

Abstract
Sulphonylureas stimulate insulin secretion by binding with high-affinity to the sulphonylurea receptor (SUR) subunit of the ATP-sensitive potassium (KATP) channel and thereby closing the channel pore (formed by four Kir6.2 subunits). In the absence of added nucleotides, the maximal block is around 60–80 %, indicating that sulphonylureas act as partial antagonists. Intracellular MgADP modulated sulphonylurea block, enhancing inhibition of Kir6.2/SUR1 (β-cell type) and decreasing that of Kir6.2/SUR2A (cardiac-type) channels. We examined the molecular basis of the different response of channels containing SUR1 and SUR2A, by recording currents from inside-out patches excised from Xenopus oocytes heterologously expressing wild-type or chimeric channels. We used the benzamido derivative meglitinide as this drug blocks Kir6.2/SUR1 and Kir6.2/SUR2A currents, reversibly and with similar potency. Our results indicate that transfer of the region containing transmembrane helices (TMs) 8–11 and the following 65 residues of SUR1 into SUR2A largely confers a SUR1-like response to MgADP and meglitinide, whereas the reverse chimera (SUR128) largely endows SUR1 with a SUR2A-type response. This effect was not specific for meglitinide, as tolbutamide was also unable to prevent MgADP activation of Kir6.2/SUR128 currents. The data favour the idea that meglitinide binding to SUR1 impairs either MgADP binding or the transduction pathway between the NBDs and Kir6.2, and that TMs 8–11 are involved in this modulatory response. The results provide a basis for understanding how β-cell KATP channels show enhanced sulphonylurea inhibition under physiological conditions, whereas cardiac KATP channels exhibit reduced block in intact cells, especially during metabolic inhibition.
Sulphonylureas are widely used to stimulate insulin secretion in type 2 diabetics. They act by binding to ATP-sensitive potassium (KATP) channels in pancreatic β-cells, and inducing them to close. As a consequence the β-cell depolarizes, which causes activation of voltage-dependent Ca2+ channels, Ca2+ influx and a rise in intracellular Ca2+ that triggers insulin secretion (Ashcroft & Rorsman, 1989). The KATP channel is a multimeric complex of four Kir6.x and four sulphonylurea (SUR) subunits (Shyng & Nichols, 1997). There are two isoforms of Kir6.x (Kir6.2, Kir6.1) and three common isoforms of SUR (SUR1, SUR2A and SUR2B): Kir6.2 associates with SUR1 to form the β-cell KATP channel, and with SUR2A to form the cardiac KATP channel (Aguilar-Bryan et al. 1995; Inagaki et al. 1995a,b, 1996; Sakura et al. 1995). SUR2B in combination with either Kir6.1 or Kir6.2 forms the smooth muscle KATP channel (Isomoto et al. 1996). Sulphonylureas bind to SUR, producing a conformational change that results in closure of the tetrameric pore formed by Kir6.2. Nucleotides interact with the KATP channel in complex ways: in particular, the channel is closed by ATP (or ADP) binding to Kir6.2 (Tucker et al. 1997; Tanabe et al. 1999), and is opened by interaction of Mg-nucleotides (MgATP, MgADP) with the two nucleotide-binding domains (NBDs) of SUR (Nichols et al. 1996; Gribble et al. 1997a, 1998a).
In the absence of added nucleotides, high-affinity block of the KATP current by sulphonylureas is not complete, but reaches a maximum of around 60–80 %, which produces a pedestal in the concentration-inhibition curve (Gribble et al. 1997b). Analysis of the single-channel currents reveals that this results because although all KATP channels bind drug with high affinity, they are still able to open when drug is bound, albeit with lower probability (Barrett-Jolley & Davies, 1997; Proks et al. 2002). Sulphonylureas therefore act as partial antagonists of the KATP channel, and the magnitude of the pedestal is determined by the equilibrium between the open-bound and closed-bound states. In this paper, we refer to this equilibrium as the efficacy of the drug (Colquhoun, 1998) and to the IC50 for current inhibition as the drug potency. At higher concentrations, the sulphonylureas produce a low-affinity block, which is independent of SUR and probably involves direct interaction with Kir6.2 (Gribble et al. 1997b).
KATP channels are characterized by complex kinetics, consisting of rapid bursts of openings separated by long closed intervals. Sulphonylureas stabilize the long closed states and reduce the frequency and duration of the bursts of openings (Gillis et al. 1989). Mutations in Kir6.2 that prolong the burst duration are associated with a reduction in the maximal extent of high-affinity sulphonylurea block (Trapp et al. 1998; Koster et al. 1999). Maximal sulphonylurea block is also reduced when the channel open probability is increased by PIP2 (Koster et al. 1999; Krauter et al. 2001). These changes in drug efficacy are a direct consequence of the change in the single-channel kinetics (Alekseev et al. 1998; Proks et al. 2002). Mg-nucleotides promote channel open probability and reduce entry into the long closed states, and are therefore expected to produce a reduction in drug efficacy. This may explain why the maximal sulphonylurea block of both native and recombinant cardiac KATP channels (Kir6.2/SUR2A) is reduced in the presence of Mg-nucleotides (Ventakesh et al. 1991; Gribble et al. 1998b).
In contrast, it is now well established that intracellular MgADP produces an apparent enhancement of tolbutamide block of KATP currents in pancreatic β-cells (Zünckler et al. 1988). Similar results are reported for inhibition of the cloned channel, Kir6.2/SUR1, by the sulphonylureas gliclazide, tolbutamide and glibenclamide and for the non-sulphonylurea insulin secretagogues meglitinide, repaglinide and mitiglinide (Gribble et al. 1998b; Dabrowski et al. 2001; Reimann et al. 2001; Proks et al. 2002). This is contrary to what is expected for an agent that increases the channel open probability.
There is evidence that the enhanced block of Kir6.2/SUR1 results because sulphonylureas prevent the stimulatory action of MgADP mediated by SUR1, thereby unmasking the inhibitory effect of the nucleotide at Kir6.2 (Gribble et al. 1997b). As a result, the blocks by MgADP and sulphonylureas summate to produce an ‘apparent’ enhancement of block. Because this effect is not observed for Kir6.2/SUR2, it seems that sulphonylureas do not prevent the stimulatory action of MgADP on this channel.
