

Abstract
In mammals, the autonomic nervous system mediates the central circadian clock oscillation from the suprachiasmatic nucleus (SCN) to the peripheral organs, and controls cardiovascular, respiratory and gastrointestinal functions. The present study was conducted in mice to address whether light signals conveyed to the SCN can control peripheral autonomic functions, and further examined the impact of centrally administered melatonin on peripheral autonomic functions via activation of melatonin receptor signalling. In vivo electrophysiological techniques were performed in anaesthetised, open-chest and artificially ventilated mice whilst monitoring the arterial blood pressure and heart rate. Light induced an increase of the renal sympathetic nerve activity, arterial blood pressure and heart rate immediately after lights on. Conversely, light rapidly suppressed the gastric vagal parasympathetic nerve activity, which was affected neither by hepatic vagotomy nor by total subdiaphragmatic vagotomy. These autonomic responses were mediated by the SCN since bilateral SCN lesion totally abolished the light-evoked neuronal and cardiovascular responses. Melatonin administered intracerebroventricularly (i.c.v.) attenuated the sympathetic and vagal nerve activities in a dose-dependent manner with a threshold of 0.1 ng and these effects were blocked by i.c.v. pre-treatment of the competitive melatonin receptor antagonist luzindole. These results suggest that light induces sympathoexcitation and vagal suppression through the SCN and that melatonin modulates the light-induced autonomic responses via activation of the central melatonin receptor signalling.
In mammals, most physiological and behavioural events are subjected to well-controlled daily oscillations, and these rhythms are generated by an internal self-sustained molecular oscillator referred to as the biological clock (Moore, 1997). It is now well established that the central oscillator of mammals resides in the hypothalamic suprachiasmatic nucleus (SCN; Hastings et al. 1997), and recent molecular dissection of the biological clock has revealed that the core oscillator of the master clock in the SCN is composed of interacting positive and negative transcription/translation feedback loops of clock genes (Dunlap, 1999; Reppert & Weaver, 2001; Young & Kay, 2001). Although these studies revealed the oscillatory mechanism at the cellular level, it is still unknown how these clock signals are transmitted from the SCN to peripheral tissues. The autonomic nervous system is a candidate that links neuronal (Inouye & Kawamura, 1979) or humoral (Silver et al. 1996) signals from the SCN to the periphery (Moore, 1996; Buijs et al. 1999; Teclemariam-Mesbah et al. 1999), and is thus suspected to play a fundamental role in the circadian homeostasis of sleep-wake cycles, as well as cardiovascular, respiratory and gastrointestinal functions (Burgess et al. 1997; Scheer et al. 2001).
The circadian-autonomic interactions are affected by light stimuli known as the most important ‘zeitgeber’ (Pittendrigh & Daan, 1976). There is accumulating evidence that light responses transmitted to the SCN through the retinohypothalamic tract are conveyed to peripheral organs via autonomic nuclei and nerves. A pioneering work on the photic regulation of the sympathetic and parasympathetic nerves in rats suggests that electrical lesion of the anterior hypothalamus can block light-induced autonomic modulation (Niijima et al. 1993). The ACTH-independent, acute suppression of corticosterone by light has also been suggested to be transmitted via the autonomic innervation to the adrenal cortex (Buijs et al. 1999).
One of the best-characterised systems involving interactions between the SCN and the autonomic system is the noradrenergic regulation of pineal function. Circadian or photic SCN signals pass through a multisynaptic noradrenergic autonomic pathway and regulate the activity of pineal rate-limiting enzyme in melatonin synthesis, the arylalkylamine N-acetyltransferase (AA-NAT), at transcriptional and post-transcriptional levels (Klein, 1985; Stehle et al. 2001). The production of melatonin is high during the dark phase and melatonin production is acutely suppressed by light stimuli presented to the animals at night (Illnerova, 1991). Secreted melatonin not only regulates peripheral organs (Cagnacci, 1996), but also transmits temporal information to the brain through melatonin receptors in the SCN and other brain regions (Vanecek, 1998) to mediate a variety of physiological responses (Hagan & Oakley, 1995; Dubocovich et al. 1999). In mammals, melatonin activates at least two distinct high-affinity membrane-bound receptors, the MT1 and MT2 receptors. Recent gene cloning of melatonin receptors has shown that these melatonin receptor subtypes, which are negatively coupled to adenylyl cyclase via Gi proteins, are encoded by separate genes (Reppert et al. 1994, 1995). Both the MT1 and MT2 melatonin receptor mRNAs are expressed within the rodent SCN (Liu et al. 1997; Dubocovich et al. 1998b) and phase-shifting effects of melatonin on the SCN circadian clock at both dusk and dawn are mediated by activation of MT2 melatonin receptor signalling through protein kinase C (PKC) (McArthur et al. 1997; Hunt et al. 2001). The presence of functional melatonin receptors in the SCN (Dubocovich et al. 1998a) provides further evidence for the existence of melatonin-mediated systemic feedback system in the brain.
The present study using a murine model in vivo was conducted to address whether light signals conveyed to the SCN can control peripheral autonomic functions, and it further examined the impact of centrally administered melatonin on peripheral autonomic functions via activation of melatonin receptor signalling. The results indicate that (1) photic stimulation through the SCN increases the renal sympathetic nerve activity (RSNA), arterial blood pressure (ABP) and heart rate (HR), but suppresses the gastric vagal parasympathetic nerve activity (GVNA) in an intensity-dependent manner and (2) central melatonin suppresses these light-induced autonomic modifications, which were totally blocked by central administration of competitive melatonin receptor antagonist luzindole or by bilateral SCN lesion.
METHODS
All experimental protocols in this work were reviewed and approved by the Institutional Animal Care and Use Committee in accordance with the Guidelines for Animal Experimentation at Kobe University.
Animals
Male BALB/c mice were purchased at 6 weeks of age from a commercial source (Japan Animal Care, Osaka, Japan). The mice were housed in groups of four animals per cage (31 cm × 26 cm cross-sectional area), supplied with mice chow and water ad libitum, and maintained at 24 °C under standard laboratory conditions with 12 h light (fluorescent light, 1.5 × 1014 photons cm−2 s−1)-12 h dark (LD) cycles (lights on 06:00 h to 18:00 h). At 9–11 weeks of age, we conducted the electrophysiological study for examining the effect of light (n = 52), melatonin (n = 14) and both light and melatonin (n = 112). For each series of trials we used 6–7 animals; the same animals were not used for each trial.
General animal preparation
Each mouse was anaesthetised with an injection of urethane (1.4 g kg−1i.p.) and supplementary doses were given as needed. Polyethylene catheters (PE-10 fused with PE-50) filled with heparinised saline (50 i.u. ml−1) were introduced into the femoral vein for administering fluids and drugs, and into the femoral artery for monitoring ABP and for withdrawing samples for arterial blood gas analyses, respectively. Heart rate was monitored with a cardiotachometer (AT-601G, Nihon Koden, Tokyo, Japan) triggered by an ECG signal (lead II). The trachea was exposed and cannulated with a tracheal tube (0.58 mm i.d.). Each mouse was prepared with bilateral pneumothoraces by incisions made in the chest wall and mechanically ventilated with oxygen-enriched humidified air with a tidal volume of 8 ml kg−1 using a Harvard ventilator (Model 687, South Natick, MA, USA) to maintain blood gases and pH within a narrow range (pH = 7.34 ± 0.01, arterial PO2 was 356 ± 7 mmHg, arterial PCO2 was 42.8 ± 0.4 mmHg) by adjusting the ventilator rate and by infusing sodium bicarbonate.
The ventilator rate was set initially at 100–120 breaths min−1, and the positive end-expiratory pressure was set at 2 cm H2O. Each animal was administered the neuromuscular blocker pancuronium bromide (0.15 mg kg−1i.v.). Additional doses were given i.v. with an infusion pump (0.05 mg kg−1 h−1 at a rate of 2 µl g−1 h−1. Before neuromuscular blockade, adequacy of anaesthesia was determined every half-hour by pinching the hind limb paw and monitoring for hind limb flinch or withdrawal or sudden fluctuation of ABP (> 5 mmHg) or HR (> 10 %). During neuromuscular blockade, adequacy of anaesthesia was tested every half-hour by determining spontaneous or paw pinch-evoked fluctuations or increases in ABP (> 5 mmHg) or in HR (> 10 %) (Mutoh et al. 2000a,b). When any one of these responses was observed, a supplemental dose of urethane (0.2–0.4 g kg−1i.p.) was given. Each mouse was placed on a servo-controlled heating blanket and body temperature was monitored via a rectal temperature probe and kept constant at 37 ± 1 °C.
The fourth cervical (C4) branch of the left phrenic nerve was isolated in the neck and cut distally, as described previously (Mutoh et al. 2000a,b). As an indicator of the frequency of central respiratory drive phrenic nerve activity (PNA) was recorded. The central end of the phrenic nerve was placed on a bipolar silver hook electrode and covered with a mixture of warm petroleum jelly and mineral oil. To maintain the neural respiratory activity of the phrenic nerve, the arterial PCO2 values were maintained at between 40 and 45 mmHg.
