

Abstract
The placenta has evolved in eutherian mammals primarily to provide nutrients for the developing fetus. The genetic control of the regulation of supply and demand for maternal nutrients is not understood. In this review we argue that imprinted genes have central roles in controlling both the fetal demand for, and the placental supply of, maternal nutrients. Recent studies on Igf2 (insulin-like growth factor 2) knockout mouse models provide experimental support for this hypothesis. These show effects on placental transport capacity consistent with a role of IGF-II in modulating both the placental supply and fetal demand for nutrients. Imprinting of genes with such functions may have coevolved with the placenta and new evidence suggests that transporter proteins, as well as the regulators themselves, may also be imprinted. These data and hypotheses are important, as deregulation of supply and demand affects fetal growth and has long term consequences for health in mammals both in the neonatal period and, as a result of fetal programming, in adulthood.
The placenta is of critical importance for the intrauterine development and growth of the embryo and fetus in eutherian mammals, and to a slightly lesser extent in marsupials. Most mammals use the placental mode of reproduction, and there are only very few extant species of monotremes (egg-laying mammals lacking the placenta), such as the duck-billed platypus. Most cell types that give rise to the placenta arise very early in development, beginning with the differentation of trophectoderm and extraembryonic endoderm cells in the blastocyst before implantation. An outline of the main tissues in the mouse placenta and their derivation during development is shown in Fig. 1 (from Rossant & Cross, 2001).
Figure 1. Placental development in the mouse.

Early development of the mouse embryo from embryonic day (E) 4 - E13, showing the origins of the extra-embryonic lineages and the components of the placenta. ICM, inner cell mass. (After Rossant & Cross, 2001.)
A number of genetic pathways have been discovered that are important for the development of different cell types in the placenta, and for their interaction (Rossant & Cross, 2001). Significantly, most mutations in genes involved with placental development and morphogenesis result in abnormal placental development and are lethal to the fetus as a consequence. There is also considerable knowledge of physiological factors and systems that are involved in nutrient transfer across the placenta, including passive diffusion and active transport mechanisms (Sibley et al. 2002). Some of the genes that encode these systems have been isolated, but there are no functional genetic investigations to date of how they are involved in nutrient transfer.
Imprinted genes are a class of genes found in placental mammals, marsupials and seed plants whose expression depends on their parental origin (Brannan & Bartolomei, 1999; Reik & Walter, 2001; Ferguson-Smith & Surani, 2001; Sleutels & Barlow, 2002). In mammals, many imprinted genes are involved in the control of fetal growth, and are expressed in both fetal and placental tissues. Recent work in mice has indicated that the roles of imprinted genes in fetal and placental tissues can be genetically separated, and that in the placenta these genes regulate both growth and specific nutrient transfer (Constância et al. 2002).
Here we examine the links between imprinted genes and placental function. We suggest that imprinted genes in the placenta control the supply of nutrients, whereas in fetal compartments they control nutrient demand by regulating the growth rate of the fetal tissues. We propose that there are interactions between demand and supply systems, which are important to fine tune mammalian growth and development. We suggest the possibility that the evolution of these systems involves imprinting and is closely linked to the evolution of placentation. In addition, there may be other links between imprinting, epigenetic gene regulation, and the development and evolution of extraembryonic tissues.
We begin this review by looking at some of the properties of imprinted genes that are involved in fetal growth control. We then examine critical issues of the physiology of placental function and look at recent evidence that this is affected by imprinted genes. From these observations we derive our central hypothesis on supply and demand regulation. Finally, we consider the implications of this hypothesis for disease situations in humans. It is important to note that we are not attempting to establish an inclusive hypothesis for all imprinted genes, and that there are likely to be imprinted genes that have different roles and whose imprinting may have evolved for different reasons. This review focuses on imprinted genes in general, and on Igf2 specifically, in order for us to develop new hypotheses in this regard. However, we would emphasise that these factors are likely to interact with a variety of other substances, including binding proteins and hormones, in the overall control of fetal nutrient supply and demand.
Imprinted genes, growth and the placenta
Approximately 60 imprinted genes have been discovered so far in mice, and for most the imprinting status is conserved in humans (http://www.mgu.har.mrc.ac.uk/imprinting/imprin-viewdatagenes.html).
Not many more than 100 imprinted genes are expected in mammalian genomes. A substantial proportion of imprinted genes are involved in the control of fetal growth, and in general paternally expressed imprinted genes enhance fetal growth, whereas maternally expressed ones suppress it (Reik & Walter, 2001; Tycko & Morison, 2002). The popular genetic conflict hypothesis explains the evolution of imprinted genes by paternally derived genes that influence nutrient acquisition being selected to extract more resources from the mother (thus being more selfish) whereas maternally derived genes have to balance the nutrient provision to the current fetus with that to future fetuses of the same mother (but potentially different fathers). Thus maternally derived genes are more conservative with regard to resource provision to the fetus (Moore & Haig, 1991; Haig & Graham, 1991).
Further support for the genetic conflict hypothesis comes from the phylogenetic distribution of imprinting. Some genes that are imprinted in eutherian mammals and in marsupials have been found not to be imprinted in chickens, and most interestingly not in monotremes (egg-laying mammals). Imprinted inactivation of the paternal X chromosome is also limited to eutherian mammals (where it occurs in the extraembryonic tissues) and to marsupials. Imprinting is thought to be absent from fishes, reptiles and amphibians, all of which are generally egg laying (although there are some sharks that have a placenta), but has independently evolved in seed plants, where the endosperm has a similar nutrient-providing role as a placenta (Alleman & Doctor, 2000). In Arabidopsis, one imprinted gene has thus far been discovered (Medea), which is maternally expressed exclusively in the endosperm and restrains its size, in agreement with the conflict hypothesis (Grossniklaus et al. 1998).
Directional effects on fetal growth of maternally and paternally expressed genes have been documented in a number of mouse knockout and transgenic experiments. For example, knockouts of the paternally expressed genes Igf2, Peg1, Peg3 and insulin result in intrauterine growth restriction (IUGR), whereas knockouts of the maternally expressed genes H19 and Igf2r, or overexpression of Igf2, result in overgrowth of the fetus (Reik et al. 2001; Tycko & Morison, 2002).
A number of imprinted genes interact with the major fetal growth-controlling IGF and insulin system of growth factors, including Igf2r, H19 and possibly others such as p57Kip2 (Reik et al. 2001). In fetal tissues these factors have roles in cell proliferation, apoptosis and the make up of extracellular space, which explains their effects on growth. However, recently it has been shown that many imprinted genes are expressed in the placenta, thus raising the possibility that they could act on development, growth and function of this organ, thereby influencing fetal growth through resource supply.
Expression and action of imprinted genes in the placenta
Of the approximately 60 imprinted genes identified so far in the mouse genome, around half have been examined for placental expression and all have been found to be expressed. Hence, there is yet an imprinted gene to be found which is not expressed in the placenta. In the following we summarise information about expression patterns of imprinted genes in the placenta and effects of knockouts on placental phenotype. Some of the knockout results have to be interpreted cautiously, since, in general, genes were knocked out both in placenta and fetus, and there is the possibility that fetal expression could have effects on placental phenotype (Gardner et al. 1999).