A key issue is the nature of the molecular mechanism underlying the different responses of KATP channels containing SUR1 and SUR2A. This question is of direct clinical relevance, because the different responses of these SUR isoforms contributes to the fact that in the intact cell sulphonylureas are much more potent on the β-cell type of KATP channel than on cardiac and smooth muscle KATP channels (Ashcroft & Gribble, 1999; Lawrence et al. 2001). We have employed a chimeric approach to explore the molecular basis of the different responses of SUR1 and SUR2A. We have used the benzamido derivative meglitinide as a tool, because this drug interacts reversibly with both SUR1- and SUR2-containing channels. Sulphonylureas such as tolbutamide and gliclazide do not block SUR2, and drugs such as glibenclamide, which interact with both types of channel, inhibit Kir6.2/SUR1 channels irreversibly.
METHODS
Molecular biology
Mouse Kir6.2 (GenBank accession no. D50581; Inagaki et al. 1995a; Sakura et al. 1995), rat SUR1 (GenBank L40624; Aguilar-Bryan et al. 1995), rat SUR2A (GenBank D83598; Inagaki et al. 1996) and rat SUR2B (GenBank D86038; Isomoto et al. 1996) cDNAs were cloned in the pBF vector. Chimeras between SUR1 and SUR2A were constructed as described previously (Ashfield et al. 1999). Capped mRNA was prepared using the mMessage mMachine large scale in vitro transcription kit (Ambion, Austin, TX, USA), as previously described (Gribble et al. 1997b).
Oocyte collection
Female Xenopus laevis were anaesthetized with MS222 (2 g l−1 added to the water). One ovary was removed via a mini-laparotomy, the incision sutured and the animal allowed to recover. All procedures used conformed with the UK Animals (Scientific Procedures) Act 1986. Immature stage V-VI oocytes were incubated for 60 min with 1.0 mg ml−1 collagenase (Roche, type A) and manually defolliculated. Oocytes were injected with ≈0.1 ng Kir6.2 mRNA and ≈2 ng of mRNA encoding either SUR1, SUR2A, SUR2B or the SUR chimera indicated. The final injection volume was 50 nl oocyte−1. Isolated oocytes were maintained in Barth's solution and studied 1–7 days after injection (Gribble et al. 1997b).
Electrophysiology
Patch pipettes were pulled from borosilicate glass and had resistances of 250–500 kΩ when filled with pipette solution. Macroscopic currents were recorded from giant excised inside-out patches at a holding potential of 0 mV and at 20–24 °C using an Axopatch 200B patch-clamp amplifier (Axon Instruments, Foster City, CA, USA; Gribble et al. 1997b). The pipette (external) solution contained (mm): 140 KCl, 1.2 MgCl2, 2.6 CaCl2, 10 Hepes (pH adjusted to 7.4 with KOH). The intracellular (bath) solution contained (mm): 107 KCl, 1.2 MgCl2, 1 CaCl2, 10 EGTA, 10 Hepes (pH 7.2 with KOH; final [K+] ≈140 mm). Meglitinide was prepared as a 10 mm stock solution in DMSO. Nucleotides were added with equivalent concentrations of MgCl2, and the pH was readjusted as required. Rapid exchange of solutions was achieved by positioning the patch in the mouth of one of a series of adjacent inflow pipes placed in the bath.
In most experiments, currents were recorded in response to repetitive 3 s voltage ramps from −110 mV to +100 mV. They were filtered at 10 kHz, digitized using a Digidata 1320A Interface, sampled at 0.4 kHz and analysed using pCLAMP software (Axon Instruments). The slope conductance was measured by fitting a straight line to the current-voltage relation between −20 mV and −100 mV: the average response to five consecutive ramps was calculated in each solution.
Concentration-response curves were fitted to the following equation (Gribble et al. 1997b):
| (1) |
where G is the conductance in the presence of meglitinide, Gc is the conductance in control solution, [Meg] is the meglitinide concentration, IC50 is the meglitinide concentration at which inhibition is half-maximal at the high-affinity site, nH is the Hill coefficient (slope factor), A represents the activation by ADP (A = 1 in the absence of nucleotide) and L is the fractional conductance remaining when the high-affinity sites are maximally occupied. Efficacy was calculated as (A - L)/A. Data were fitted using Axon Clampfit and Microcal Origin software.
Data are presented as means ± 1 s.e.m. Statistical significance was tested initially by ANOVA, and subsequently, when appropriate, by Student's t test.
RESULTS
Figure 1 compares the effect of 10 µm meglitinide on Kir6.2/ SUR1, Kir6.2/SUR2A and Kir6.2/SUR2B currents in the absence and presence of 100 µm MgADP. It is evident that in the absence of nucleotide all three types of recombinant KATP channel are rapidly and reversibly blocked by meglitinide, and that the extent of block is similar (≈75 %). In contrast, there is a marked difference in the inhibitory action of the drug in the presence of 100 µm MgADP. Thus, Kir6.2/SUR1 currents are blocked more strongly (93 ± 1 %, n = 21), whereas Kir6.2/SUR2A and Kir6.2/SUR2B currents are inhibited to a lesser extent (40 ± 6 %, n = 23, and 54 ± 8 %, n = 8, respectively), than in the absence of nucleotide. This is consistent with the idea that meglitinide, like glibenclamide (Gribble et al. 1998b), ablates the stimulatory effect of MgADP on Kir6.2/ SUR1 channels but not on Kir6.2/SUR2 channels.
Figure 1. MgADP enhances meglitinide block of Kir6.2/SUR1 but impairs that of Kir6.2/SUR2.
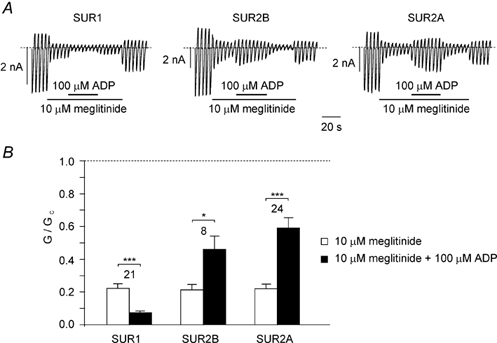
A, macroscopic currents recorded from oocytes coexpressing Kir6.2 and either SUR1, SUR2A or SUR2B in response to a series of voltage ramps from −110 to +100 mV. Meglitinide and MgADP were added to the intracellular solution as indicated by the bars. B, mean Kir6.2/SUR1, Kir6.2/SUR2A or Kir6.2/SUR2B conductance in the presence of 10 µm meglitinide or 10 µm meglitinide plus 100 µm MgADP. Conductance (G) is expressed relative to the mean of the conductances in control solution before and after addition of drug or nucleotide (Gc). The number of patches is given above each bar. *P < 0.05, ***P < 0.001, by paired t test.