Ventricular cannulation
In all animals, at least 2 weeks before the experiments, a stainless steel guide was implanted for intracerebroventricular (i.c.v.) drug application (see Akiyama et al. 1999). Briefly, animals were anaesthetised with a combination of xylazine (20 mg kg−1i.m.) and ketamine (50 mg kg−1i.m.). The animals were then placed in a stereotaxic head frame (SR-6N, Narishige, Tokyo, Japan) and a stainless steel guide cannula (22 gauge, 6.0 mm) was implanted in the left lateral cerebral ventricle (0.5 mm caudal and 1.1 mm lateral to the bregma at a depth of 2.1 mm below the skull surface). The cannula was fixed to the skull with two screws and dental cement. The animals were treated with an antibiotic (enrofloxacin, 10 mg kg−1s.c.) for at least 7 days after the surgery.
SCN lesion
A bilateral thermal lesion of the SCN was performed stereotaxically under xylazine (20 mg kg−1i.m.) and ketamine (50 mg kg−1i.m.) anaesthesia as described previously (Hara et al. 2001). A stainless steel electrode (0.35 mm i.d.) was inserted into the SCN (0.5 mm posterior and 0 mm lateral to the bregma at a depth of 5.3 mm below the skull surface) using a thermal lesion device (RFG-4A, Radionics, MA, USA). A lesion was created by maintaining the temperature at 55 °C for 15 s using a current path, and sham-operated animals were created using a non-current path. After surgery, the animals were moved to a locomotor activity device. For an assessment of their locomotor activity, the mice were individually housed in transparent plastic cages (31 cm × 20cm × 13 cm) and their locomotor activity rhythms under LD cycle were measured by area sensors (FA-05 F5B, Omron, Tokyo, Japan) with a thermal radiation detector system. Data were stored on a personal computer. One month after surgery, we selected animals with complete SCN lesions. Complete lesions were assessed by determinations over 24 h period using both a χ2 periodogram in the range of 20–28 h. In this study all SCN-lesioned animals were used which showed a loss of rhythmic locomotor activity but displayed a normal light-induced pupillary reflex, and also normal palpebral and corneal reflexes. The SCN lesion sites were confirmed histologically by Nissl staining after the termination of experiments. The lesion expands at most to the anterior hypothalamus surrounding the SCN but never damages the optic chiasm and other brain regions.
Renal sympathetic and gastric vagal efferent nerve recordings
Efferent units were recorded in a strand of the renal branch of the splanchnic sympathetic nerve and the gastric branch of the ventral subdiaphragmatic vagus. For recording the RSNA, the left renal nerve was exposed by the retroperitoneal approach and identified near the renal artery. For recording the GVNA, the gastric branch of the ventral subdiaphragmatic vagal nerve was identified on the oesophagus after an incision in the midline of the abdomen. To explore the afferent vagal-mediated mechanisms of the GVNA, experiments were performed under three conditions: (1) the dorsal subdiaphragmatic vagal nerve and the hepatic and accessory coeliac branches of the ventral subdiaphragmatic vagal nerve were left intact, (2) they were cut beneath the diaphragm and (3) only the hepatic branches were lesioned.
The nerves were separated from the surrounding tissue with the aid of a dissecting microscope and cut just proximal to the entrance of the left kidney and the stomach (Niijima, 1975; Niijima et al. 1993). Then a small bundle isolated from the central stump of the nerve was placed on a bipolar silver hook electrode and covered with a mixture of warm petroleum jelly and mineral oil. The signal recorded via the electrode was amplified, filtered (0.3–3 kHz), counted every second as population activity in nerves by a pulse counter after passing through a window discriminator (i.e. above threshold event counting), and fed in parallel to an oscilloscope, thermal chart recorder, audio monitor, and a digital tape-recorder with a sampling rate of 10 kHz per channel for off-line analysis. The threshold levels for the standard pulses were confirmed post mortem.
Experimental protocol
The RSNA or GVNA was monitored in separated series of experiments in conjunction with the PNA (as an index of central respiratory drive) and the cardiovascular indexes of ABP and HR. The preparation was allowed to stabilise for 30 min under a dim red safelight with illuminance of less than 1 × 1011 photons cm−2 s−1 before starting experiments. All experiments were basically conducted 6 h after lights on (zeitgeber time (ZT) 6). This time point was also selected for in vivo recordings of the sympathetic and vagal efferent nerve activities in rats which showed no diurnal variations in the responsiveness of the autonomic nerve activities to acute light exposure (2000 lx) (Niijima et al. 1993). The observation that the autonomic neuronal sensitivities to light do not vary at different time points of the day has been confirmed for mice in this study. We compared the responsiveness of RSNA and GVNA to standard light (fluorescent light, 1.5 × 1014 photons cm−2 s−1) and bright light (2.1 × 1014 photons cm−2 s−1, similar to 2000 lx white light; an intensity little weaker than indirect sunlight measured 2.5 cm from a window on a clear spring day (Lewy et al. 1980)) pulses (10 min) starting the experiments at different time points: ZT 6 (biological day), ZT 14 (biological dusk), ZT 18 (biological night) and ZT 22 (biological dawn). Light stimuli were applied to the left eye by exposing a white light beam of a glass fibre illumination apparatus with a heat-absorbing filter (LGPS, Olympus, Tokyo, Japan). Stimulus irradiance of light level was measured by a light meter (LI-250, LI-COR, Lincoln, NE, USA).
Mice were kept in complete darkness for at least 15 min of baseline recordings prior to the start of each series of experiments. Then we determined the dose-related effects of light (intensity) or melatonin (dose for i.c.v. injection) on the autonomic functions when administered separately. In one series of experiments, photic intensity of light pulse (10 min) was varied from 1.5 × 1013 to 6.1 × 1017 photons cm−2 s−1. In another series, increasing concentrations of melatonin (0.01–100 ng, increased by logs) were sequentially administered i.c.v. in a 5 µl solution at a rate of 1 µl min−1 separated by 30 min intervals. In a third series of experiments we treated animals with of the threshold dose of i.c.v. melatonin followed by stimulation with light of the most effective intensity. After recording the baseline activity for 15 min, 5 µl of melatonin or vehicle were given i.c.v. at a rate of 1 µl min−1. After 10 min, light stimuli were applied to the left eye using the glass fibre illumination apparatus and the responses were followed for at least 90 min. For melatonin receptor blockade, 5 µl of luzindole (100 µm) were administered 10 min prior to melatonin injection through the i.c.v. cannula at a rate of 1 µl min−1. This i.c.v. dose of luzindole, which did not change the baseline values of the neuronal activities and cardiorespiratory indexes by itself, was shown to block any autonomic response to high dose melatonin (100 ng). Animals were killed after the experiment by injection of a lethal dose of pentobarbital.
Central drug application and plasma melatonin levels
Melatonin (N-acetyl-5-methoxytryptamine) purchased from Sigma Chemical (St Louis, MO, USA) was freshly dissolved in 100 % dimethylsulfoxide (DMSO) and finally diluted in a vehicle consisting of 2 % DMSO in artificial cerebrospinal fluid (aCSF; Sigma). Luzindole (N-acetyl-2-benzyltryptamine; Sigma) was dissolved in a minimum amount of ethanol (95 %) and then in aCSF to obtain a stock solution of 10 mm. The desired final concentration was prepared by further dilution of this concentrated stock with aCSF on the day of the experiment. All solutions were administered at a rate of 1 µl min−1 through an i.c.v. injection cannula (27 gauge, extending 0.5 mm below the tip of the guide cannula) attached to a 10 µl Hamilton syringe via a polyethylene tube. The syringe was left in place for 5 min before slow retraction. The position of the i.c.v. cannula was confirmed by administration of 5 µl of Fast Green at the end of the experiment and histological examination.
Melatonin is a non-polar, lipid-soluble indole and may thus readily cross the blood-brain barrier. To examine whether central administration of melatonin causes an increase in blood melatonin levels, changes in plasma melatonin concentrations were measured in pilot studies after i.c.v. infusion of melatonin at ZT 6. Sequential blood samples (80–100 µl) were taken through a catheter (0.58 mm i.d.) inserted into the right atrium at 15 and 60 min after i.c.v. administration of melatonin (0.1 ng) or vehicle. Plasma melatonin concentrations were determined by radioimmunoassay (RIA) using Sep-pak C18 cartridge (Waters Associates, Milford, MA, USA) as previously described (Chiba et al. 1998). The plasma melatonin levels were below the detection limit in all samples (< 3.2 pg ml−1), irrespective of whether they were taken from mice after i.c.v. administration of melatonin (n = 3) or vehicle (n = 3). We thus consider that i.c.v. melatonin application has negligible direct effects on peripheral organs.
In the present study, we used BALB/c mice that show a melatonin deficiency (Vivien-Roels et al. 1998). Our preliminary results have demonstrated that C3H/HeN mice that exhibit a normal melatonin secretion pattern did not show any significant difference in the autonomic neuronal responsiveness of melatonin and/or light with melatonin-deficient BALB/c mice (T. Mutoh and H. Okamura, unpublished observation).