Imprinted genes have been discovered to affect the growth of several cell types in the placenta. Paternally expressed gene transcripts are found in labyrinthine trophoblast and spongiotrophoblast including glycogen cells (Igf2), the labyrinthine blood vessels (Peg1), and the labyrinth and spongiotrophoblast (Peg3) (Redline et al. 1993; Li et al. 1999; Mayer et al. 2000; Constância et al. 2002). For all three genes the knockouts result in a smaller size of the placenta (65, 85 and 72 %, respectively, at late gestation) (Efstratiadis, 1998; Lefebvre et al. 1998; Li et al. 1999). The Igf2 gene is remarkable in that it possesses a placenta-specific promoter (P0), which results in an IGF-II-encoding transcript specifically in the labyrinthine trophoblast cells (Moore et al. 1997; Constância et al. 2000, 2002). Expression in other cell types in the placenta is from the ‘fetal’ promoters P1-P3. Knocking out the placenta-specific Igf2 transcript results in a placental size deficit close to that in the Igf2 null knockout (68 %), raising the question of what role, if any, transcripts other than P0 play in the placenta. The relative growth deficit of individual placental tissues has only been measured in some knockouts. Both the P0 (Constância et al. 2002) and the Peg3 (N. Anderson, unpublished observations) knockouts apparently affect all tissues of the placenta, resulting in a proportionate reduction in size, although the transcripts are more narrowly localised. This may indicate an early action during development (during which expression may be more widespread) or alternatively interactions between individual placental tissues which affect and coordinate their growth.
Genes which are maternally expressed in the placenta have also been shown to affect growth, and in one case development, of the placenta. H19, Igf2r and p57Kip2 are widely expressed in placenta in a similar pattern to that of Igf2 itself, whereas expression of Ipl is limited to the labyrinth, and that of Mash2 to the spongiotrophoblast and labyrinth (Leighton et al. 1995; Wutz et al. 1998; Takahashi et al. 2000). Deletion of H19, Igf2r and p57Kip2 all result in hyperplasia of all layers of the placenta (to 140, 140 and 144 %, respectively) (Eggenschwiler et al. 1997; Takahashi et al. 2000). Interestingly, while expression of Ipl is limited to the labyrinth, its knockout again results in global hyperplasia of placental tissues, with a disproportionate expansion of the spongiotrophoblast (Frank et al. 2002). It may be the case, for example, that growth of the placenta might be primarily controlled through the labyrinthine trophoblast.
It is likely that there are other maternally expressed imprinted genes (for example those on chromosome 12) with functions in placental growth, since a number of paternal uniparental disomies result in placental overgrowth (Hurst & McVean, 1997; Georgiades et al. 2001). The knockout of the Mash2 gene has shown that this gene plays an essential role in the development of the spongiotrophoblast. Embryos die at E10.5 and lack a spongiotrophoblast and chorio-allantoic fusion. This defect is autonomous to the placenta; lack of Mash2 does not affect the fetus if the placental defect is rescued (Tanaka et al. 1997). The effect of Mash2 as assessed in the knockout has been interpreted as contradicting the conflict hypothesis, but other interpretations are possible (Hurst & McVean, 1997; Iwasa, 1998).
Imprinted genes may also affect the supply of resources to the fetus via effects on specific transporters and channels involved in transplacental solute exchange. Genes in this category include the maternally expressed Slc22a2 and Slc22a3 genes, which are linked to Igf2r, and encode organic cation transporters with a broad substrate specificity (which are possibly involved in clearing extracellular monoamines), and the maternally expressed Impt1/Slc22a1l gene which is linked to Igf2 and p57Kip2 and also encodes an organic cation transporter (Dao et al. 1998; Zwart et al. 2001a). A knockout of the Slc22a3 gene has indeed revealed effects on placental transfer functions (as measured by the transfer of the neurotoxin MPP+), even though placental and fetal growth were unaffected (Zwart et al. 2001b). Finally, the paternally expressed Ata3 gene (Mizuno et al. 2002) encodes a component of the system A amino acid transport system of the placenta (and other organs). This gene (and its neighbours Ata1 and 2) is of special interest since the system A is upregulated in the Igf2 P0 knockout (Constância et al. 2002; see below).
Of additional interest are a seemingly heterogeneous class of genes that are imprinted (all so far with maternal expression) exclusively or predominantly in the placenta. These include the Slc22a2 and Slc22a3 genes mentioned above and the Obph1, Nap1l4, Tnfrh1 and Tssc4 genes, which are linked to Igf2 and p57Kip2 (Engemann et al. 2000; Paulsen et al. 2000; Zwart et al. 2001a), and the Dcn gene, which is located on chromosome 10 although not obviously in association with other imprinted genes (Mizuno et al. 2002). The Tnfrh1 gene is interesting as it encodes a decoy receptor in the tumour necrosis factor (TNF) receptor family, and it is tempting to speculate that it may dampen the maternal immune response to the invasive placenta, which is essentially a hemiallograft (Clark et al. 2002). It remains to be seen whether this heterogeneous class of genes is larger than hitherto suspected and what precise functions these genes have in the placenta.
Thus imprinted genes could affect the function of the placenta by regulating nutrient supply in two principal ways: either by affecting overall growth of the placenta or of particular structures (such as the labyrinthine trophoblast), or by affecting specific transport systems. These possibilities are being addressed in detail in studies on the Igf2 gene. Before considering this, however, we need to review briefly the physiology of placental exchange mechanisms and the factors which modulate solute flux.
Factors controlling nutrient transfer across the placenta
Current data suggest that there are four principal mechanisms by which solutes cross the placenta (Sibley & Boyd, 1998): bulk flow/solvent drag, endocytosis/exocytosis, diffusion, and transporter-protein mediated transfer. Here we focus on the latter two mechanisms.
Diffusion
In the absence of an electrical potential difference (PD) across the barrier (but see below), the rate of transfer (Jnet, mol s−1 g−1) of any solute across the placenta is determined by Fick’ s Law (see Sibley & Boyd, 1988):
Jnet = PS (Cm – Cf),
where P is placental permeability (cm2 s−1), S is surface area available for diffusion within 1 g placenta (cm2 g−1), and Cm and Cf are mean plasma solute concentrations of the unbound solute in plasma water in maternal and fetal blood flowing past the exchange area. There are two pathways available for diffusion: a lipophilic route for fat-soluble molecules and a hydrophilic route for fat-insoluble molecules. Their relative importance in determining Jnet is determined by the degree of solubility in lipid of the molecule in question. For a fat-soluble molecule, which can diffuse across the plasma membrane, the entire exchange barrier is available for diffusion. The rate of transfer of such molecules (e.g. oxygen, carbon dioxide) is therefore principally determined by the concentration gradient (Cm - Cf), which is in turn dependent on the rate of delivery or extraction from the exchange site, i.e. the blood flow. There are many hormones and other factors contributing to the control of uterine and umbilical blood flow and therefore of transfer of such substances (Carter & Myatt, 1995).