Effect of MgADP on concentration-response relationships
The effects of MgADP on channel inhibition by a single dose of meglitinide could result from alterations in either the efficacy or the potency of the drug. To distinguish between these possibilities, we measured the concentration- response relationships for meglitinide block of Kir6.2/ SUR1 and Kir6.2/SUR2A currents in the presence and absence of nucleotide (Fig. 2). As described previously (Gribble et al. 1998b), meglitinide blocked Kir6.2/SUR1 and Kir6.2/ SUR2A currents with similar IC50 values (0.3 µm and 0.6 µm, respectively), and in both cases the maximum degree of inhibition was ≈75 %. MgADP (100 µm) enhanced the efficacy of meglitinide on Kir6.2/SUR1 but not Kir6.2/ SUR2A: the maximal inhibition of Kir6.1/SUR1 currents increased to 95 ± 3 % (n = 5; P < 0.001 compared with the efficacy in the absence of nucleotide) whereas that of Kir6.2/SUR2A was 65 ± 5 % (n = 6; n.s.). By contrast, MgADP had only a small effect on the drug potency, increasing the IC50 of meglitinide to 0.7 ± 0.1 µm (n = 5; P < 0.05) in the case of Kir6.2/SUR1 currents, and to 1.6 ± 0.3 µm (n = 6; P < 0.05) in the case of Kir6.2/SUR2A currents. These results support the idea that meglitinide abolishes the stimulatory effect of MgADP on SUR1 but not SUR2A, whereas the affinity of drug binding is only marginally affected by the nucleotide.
Figure 2. Relationship between meglitinide concentration and inhibition of Kir6.2/SUR1 (A) or Kir6.2/SUR2A (B) conductance.
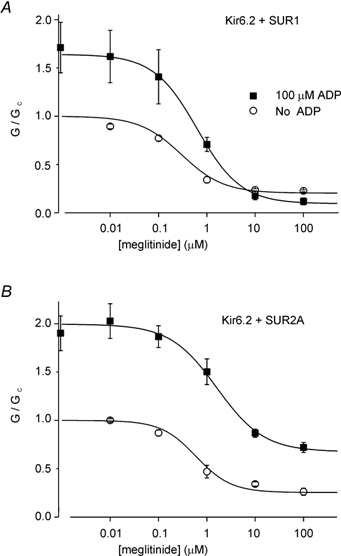
Measurements were made in the absence (○) or presence (▪) of 100 µm MgADP. The conductance in the presence of meglitinide (G) is expressed as a fraction of its mean amplitude in the absence of both drug and nucleotide (Gc). The lines are the best fit of the mean data to eqn (1) using the following values: A, Kir6.2/SUR1; no nucleotide (n = 6): A = 1, IC50= 0.3 µm, nH= 1.0, L = 0.20. MgADP (n = 5–10): A = 1.64, IC50= 0.6 µm, nH= 1.0, L = 0.09. B, Kir6.2/SUR2A; no nucleotide (n = 5): A = 1, IC50= 0.6 µm, nH= 1.2, L = 0.25. MgADP (n = 6–8): A = 2.0, IC50= 1.7 µm, nH= 1.0, L = 0.70.
Chimeric and mutational studies
A chimeric approach was used to explore the structural basis underlying the different responses of Kir6.2/SUR1 and Kir6.2/SUR2A channels. Chimeric SURs, in which different domains were swapped between SUR1 and SUR2A, were coexpressed with Kir6.2, and the effect of 10 µm meglitinide on the resulting channel currents was examined in the absence or presence of 100 µm MgADP. Meglitinide block was similar for all wild-type and chimeric channels in the absence of nucleotide (Fig. 3B; n.s. by ANOVA). As described above, MgADP produced an apparent enhancement of inhibition of Kir6.2/SUR1 currents, but an apparent reduction in the inhibition of Kir6.2/SUR2A currents.
Figure 3. Regions of SUR associated with meglitinide-MgADP interactions.
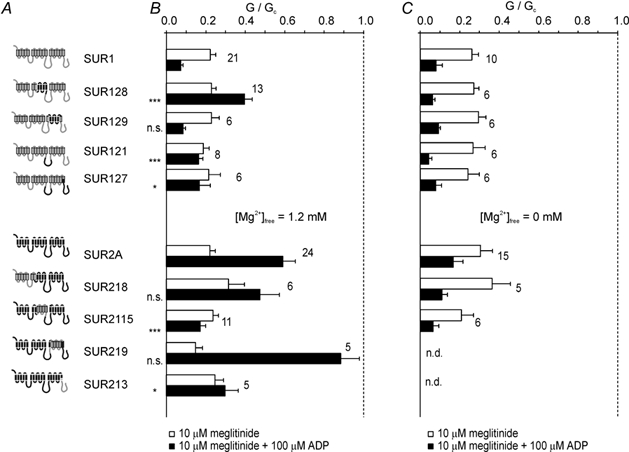
A, schematic diagram of SUR1 and SUR2A chimeras. Regions of SUR1 are shown in grey and those of SUR2A in black. B, mean conductance recorded for channels comprising Kir6.2 and either wild-type or chimeric SUR as indicated, in the presence of 10 µm meglitinide (open bars) or 10 µm meglitinide plus 100 µm MgADP (filled bars). All solutions contained 1.2 mm free Mg2+. Conductance (G) is expressed relative to the mean of the conductances in control solution before and after addition of drug or nucleotide (Gc). The number of patches is given beside each bar. All groups were initially compared by ANOVA. Significant differences were followed up by t tests, as indicated, comparing SUR1-based chimeras with SUR1, and SUR2-based chimeras with SUR2. Significance levels are indicated next to the bars. n.s., not significant, *P < 0.05, ***P < 0.001. C, mean conductance recorded for channels comprising Kir6.2 and either wild-type or chimeric SUR as indicated, in the presence of 10 µm meglitinide (open bars) or 10 µm meglitinide plus 100 µm ADP (filled bars). All solutions contained 10 mm EDTA to remove free Mg2+. Conductance (G) is expressed relative to the mean of the conductances in control solution before and after addition of drug or nucleotide (Gc). The number of patches is given beside each bar. There were no significant differences between channel types by ANOVA.