Data analysis
Collected data included the light-evoked changes in neuronal impulse activity, ABP and HR. For light-evoked responses, the efferent impulse activity of the renal sympathetic nerve or the gastric vagus was analysed at 10 s intervals. The baseline impulse activity was determined over a 15 min period. The peak response was defined as the average number of event counts per second during the most active 10 s of the initial 30 min after the light exposure. The onset latency for the peak increase in the unit activity was defined as the time between the beginning of light treatment and the first detectable increase in the unit activity. The peak changes in neuronal activity, ABP and HR were defined as the 10 s bin with the biggest change within the initial 30 min after the light exposure. To determine whether the light-evoked changes (Δ) from the baseline value to the peak response for neuronal activity, ABP and HR were significantly different among trials, one-way ANOVA was used with treatment as a within subject effect followed by a series of Tukey's contrast tests between the treatment groups, following significant F tests. To determine whether the light-evoked peak responses of neuronal activity, ABP and HR were significantly different from baseline, Student's paired t test was used. Statistical significance was assured at P < 0.05. All values were expressed as means ± s.e.m., unless otherwise indicated.
RESULTS
Light enhances sympathetic nerve activity but suppresses vagal parasympathetic nerve activity independent of the time of treatment
We first compared the sensitivity of RSNA to light at four different times (ZT 6, 14, 18 and 22; Fig. 1A). There were no statistically significant differences in the light-induced responses (Δ) of RSNA among the different time points (P > 0.05, ANOVA), although the baseline activities at ZT 18 (biological night) were significantly higher (P = 0.037, Tukey's test) than those at ZT 6 (biological day; Fig. 1B). For the baseline activity and light sensitivity of GVNA, we did not detect any significant difference among the different time points (P > 0.05, ANOVA; data not shown). Since the responsiveness of the sympathetic and parasympathetic nerve activities to light did not vary with the time of the day, we performed all following experiments of this study at ZT 6.
Figure 1. Responses of the renal sympathetic nerve activity (RSNA) to light at different times of the day.
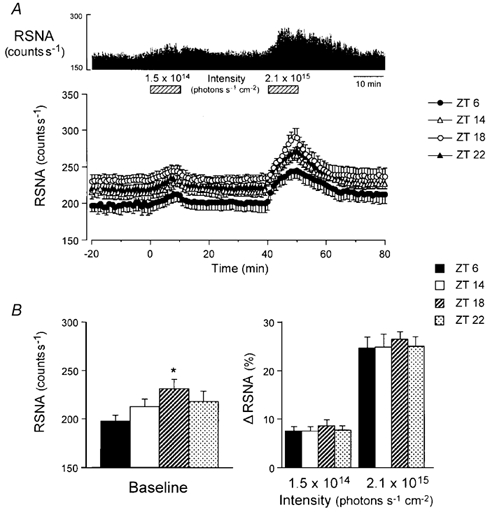
A, an example of the effects of light on RSNA at zeitgeber time (ZT) 6 (upper panel) and grouped data (lower panel) showing the time course of light-induced RSNA responses in mice starting experiments at ZT 6 (filled circles, n = 6), ZT 14 (open circles, n = 6), ZT 18 (open triangles, n = 6) and ZT 22 (filled triangles, n = 6). In all groups, exposure to 2.1 × 1014 photons cm−2 s−1 light pulse (10 min) induced apparent increase in RSNA immediately after lights on, while exposure to 1.5 × 1014 photons cm−2 s−1 light caused only a small and transient increase. B, group data showing the baseline count levels under complete darkness (left panel) and the peak responses to light (1.5 × 1014 and 2.1 × 1014 photons cm−2 s−1, 10 min; right panel) of RSNA at ZT 6, 14, 18 and 22. No statistically significant differences in the light-induced RSNA responsiveness were observed between the different time points, with the exception of the baseline impulse activity between ZT 6 and ZT 18 (P = 0.037, Tukey's test). *P < 0.05vs. ZT 6.
Secondly, we determined the effects of light pulses (10 min) of varying intensities (1.5 × 1013 to 6.1 × 1017 photons cm−2 s−1) on RSNA and GVNA. Light dose-dependently enhanced the RSNA (P < 0.001, dose effect, ANOVA; Fig. 2A). It should be noted that the RSNA was increased in all animals after treatment with light (10 min) of 2.1 × 1014 photons cm−2 s−1, which also increased HR and ABP (Fig. 2B). Conversely, the light (2.1 × 1014 photons cm−2 s−1, 10 min) suppressed the activity of the gastric and hepatic branches of the vagal nerve (Fig. 2C and D). This suppression of the vagal parasympathetic nerve activity was inversely proportional to the light intensity (P < 0.001, dose effect, ANOVA; Fig. 2A, right panel). The latencies for the onset, peak and recovery of the light-evoked neuronal responses were 16.0 ± 4.0 s for RSNA and 20.0 ± 3.2 s for the vagal nerve activity; 11.6 ± 2.9 min for RSNA and 14.4 ± 1.9 min for the vagal nerve activity; and 33.8 ± 6.6 min for RSNA and 49.6 ± 5.8 min for the vagal nerve activity, respectively.
Figure 2. Effects of light on RSNA and vagal parasympathetic nerve activity (VNA).
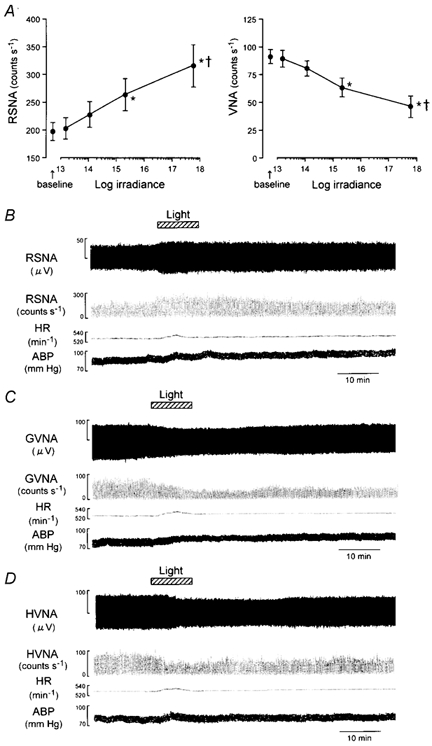
A, group data showing the effects of light pulses (10 min) of varying intensities (1.5 × 1013 to 6.1 × 1017 photons cm−2 s−1) on RSNA (left panel, n = 12) and VNA (right panel, n = 16). Light enhanced the RSNA in an intensity-dependent manner (P < 0.0001, dose effect, ANOVA). B, an example of the effects of light (2.1 × 1014 photons cm−2 s−1, 10 min) on RSNA, event counts of the RSNA generated by window discriminator, heart rate (HR), and arterial blood pressure (ABP). C and D, examples of the effects of light (2.1 × 1014 photons cm−2 s−1, 10 min) on the activity of gastric (GVNA; C) and hepatic (HVNA; D) branches of the vagal parasympathetic nerve. *P < 0.05vs. baseline; †P < 0.05 vs. 1.5 × 1013 photons cm−2 s−1.
Central melatonin suppresses both sympathetic and vagal parasympathetic nerve activities
To explore the role of melatonin on the autonomic functions, we measured the changes of peripheral autonomic nerve activities elicited by central application of various doses of melatonin. The i.c.v. injection of melatonin (0.01–100 ng) dose-dependently decreased the RSNA (P < 0.0001, dose effect, ANOVA; Fig. 3A). Melatonin also attenuated the GVNA in a dose-dependent manner (P < 0.0001, dose effect, ANOVA; Fig. 3B). Based on these results, we selected a dose of 0.1 ng melatonin that had a threshold effect on RSNA and GVNA.
Figure 3. Effects of intracerebroventricular (i.c.v.) administration of melatonin on RSNA and GVNA.
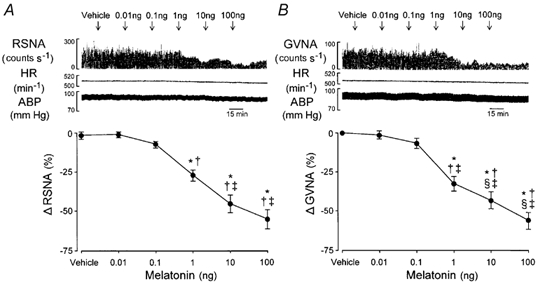
Melatonin (i.c.v.) suppressed the RSNA (A) and GVNA (B) in a dose-dependent manner (P < 0.0001, dose effect, ANOVA). Upper traces in A and B show an example of the effects of consecutive i.c.v. injections of melatonin (0.01–100 ng) on event counts of RSNA and GVNA, HR and ABP. Lower graphs show group data with the peak RSNA (n = 7) and GVNA (n = 7) responses to i.c.v. melatonin. Arrows indicate time of i.c.v. microinjection. *P < 0.05vs. baseline; †P < 0.05vs. vehicle; ‡P < 0.05vs.i.c.v. melatonin (0.01 ng).
In the following experiments, we analysed the effects of light stimuli (2.1 × 1014 photons cm−2 s−1, 10 min) applied in combination with 0.1 ng i.c.v. melatonin on RSNA and GVNA and cardiorespiratory variables and determined the interactions of these two treatments.
