016/s0306-4522(01)00070-7. [DOI] [PubMed] [Google Scholar]
- Jahn R, De Camilli P. Membrane proteins of synaptic vesicles: markers for neurons and endocrine cells; tools for the study of neurosecretion. In: Gratzl M, Langley K, editors. Markers for Neural and Endocrine Cells Molecular and Cell Biology Diagnostic Applications. Weinheim: VCH; 1991. pp. 25–92. [Google Scholar]
- Johnston PA, Jahn R, Südhof TC. Transmembrane topography and evolutionary conservation of synaptophysin. J Biol Chem. 1989;264:1268–1273. [PubMed] [Google Scholar]
- Leclerc N, Beesley PW, Brown I, Colonnier M, Gurd JW, Paladino T, Hawkes R. Synaptophysin expression during synaptogenesis in the rat cerebellar cortex. J Comp Neurol. 1989;280:197–212. doi: 10.1002/cne.902800204. [DOI] [PubMed] [Google Scholar]
- Liggins GC, Howie RN. A controlled trial of antepartum glucocorticoid treatment for the prevention of the respiratory distress syndrome in premature infants. Pediatrics. 1972;50:515–525. [PubMed] [Google Scholar]
- Malinak C, Silverstein FS. Hypoxic-ischemic injury acutely disrupts microtubule-associated protein 2 immunostaining in neonatal rat brain. Biol Neonate. 1996;69:257–267. doi: 10.1159/000244319. [DOI] [PubMed] [Google Scholar]
- Matesic DF, Lin R. Microtubule-associated protein 2 as an early indicator of ischemia-induced neurodegeneration in the gerbil forebrain. J Neurochem. 1994;63:1012–1020. doi: 10.1046/j.1471-4159.1994.63031012.x. [DOI] [PubMed] [Google Scholar]
- Matus A. MAP2. In: Hyams JS, Lloyd CW, editors. Microtubules. New York: Wiley-Liss; 1994. pp. 155–166. [Google Scholar]
- Mehra RD, Hendrickson AE. A comparison of the development of neuropeptide and MAP2 immunocytochemical labeling in the macaque visual cortex during pre- and postnatal development. J Neurobiol. 1993;24:101–124. doi: 10.1002/neu.480240109. [DOI] [PubMed] [Google Scholar]
- Meyer JS. Biochemical effects of corticosteroids on neural tissues. Physiol Rev. 1985;65:946–1020. doi: 10.1152/physrev.1985.65.4.946. [DOI] [PubMed] [Google Scholar]
- Mulder EJ, Derks JB, Visser GH. Antenatal corticosteroid therapy and fetal behaviour: a randomised study of the effects of betamethasone and dexamethasone. Brit J Obstet Gynaecol. 1997;104:1239–1247. doi: 10.1111/j.1471-0528.1997.tb10969.x. [DOI] [PubMed] [Google Scholar]
- Navone F, Jahn R, DiGioia G, Stukenbrok H, Greengard P, De Camilli P. Protein p38: an integral membrane protein specific for small vesicles of neurons and neuroendocrine cells. J Cell Biol. 1986;103:2511–2527. doi: 10.1083/jcb.103.6.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newnham JP, Evans SF, Godfrey M, Huang W, Ikegami M, Jobe A. Maternal but not fetal administration of corticosteroids restricts fetal growth. J Mat Fet Med. 1999;8:81–87. doi: 10.1002/(SICI)1520-6661(199905/06)8:3<81::AID-MFM3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Packan DR, Sapolsky RM. Glucocorticoid endangerment of the hippocampus: tissue steroid and receptor specificity. Neuroendocrinology. 1990;51:613–618. doi: 10.1159/000125400. [DOI] [PubMed] [Google Scholar]
- Rakic P, Bourgeois JP, Eckenhoff MF, Zecevic N, Goldman-Rakic PS. Concurrent overproduction of synapses in diverse regions of the primate cerebral cortex. Science. 1986;232:232–235. doi: 10.1126/science.3952506. [DOI] [PubMed] [Google Scholar]
- Riederer BM, Guadano-Ferraz A, Innocenti GM. Difference in distribution of microtubule-associated proteins 5a and 5b during the development of cerebral cortex and corpus callosum in cats: dependence on phosphorylation. Dev Brain Res. 1990;56:235–243. doi: 10.1016/0165-3806(90)90088-g. [DOI] [PubMed] [Google Scholar]
- Riederer B, Matus A. Differential expression of distinct microtubule-associated proteins during brain development. Proc Natl Acad Sci U S A. 1985;82:6006–6009. doi: 10.1073/pnas.82.17.6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM. Glucocorticoids, hippocampal damage and the glutamatergic synapse. Prog Brain Res. 1990;86:13–23. doi: 10.1016/s0079-6123(08)63163-5. [DOI] [PubMed] [Google Scholar]