Reduced channel inhibition was also observed for the SUR1-based chimera, SUR128, which contained transmembrane helices (TMs) 8–11 and the following 65 residues of SUR2A. Consistent with the idea that this region is involved in the interaction between meglitinide and MgADP, the reverse SUR2A-based chimera (SUR2115) exhibited enhanced meglitinide block in the presence of MgADP.
Although the most effective chimeras in swapping the meglitinide/MgADP response were those involving TMs 8–11 and the following 65 residues, exchanging the NBDs between SUR1 and SUR2A also had partial effects (Fig. 3B). Thus, transfer of NBD1 or both NBDs of SUR2A into SUR1 (SUR121 and SUR127, respectively) impaired the ability of MgADP to enhance meglitinide block, whereas transferring NBD2 of SUR1 into SUR2A (SUR213) reduced the stimulatory effect of MgADP in the presence of meglitinide. Because the chimera SUR2115 (containing TMs 8–11 of SUR1) did not completely mimic the response of SUR1 to MgADP and meglitinide, we also tested a chimera that contained NBD2 of SUR1 in addition to TMs 8–11 (SUR2115/213). This chimera was blocked by MgADP in the presence of meglitinide to a similar extent as SUR1 (Fig. 5A). The most likely explanation for this effect, however, is that Kir6.2/SUR2115/213 channels cannot be activated by MgADP, because the nucleotide blocked the currents even in the absence of the drug (see below and Fig. 5B).
Figure 5. Subdefining regions and key residues associated with meglitinide-MgADP interactions.
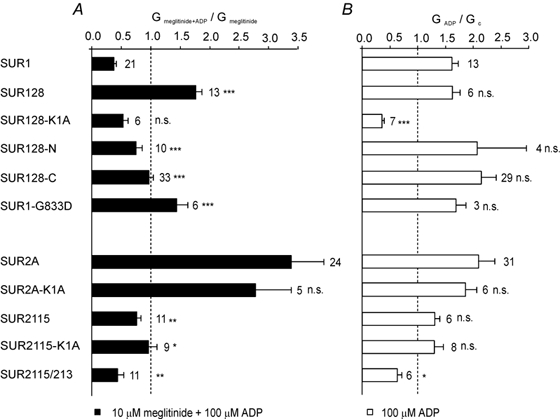
A, mean conductance recorded for channels comprising Kir6.2 and either wild-type or mutant SUR as indicated, in the presence of 10 µm meglitinide and 100 µm MgADP. All solutions contained 1.2 mm free Mg2+. Conductance (Gmeglitinide + ADP) is expressed as a mean of that in the presence 10 µm meglitinide alone before and after addition of the nucleotide (Gmeglitinide). The number of patches is given beside each bar. B, mean conductance recorded for channels comprising Kir6.2 and either wild-type or mutant SUR as indicated, in the presence of 100 µm ADP. Conductance (GADP) is expressed relative to the mean of the conductances in control solution before and after addition of nucleotide (Gc). The number of patches is given beside each bar. In both A and B, all groups were initially compared by ANOVA. Significant differences were followed up by t tests, as indicated, comparing SUR1-based chimeras with SUR1, and SUR2-based chimeras with SUR2. Significance levels are indicated next to the bars. n.s., not significant, *P < 0.05, **P < 0.01, ***P < 0.001.
To exclude the possibility that the chimeras affected the stimulatory effect of MgADP on SUR or its inhibitory effect on Kir6.2, rather than the interaction between the nucleotide and drug, we examined both the effects of ADP (100 µm) in the absence and presence of Mg2+ (Fig. 4), and the effect of ADP on meglitinide block in the absence of Mg2+ (Fig. 3C). No significant differences in ADP block were observed between the chimeras and SUR1 in the absence of both meglitinide and Mg2+. Although Kir6.2/ SUR2A currents were slightly less blocked by Mg-free ADP than Kir6.2/SUR1 currents (Fig. 4; P < 0.001), this difference was not preserved in the concomitant presence of meglitinide (Fig. 3C) Most of the chimeric channels, with the exception of SUR127 and SUR2115/213, were also activated to a similar extent by MgADP (≈1.5- to 2-fold), suggesting that these chimeras do not markedly alter the stimulatory effect of MgADP in the absence of meglitinide. These results therefore suggest that MgADP activation is only modified in the presence of meglitinide, and that TMs 8–11 and the following 65 residues of NBD1 are directly involved in the interaction between meglitinide and MgADP.
Figure 4. ADP modulation of channels containing SUR1/SUR2 chimeras.
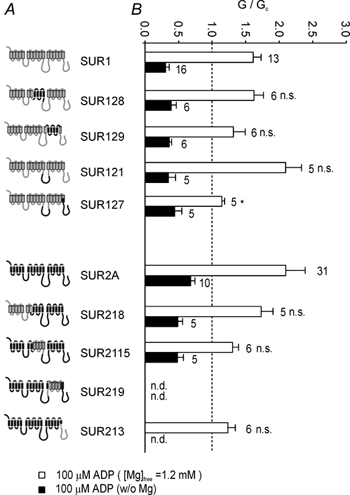
A, schematic diagram of SUR1 and SUR2A chimeras. Regions of SUR1 are shown in grey and those of SUR2A in black. B, mean conductance recorded for channels comprising Kir6.2 and either wild-type or chimeric SUR as indicated, in 100 µm ADP and either the presence of 1.2 mm free Mg2+ (open bars) or in the absence of Mg2+ (filled bars). Conductance (G) is expressed relative to the mean of the conductances in control solution before and after addition of nucleotide (Gc). The number of patches is given adjacent to each bar. n.d., not determined. All groups were initially compared by ANOVA. Significant differences were followed up by t tests, as indicated, comparing SUR1-based chimeras with SUR1, and SUR2-based chimeras with SUR2. Significance levels are indicated next to the bars. n.s., not significant, *P < 0.05.