On the other hand, the rate of transfer of hydrophilic molecules (glucose, amino acids, etc.), which cannot diffuse easily across the plasma membranes, will be much slower and dependent on the permeability properties of the placenta. A variety of studies have provided physiological evidence of a water-filled paracellular route across the placenta (Sibley & Boyd, 1998); this underlies the permeability of the placenta and provides a pathway for diffusion of hydrophilic solutes. Factors that alter the dimensions of this pathway will affect the rate of transfer of these molecules. Our recent data in the mouse suggest that placenta-specific IGF-II might be one such factor; mice lacking the placenta-specific Igf2 transcript had a lower permeability to 51Cr-EDTA (an inert hydrophilic tracer) than did the intact animals (Constância et al. 2002). We are currently investigating the mechanisms underlying this effect.
Any electrical PD across the placental exchange barrier will drive net diffusion of charged molecules. Whether or not such a PD exists remains somewhat controversial. However, recent data from in vivo and in vitro studies do suggest that there is a small PD, fetal side negative, across the human placenta (Greenwood et al. 1993; Ward et al. 1998). It is possible that this PD might be modulated in relation to the osmotic effect of nutrient transfer (Birdsey et al. 1999) and so alter the rate of diffusional transfer of charged molecules.
Transporter protein-mediated transfer
Bulk flow/solvent drag and diffusion provide mechanisms of transfer across the placenta for all solutes, with rates dependent on the physicochemical characteristics of the molecules themselves and the other factors described above. However, a wealth of data show that, in addition, there is a wide range of selective transporter proteins in the transporting epithelium of the placenta, the syncytiotrophoblast. As in other epithelia, these transporter proteins are highly substrate specific and include ion channels (e.g. Kir2.1 for K+; Clarson et al. 2001), exchangers (e.g. Na+-H+ exchanger; Sibley et al. 2002), co-transporters (e.g. system A amino acid transporter; Johnson & Smith, 1988) and pumps (e.g. plasma membrane Ca2+-ATPase, PMCA; Fisher et al. 1987). The rate of transfer of the specific substrates via the transporters is dependent on the concentration gradients for channels and systems operating by facilitated diffusion (e.g. glucose transfer on the GLUT1 transporter; Jansson et al. 2002) and, for secondary (system A) and primary (PMCA) active transporters, the availability and utilisation of ATP. Hormones and other factors controlling the rate of transfer via these systems mainly operate by either affecting the affinity of the transporter for substrate (Km) or the number of transporter molecules in the plasma membrane (Vmax).
As vectorial transport across the placenta will be dependent on polarisation of the syncytiotrophoblast, hormones might also act by affecting the polarity of insertion of the transporters into either the microvillous (maternal-facing) plasma membrane or the basal (fetal-facing) plasma membrane. There is a wide range of hormones and other factors likely to control transporter protein-mediated transfer (Sibley & Boyd, 1988), although the relative importance of these in vivo is largely unknown. Of particular relevance to this article are the in vitro data showing that insulin and IGF-I and IGF-II can modulate the uptake of glucose and of the non-metabolisable amino acid analogue α-aminoisobutyric acid by primary cytotrophoblast cells derived from the human term placenta (Kniss et al. 1994; Bloxam et al. 1994; Karl, 1995).
Placental transfer in relation to fetal growth
The factors described above will control the capacity of the placenta to supply nutrients to the fetus. The solute and water composition of the fetus at term must equal the net transfer of solute and water across the placenta over the course of gestation, plus the relatively small amount of transfer across amnion and other membranes. Therefore modulation of placental transfer will directly affect fetal growth.
Blood flow to the uterine and umbilical circulations of the placenta has been considered to be the prime determinant of transfer rate, and therefore of the supply of nutrients to the fetus for growth. This is both because of the importance of blood flow for exchange of the respiratory gases, as described above, and because it is clear that flows through both circulations of the placenta are altered in IUGR (Harrington et al. 1991). Furthermore, there has been no direct evidence previously that the transfer capacity of the placental exchange barrier itself to supply nutrients can be rate limiting for fetal growth. However, this has now been demonstrated clearly in mice lacking the placenta-specific Igf2 transcript (Constância et al. 2002). These animals have smaller than normal placentas at day 16 of gestation, with a lower passive permeability, but appear to maintain fetal growth close to normal by upregulating amino acid transfer capacity (see next section). There is a direct correlate to this in human studies, where an inverse relationship between the activity of the system A amino acid transporter in the microvillous plasma membrane of the placenta (per milligram membrane protein) and the size of the baby at birth has been found (Godfrey et al. 1998). This suggests that in the proportionately small placentas of small normal babies, there is an upregulation of the expression of the system A amino acid transporter in order to maintain transfer capacity, as in the placenta-specific Igf2 knockout mice. It has also been shown recently that inhibiting the system A transporter in rats leads to fetal growth restriction (Cramer et al. 2002). Furthermore system A activity in the microvillous membrane is lower, per milligram membrane protein, in human placentas from IUGR babies (reviewed in Sibley et al. 2002).
These data suggest that controlling the capacity of the placental exchange barrier to supply amino acids, particularly those such as glycine, which are transported selectively by system A, does directly modulate fetal growth above and beyond the effects of blood flow. This is supported by human in vivo studies of the transfer of amino acids labelled with stable isotopes, which suggest that amino acid transfer is, normally, only just sufficient for fetal growth requirements (reviewed in Cetin, 2001). There is additional evidence that the expression and activity of several other transporters in the syncytiotrophoblast are altered in human IUGR (Sibley et al. 2002). The pathophysiology is complex, as not all activities are decreased: PMCA activity in the basal membrane is increased (Strid & Powell, 2001) whereas, for example, the system y+ cationic amino acid transporter activity is unaltered (Jansson et al. 1998; Ayuk et al. 2002). Nevertheless it is clear that this syncytiotrophoblast transport anomaly is an important feature of human IUGR, further emphasising the importance of controlling the supply of nutrients by modulation of the placental barrier transport capacity, as well as by modulating blood flow.
Function of placental IGF-II in nutrient transfer
The actions of imprinted genes in regulating nutrient transfer can be diverse: they can affect the growth and transport capacity of the placenta, or they may modulate the requirements for nutrients by the fetus, mainly through the control of fetal growth. These functions are difficult to discern experimentally because most imprinted genes are expressed in fetal tissues as well as in the placenta, where they may play additional roles. Thus most knockouts of imprinted genes described above affect fetal growth as well as placental growth, but the effect of altered placental growth and function on fetal growth when fetal expression of imprinted genes remained normal has not been tested. Recently, we tested the hypothesis that a major imprinted growth control gene, Igf2, exerts a crucial role in the supply of nutrients by the placenta.