Central melatonin application suppresses light-induced sympathetic and cardiovascular responses
Injection of melatonin i.c.v. attenuated the light-induced augmentation of the RSNA (Fig. 4A and B). The increase in activity over baseline (Δ) in the vehicle-administered group (34.6 ± 10.4 %) was reduced nearly one-fifth in the melatonin-treated group (7.2 ± 2.8 %; P = 0.015, Tukey's test; Fig. 4C). The light-evoked changes in ABP and HR from the baseline values were statistically significant for the vehicle-administered group (P < 0.006, paired t test), and the changes were significantly attenuated by prior i.c.v. melatonin injection (vehicle vs. melatonin; P = 0.015, Tukey's test). By contrast, the light treatment did not alter bursting rate of the PNA (P > 0.05, paired t test) in either the melatonin- or vehicle-administered animals.
Figure 4. Effects of i.c.v. administration of melatonin on light-induced RSNA and cardiorespiratory responses.
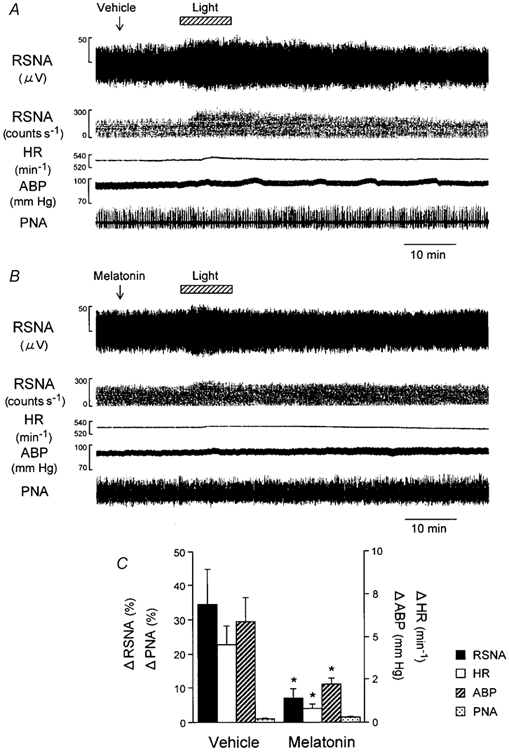
Melatonin (i.c.v.) significantly attenuated the light-induced increase in RSNA, HR and ABP but had no significant effect on PNA. Traces in A and B show examples of the effects of light (2.1 × 1014 photons cm−2 s−1, 10 min) on RSNA, event counts of the RSNA, HR, ABP and phrenic nerve activity (PNA) following i.c.v. injection of vehicle (A) or melatonin (0.1 ng; B). C, group data showing the effects of i.c.v. melatonin (n = 14) on peak responses of the RSNA, HR, ABP and PNA to light, compared with those of i.c.v. vehicle (n = 14). *P < 0.05vs. vehicle.
Time courses of the light-induced RSNA, ABP, HR and PNA responses in the melatonin- and vehicle-administered groups are shown in Fig. 5. In both groups of animals, i.c.v. microinjection of melatonin or vehicle (5 µl, at a rate of 1 µl min−1) did not evoke any significant changes in the baseline RSNA, ABP, HR or PNA. Exposure to light, which was administered 10 min after the i.c.v. vehicle injection, increased the RSNA immediately (≈30 s) after lights on, peaked within 5–8 min, and returned to the baseline after 75 min. The light treatment also increased ABP and HR. Maximal values were seen 5–10 min after lights on and then began to decline, returning to the baseline level within 30 min.
Figure 5. Time course of the light-induced responses of RSNA and cardiorespiratory responses following i.c.v. administration of melatonin and vehicle.
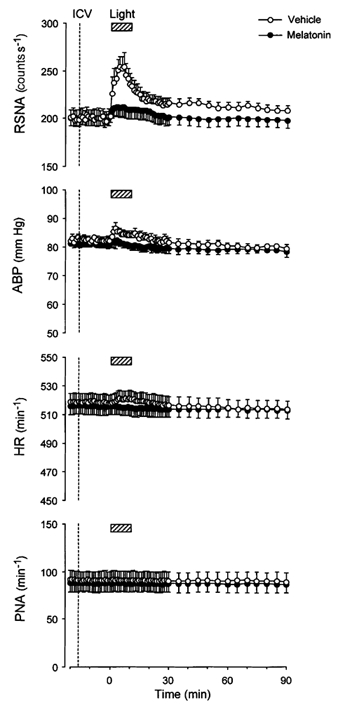
Grouped data showing time course of the effects of melatonin (filled circles, n = 14) and vehicle (open circles, n = 14) i.c.v. injections on light (2.1 × 1014 photons cm−2 s−1, 10 min)-induced changes in RSNA, HR, ABP and PNA.
After i.c.v. injection of melatonin, the light-evoked increase in RSNA became smaller and lasted for a shorter time, returning to near baseline within 25 min (Fig. 5, top panel). The light-evoked increases in ABP and HR were completely suppressed by i.c.v. melatonin injection. Bursting rate of the PNA remained stable over the duration of the experiment in both the melatonin- and vehicle-administered animals.
Central melatonin augments light-induced suppression of vagal parasympathetic responses
Melatonin administered i.c.v. enhanced the light-evoked decrease of the GVNA (Fig. 6A and B). The decrease in activity over baseline (Δ) in the melatonin-administered group (-39.3 ± 1.5 %) was significantly greater in degree than that in the vehicle-administered group (-24.9 ± 1.8 %; P = 0.001, Tukey's test).
Figure 6. Effects of i.c.v. administration of melatonin on light-induced GVNA responses.
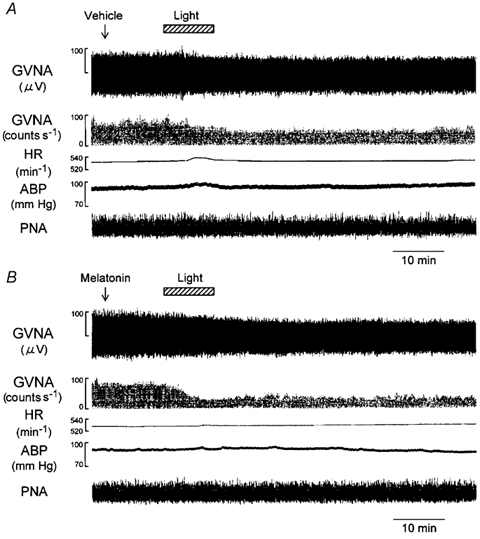
Traces in A and B show examples of the effects of light (2.1 × 1014 photons cm−2 s−1, 10 min) on GVNA, event counts of the GVNA, HR, ABP and PNA following i.c.v. injection of vehicle (A) or melatonin (0.1 ng; B). The light-evoked GVNA suppression was greater in melatonin-administered mouse than in the vehicle-administered mouse.
The time courses of the light-evoked changes in GVNA in the melatonin- and vehicle-administered mice are shown in Fig. 7A (top panel). In both groups of animals, i.c.v. microinjection of melatonin or vehicle did not evoke any significant change in the baseline GVNA. Light treatment following the i.c.v. vehicle injection suppressed the GVNA immediately (≈30 s) after lights on. Maximal inhibitory effects were seen within 10–15 min, returning within 10 % of baseline at 50 min. Melatonin (i.c.v.) injection augmented the light-evoked suppression of GVNA that returned to within 15 % of baseline after 80 min.
Figure 7. Effects of hepatic or subdiaphragmatic vagotomy on light-induced GVNA responses.
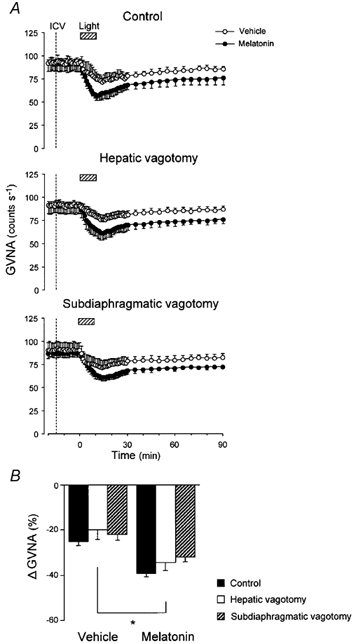
A, group data showing time course of the effects of melatonin (0.1 ng; filled circles) and vehicle (open circles) i.c.v. injections on light (2.1 × 1014 photons cm−2 s−1, 10 min)-induced decrease in GVNA in intact control, hepatic vagotomised and total subdiaphragmatic vagotomised mice. B, group data showing the effects of i.c.v. melatonin on peak responses of the GVNA to light, compared with those of i.c.v. vehicle, in intact control (n = 14), hepatic vagotomised (n = 7) and total subdiaphragmatic vagotomised (n = 7) animals. Melatonin (i.c.v.) significantly enhanced the light-induced decrease in GVNA. The i.c.v. melatonin-induced suppression of the GVNA was observed in all cases (P > 0.05, treatment effect, ANOVA) even after the total subdiaphragmatic vagotomy. *P < 0.05, vehicle vs. melatonin.
To determine whether the melatonin action was conveyed by vagal afferents, the light-induced GVNA responses were studied further in animals with hepatic vagotomy or total subdiaphragmatic vagotomy. In both melatonin- and vehicle-administered groups, hepatic vagotomy or total subdiaphragmatic vagotomy did not alter the baseline activity or the light-evoked responsiveness of GVNA as recorded from animals in which these nerves were left intact (P = 0.476, treatment effect, ANOVA; Fig. 7A and B).