To investigate whether the effects of chimeras SUR128 and SUR2115 are due to TMs 8–11 or the following 65 residues, we made further chimeras and mutations in the TM region and in NBD1 (Fig. 5). To determine the importance of the 65 residues at the start of NBD1, we constructed an SUR1 chimera in which only these amino acids were replaced by their SUR2 equivalents (SUR128-C). Although MgADP no longer enhanced inhibition of Kir6.2/SUR128-C currents in the presence of meglitinide, the response was only partially converted into an SUR2A-like response (Fig. 5A). A similar effect was observed on swapping only TMs 8–11 of SUR2 into SUR1 (SUR128-N). Thus both TMs 8–11 and the loop residues appear to be required for a SUR2A-type response. As TM 11 of SUR1 differs from that of SUR2 by a single amino acid, we also mutated S551 in SUR1 to its counterpart in SUR2 (T), and mutated the corresponding T in SUR2A to S. Neither mutation affected the degree of meglitinide inhibition in the presence of MgADP (data not shown), ruling out an important role for TM 11 in the differential response of SUR1 and SUR2A.
We also investigated the effect of mutating the Walker A lysine residue in NBD1 to alanine (K1A), which abolishes MgADP activation of SUR1-, but not SUR2A-containing channels (Gribble et al. 1997a; 2000). This mutation prevented MgADP activation of SUR128 in both the presence and absence of meglitinide (Fig. 5). Furthermore, the K1A mutation in SUR2A did not affect the ability of MgADP to reduce meglitinide block, consistent with the fact that it does not abolish MgADP activation in the absence of the drug (Fig. 3A; Reimann et al. 2000). It also had no effect on the response of the 2115 chimera (Fig. 5). Surprisingly, however, mutation of G833D in SUR1, which lies within the linker region of NBD1, converted MgADP enhancement of meglitinide inhibition into a reduction in block (Fig. 5A).
Comparison between meglitinide and tolbutamide
Tolbutamide, like meglitinide, abolishes the activation of Kir6.2/SUR1 channels by MgADP (Gribble et al. 1997b). The binding sites for these drugs are not, however, believed to be identical, as the mutation S1237Y in SUR1 abolishes tolbutamide, but not meglitinide, inhibition (Ashfield et al. 1999). To investigate whether interaction of these drugs with MgADP involves a common mechanism, we examined the effect of tolbutamide on Kir6.2/SUR128 channels in the absence and presence of nucleotide. As shown in Fig. 6, whereas tolbutamide block of Kir6.2/SUR1 currents was enhanced by MgADP, that of Kir6.2/SUR128 currents was reduced. Consistent with the lack of tolbutamide block of Kir6.2/SUR2A currents reported previously (Gribble et al. 1998b), MgADP also activated these currents in the presence of tolbutamide. Thus, although tolbutamide and meglitinide bind to non-identical sites, they mediate their effects on MgADP activation via a common mechanism.
Figure 6. Interactions between MgADP and tolbutamide.
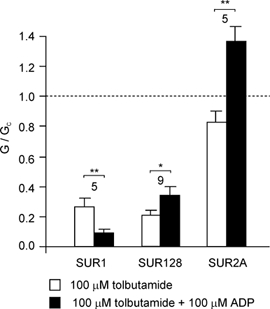
Mean Kir6.2/SUR1, Kir6.2/SUR128 or Kir6.2/SUR2A conductance in the presence of 100 µm tolbutamide or 100 µm tolbutamide plus 100 µm MgADP. Conductance (G) is expressed relative to the mean of the conductances in control solution before and after addition of drug or nucleotide (Gc). The number of patches is given above each bar. *P < 0.05, **P < 0.01, by paired t test.
DISCUSSION
As found previously for several sulphonylureas, and related drugs, we show that MgADP enhances the apparent block of Kir6.2/SUR1 currents by meglitinide, but reduces that of Kir6.2/SUR2A currents. The results are consistent with studies on native β-cell and cardiac KATP channels (Zünckler et al. 1988; Venkatesh et al. 1991; Findlay, 1993). Previous experiments on Kir6.2/SUR1 channels showed that the ability of tolbutamide to prevent Mg-nucleotide activation is distinct from its ability to induce channel closure (Reimann et al. 1999). Thus, when SUR1 was coexpressed with an N-terminally truncated form of Kir6.2 (Kir6.2ΔN14), tolbutamide no longer inhibited Kir6.2/SUR1 currents, although it still prevented channel activation by Mg-nucleotides. This suggests that different mechanisms mediate the transduction of drug binding into channel inhibition (on the one hand) and prevent channel activation by MgADP (on the other hand). This idea is supported by the fact that the inhibitory effect of meglitinide is retained in Kir6.2/SUR2A channels, although its ability to prevent MgADP activation is absent.
Regions of SUR associated with meglitinide-MgADP interactions
The chimeric studies identify domains of SUR involved in the different responses of Kir6.2/SUR1 and Kir6.2/SUR2A channels to the concomitant application of MgADP and meglitinide. They indicate that transfer of TMs 8–11 and the following 65 residues of SUR1 into SUR2A confers a largely SUR1-like response to MgADP and meglitinide, whereas the reverse chimera (SUR128) largely endows SUR1 with a SUR2A-type response. This effect was not specific for meglitinide, as tolbutamide was also unable to prevent MgADP activation of Kir6.2/SUR128 currents. Further dissection of these regions using additional chimeras revealed that both TMs 8–10 and the first 65 residues of the TM 11–12 linker are required for a SUR2A-like response, but that differences in TM 11 do not play a role.
Neither the SUR128 nor the SUR2115 chimeras fully mimicked the response of wild-type SUR2A or SUR1, suggesting that other residues may be involved in meglitinide- nucleotide interactions. It has, however, proved difficult to identify these regions unambiguously. There is some evidence that residues in NBD1 of SUR1, in addition to the first 65, may be involved, as channels containing the G833D mutation in the ABC signature sequence exhibited a SUR2A-like response, with reduced meglitinide block in the presence of MgADP. Furthermore, swapping either NBD1 alone, or both NBDs of SUR2 into SUR1 partially impaired the ability of meglitinide to prevent MgADP activation.
Although it is most likely that the effect of meglitinide on TMs 8–11 is mediated allosterically, it is difficult to exclude the possibility that this region encompasses part of the sulphonylurea-binding pocket. This pocket is believed to contain residues from more than one region of SUR, and the precise residues involved in drug binding may vary from agent to agent. The cytosolic loop linking TMs 15 and 16 has been shown to play a role in sulphonylurea binding to SUR1, as mutation of S1237Y abolished tolbutamide block and [3H]glibenclamide binding, whilst inhibition by meglitinide (which resembles the non-sulphonylurea moiety of glibenclamide) was unaffected (Ashfield et al. 1999). However, the homologous cytoplasmic loop between TMs 9 and 10 is unlikely to play a role in drug binding, as deletion of TMs 9–10 of SUR1 did not abolish [3H]glibenclamide binding in the baculovirus expression system (Mikhailov et al. 2001).