IGF-II and placental growth
The surface area for exchange of nutrients correlates directly with placental size. Several studies have now shown that IGF-II is a major modulator of placental size. Igf2 is abundantly expressed in all placental cell types that are derived from the zygote (Lee et al. 1990; Redline et al. 1993). A significant proportion of IGF-II peptide that is produced in the labyrinthine trophoblast is encoded in the P0 transcript, which is only found in these cells (Constância et al. 2002). Mouse models that lack IGF-II in all placental cell types (De Chiara et al. 1991) or that have reduced levels specifically in the labyrinthine trophoblast cells (Constância et al. 2002) both result in severe growth impairment of the placenta (30–40 % smaller than normal). It is remarkable that the placental growth restriction of the P0-deficient mice is of the same magnitude as in Igf2 null allele mice. However, it is not known if all transcripts of Igf2 in the placenta are translated with the same efficiency. Placentomegaly (ranging from 140 % to 230 % of normal) is observed in IGF-II overexpressing mouse models (Leighton et al. 1995; Eggenschwiler et al. 1997; Sun et al. 1997). The mechanisms that lead to the observed changes in placenta size are unknown but are likely to be the results of modifications in cell survival and proliferation as shown for embryo size (Burns & Hassan, 2001). The transduction of the proliferative IGF-II signal may, however, be distinct in fetal organs and placenta. Genetic evidence suggests that a placenta-specific receptor, which is distinct from fetal receptors (IGF-IR and INS-R), mediates the IGF-II growth-promoting role in this organ (Efstratiadis, 1998).
Placental growth can also be indirectly modulated by the action of IGF-II produced in fetal tissues. Gardner et al. (1999) reported that chimeric embryos with IGF-II levels normal in placenta but deficient in all fetal organs had smaller placentas. The interplay between placental IGF-II and fetal IGF-II and the endocrine action of this growth factor remains largely elusive, but it is possible that low levels of circulating IGF-II in serum (caused by the lack of fetal IGF-II) leads to failure in endocrine stimulation of placental growth. The placenta is likely to be particularly sensitive to endocrine stimulation since it must respond in an effective manner to nutrient demand and other requirements emanating from the fetus. This raises the interesting possibility that levels of circulating IGF-II are correlated with the regulation of nutrient supply (see below).
IGF-II and placental transfer capacity
In the Igf2 null knockout, placental and fetal size deficits arise at a similar stage in development, early after midgestation (Baker et al. 1993). In the P0 mouse model, the size deficit of the placenta arises at a similar time, but that of the fetus occurs much later in gestation (Constância et al. 2002). At E12 and E14 the P0 placenta is ≈20 % smaller than wild-type, but the fetuses are normal size. At E16, P0-deficient fetuses are marginally smaller (4 %) while the placenta is 32 % smaller than wild-type. The lack of a positive correlation between placental size and fetal size at these stages was surprising and suggested the possibility that placental transfer capacity was not reduced in proportion with placental size reduction. However, measuring the passive permeability to hydrophilic solute we observed (both at E16 and at E19) that the P0-deficient fetuses received even less of the EDTA radioactive tracer than would be expected from just a reduction in placental size (Constância et al. 2002). Thus low levels of IGF-II in the murine labyrinthine trophoblast result not only in a reduction in the size of all placental tissues, but also apparently in a reduction in the rate of transfer by passive diffusion, per unit surface area of the placenta. The mechanisms of the reduction of nutrient diffusion capacity of the P0 placentas are at present unknown. As discussed in previous sections, the rate of diffusional transfer of nutrients is mainly determined by the width of the exchange barrier, the dimensions of the channels and blood flow. These parameters need to be studied in detail to provide an explanation for the passive diffusion defect observed in the P0 mutants.
The permeability defect of the P0-deficient placenta at E16, coupled to the reduction of overall surface area for exchange of nutrients, should have greatly compromised the growth of the fetus at this gestational stage. However, we found a concomitant increase in 14C-MeAIB (transported by the system A amino acid transporter in the placenta and used as an indicator of secondary active transport) transfer suggesting compensation for the decreased placental size and passive permeability (Constância et al. 2002). At E19, when fetal growth in the knockout is reduced to 78 % of control, we found that 14C-MeAIB transfer across the small mutant placenta, although still increased per gram of placenta, provided significantly less analogue to the mutant than wild-type fetus. Hence, late in gestation, the increase in system A activity failed to compensate for the small size and reduced passive permeability of the P0 placenta with the result that fetal growth retardation ensued from a lack of nutrients. This early compensatory mechanism seems to be specific for amino acid transfer as we found that glucose transfer is not altered in P0 mutant mice (M. Constância, unpublished observations).
The upregulation of amino acid transport (via system A) may be a direct consequence of an IGF-II deficiency in the labyrinthine trophoblast, and thus local placental production of IGF-II by the P0 transcript may negatively regulate amino acid transport systems. Alternatively fetal IGF-II may act as an endocrine signal of fetal nutrient requirements and upregulate transporters in the placenta. Certainly, in vitro experiments (mentioned in the previous section) suggest that exogenous IGF-II acting like an endocrine signal normally stimulates amino acid transport by trophoblast cells. Fetal IGF-II may therefore be an important demand signal for nutrients from the fetus to the placenta, either acting directly through serum levels or indirectly through intermediary signalling molecules. This suggests that the placenta discriminates between IGF-II produced within the organ and that produced by the fetus: it will be important to elucidate the mechanisms of such discrimination.
If the system A transporter is upregulated in response to fetal IGF-II signalling to the placenta, this upregulation may not occur in mice that lack IGF-II in all placental and fetal tissues. The lack of IGF-II in fetal tissues reduces general growth and nutrient requirements and hence a smaller placental supply of nutrients is required to support the demands of fetal growth. This hypothesis is consistent with the findings by Matthews et al. (1999) that showed a reduction of expression of several cationic and anionic amino acid transport proteins in mouse placental tissue derived from Igf2 null mice.
The supply and demand hypothesis of imprinting
We have argued from the above experiments that the Igf2 gene combines and balances the genetic control of supply (through expression in the placenta) with the genetic control of demand (through expression in fetus) for nutrients. This hypothesis can be extended to the imprinted gene cluster surrounding Igf2 and to other imprinting clusters, and may provide an additional reason for their clustering in the genome.
The Igf2 gene lies in a large cluster of imprinted genes most of which are maternally expressed and some of which are only imprinted in the placenta. We propose that most of these genes control supply negatively (Fig. 2). Indeed the knockout of the maternally expressed Ipl gene results in a large placenta but normal-sized fetus (Frank et al. 2002) (increase of supply but not demand) and so does the knockout of the maternally expressed p57Kip2 gene (Takahashi et al. 2000) (and intriguingly the placental hyperplasia phenotype is abolished by IGF-II deficiency in the placenta, suggesting an antagonistic interaction between p57Kip2 and IGF-II; Caspary et al. 1999). Thus this imprinting cluster contains a strong demand promoter (IGF-II acting in fetal tissues), a strong supply promoter (IGF-II acting in placental tissues), and many supply suppressors whose strengths need to be determined (Fig. 2). The positive effects on supply and demand (paternally expressed Igf2 gene) are thus balanced by negative effects on supply (maternally expressed genes). The fine balance between these systems may arise by coevolution, which is facilitated by clustering.