Luzindole antagonises the effects of central melatonin injections on sympathetic and vagal parasympathetic responsiveness to light
To evaluate the nature of the receptor that mediates the i.c.v. melatonin-induced autonomic modifications, we examined the effects of the melatonin receptor-specific competitive antagonists luzindole on the melatonin-mediated responses. Application (i.c.v.) of the non-selective MT1/MT2 melatonin receptor antagonist luzindole (100 µm) did not alter the baseline RSNA or GVNA (Fig. 8A). However, 100 µm luzindole blocked the effects of i.c.v. melatonin on light-induced changes in RSNA, HR and ABP (Fig. 4C) and in GVNA (Fig. 7B) (P < 0.05, treatment effect, ANOVA; Fig. 8B). The i.c.v. luzindole recovered the light-induced autonomic and cardiovascular responsiveness by 84 ± 9 % (RSNA), 89 ± 10 % (GVNA), 92 ± 11 % (HR) and 84 ± 7 % (ABP) of the peak responses (P > 0.05vs. light exposure following i.c.v. vehicle pretreatment).
Figure 8. Effects of luzindole on central melatonin-mediated autonomic and cardiorespiratory responsiveness to light.
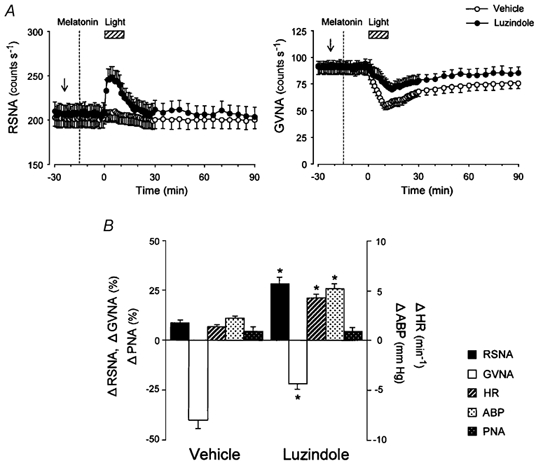
A, group data showing time course of the effects of the competitive melatonin receptor antagonist luzindole (100 µm; filled circles, n = 14) and vehicle (open circles, n = 14) i.c.v. pretreatments on RSNA (left panel) and GVNA (right panel) responses to light (2.1 × 1014 photons cm−2 s−1, 10 min) following i.c.v. melatonin (0.1 ng) injection. The i.c.v. melatonin-mediated RSNA and GVNA modulations (see Figs 5 and 7A, top panel) were no longer observed in mice pretreated with i.c.v. luzindole. Arrow indicates time of i.c.v. luzindole microinjection. B, group data showing the effects of i.c.v. luzindole on peak responses of the RSNA, GVNA, HR, ABP and PNA to light in animals treated with vehicle (left, n = 14) and melatonin (right, n = 14) i.c.v. injections. Luzindole (i.c.v.) significantly inhibited the i.c.v. melatonin-mediated decreases in RSNA, HR and ABP and increase in GVNA for the responsiveness to light (compare these results with those in Figs 4C and 7B). *P < 0.05vs. vehicle.
SCN lesion abolishes the effects of central melatonin injections on light-induced autonomic responses
To investigate whether the SCN is involved in the light- or melatonin-induced autonomic and cardiorespiratory responsiveness we examined SCN-lesioned and sham-operated mice.
Initially, we performed separate analyses of the effects of changes in light intensity and melatonin doses in SCN-lesioned and sham-operated mice and found that even a maximum dose of light (20 000 lx; data not shown) or melatonin (100 ng; Fig. 9A) did not alter the RSNA, GVNA or associated cardiovascular responsiveness in SCN-lesioned animals.
Figure 9. Effects of bilateral SCN lesion on light-induced autonomic and cardiorespiratory responses.
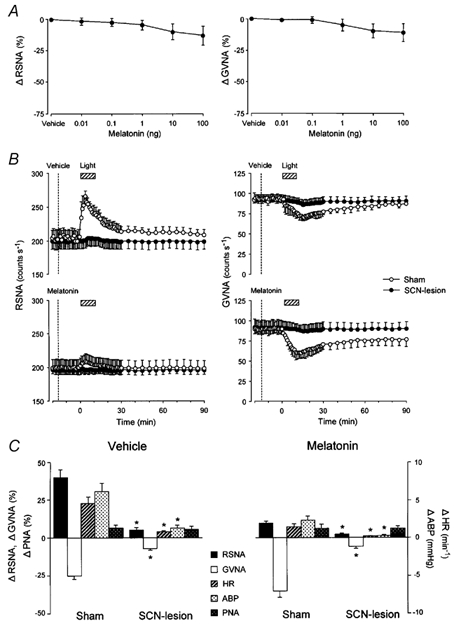
A, group data showing the effects of consecutive i.c.v. injections of melatonin (0.01–100 ng) on RSNA (right, n = 7) and GVNA (left, n = 7) in SCN-lesioned mice. Melatonin (i.c.v.)-induced dose-dependent suppressions of RSNA and GVNA were no longer observed in mice with bilateral SCN lesion (compare these results with those in Fig. 3A and B). B, group data showing time course of the effects of melatonin (0.1 ng) and vehicle i.c.v. injections on light (2.1 × 1014 photons cm−2 s−1, 10 min)-induced change of RSNA (left panel) and GVNA (right panel) in SCN-lesioned (filled circles, n = 14) and sham-operated (open circles, n = 14) mice. The light-induced RSNA and GVNA responsiveness following i.c.v. injection of melatonin or vehicle observed in sham-operated animals disappeared in SCN-lesioned animals. C, group data showing the effects of SCN lesion on light-induced peak responses of RSNA, GVNA, HR, ABP and PNA in animals pretreated with i.c.v. vehicle (left panel, n = 14) and melatonin (right panel, n = 14). SCN lesion significantly reduced the light-induced increases in RSNA, HR and ABP and decrease in GVNA. *P < 0.05vs. sham-operation.
The light-induced increase in RSNA and decrease in GVNA, which were evident in sham-operated animals, were no longer observed in SCN-lesioned animals (Fig. 9B). No statistically significant differences from the baseline values were observed in RSNA, GVNA, ABP, HR or PNA for SCN-lesioned animals (P > 0.05, paired t test), while significant increases in these variables were observed for sham-operated animals (P < 0.05, paired t test; Fig. 9C). The light-evoked changes in the autonomic neuronal activities, ABP and HR over baseline (Δ) in SCN-lesioned animals were significantly less than those in sham-operated animals (P < 0.05, treatment effect, ANOVA; Fig. 9C).
DISCUSSION
The present study has revealed four main results. (1) Light increases arterial blood pressure and heart rate along with an increase of the renal sympathetic nerve activity immediately after lights on. (2) Light rapidly suppresses the gastric vagal parasympathetic nerve activity, which was not affected by hepatic vagotomy or by total subdiaphragmatic vagotomy. (3) Via activation of central melatonin receptors, melatonin injections into the lateral ventricle dose dependently attenuate the sympathetic and vagal parasympathetic nerve activities with a threshold of 0.1 ng. (4) Bilateral SCN lesions totally abolish the neuronal and cardiovascular responsiveness to light and/or melatonin. These results suggest that light stimulation induces sympathoexcitation and vagal suppression through the SCN and that melatonin modulates the light-induced autonomic responses via activation of the central melatonin receptor signalling.
The present results indicate that photic signals entering the SCN have a physiologically relevant effect on regulation of the cardiovascular function by modulating the sympathetic outflow without affecting the central respiratory drive determined by recording phrenic nerve activity. We measured RSNA which represents the activity of postganglionic sympathetic vasoconstrictor fibres which innervate the renal vascular bed (DiBona, 1982; van Tilborg et al. 1994). In this study, light stimulation was shown to increase RSNA, ABP and HR with similar latencies immediately after its onset. It seems unlikely that the renal sympathoexcitation and brief tachycardiac response which occurred immediately after lights on were baroreceptor-mediated responses to a drop in ABP because the enhancement of the RSNA was not associated with a decrease in ABP (at least during the 90 min recording period after lights on). The fact that central melatonin microinjection into the lateral ventricle or bilateral SCN lesion can suppress the light-evoked autonomic responsiveness indicates that the effects of light on the sympathetic and cardiovascular systems are due to a central action. Given that light stimuli with the same intensity (≈2000 lx) can also enhance the sympathetic efferent activity of the adrenal, hepatic and pancreatic nerves in rats (Niijima et al. 1993), light may have a uniform excitatory effect on the sympathetic nervous system innervating various peripheral organs.