Possible mechanisms underlying the differences between SUR1 and SUR2-like responses
The idea that sulphonylureas and benzamido derivatives impair the ability of MgADP to stimulate activation of Kir6.2/SUR1 channels has been suggested previously (Gribble et al. 1997; Schwanstecher et al. 1999). At least two possible mechanisms can be suggested: the drug might displace MgADP from one or both NBDs of SUR1, or it might impair the transduction of MgADP binding into channel activation while leaving nucleotide binding unchanged. Whichever the mechanism, it appears that this does not operate in SUR2. The transduction and displacement models are considered in further detail below.
Concurrent binding of meglitinide and MgADP, with differential transduction
One possible role for TMs 8–11 is in modulating how MgADP activation is transduced to the channel pore formed by Kir6.2. Meglitinide binding, even at a distant site, might prevent this transduction in SUR1 but not SUR2, due to differences in the properties of TMs 8–11. The reduced efficacy of meglitinide on Kir6.2/SUR2A currents in the presence of MgADP can be simply explained by the increased channel open probability in the presence of the nucleotide, as discussed in the Introduction (Alekseev et al. 1998; Proks et al. 2002). This model is compatible with all our data with the exception of the SUR1 G833D mutation, which caused an SUR2-like response despite the presence of TMs 8–11 of SUR1. As the role of the ABC signature sequence in SUR is not understood, however, it is possible that the G833D mutation could modify the interaction of the NBDs with the TM domains.
Interestingly, TMs 8–11 and NBD2 of SUR1 have also been implicated in the ability of the diazoxide analogue NNC-55-9216 (3-isopropylamino-7-methoxy-4H-1,2,4-benzathiadiazine1,1,dioxide) to selectively activate Kir6.2/SUR1 channels (Dabrowski et al. 2002), and TMs 6–11 and NBD1 of SUR1 were also reported to confer the differential response of SUR1 and SUR2A-type channels to diazoxide (Babenko et al. 2000). As the activity of KATP channel openers is critically dependent on nucleotide binding to the NBDs (Gribble & Ashcroft, 2000; D'hahan et al. 1999), these findings are consistent with the idea that TMs 8–11 form an important site in the transduction pathway between the NBDs of SUR and the channel pore.
Interactions between binding of nucleotides and meglitinide
Impaired MgADP activation of Kir6.2/SUR1 channels by sulphonylureas and benzamido derivatives might also arise if the drug displaces nucleotide from one or both NBDs of SUR. In this case, we speculate that either activation of SUR2 is unaffected by the nucleotide displacement or that displacement of nucleotides does not occur. In the latter case, the nucleotide might even impair sulphonylurea binding.
The idea that sulphonylureas displace nucleotide binding from the NBDs of SUR1 is supported by 8-azido ATP binding studies, which showed that glibenclamide could enhance the dissociation of prebound 8-azido ATP from NBD1 of SUR1 in the presence of Mg-nucleotides (Ueda et al. 1999). Similar data for SUR2 are not, unfortunately, available. The critical role of nucleotide binding to NBD1 for MgADP activation of channels containing SUR1, but not SUR2, has been demonstrated previously by mutation of the Walker A lysine residue in NBD1. Mutating this lysine to alanine (K1A) prevented MgADP activation of Kir6.2/SUR1, but not Kir6.2/SUR2A channels (Gribble et al. 1997a; Reimann et al. 2000). Although this suggested that the ability of MgADP to activate Kir6.2/SUR2A in the presence of meglitinide could be a consequence of the fact that MgADP binding at NBD1 was not required for nucleotide activation, further experiments did not support this idea. Thus, chimera SUR2115 exhibited an SUR1-like response in the presence of meglitinide and MgADP but was still activated by MgADP following mutation of the Walker A lysine.
Effects of nucleotides on sulphonylurea binding to native KATP channels have been recognized for many years. In a recent study on heterologously expressed SUR1 and SUR2A-Y1206S (a mutant form of SUR2 with enhanced glibenclamide affinity), it was demonstrated that nucleotide modulation of [3H]glibenclamide binding was not identical between the two types of SUR subunit. Thus, whereas MgATP homogeneously reduced the affinity of [3H]glibenclamide binding to SUR1 without affecting the maximal binding (Bmax), the nucleotide reduced the Bmax for [3H]glibenclamide binding to Kir6.2/SUR2A-Y1206S without altering the affinity of the remaining sites (Hambrock et al. 2002; Löffler-Walz et al. 2002). It was further suggested that the reduced Bmax of SUR2-type subunits might arise from a tetrameric reorganization of KATP channel complexes in the presence of MgATP, resulting in the unavailability of a proportion of the high affinity sulphonylurea binding sites (Löffler-Walz et al. 2002). Although MgADP was less effective in the binding studies, it was not without effect, suggesting that reduced meglitinde binding to SUR2A in the presence of MgADP might contribute to the impaired drug efficacy in our experiments. The hypothesis that MgATP induces a tetrameric conformational change in SUR2, reducing the number of glibenclamide binding sites, is also compatible with reports that MgATP alters the operating state of native cardiac KATP channels, resulting in impaired glibenclamide inhibition in the presence of MgUDP (Brady et al. 1998).
Conclusion
We show here that both TMs 8–11 and the nucleotide binding domains of SUR are involved in the interactive effects of sulphonylureas and MgADP on KATP channel activity. These regions of SUR might mediate communication between different domains of the same SUR subunit, or between one SUR subunit and other subunits (SUR or Kir6.2) within the octameric KATP channel complex. Indeed, a functional link between Kir6.2 and the sulphonylurea binding site has been suggested from binding studies, which showed that coexpression of Kir6.2 with either SUR1 or SUR2 modulates [3H]glibenclamide binding characteristics (Hambrock et al. 2002; Proks et al. 2002). Evidence for structural interactions between the TM domains of SUR and Kir6.2 subunits has also been provided by trafficking studies (Schwappach et al. 1999).
Our results provide an underlying basis for the important pathophysiological findings that sulphonylurea block is almost complete in intact pancreatic β-cells despite the incomplete inhibition observed in excised patches, and that sulphonylurea block of cardiac KATP channels is impaired under conditions of metabolic inhibition (Zünkler et al. 1988; Venkatesh et al.1991; Findlay, 1993; Lawrence et al. 2001).