Figure 2. Proposed functions of imprinted supply and demand genes in the distal chromosome 7 cluster in the mouse.
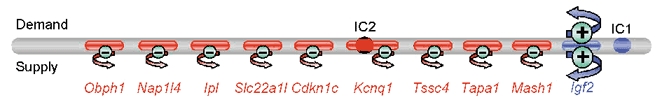
The cluster contains a positive regulator of supply and demand for maternal nutrients (+ sign and large arrows), the paternally expressed Igf2 gene. We propose that most of the genes in the cluster that are maternally expressed counteract negatively the dominant signals from IGF-II by reducing the supply of nutrients by the placenta (- sign and small arrows). A negative effect of the maternally expressed genes on nutrient demand by the fetus cannot be excluded (not represented). This functional antagonism may or may not involve a direct interaction with the Igf2 gene product. The genes represented here are expressed and imprinted in the placenta. The positive effects on supply and demand are controlled by paternal germ line epigenetic modifications at Imprinting Centre 1 (IC1) and balanced by the negative effects on supply controlled by maternal germ line epigenetic modifications at IC2. Note that not all of the genes within the imprinted cluster are represented. The genes shown here are: Obph1 (oxysterol-binding protein), Nap1l4 (nucleosome assembly protein), Ipl (cytoplasmic protein with a pleckstrin-homology domain), Slc22a1l (organic cation transporter), Cdkn1c (cyclin-dependent kinase inhibitor), Kcnq1 (voltage-gated potassium channel protein KQT-like1), Tssc4 (tumor-suppressing subchromosomal transferable fragment 4), Tapa1 (CD81 antigen), Mash2 (BHLH transcription factor), Igf2 (insulin-like growth factor). Red: maternally expressed genes; Blue: paternally expressed genes.
There may be additional imprinting clusters that also regulate the supply and demand balance. In the imprinting cluster on proximal chromosome 17, a positive demand and supply system (the silent paternal allele of Igf2r in fetus and placenta, respectively) is balanced by the negative supply and demand system of the maternally active allele (in the case of Igf2 this corresponds to the effect of the inactive maternal allele). The absence or presence of IGF-IIR will have these effects simply because its function is to regulate IGF-II levels negatively (Haig & Graham, 1991). Intriguingly, the Igf2r gene is flanked by two maternally expressed genes (Slc22a2 and Slc22a3) whose imprinting is placenta specific and which encode major facilitator-family transporters with predicted effects on placental organic cation transport capacity (Zwart et al. 2001a) suggesting that these two genes may be supply suppressors. Indeed a knockout of one of these genes has revealed effects on placental transfer functions (Zwart et al. 2001b). In addition to the balancing between genes within the clusters there is of course also balancing between different imprinting clusters, as exemplified by the Igf2/Igf2r functional antagonism. In this respect it is striking that another organic cation transporter gene which is imprinted in placenta, Impt1/Slc22a1l, is located in the chromosome 7 imprinting cluster (Dao et al. 1998) (Fig. 2).
Our observation that amino acid transport is upregulated to meet demand (determined by fetal IGF-II) suggests that there could be selection on such transporter genes to become imprinted. Indeed it has recently been shown that one of the components of the active amino acid transport system A, the Ata3 gene, is paternally expressed and that it lies in a gene cluster on chromosome 15 together with two other components of the same system, Ata1 and Ata2 (Mizuno et al. 2002).
While either reduced supply or reduced demand can lead to small offspring (placental or fetal lack of supply or demand promoter), increased demand has to be met with increased supply to lead to enhanced growth (overexpression of growth/demand promoter in fetus leads to upregulation of active transport). However, importantly, an increased capacity for supply alone (overexpression of supply promoter in placenta or lack of supply suppressor) does not need to lead to overgrowth if demand remains the same, as in the case of the placental overgrowth seen in the Ipl gene knockout (Frank et al. 2002). This can lead to situations in which imbalance of imprinted genes in knockouts or in monoparental paternal disomies results in large placentas but normal-sized fetuses, an observation that has been interpreted as contradicting the genetic conflict hypothesis (Hurst & McVean, 1997). It can also lead to the birth of normal-looking disomic offspring, despite the presence of imprinted genes on the tested chromosomes (http://www.mgu.har.mrc.ac.uk/imprinting/imprin-viewdatagenes.html). While additional physiological data are needed, these anomalies may ultimately be explained by our supply and demand hypothesis. We therefore propose that the regulation of the pivotal balance between supply and demand of nutrients in mammals with a placenta is a specifically selected function of imprinted genes, which may have coevolved with placentation.
Implications for human health
Poor intrauterine growth has an important role in determining life quality and expectancy. When fetal growth is slowed sufficiently to result in clinical IUGR there is a significantly increased risk of cerebral palsy, short stature and sub-normal intellectual and psychological performance during later childhood (Gray et al. 2001; Lundgren et al. 2001; Strauss, 2000). Even within the normal range, smaller size at birth is associated with an increased incidence of cardiovascular and metabolic diseases in later life (Barker, 1999).
Human epidemiological studies have linked the association between impaired intrauterine growth and adult disease to poor nutrition during pregnancy and early postnatal life (Barker, 1999). Studies in experimental animals have supported this concept and shown that the prenatal nutritional environment can have significant effects on postnatal cardiovascular and metabolic function, within the normal birthweight range (Bertram & Hanson, 2001). Together, the epidemiological and experimental findings have led to the hypothesis that variations in maternal diet and body composition result in restructuring of fetal tissues and organ systems in a way that predisposes them to subsequent pathophysiology. However, in humans, fetal growth restriction can result from chromosomal anomalies and from alterations in expression of imprinted genes (Kingdom et al. 2000; Miozzo & Simoni, 2002). Indeed, the recent data from the mouse presented here suggest that changes in expression of imprinted genes could provide a genetic link between the provision of nutrients, the intrauterine growth rate, and the programming of fetal systems determining the risk of disease in adulthood.
This interaction between imprinted genes and nutrition could take place in at least two ways. First, inheritance of specific transcripts of imprinted genes, such as the placenta-specific Igf2 transcript, could control the placental supply of nutrients available for fetal growth, irrespective of the maternal nutritional state. Indeed, once the placental constraint on growth is lifted in the small P0 knockout mice at birth, postnatal catch-up growth is rapid (Constância et al. 2002), a scenario with the highest risk of coronary heart disease in human studies (Eriksson et al. 1999). It is therefore likely that genetic determinants for diseases resulting from fetal programming may be found in imprinted genes. Secondly, imprinting of the genes themselves may be affected by environmental factors such as nutrition. Imprinting is controlled epigenetically by differential DNA methylation and chromatin modifications, which in turn can be modified by environmental factors and perhaps nutrition (Reik et al. 1993; Dean et al. 1998; Khosla et al. 2001). This would result in a direct relationship between maternal nutritional state and feto-placental growth at the earliest stages of development. This could then be amplified or ameliorated later in gestation by a differential pattern of gene expression in fetal and placental tissues resulting from the earlier nutritionally induced changes in imprinting. Hence, imprinted genes may provide a system for nutrient-gene interactions which modify the balance between the supply and demand of nutrients in utero with major implications for fetal growth and the intrauterine programming of adult disease.