Colwell et al. (1993) have shown that general anaesthesia, with the exception of urethane, can suppress or completely block the effects of light on the clock in the SCN. This raises the possibility that some anaesthetics suppress the light-induced neuronal activity in the SCN. Indeed, in urethane-anaesthetised animals responses of SCN neurons to light were sustained, but blocked with pentobarbital (Aggelopoulos & Meissl, 2000). Moreover, urethane is known to exert minimal effects on the cardiorespiratory system and barely affects hypothalamic function which could indirectly alter the autonomic function via effects on a number of reflexes (Maggi & Meli, 1986a,b), suggesting that the CNS-mediated cardiorespiratory control is little affected by this anaesthetic per se. It should be noted here that under light urethane anaesthesia, animals exhibit patterns of cortical activity (EEG) states similar to those seen in wake, drowsiness and slow-wave sleep in non-anaesthetised animals (Hunter & Milsom, 1998), and such animals would therefore be expected to have an arousal response to bright light. However in the present study, the depth of urethane anaesthesia was evaluated by estimating the loss of spontaneous or paw pinch-evoked movements or sudden fluctuation of ABP or HR (Mutoh et al. 2000a,b) to ensure the maintenance of an adequate plane of general anaesthesia without arousal. Thus, the excitation of the sympathetic nerve activity and the vasomotor reactions obtained in this study will be caused by a bright light exposure rather than by a simple stress evoked by the arousal.
An important question is through which pathway light modulates peripheral autonomic functions. Since bilateral lesion of the SCN totally abolished the effect of light on the autonomic and cardiovascular functions, the SCN appears to play a crucial role for transmission of the photic signals to the autonomic nervous system innervating peripheral organs. Recently, evidence has been provided for the existence of multisynaptic pathways from the SCN to various visceral organs (e.g. adrenal gland, heart and liver) by using virus tracers that are transported retrogradely and transneuronally (Buijs et al. 1999; Scheer et al. 2001). In mammals, environmental light-dark information is transformed to an electrical activity in the retina, and conveyed to the SCN directly via the retinohypothalamic tract (Moore & Lenn, 1972) or indirectly via the geniculohypothalamic tract (Swanson et al. 1974; Krout et al. 2002). The projections from the SCN to the subparaventricular zone (Watts et al. 1987) may be the first link in a multisynaptic pathway to the autonomic preganglionic cells in the intermediolateral cell column (IML) of the spinal cord, or to the rostral ventrolateral medulla (RVLM) containing cell bodies of the sympathetic pathways to regulate the cardiovascular functions (Vrang et al. 1995; Shafton et al. 1998). Photic stimulation of the retinohypothalamic tract results in glutamate release in the SCN and triggers a complex intracellular cascade similar to that described for induction of long-term potentiation (LTP) by glutamate (van den Pol et al. 1996). A long-lasting autonomic neuronal responsiveness to light (≈80 min) observed in this study may be the result of the signal transduction pathways for light-evoked LTP originating in the SCN.
In contrast to the enhancement of the sympathetic nerve activity, light suppressed the activity in the gastric and hepatic branches of the vagal parasympathetic nerve in an intensity-dependent manner. The vagal parasympathetic suppression showed a clear reciprocity to the sympathoexcitation with regard to latencies (≈30 s) and time courses after lights on (see Fig. 5 and Fig. 7A). These effects are strikingly similar to those observed in rats (Niijima et al. 1993).
It is known that the vagal afferent nerves convey signals to the brain thereby participating in the reflex regulation of gastric motility and emptying as well as gastric acid secretion via the vagal efferent pathways (Raybould & Lloyd, 1994). However, the present study shows that the effect of light on the GVNA is originally driven from the CNS and does not involve mechanisms mediated by vagal afferents, since neither hepatic vagotomy nor total subdiaphragmatic vagotomy changed the light-induced GVNA responsiveness. Similar to the light-induced activation of RSNA, the SCN appears to play a crucial role in the inhibitory response of the vagal efferents to light, since light-induced vagal suppression was no longer found in animals with SCN lesion. For these vagal responses, the connection of the paraventricular nucleus, which is a primary projecting area of the SCN neurones, to the dorsal motor nucleus (DMN) of the vagus, may have an important role, since electrical/chemical stimulation of these nuclei inhibits the gastric acid responses (Rogers & Hermann, 1987; Flanagan et al. 1992; Beltran et al. 1999).
In the present study performed at ZT 6, melatonin microinjections (0.01–100 ng) into the lateral ventricle rapidly suppressed RSNA and GVNA with a threshold dose of 0.1 ng. It is well known that melatonin induces concentration-dependent phase advances of SCN neuronal firing rate when administered at dusk and dawn (McArthur et al. 1997; Hunt et al. 2001). Moreover, melatonin was shown to inhibit single-unit activity and 2-deoxy-[1-14C]glucose (2-DG) uptake in the rat SCN immediately after its application, with maximum effect between CT 6 (CT = circadian time; CT0 is subjective dawn and CT12 is subjective dusk) and CT 10 (Cassone et al. 1987, 1988; Shibata et al. 1989). Since melatonin reduces neuronal excitability of the SCN neurones independent of the time of application in the circadian cycle even with physiological concentrations (1 nm;van den Top et al. 2001), the threshold dose (4–6 nm calculated) of melatonin used in this study might be sufficient for inhibition of the SCN neuronal firing, thereby facilitating the autonomic suppression.
The melatonin effects on autonomic and cardiovascular responsiveness were blocked by the melatonin receptor antagonist luzindole and they could not be elicited in SCN-lesioned mice. These results suggest that melatonin mediates the light-induced changes in autonomic nerve activities via activation of melatonin receptors within the SCN. Melatonin receptors are also distributed in various peripheral organs such as the superior cervical ganglia, caudal artery, kidney, adrenal gland, stomach and heart (Vanecek, 1998; Drew et al. 2001). It seems, however, unlikely that the effects of i.c.v. melatonin injections described here were mediated via peripheral melatonin receptors, since the i.c.v. melatonin injections performed in our study did not increase plasma melatonin levels which were below the detection level in both melatonin- and vehicle-treated animals. The fact that, in sheep, melatonin levels are 20 times higher in the CSF than in the peripheral circulation (Skinner & Malpaux, 1999) may support the potential importance of central melatonin receptor signalling for neurally mediated physiological control mechanisms. The melatonin receptor subtype(s) mediating the effects of melatonin described in this study remain to be identified by use of the specific and selective melatonin receptor antagonists such as the MT2 melatonin receptor antagonist, 4-phenyl-2-propionamidotetraline (4P-PDOT) (Dubocovich et al. 1998a; Hunt et al. 2001), since luzindole shows affinity for both the MT1 and MT2 melatonin receptors (Dubocovich et al. 1998b; Nonno et al. 1999).
A final point of consideration is the physiological relevance of the melatonin-mediated modulation of light-induced renal sympathoexcitation and vagal parasympathetic suppression. Since light is known as the strong entraining signal, or zeitgeber of the circadian clock, bright light has been used to treat rhythm-related sleep/wake disturbances in humans (Arendt, 2000). Furthermore, the bright light-induced phase shifts of circadian rhythms are enhanced by co-administration of the ‘non-photic’ zeitgeber melatonin (Benloucif et al. 1999). Sleeplessness associated with frequent rhythm disruption in shift workers is also reported to increase the incidence of the development of cardiovascular and gastrointestinal diseases (e.g. atherosclerosis, hypertension and gastric ulceration; Cagnacci, 1996; Richardson & Tate, 2000). Given that the bright light treatment can help adaptation to an extended night-work period by resetting the human circadian pacemaker to reach daytime sleep (Baehr et al. 1999), our present data suggest that the therapeutic regimen may also provide some negative aspects associated with the hypertensive and tachycardiac responses to light exposure. In the present study, central melatonin suppressed the light-induced sympathetic and cardiovascular responsiveness. Interestingly, melatonin also enhanced the gastric parasympathetic inhibitory responsiveness which is thought to prevent the induction of gastric lesions via inhibition of the secretion of gastric acid and pepsin (Kato et al. 1998). The role of melatonin in the autonomic nervous system revealed by the present study may provide a mechanism to explain some of the long-discussed protective and anti-stress effects of melatonin on the cardiovascular and gastrointestinal systems.
In summary, the present data obtained in mice demonstrate that light induces sympathoexcitation and vagal suppression and associated cardiovascular responses through the SCN and that melatonin modulates the light-induced autonomic responsiveness via activation of the central melatonin receptor signalling. The successful recording of autonomic neuronal activities and associated cardiovascular and respiratory parameters in mice is of particular neurobiological significance, since in view of the potential for genomic manipulation in mice this in vivo experimental model will help to develop an understanding of the links between the CNS, the autonomic nervous system and rhythmic gene expression.
Acknowledgments
This work was supported in part by grants from the Special Coordination Funds and the Grant-in-Aid for the Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan. The authors gratefully acknowledge the excellent support provided by Drs Akira Niijima, Toshiya Manabe and Toshio Terashima.