Acknowledgments
We thank the Wellcome Trust and Diabetes UK for support. F.R. is a Diabetes UK RD Lawrence Fellow, M.D. is a Robert Turner Visiting Scholar. F.M.G. is a Wellcome Clinician Scientist Fellow. F.M.A. is the Royal Society GlaxoSmithKline Research Professor.
REFERENCES
- Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP, IV, Boyd AE, III, Gonzalez G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA. Cloning of the beta-cell high-affinity sulphonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–425. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- Alekseev AE, Brady PA, Terzic A. Ligand-insensitive state of cardiac ATP-sensitive K+ channels: basis for channel opening behaviour. J Gen Physiol. 1998;111:381–394. doi: 10.1085/jgp.111.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, Gribble FM. ATP-sensitive K+ channels and insulin secretion; their role in health and disease. Diabetologia. 1999;42:903–919. doi: 10.1007/s001250051247. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic beta-cell. Prog Biophys Mol Biol. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- Ashfield R, Gribble FM, Ashcroft SjH, Ashcroft FM. Identification of the high-affinity tolbutamide site on the SUR1 subunit of the KATP channel. Diabetes. 1999;48:1341–1347. doi: 10.2337/diabetes.48.6.1341. [DOI] [PubMed] [Google Scholar]
- Babenko AP, Gonzalez G, Bryan J. Pharmaco-topology of sulfonylurea receptors. Separate domains of the regulatory subunits of K(ATP) channel isoforms are required for selective interaction with K(+) channel openers. J Biol Chem. 2000;275:717–720. doi: 10.1074/jbc.275.2.717. [DOI] [PubMed] [Google Scholar]
- Barrett-Jolley R, Davies NW. Kinetic analysis of the inhibitory effect of glibenclamide on KATP channels of mammalian skeletal muscle. J Membr Biol. 1997;155:257–262. doi: 10.1007/s002329900178. [DOI] [PubMed] [Google Scholar]
- Brady PA, Alekseev AE, Terzic A. Operative condition-dependent response of cardiac ATP-sensitive K+ channels toward sulfonylureas. Circ Res. 1998;82:272–278. doi: 10.1161/01.res.82.2.272. [DOI] [PubMed] [Google Scholar]
- Colquhoun D. Binding, gating and efficacy. Br J Pharmacol. 1998;125:923–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowski M, Ashcroft FM, Ashfield R, Lebrun P, Pirotte B, Egebjerg J, Hansen JB, Wahl P. The novel diazoxide analogue, 3-isopropylamino-7-methoxy-4H-1,2-benzothiadiazine 1,1-dioxide, is a selective SUR1/Kir6. 2 channel opener. Diabetes. 2002;51:1896–1906. doi: 10.2337/diabetes.51.6.1896. [DOI] [PubMed] [Google Scholar]
- Dabrowski M, Wahl P, Holmes WE, Ashcroft FM. Effect of repaglinide on cloned beta cell, cardiac and smooth muscle types of ATP-sensitive potassium channel. Diabetologia. 2001;44:747–756. doi: 10.1007/s001250051684. [DOI] [PubMed] [Google Scholar]
- D'hahan N, Moreau C, Prost AL, Jacquet H, Alekseev AE, Terzic A, Vivaudou M. Pharmacological plasticity of cardiac ATP-sensitive potassium channels toward diazoxide revealed by ADP. Proc Natl Acad Sci U S A. 1999;96:12162–12167. doi: 10.1073/pnas.96.21.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörschner H, Brekardin E, Uhde I, Schwanstecher C, Schwanstecher M. Stoichiometry of sulfonylurea-induced ATP-sensitive potassium channel closure. Mol Pharmacol. 1999;55:1060–1066. doi: 10.1124/mol.55.6.1060. [DOI] [PubMed] [Google Scholar]
- Findlay I. Sulphonylurea drugs no longer inhibit ATP-sensitive K+ channels during metabolic stress in cardiac muscle. J Pharmacol Exp Ther. 1993;266:456–467. [PubMed] [Google Scholar]
- Gillis KD, Gee WM, Hammoud A, Mcdaniel ML, Falke LC, Misler S. Effects of sulfonamides on a metabolite-regulated ATP-sensitive K+ channel in rat pancreatic B-cells. Am J Physiol. 1989;257:C1119–1127. doi: 10.1152/ajpcell.1989.257.6.C1119. [DOI] [PubMed] [Google Scholar]
- Gribble F, Ashcroft FM. New windows on the mechanism of action of potassium channel openers. Trends Pharmacol Sci. 2000;21:439–445. doi: 10.1016/s0165-6147(00)01563-7. [DOI] [PubMed] [Google Scholar]
- Gribble FM, Reimann F, Ashfield R, Ashcroft FM. Nucleotide modulation of pinacidil stimulation of the cloned K(ATP) channel Kir6. 2/SUR2A. Mol Pharmacol. 2000;57:1256–1261. [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Ashcroft FM. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO J. 1997a;16:1145–1152. doi: 10.1093/emboj/16.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Ashcroft FM. The interaction of nucleotides with the tolbutamide block of K-ATP currents: a reinterpretation. J Physiol. 1997b;504:35–45. doi: 10.1111/j.1469-7793.1997.00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Haug T, Ashcroft FM. MgATP activates the beta-cell KATP channel by interaction with its SUR1 subunit. Proc Natl Acad Sci U S A. 1998a;95:7185–7190. doi: 10.1073/pnas.95.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Seino S, Ashcroft FM. Tissue specificity of sulphonylureas: studies on cloned cardiac and beta-cell KATP channels. Diabetes. 1998b;47:1412–1418. doi: 10.2337/diabetes.47.9.1412. [DOI] [PubMed] [Google Scholar]
- Hambrock A, Löffler-Walz C, Quast U. Glibenclamide binding to sulphonylurea receptor subtypes: dependence on adenine nucleotides. Br J Pharmacol. 2002;136:995–1004. doi: 10.1038/sj.bjp.0704801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, IV, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995a;270:1166–1169. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, IV, Wang CZ, Aguilar-Bryan L, Bryan J, Minoru H, Seino S. A family of sulfonylurea receptors determines the properties of ATP-sensitive K+ channels. Neuron. 1996;16:1011–1017. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Tsuura Y, Namba N, Masuda K, Gonoi T, Seino Y, Mizuta M, Seino S. Cloning and functional characterization of a novel ATP-sensitive potassium channel ubiquitously expressed in rat tissues, including pancreatic islets, pituitary, skeletal muscle, and heart. J Biol Chem. 1995b;270:5691–5694. doi: 10.1074/jbc.270.11.5691. [DOI] [PubMed] [Google Scholar]