Acknowledgments
The authors’ work is funded by the MRC and the BBSRC.
REFERENCES
- Alleman M, Doctor J. Genomic imprinting in plants: observations and evolutionary implications. Plant Mol Biol. 2000;43:147–161. doi: 10.1023/a:1006419025155. [DOI] [PubMed] [Google Scholar]
- Ayuk PT, Theophanous D, D'Souza SW, Sibley CP, Glazier JD. L-arginine transport by the microvillous plasma membrane of the syncytiotrophoblast from human placenta in relation to nitric oxide production: effects of gestation, preeclampsia, and intrauterine growth restriction. J Clin Endocrinol Metab. 2002;87:747–751. doi: 10.1210/jcem.87.2.8204. [DOI] [PubMed] [Google Scholar]
- Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- Barker DJP. Fetal programming and public health. In: O'Brien PMS, Wheeler T, Barker DJP, editors. Fetal Programming: Influences on Development and Disease in Later Life. London: RCOG Press; 1999. pp. 3–11. [Google Scholar]
- Bertram CE, Hanson MA. Animal models and programming of the metabolic syndrome. Br Med Bull. 2001;60:103–121. doi: 10.1093/bmb/60.1.103. [DOI] [PubMed] [Google Scholar]
- Birdsey TJ, Boyd RD, Sibley CP, Greenwood SL. Effect of hyposmotic challenge on microvillous membrane potential in isolated human placental villi. Am J Physiol. 1999;276:R1479–1488. doi: 10.1152/ajpregu.1999.276.5.R1479. [DOI] [PubMed] [Google Scholar]
- Bloxam DL, Bax BE, Bax CM. Epidermal growth factor and insulin-like growth factor I differently influence the directional accumulation and transfer of 2- aminoisobutyrate (AIB) by human placental trophoblast in two-sided culture. Biochem Biophys Res Commun. 1994;199:922–929. doi: 10.1006/bbrc.1994.1317. [DOI] [PubMed] [Google Scholar]
- Brannan CI, Bartolomei MS. Mechanisms of genomic imprinting. Curr Opin Genet Dev. 1999;9:164–170. doi: 10.1016/S0959-437X(99)80025-2. [DOI] [PubMed] [Google Scholar]
- Burns JL, Hassan AB. Cell survival and proliferation are modified by insulin-like growth factor 2 between days 9 and 10 of mouse gestation. Development. 2001;128:3819–3830. doi: 10.1242/dev.128.19.3819. [DOI] [PubMed] [Google Scholar]
- Carter AM, Myatt L. Control of placental blood flow: workshop report. Reprod Fertil Dev. 1995;7:1401–1406. doi: 10.1071/rd9951401. [DOI] [PubMed] [Google Scholar]
- Caspary T, Cleary MA, Perlman EJ, Zhang P, Elledge SJ, Tilghman SM. Oppositely imprinted genes p57(Kip2) and Igf2 interact in a mouse model for Beckwith-Wiedemann syndrome. Genes Dev. 1999;13:3115–3124. doi: 10.1101/gad.13.23.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetin I. Amino acid interconversions in the fetal-placental unit: the animal model and human studies in vivo. Pediatr Res. 2001;49:148–154. doi: 10.1203/00006450-200102000-00004. [DOI] [PubMed] [Google Scholar]
- Clark L, Wei M, Cattoretti G, Mendelsohn C, Tycko B. The Tnfrh1 (Tnfrsf23) gene is weakly imprinted in several organs and expressed at the trophoblast-decidua interface. BMC Genet. 2002;3:11. doi: 10.1186/1471-2156-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarson LH, Greenwood SL, Mylona P, Sibley CP. Inwardly rectifying K(+) current and differentiation of human placental cytotrophoblast cells in culture. Placenta. 2001;22:328–336. doi: 10.1053/plac.2000.0622. [DOI] [PubMed] [Google Scholar]
- Constância M, Dean W, Lopes S, Moore T, Kelsey G, Reik W. Deletion of a silencer element in Igf2 results in loss of imprinting independent of H19. Nat Genet. 2000;26:203–206. doi: 10.1038/79930. [DOI] [PubMed] [Google Scholar]
- Constância M, Hemberger M, Hughes J, Dean W, Ferguson-Smith A, Fundele R, Stewart F, Kelsey G, Fowden A, Sibley C, Reik W. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature. 2002;417:945–948. doi: 10.1038/nature00819. [DOI] [PubMed] [Google Scholar]
- Cramer S, Beveridge M, Kilberg M, Novak D. Physiological importance of system A-mediated amino acid transport to rat fetal development. Am J Physiol Cell Physiol. 2002;282:C153–160. doi: 10.1152/ajpcell.2002.282.1.C153. [DOI] [PubMed] [Google Scholar]
- Dao D, Frank D, Qian N, O'Keefe D, Vosatka RJ, Walsh CP, Tycko B. IMPT1, an imprinted gene similar to polyspecific transporter and multi-drug resistance genes. Hum Mol Genet. 1998;7:597–608. doi: 10.1093/hmg/7.4.597. [DOI] [PubMed] [Google Scholar]
- Dean W, Bowden L, Aitchison A, Klose J, Moore T, Meneses JJ, Reik W, Feil R. Altered imprinted gene methylation and expression in completely ES cell-derived mouse fetuses: association with aberrant phenotypes. Development. 1998;125:2273–2282. doi: 10.1242/dev.125.12.2273. [DOI] [PubMed] [Google Scholar]
- De Chiara TM, Robertson EJ, Efstratiadis A. Parental imprinting of the mouse insulin-like growth factor II gene. Cell. 1991;64:849–859. doi: 10.1016/0092-8674(91)90513-x. [DOI] [PubMed] [Google Scholar]
- Engemann S, Strodicke M, Paulsen M, Franck O, Reinhardt R, Lane N, Reik W, Walter J. Sequence and functional comparison in the Beckwith-Wiedemann region: implications for a novel imprinting centre and extended imprinting. Hum Mol Genet. 2000;9:2691–2706. doi: 10.1093/hmg/9.18.2691. [DOI] [PubMed] [Google Scholar]
- Efstratiadis A. Genetics of mouse growth. Int J Dev Biol. 1998;42:955–976. [PubMed] [Google Scholar]
- Eggenschwiler J, Ludwig T, Fisher P, Leighton PA, Tilghman SM, Efstratiadis A. Mouse mutant embryos overexpressing IGF-II exhibit phenotypic features of the Beckwith-Wiedemann and Simpson-Golabi-Behmel syndromes. Genes Dev. 1997;11:3128–3142. doi: 10.1101/gad.11.23.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson JG, Forsen T, Tuomilehto J, Winter PD, Osmond C, Barker DJP. Catch-up growth in childhood and death from coronary heart disease: longitudinal study. B M J. 1999;318:427–431. doi: 10.1136/bmj.318.7181.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson-Smith AC, Surani MA. Imprinting and the epigenetic asymmetry between parental genomes. Science. 2001;293:1086–1089. doi: 10.1126/science.1064020. [DOI] [PubMed] [Google Scholar]