REFERENCES
- Aggelopoulos NC, Meissl H. Responses of neurones of the rat suprachiasmatic nucleus to retinal illumination under photopic and scotopic conditions. J Physiol. 2000;523:211–222. doi: 10.1111/j.1469-7793.2000.t01-1-00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama M, Kouzu Y, Takahashi S, Wakamatsu H, Moriya T, Maetani M, Watanabe S, Tei H, Sakaki Y, Shibata S. Inhibition of light- or glutamate-induced mPer1 expression represses the phase shifts into the mouse circadian locomotor and suprachiasmatic firing rhythms. J Neurosci. 1999;19:1115–1121. doi: 10.1523/JNEUROSCI.19-03-01115.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt J. Melatonin, circadian rhythms, and sleep. N Engl J Med. 2000;343:1114–1116. doi: 10.1056/NEJM200010123431510. [DOI] [PubMed] [Google Scholar]
- Baehr EK, Fogg LF, Eastman CI. Intermittent bright light and exercise to entrain human circadian rhythms to night work. Am J Physiol. 1999;277:R1598–1604. doi: 10.1152/ajpregu.1999.277.6.R1598. [DOI] [PubMed] [Google Scholar]
- Beltran B, Barrachina MD, Mendez A, Quintero E, Esplugues JV. Synthesis of nitric oxide in the dorsal motor nucleus of the vagus mediates the inhibition of gastric acid secretion by central bombesin. Br J Pharmacol. 1999;127:1603–1610. doi: 10.1038/sj.bjp.0702717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benloucif S, Masana MI, Yun K, Dubocovich ML. Interactions between light and melatonin on the circadian clock of mice. J Biol Rhythms. 1999;14:281–289. doi: 10.1177/074873099129000696. [DOI] [PubMed] [Google Scholar]
- Buijs RM, Wortel J, Van Heerikhuize JJ, Feenstra MG, Ter Horst GJ, Romijn HJ, Kalsbeek A. Anatomical and functional demonstration of a multisynaptic suprachiasmatic nucleus adrenal (cortex) pathway. Eur J Neurosci. 1999;11:1535–1544. doi: 10.1046/j.1460-9568.1999.00575.x. [DOI] [PubMed] [Google Scholar]
- Burgess HJ, Trinder J, Kim Y, Luke D. Sleep and circadian influences on cardiac autonomic nervous system activity. Am J Physiol. 1997;273:H1761–1768. doi: 10.1152/ajpheart.1997.273.4.H1761. [DOI] [PubMed] [Google Scholar]
- Cagnacci A. Melatonin in relation to physiology in adult humans. J Pineal Res. 1996;21:200–213. doi: 10.1111/j.1600-079x.1996.tb00287.x. [DOI] [PubMed] [Google Scholar]
- Cassone VM, Roberts MH, Moore RY. Melatonin inhibits metabolic activity in the rat suprachiasmatic nuclei. Neurosci Lett. 1987;81:29–34. doi: 10.1016/0304-3940(87)90335-1. [DOI] [PubMed] [Google Scholar]
- Cassone VM, Roberts MH, Moore RY. Effects of melatonin on 2-deoxy-[1-14C]glucose uptake within rat suprachiasmatic nucleus. Am J Physiol. 1988;255:R332–337. doi: 10.1152/ajpregu.1988.255.2.R332. [DOI] [PubMed] [Google Scholar]
- Chiba A, Akema T, Iigo M, Nagami Y, Kimura F, Toyoda J. A possible role of the pineal gland in acute immobilization-related suppression of naloxone-induced LH release in ovariectomized estrogen-primed rats. J Neuroendocrinol. 1998;10:79–84. doi: 10.1046/j.1365-2826.1998.00631.x. [DOI] [PubMed] [Google Scholar]
- Colwell CS, Kaufman CM, Menaker M, Ralph MR. Light-induced phase shifts and Fos expression in the hamster circadian system: the effects of anesthetics. J Biol Rhythms. 1993;8:179–188. doi: 10.1177/074873049300800301. [DOI] [PubMed] [Google Scholar]
- Dibona GF. The functions of the renal nerves. Rev Physiol Biochem Pharmacol. 1982;94:75–181. [Google Scholar]
- Drew JE, Barrett P, Mercer JG, Moar KM, Canet E, Delagrange P, Morgan PJ. Localization of the melatonin-related receptor in the rodent brain and peripheral tissues. J Neuroendocrinol. 2001;13:453–458. doi: 10.1046/j.1365-2826.2001.00651.x. [DOI] [PubMed] [Google Scholar]
- Dubocovich ML, Cardinali DP, Guardiola-Lemaitre B, Hagan RM, Krause DN, Sugden D, Vanhoutte PM, Yocca FD. Melatonin receptors. In: Girdlestone D, editor. The IUPHAR Compendium of Receptor Characterization and Classification. London, UK: IUPHAR Media; 1998a. [Google Scholar]
- Dubocovich ML, Masana MI, Benloucif S. Molecular pharmacology and function of melatonin receptor subtypes. Adv Exp Med Biol. 1999;460:181–190. [PubMed] [Google Scholar]
- Dubocovich ML, Yun K, Al-Ghoul WM, Benloucif S, Masana MI. Selective MT2 melatonin receptor antagonists block melatonin-mediated phase advances of circadian rhythms. FASEB J. 1998b;12:1211–1220. doi: 10.1096/fasebj.12.12.1211. [DOI] [PubMed] [Google Scholar]
- Dunlap JC. Molecular bases for circadian clocks. Cell. 1999;96:271–290. doi: 10.1016/s0092-8674(00)80566-8. [DOI] [PubMed] [Google Scholar]
- Flanagan LM, Olson BR, Sved AF, Verbalis JG, Stricker EM. Gastric motility in conscious rats given oxytocin and an oxytocin antagonist centrally. Brain Res. 1992;578:256–260. doi: 10.1016/0006-8993(92)90255-8. [DOI] [PubMed] [Google Scholar]
- Hagan RM, Oakley NR. Melatonin comes of age? Trends Pharmacol Sci. 1995;16:81–83. doi: 10.1016/s0165-6147(00)88985-3. [DOI] [PubMed] [Google Scholar]
- Hara R, Wan K, Wakamatsu H, Aida R, Moriya T, Akiyama M, Shibata S. Restricted feeding entrains circadian clock in the mouse liver without participation of suprachiasmatic nucleus. Genes Cells. 2001;6:269–278. doi: 10.1046/j.1365-2443.2001.00419.x. [DOI] [PubMed] [Google Scholar]
- Hastings MH, Duffield GE, Ebling FJ, Kidd A, Maywood ES, Schurov I. Non-photic signalling in the suprachiasmatic nucleus. Biol Cell. 1997;89:495–503. doi: 10.1016/s0248-4900(98)80005-1. [DOI] [PubMed] [Google Scholar]
- Hunt AE, Al-Ghoul WM, Gillette MU, Dubocovich ML. Activation of MT2 melatonin receptors in rat suprachiasmatic nucleus phase advances the circadian clock. Am J Physiol Cell Physiol. 2001;280:C110–118. doi: 10.1152/ajpcell.2001.280.1.C110. [DOI] [PubMed] [Google Scholar]
- Hunter JD, Milsom WK. Cortical activation states in sleep and anesthesia. I: Cardio-respiratory effects. Respir Physiol. 1998;112:71–81. doi: 10.1016/s0034-5687(98)00018-8. [DOI] [PubMed] [Google Scholar]
- Illnerova H. The suprachiasmatic nucleus and rhythmic pineal melatonin production. In: Klein DC, Moore RY, Reppert SM, editors. Suprachiasmatic Nucleus. The Mind's Clock. New York: Oxford University Press; 1991. pp. 197–216. [Google Scholar]
- Inouye ST, Kawamura H. Persistence of circadian rhythmicity in a mammalian hypothalamic ‘island’ containing the suprachiasmatic nucleus. Proc Natl Acad Sci U S A. 1979;76:5962–5966. doi: 10.1073/pnas.76.11.5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Murai I, Asai S, Takahashi Y, Matsuno Y, Komuro S, Kurosaka H, Iwasaki A, Ishikawa K, Arakawa Y. Central nervous system action of melatonin on gastric acid and pepsin secretion in pylorus-ligated rats. NeuroReport. 1998;9:3989–3992. doi: 10.1097/00001756-199812010-00040. [DOI] [PubMed] [Google Scholar]
- Klein DC. Photoperiodism, Melatonin and the Pineal Gland. Pitman, London: Ciba Foundation Symposium 117; 1985. Photoneural regulation of the mammalian rhythm in melatonin production; pp. 38–56. [DOI] [PubMed] [Google Scholar]
- Krout KE, Kawano J, Mettenleiter TC, Loewy AD. CNS inputs to the suprachiasmatic nucleus of the rat. Neuroscience. 2002;110:73–92. doi: 10.1016/s0306-4522(01)00551-6. [DOI] [PubMed] [Google Scholar]
- Lewy AJ, Wehr TA, Goodwin FK, Newsome DA, Markey SP. Light suppresses melatonin secretion in humans. Science. 1980;210:1267–1269. doi: 10.1126/science.7434030. [DOI] [PubMed] [Google Scholar]
- Liu C, Weaver DR, Jin X, Shearman LP, Pieschl RL, Gibkoff VK, Reppert SM. Molecular dissection of two distinct actions of melatonin on the suprachiasmatic circadian clock. Neuron. 1997;19:91–102. doi: 10.1016/s0896-6273(00)80350-5. [DOI] [PubMed] [Google Scholar]
- McArthur AJ, Hunt AE, Gillette MU. Melatonin action and signal transduction in the rat suprachiasmatic circadian clock: activation of protein kinase C at dusk and dawn. Endocrinology. 1997;138:627–634. doi: 10.1210/endo.138.2.4925. [DOI] [PubMed] [Google Scholar]
- Maggi CA, Meli A. Suitability of urethane anesthesia for physiopharmacological investigations in various systems. Part II: Cardiovascular systems. Experientia. 1986a;42:292–297. doi: 10.1007/BF01942510. [DOI] [PubMed] [Google Scholar]