- Isomoto S, Kondo C, Yamada M, Matsumoto S, Horio Y, Matsuzawa Y, Kurachi Y. A novel sulphonylurea receptor forms with BIR (Kir6. 2) a smooth muscle type of ATP-sensitive K+ channel. J Biol Chem. 1996;271:24321–24325. doi: 10.1074/jbc.271.40.24321. [DOI] [PubMed] [Google Scholar]
- Koster JC, Sha Q, Nichols CG. Sulfonylurea and K+-channel opener sensitivity of KATP channels. Functional coupling of Kir6.2 and SUR1 subunits. J Gen Physiol. 1999;114:203–213. doi: 10.1085/jgp.114.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauter T, Ruppersberg JP, Baukrowitz T. Phospholipids as modulators of KATP channels:distinct mechanisms for control of sensitivity to sulphonylureas, K+ channel openers, and ATP. Mol Pharmacol. 2001;59:1086–1093. [PubMed] [Google Scholar]
- Lawrence CL, Proks P, Rodrigo GC, Jones P, Hayabuchi Y, Standen NB, Ashcroft FM. Gliclazide produces high-affinity block of KATP channels in mouse isolated pancreatic β-cells but not rat heart or arterial smooth muscle cells. Diabetologia. 2001;44:1019–1025. doi: 10.1007/s001250100595. [DOI] [PubMed] [Google Scholar]
- Löffler-Walz C, Hambrock A, Quast U. Interaction of K(ATP) channel modulators with sulfonylurea receptor SUR2B: implication for tetramer formation and allosteric coupling of subunits. Mol Pharmacol. 2002;61:407–414. doi: 10.1124/mol.61.2.407. [DOI] [PubMed] [Google Scholar]
- Mikhailov MV, Mikhailova EA, Ashcroft SJ. Molecular structure of the glibenclamide binding site of the beta-cell KATP channel. FEBS Lett. 2001;499:154–160. doi: 10.1016/s0014-5793(01)02538-8. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JP, IV, Gonzalez G, Aguilar-Bryan L, Permutt MA, Bryan J. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- Proks P, Reimann F, Greene N, Gribble FM, Ashcroft FM. Sulphonylurea stimulation of insulin secretion. Diabetes. 2002;51:S368–S376. doi: 10.2337/diabetes.51.2007.s368. [DOI] [PubMed] [Google Scholar]
- Reimann F, Gribble FM, Ashcroft FM. Differential response of KATP channels containing SUR2A or SUR2B subunits to nucleotides and pinacidil. Mol Pharmacol. 2000;58:1318–1325. doi: 10.1124/mol.58.6.1318. [DOI] [PubMed] [Google Scholar]
- Reimann F, Proks P, Ashcroft FM. Effects of mitiglinide (S 21403) on Kir6. 2/SUR1, Kir6.2/SUR2A and Kir6.2/SUR2B types of ATP-sensitive potassium channel. Br J Pharmacol. 2001;132:1542–1548. doi: 10.1038/sj.bjp.0703962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann F, Tucker SJ, Proks P, Ashcroft FM. Involvement of the N-terminus of Kir6. 2 in coupling to the sulphonylurea receptor. J Physiol. 1999;518:325–336. doi: 10.1111/j.1469-7793.1999.0325p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakura H, Ämmälä C, Smith PA, Gribble FM, Ashcroft FM. Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel subunit expressed in pancreatic β-cells, brain, heart and skeletal muscle. FEBS Lett. 1995;377:338–344. doi: 10.1016/0014-5793(95)01369-5. [DOI] [PubMed] [Google Scholar]
- Schwanstecher M, Schwanstecher C, Chudziak F, Panten U, Clement JP, IV, Gonzalez G, Aguilar-Bryan L, Bryan J. ATP-sensitive potassium channels. Meth Enzymol. 1999;294:445–458. doi: 10.1016/s0076-6879(99)94026-0. [DOI] [PubMed] [Google Scholar]
- Schwappach B, Zerangue N, Jan YN, Jan LY. Molecular basis for KATP assembly: transmembrane interactions mediate association of a K+ channel with an ABC transporter. Neuron. 2000;26:155–167. doi: 10.1016/s0896-6273(00)81146-0. [DOI] [PubMed] [Google Scholar]
- Shyng SL, Nichols CG. Octameric stochiometry of the KATP channel complex. J Gen Physiol. 1997;110:655–664. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe K, Tucker SJ, Matsuo M, Proks P, Ashcroft FM, Seino S, Amachi T, Ueda K. Direct photoaffinity labeling of the Kir6. 2 subunit of the ATP-sensitive K+ channel by 8-azido-ATP. J Biol Chem. 1999;274:3931–3933. doi: 10.1074/jbc.274.7.3931. [DOI] [PubMed] [Google Scholar]
- Trapp S, Proks P, Tucker SJ, Ashcroft FM. Molecular analysis of ATP-sensitive K channel gating and implications for channel inhibition by ATP. J Gen Physiol. 1998;112:333–349. doi: 10.1085/jgp.112.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6. 2 produces ATP-sensitive K-channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–181. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- Ueda K, Komine J, Matsuo M, Seino S, Amachi T. Cooperative binding of ATP and MgADP in the sulphonylurea receptor is modulated by glibenclamide. Proc Natl Acad Sci U S A. 1999;96:1268–1272. doi: 10.1073/pnas.96.4.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh N, Lamp ST, Weiss JN. Sulfonylureas, ATP-sensitive K+ channels, and cellular K+ loss during hypoxia, ischemia, and metabolic inhibition in mammalian ventricle. Circ Res. 1991;69:623–637. doi: 10.1161/01.res.69.3.623. [DOI] [PubMed] [Google Scholar]
- Zünckler BJ, Lins S, Ohno-Shosaku T, Trube G, Panten U. Cytosolic ADP enhances the sensitivity of tolbutamide of ATP-dependent K+ channels from pancreatic β-cells. FEBS Lett. 1988;239:241–244. doi: 10.1016/0014-5793(88)80925-6. [DOI] [PubMed] [Google Scholar]