- Fisher GJ, Kelley LK, Smith CH. ATP-dependent calcium transport across basal plasma membranes of human placental trophoblast. Am J Physiol. 1987;252:C38–46. doi: 10.1152/ajpcell.1987.252.1.C38. [DOI] [PubMed] [Google Scholar]
- Frank D, Fortino W, Clark L, Musalo R, Wang W, Saxena A, Li CM, Reik W, Ludwig T, Tycko B. Placental overgrowth in mice lacking the imprinted gene Ipl. Proc Natl Acad Sci U S A. 2002;99:7490–7495. doi: 10.1073/pnas.122039999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RL, Squire S, Zaina S, Hills S, Graham CF. Insulin-like growth factor-2 regulation of conceptus composition: effects of the trophectoderm and inner cell mass genotypes in the mouse. Biol Reprod. 1999;60:190–195. doi: 10.1095/biolreprod60.1.190. [DOI] [PubMed] [Google Scholar]
- Georgiades P, Watkins M, Burton GJ, Ferguson-Smith AC. Roles for genomic imprinting and the zygotic genome in placental development. Proc Natl Acad Sci U S A. 2001;98:4522–4527. doi: 10.1073/pnas.081540898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey KM, Matthews N, Glazier J, Jackson A, Wilman C, Sibley CP. Neutral amino acid uptake by the microvillous plasma membrane of the human placenta is inversely related to fetal size at birth in normal pregnancy. J Clin Endocrinol Metab. 1998;83:3320–3326. doi: 10.1210/jcem.83.9.5132. [DOI] [PubMed] [Google Scholar]
- Gray PH, Jones P, O' Callaghan MJ. Maternal antecedents for cerebral palsy in extremely preterm babies: a case-control study. Develop Med Child Neurol. 2001;43:580–585. doi: 10.1017/s0012162201001074. [DOI] [PubMed] [Google Scholar]
- Greenwood SL, Boyd RD, Sibley CP. Transtrophoblast and microvillus membrane potential difference in mature intermediate human placental villi. Am J Physiol. 1993;265:C460–466. doi: 10.1152/ajpcell.1993.265.2.C460. [DOI] [PubMed] [Google Scholar]
- Grossniklaus U, Vielle-Calzada JP, Hoeppner MA, Gagliano WB. Maternal control of embryogenesis by MEDEA, a polycomb group gene in Arabidopsis. Science. 1998;280:446–450. doi: 10.1126/science.280.5362.446. [DOI] [PubMed] [Google Scholar]
- Haig D, Graham C. Genomic imprinting and the strange case of the insulin-like growth factor II receptor. Cell. 1991;64:1045–1046. doi: 10.1016/0092-8674(91)90256-x. [DOI] [PubMed] [Google Scholar]
- Harrington KF, Campbell S, Bewley S, Bower S. Doppler velocimetry studies of the uterine artery in the early prediction of pre-eclampsia and intra-uterine growth retardation. Eur J Obstet Gynecol Reprod Biol. 1991;42(suppl.):S14–20. [PubMed] [Google Scholar]
- Hurst LD, McVean GT. Growth effects of uniparental disomies and the conflict theory of genomic imprinting. Trends Genet. 1997;13:436–443. doi: 10.1016/s0168-9525(97)01273-0. [DOI] [PubMed] [Google Scholar]
- Iwasa Y. The conflict theory of genomic imprinting: how much can be explained? Curr Top Dev Biol. 1998;40:255–293. doi: 10.1016/s0070-2153(08)60369-5. [DOI] [PubMed] [Google Scholar]
- Jansson T, Ylven K, Wennergren M, Powell TL. Glucose transport and system A activity in syncytiotrophoblast microvillous and basal plasma membranes in intrauterine growth restriction. Placenta. 2002;23:392–399. doi: 10.1053/plac.2002.0826. [DOI] [PubMed] [Google Scholar]
- Johnson LW, Smith CH. Neutral amino acid transport systems of microvillous membrane of human placenta. Am J Physiol. 1988;254:C773–780. doi: 10.1152/ajpcell.1988.254.6.C773. [DOI] [PubMed] [Google Scholar]
- Karl PI. Insulin-like growth factor-1 stimulates amino acid uptake by the cultured human placental trophoblast. J Cell Physiol. 1995;165:83–88. doi: 10.1002/jcp.1041650111. [DOI] [PubMed] [Google Scholar]
- Khosla S, Dean W, Brown D, Reik W, Feil R. Culture of preimplantation mouse embryos affects fetal development and the expression of imprinted genes. Biol Reprod. 2001;64:918–926. doi: 10.1095/biolreprod64.3.918. [DOI] [PubMed] [Google Scholar]
- Kingdom J, Baker P, Blair E. Definitions of intrauterine growth restriction. In: Kingdom J, Baker P, editors. Intrauterine Growth Restriction: Aetiology and Management. London: Springer-Verlag; 2000. pp. 1–4. [Google Scholar]
- Kniss DA, Shubert PJ, Zimmerman PD, Landon MB, Gabbe SG. Insulin like growth factors. Their regulation of glucose and amino acid transport in placental trophoblasts isolated from first-trimester chorionic villi. J Reprod Med. 1994;39:249–256. [PubMed] [Google Scholar]
- Lee JE, Pintar J, Efstratiadis A. Pattern of the insulin-like growth factor II gene expression during early mouse embryogenesis. Development. 1990;110:151–159. doi: 10.1242/dev.110.1.151. [DOI] [PubMed] [Google Scholar]
- Lefebvre L, Viville S, Barton SC, Ishino F, Keverne EB, Surani MA. Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest. Nat Genet. 1998;20:163–169. doi: 10.1038/2464. [DOI] [PubMed] [Google Scholar]
- Leighton PA, Ingram RS, Eggenschwiler J, Efstratiadis A, Tilghman SM. Disruption of imprinting caused by deletion of the H19 gene region in mice. Nature. 1995;375:34–39. doi: 10.1038/375034a0. [DOI] [PubMed] [Google Scholar]
- Li L, Keverne EB, Aparicio SA, Ishino F, Barton SC, Surani MA. Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science. 1999;284:330–333. doi: 10.1126/science.284.5412.330. [DOI] [PubMed] [Google Scholar]
- Lundgren EM, Cnattingius S, Jonsson B, Tuvemo T. Intellectual and psychological performance in males born small for gestational age with and without catch-up growth. Pediatr Res. 2001;50:91–96. doi: 10.1203/00006450-200107000-00017. [DOI] [PubMed] [Google Scholar]
- Matthews JC, Beveridge MJ, Dialynas E, Bartke A, Kilberg MS, Novak DA. Placental anionic and cationic amino acid transporter expression in growth hormone overexpressing and null IGF-II or null IGF-I receptor mice. Placenta. 1999;20:639–650. doi: 10.1053/plac.1999.0421. [DOI] [PubMed] [Google Scholar]