- Maggi CA, Meli A. Suitability of urethane anesthesia for physiopharmacological investigations in various systems. Part III: Other systems and conclusions. Experientia. 1986b;42:531–537. doi: 10.1007/BF01946692. [DOI] [PubMed] [Google Scholar]
- Matsukawa K, Ninomiya I. Anesthetic effects on tonic and reflex renal sympathetic nerve activity in awake cats. Am J Physiol. 1989;256:R371–378. doi: 10.1152/ajpregu.1989.256.2.R371. [DOI] [PubMed] [Google Scholar]
- Moore RY. Neural control of the pineal gland. Behav Brain Res. 1996;73:125–130. doi: 10.1016/0166-4328(96)00083-6. [DOI] [PubMed] [Google Scholar]
- Moore RY. Circadian rhythms: Basic neurobiology and clinical applications. Annu Rev Med. 1997;48:253–266. doi: 10.1146/annurev.med.48.1.253. [DOI] [PubMed] [Google Scholar]
- Moore RY, Lenn NJ. A retinohypothalamic projection in the rat. J Comp Neurol. 1972;146:1–14. doi: 10.1002/cne.901460102. [DOI] [PubMed] [Google Scholar]
- Mutoh T, Bonham AC, Joad JP. Substance P in the nucleus of the solitary tract augments bronchopulmonary C fiber reflex output. Am J Physiol Regul Integr Comp Physiol. 2000a;279:R1215–1223. doi: 10.1152/ajpregu.2000.279.4.R1215. [DOI] [PubMed] [Google Scholar]
- Mutoh T, Joad JP, Bonham AC. Chronic passive cigarette smoke exposure augments bronchopulmonary C-fibre inputs to nucleus tractus solitarii neurones and reflex output in young guinea-pigs. J Physiol. 2000b;523:223–233. doi: 10.1111/j.1469-7793.2000.00223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niijima A. Observation on the localization of mechanoreceptors in the kidney and afferent nerve fibres in the renal nerves in the rabbit. J Physiol. 1975;245:81–90. doi: 10.1113/jphysiol.1975.sp010836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niijima A, Nagai K, Nagai N, Akagawa H. Effects of light stimulation on the activity of the autonomic nerves in anesthetized rats. Physiol Behav. 1993;54:555–561. doi: 10.1016/0031-9384(93)90249-f. [DOI] [PubMed] [Google Scholar]
- Nonno R, Pannacci M, Lucini V, Angeloni D, Fraschini F, Stankov BM. Ligand efficacy and potency at recombinant human MT2 melatonin receptors: evidence for agonist activity of some mt1-antagonists. Br J Pharmacol. 1999;127:1288–1294. doi: 10.1038/sj.bjp.0702658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittendrigh CS, Daan S. A functional analysis of circadian pacemakers in nocturnal rodents IV. Entrainment: Pacemaker as clock. J Comp Physiol. 1976;106:291–331. [Google Scholar]
- Raybould HE, Lloyd KC. Integration of postprandial function in the proximal gastrointestinal tract. Role of CCK and sensory pathways. Ann NY Acad Sci. 1994;713:143–156. doi: 10.1111/j.1749-6632.1994.tb44061.x. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Godson C, Mahle CD, Weaver DR, Slaugenhaupt SA, Gusella JF. Molecular characterization of a second melatonin receptor expressed in human retina and brain: the Mel1b melatonin receptor. Proc Natl Acad Sci U S A. 1995;92:8734–8738. doi: 10.1073/pnas.92.19.8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. Molecular analysis of mammalian circadian rhythms. Annu Rev Physiol. 2001;63:647–676. doi: 10.1146/annurev.physiol.63.1.647. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR, Ebisawa T. Cloning and characterization of a mammalian melatonin receptor that mediates reproductive and circadian responses. Neuron. 1994;13:1177–1185. doi: 10.1016/0896-6273(94)90055-8. [DOI] [PubMed] [Google Scholar]
- Richardson G, Tate B. Hormonal and pharmacological manipulation of the circadian clock: recent developments and future strategies. Sleep. 2000;1:S77–85. [PubMed] [Google Scholar]
- Rogers RC, Hermann GE. Oxytocin, oxytocin antagonist, TRH, and hypothalamic paraventricular nucleus stimulation effects on gastric motility. Peptides. 1987;8:505–513. doi: 10.1016/0196-9781(87)90017-9. [DOI] [PubMed] [Google Scholar]
- Scheer FA, Ter Horst GJ, van der Vliet J, Buijs RM. Physiological and anatomic evidence for regulation of the heart by suprachiasmatic nucleus in rats. Am J Physiol Heart Circ Physiol. 2001;280:H1391–1399. doi: 10.1152/ajpheart.2001.280.3.H1391. [DOI] [PubMed] [Google Scholar]
- Shafton AD, Ryan A, Badoer E. Neurons in the hypothalamic paraventricular nucleus send collaterals to the spinal cord and to the rostral ventrolateral medulla in the rat. Brain Res. 1998;801:239–243. doi: 10.1016/s0006-8993(98)00587-3. [DOI] [PubMed] [Google Scholar]
- Shibata S, Cassone VM, Moore RY. Effects of melatonin on neuronal activity in the rat suprachiasmatic nucleus in vitro. Neuroscience Lett. 1989;97:140–144. doi: 10.1016/0304-3940(89)90153-5. [DOI] [PubMed] [Google Scholar]
- Silver R, Lesauter J, Tresco PA, Lehman MN. A diffusible coupling signal from the transplanted suprachiasmatic nucleus controlling circadian locomotor rhythms. Nature. 1996;382:810–813. doi: 10.1038/382810a0. [DOI] [PubMed] [Google Scholar]
- Skinner DC, Malpaux B. High melatonin concentrations in third ventricular cerebrospinal fluid are not due to Galen blood recirculating through the choroid plexus. Endocrinology. 1999;140:4399–4405. doi: 10.1210/endo.140.10.7074. [DOI] [PubMed] [Google Scholar]
- Sokolove PG, Bushell WN. The chi square periodogram: its utility for analysis of circadian rhythms. J Theor Biol. 1978;72:131–160. doi: 10.1016/0022-5193(78)90022-x. [DOI] [PubMed] [Google Scholar]
- Stehle JH, Von Gall C, Schomerus C, Korf HW. Of rodents and ungulates and melatonin: creating a uniform code for darkness by different signaling mechanisms. J Biol Rhythms. 2001;16:312–325. doi: 10.1177/074873001129002033. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Cowan WM, Jones EG. An autoradiographic study of the efferent connections of the ventral geniculate nucleus in the albino rat and the cat. J Comp Neurol. 1974;156:143–164. doi: 10.1002/cne.901560203. [DOI] [PubMed] [Google Scholar]
- Teclemariam-Mesbah R, Ter-Horst GJ, Postema F, Wortel J, Buijs RM. Anatomical demonstration of the suprachiasmatic nucleus-pineal pathway. J Comp Neurol. 1999;406:171–182. [PubMed] [Google Scholar]
- van den Pol AN, Obrietan K, Chen G, Belousov AB. Neuropeptide Y-mediated long-term depression of excitatory activity in suprachiasmatic nucleus neurons. J Neurosci. 1996;16:5883–5895. doi: 10.1523/JNEUROSCI.16-18-05883.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Top M, Buijs RM, Ruijter JM, Delagrange P, Spanswick D, Hermes ML. Melatonin generates an outward potassium current in rat suprachiasmatic nucleus neurones in vitro independent of their circadian rhythm. Neuroscience. 2001;107:99–108. doi: 10.1016/s0306-4522(01)00346-3. [DOI] [PubMed] [Google Scholar]
- van Tilborg KA, Rabelink TJ, van Rijn HJ, Boomsma F, Koomans HA. Arterial baroreflex control of renal hemodynamics in humans. Circulation. 1994;90:1883–1890. doi: 10.1161/01.cir.90.4.1883. [DOI] [PubMed] [Google Scholar]
- Vanecek J. Cellular mechanisms of melatonin action. Physiol Rev. 1998;78:687–721. doi: 10.1152/physrev.1998.78.3.687. [DOI] [PubMed] [Google Scholar]
- Vivien-Roels B, Malan A, Rettori MC, Delagrange P, Jeanniot JP, Pevet P. Daily variations in pineal melatonin concentrations in inbred and outbred mice. J Biol Rhythms. 1998;13:403–409. doi: 10.1177/074873098129000228. [DOI] [PubMed] [Google Scholar]
- Vrang N, Larsen PJ, Møller M, Mikkelsen JD. Topographical organization of the rat suprachismatic-paraventricular projection. J Comp Neurol. 1995;353:585–603. doi: 10.1002/cne.903530409. [DOI] [PubMed] [Google Scholar]
- Watts AG, Swanson LW, Sanchez-Watts G. Efferent projections of the suprachiasmatic nucleus: I. Studies using anterograde transport of Phaseolus vulgaris leucoagglutinin in the rat. J Comp Neurol. 1987;258:204–229. doi: 10.1002/cne.902580204. [DOI] [PubMed] [Google Scholar]
- Young MW, Kay SA. Time zones: a comparative genetics of circadian clocks. Nat Rev Genet. 2001;2:702–715. doi: 10.1038/35088576. [DOI] [PubMed] [Google Scholar]