- Schwab M, Antonow-Schlorke I, Kühn B, Müller T, Schubert H, Walter B, Sliwka U, Nathanielsz PW. Effect of antenatal betamethasone treatment on microtubule associated proteins MAP1B and MAP2 in fetal sheep. J Physiol. 2001a;530:497–506. doi: 10.1111/j.1469-7793.2001.0497k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M, Roedel M, Anwar MA, Müller T, Schubert H, Buchwalder LF, Walter B, Nathanielsz PW. Effects of betamethasone administration to the fetal sheep in late gestation on fetal cerebral blood flow. J Physiol. 2000;528:619–632. doi: 10.1111/j.1469-7793.2000.00619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M, Schmidt K, Roedel M, Müller T, Schubert H, Anwar MA, Nathanielsz PW. Non-linear changes of electrocortical activity after antenatal betamethasone treatment in fetal sheep. J Physiol. 2001b;531:535–543. doi: 10.1111/j.1469-7793.2001.0535i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senat MV, Minoui S, Multon O, Fernandez H, Frydman R, Ville Y. Effects of dexamethasone and betamethasone on fetal heart rate variability in preterm labour: a randomised study. Br J Obstet Gynaecol. 1998;105:749–755. doi: 10.1111/j.1471-0528.1998.tb10206.x. [DOI] [PubMed] [Google Scholar]
- Servier AC, Munger BL. A silver method for paraffin sections of neural tissue. J Neuropathol Exp Neurol. 1965;24:130–135. doi: 10.1097/00005072-196501000-00012. [DOI] [PubMed] [Google Scholar]
- Takahashi LK. Prenatal stress: consequences of glucocorticoids on hippocampal development and function. Int J Dev Neurosci. 1998;16:199–207. doi: 10.1016/s0736-5748(98)00020-3. [DOI] [PubMed] [Google Scholar]
- Thomas L, Hartung K, Langosch D, Rehm H, Bamberg E, Franke WW, Betz H. Identification of synaptophysin as a hexameric channel protein of the synaptic vesicle membrane. Science. 1988;242:1050–1053. doi: 10.1126/science.2461586. [DOI] [PubMed] [Google Scholar]
- Tucker RP. The roles of microtubule-associated proteins in brain morphogenesis: a review. Brain Res Rev. 1990;15:101–120. doi: 10.1016/0165-0173(90)90013-e. [DOI] [PubMed] [Google Scholar]
- Tucker RP, Binder LI, Matus AI. Neuronal microtubule-associated proteins in the embryonic avian spinal cord. J Comp Neurol. 1988;27:44–55. doi: 10.1002/cne.902710106. [DOI] [PubMed] [Google Scholar]
- Uno H, Lohmiller L, Thieme C, Kemnitz JW, Engle MJ, Roecker EB, Farrell PM. Brain damage induced by prenatal exposure to dexamethasone in fetal rhesus macaques. I Hippocampus Dev Brain Res. 1990;53:157–167. doi: 10.1016/0165-3806(90)90002-g. [DOI] [PubMed] [Google Scholar]
- Vallée M, Mayo W, Dellu F, Le Moal M, Simon H, Maccari S. Prenatal stress induces high anxiety and postnatal handling induces low anxiety in adult offspring: correlation with stress-induced corticosterone injection. J Neurosci. 1997;17:2626–2636. doi: 10.1523/JNEUROSCI.17-07-02626.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh SW, Ducsay CA, Novy MJ. Circardian hormonal interactions among the mother fetus and amniotic fluid. Am J Obstet Gynecol. 1984;150:745–753. doi: 10.1016/0002-9378(84)90679-3. [DOI] [PubMed] [Google Scholar]
- Weinstock M. Alterations induced by gestational stress in brain morphology and behaviour of the offspring. Prog Neurobiol. 2001;65:427–451. doi: 10.1016/s0301-0082(01)00018-1. [DOI] [PubMed] [Google Scholar]
- Welberg LA, Seckl JR, Holmes MC. Prenatal glucocorticoid programming of brain corticosteroid receptors and corticotrophin-releasing hormone: possible implications for behaviour. Neuroscience. 2001;104:71–79. doi: 10.1016/s0306-4522(01)00065-3. [DOI] [PubMed] [Google Scholar]
- Wiedenmann B, Franke WW. Identification and localization of synaptophysin an integral membrane glycoprotein of Mr 38000 characteristic of presynaptic vesicles. Cell. 1985;41:1017–1028. doi: 10.1016/s0092-8674(85)80082-9. [DOI] [PubMed] [Google Scholar]
- Yang K, Jones SA, Challis JR. Changes in glucocorticoid receptor number in the hypothalamus and pituitary of the sheep fetus with gestational age and after adrenocorticotropin treatment. Endocrinology. 1990;126:11–17. doi: 10.1210/endo-126-1-11. [DOI] [PubMed] [Google Scholar]