- Mayer W, Hemberger M, Frank HG, Grummer R, Winterhager E, Kaufmann P, Fundele R. Expression of the imprinted genes MEST/Mest in human and murine placenta suggests a role in angiogenesis. Dev Dyn. 2000;217:1–10. doi: 10.1002/(SICI)1097-0177(200001)217:1<1::AID-DVDY1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Miozzo M, Simoni G. The role of imprinted genes in fetal growth. Biol Neonate. 2002;81:217–228. doi: 10.1159/000056752. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Sotomaru Y, Katsuzawa Y, Kono T, Meguro M, Oshimura M, Kawai J, Tomaru Y, Kiyosawa H, Nikaido I, Amanuma H, Hayashizaki Y, Okazaki Y. Asb4, Ata3, and Dcn are novel imprinted genes identified by high-throughput screening using RIKEN cDNA microarray. Biochem Biophys Res Commun. 2002;290:1499–1505. doi: 10.1006/bbrc.2002.6370. [DOI] [PubMed] [Google Scholar]
- Moore T, Constância M, Zubair M, Bailleul B, Feil R, Sasaki H, Reik W. Multiple imprinted sense and antisense transcripts, differential methylation and tandem repeats in a putative imprinting control region upstream of mouse Igf2. Proc Natl Acad Sci U S A. 1997;94:12509–12514. doi: 10.1073/pnas.94.23.12509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore T, Haig D. Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet. 1991;7:45–49. doi: 10.1016/0168-9525(91)90230-N. [DOI] [PubMed] [Google Scholar]
- Paulsen M, El-Maarri O, Engemann S, Strodicke M, Franck O, Davies K, Reinhardt R, Reik W, Walter J. Sequence conservation and variability of imprinting in the Beckwith-Wiedemann syndrome gene cluster in human and mouse. Hum Mol Genet. 2000;9:1829–1841. doi: 10.1093/hmg/9.12.1829. [DOI] [PubMed] [Google Scholar]
- Redline RW, Chernicky CL, Tan HQ, Ilan J, Ilan J. Differential expression of insulin-like growth factor-II in specific regions of the late (post day 9. 5) murine placenta. Mol Reprod Dev. 1993;36:121–129. doi: 10.1002/mrd.1080360202. [DOI] [PubMed] [Google Scholar]
- Reik W, Davies K, Dean W, Kelsey G, Constância M. Imprinted genes and the coordination of fetal and postnatal growth in mammals. Novartis Found Symp. 2001;237:19–35. doi: 10.1002/0470846666.ch3. [DOI] [PubMed] [Google Scholar]
- Reik W, Romer I, Barton SC, Surani MA, Howlett SK, Klose J. Adult phenotype in the mouse can be affected by epigenetic events in the early embryo. Development. 1993;119:933–942. doi: 10.1242/dev.119.3.933. [DOI] [PubMed] [Google Scholar]
- Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- Rossant J, Cross JC. Placental development: lessons from mouse mutants. Nat Rev Genet. 2001;2:538–548. doi: 10.1038/35080570. [DOI] [PubMed] [Google Scholar]
- Sibley CP, Boyd RDH. Mechanisms of transfer across the human placenta. In: Polin RA, Fox WW, editors. Fetal and Neonatal Physiology. 2. Philadelphia: W.B. Saunders; 1998. pp. 77–89. [Google Scholar]
- Sibley CP, Boyd RDH. Control of transfer across the mature placenta. In: Clarke J, editor. Oxford Reviews of Reproductive Biology. Vol. 10. Oxford: Oxford University Press; 1988. pp. 382–435. [PubMed] [Google Scholar]
- Sibley CP, Glazier JD, Greenwood SL, Lacey H, Mynett K, Speake P, Jansson T, Johansson M, Powell TL. Regulation of placental transfer: the na(+)/h(+) exchanger - a review. Placenta. 2002;23(suppl. A):S39–S46. doi: 10.1053/plac.2002.0790. [DOI] [PubMed] [Google Scholar]
- Sleutels F, Barlow DP. The origins of genomic imprinting in mammals. Adv Genet. 2002;46:119–163. doi: 10.1016/s0065-2660(02)46006-3. [DOI] [PubMed] [Google Scholar]
- Strauss RS. Adult functional outcome of those born small-for-gestational age: twenty-six-year follow-up of the 1970 British Birth Cohort. JAMA. 2000;283:625–632. doi: 10.1001/jama.283.5.625. [DOI] [PubMed] [Google Scholar]
- Strid H, Powell TL. ATP-dependent Ca2+ transport in pregnancy is complicated by diabetes or intrauterine growth restriction. Placenta. 2001;22:A66. doi: 10.1053/plac.2002.0941. [DOI] [PubMed] [Google Scholar]
- Sun FL, Dean WL, Kelsey G, Allen ND, Reik W. Transactivation of Igf2 in a mouse model of Beckwith-Wiedemann syndrome. Nature. 1997;389:809–815. doi: 10.1038/39797. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Kobayashi T, Kanayama N. p57(Kip2). regulates the proper development of labyrinthine and spongiotrophoblasts. Mol Hum Reprod. 2000;6:1019–1025. doi: 10.1093/molehr/6.11.1019. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Gertsenstein M, Rossant J, Nagy A. Mash2 acts cell autonomously in mouse spongiotrophoblast development. Dev Biol. 1997;190:55–65. doi: 10.1006/dbio.1997.8685. [DOI] [PubMed] [Google Scholar]
- Tycko B, Morison IM. Physiological functions of imprinted genes. J Cell Physiol. 2002;192:245–258. doi: 10.1002/jcp.10129. [DOI] [PubMed] [Google Scholar]
- Ward S, Jauniaux E, Shannon C, Rodeck C, Boyd R, Sibley C. Electrical potential difference between exocelomic fluid and maternal blood in early pregnancy. Am J Physiol. 1998;274:R1492–1495. doi: 10.1152/ajpregu.1998.274.5.R1492. [DOI] [PubMed] [Google Scholar]
- Wutz A, Smrzka OW, Barlow DP. Making sense of imprinting the mouse and human IGF2R loci. Novartis Found Symp. 1998;214:251–259. doi: 10.1002/9780470515501.ch15. [DOI] [PubMed] [Google Scholar]
- Zwart R, Sleutels F, Wutz A, Schinkel AH, Barlow DP. Bidirectional action of the Igf2r imprint control element on upstream and downstream imprinted genes. Genes Dev. 2001a;15:2361–2366. doi: 10.1101/gad.206201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart R, Verhaagh S, Buitelaar M, Popp-Snijders C, Barlow DP. Impaired activity of the extraneuronal monoamine transporter system known as uptake-2 in Orct3/Slc22a3-deficient mice. Mol Cell Biol. 2001b;21:4188–4196. doi: 10.1128/MCB.21.13.4188-4196.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]