

Abstract
Subanaesthetic doses of the N-methyl-d-aspartate (NMDA) antagonist ketamine have been shown to determine a dual modulating effect on glutamatergic transmission in experimental animals, blocking NMDA receptor activity and enhancing non-NMDA transmission through an increase in the release of endogenous glutamate. Little is known about the effects of ketamine on the excitability of the human central nervous system. The effects of subanaesthetic, graded incremental doses of ketamine (0.01, 0.02 and 0.04 mg kg−1 min−1, i.v.) on the excitability of cortical networks of the human motor cortex were examined with a range of transcranial magnetic and electric stimulation protocols in seven normal subjects. Administration of ketamine at increasing doses produced a progressive reduction in the mean resting motor threshold (RMT) (F(3, 18) = 22.33, P < 0.001) and active motor threshold (AMT) (F(3, 18) = 12.17, P < 0.001). Before ketamine administration, mean RMT ±s.d. was 49 ± 3.3 % of maximum stimulator output and at the highest infusion level it was 42.6 ± 2.6 % (P < 0.001). Before ketamine administration, AMT ±s.d. was 38 ± 3.3 % of maximum stimulator output and at the highest infusion level it was 33 ± 4.4 % (P < 0.002). Ketamine also led to an increase in the amplitude of EMG responses evoked by magnetic stimulation at rest; this increase was a function of ketamine dosage (F(3, 18) = 5.29, P= 0.009). In contrast to responses evoked by magnetic stimulation, responses evoked by electric stimulation were not modified by ketamine. The differential effect of ketamine on responses evoked by magnetic and electric stimulation demonstrates that subanaesthetic doses of ketamine enhance the recruitment of excitatory cortical networks in motor cortex. Transcranial magnetic stimulation produces a high-frequency repetitive discharge of pyramidal neurones and for this reason probably depends mostly on short-lasting AMPA transmission. An increase in this transmission might facilitate the repetitive discharge of pyramidal cells after transcranial magnetic stimulation which, in turn, results in larger motor responses and lower thresholds. We suggest that the enhancement of human motor cortex excitability to transcranial magnetic stimulation is the effect of an increase in glutamatergic transmission at non-NMDA receptors similar to that described in experimental studies.
Glutamate is the main excitatory neurotransmitter in the central nervous system that mediates synaptic transmission primarily by activation of ionotropic AMPA and NMDA receptors. Ketamine is a non-competitive antagonist of NMDA receptor (Anis et al. 1983; Thomson et al. 1985; Harrison & Simmonds, 1985; MacDonald et al. 1987; Irifune et al. 1992; Miller, 2000), which readily penetrates the blood-brain barrier and, at subanaesthetic doses, is well known to produce psychosis-like effects in healthy humans with a broad range of symptoms, behaviours and cognitive deficits that resemble schizophrenia (Ghoneim et al. 1985; Krystal et al. 1994; Miller, 2000). Psychosis-like behavioural effects produced by ketamine have been primarily attributed to postsynaptic reduction of NMDA glutamatergic activity. However, several studies suggest that the action of ketamine on glutamatergic neurotransmission may be more complex than a pure reduction in NMDA activity. Indeed, there is experimental evidence of excitatory effects of ketamine that, at least at certain dosages, can increase cortical cell spiking and can induce seizures (Ferrer-Allado et al. 1973; Bowyer et al. 1983). Moreover, recent studies suggest that subanaesthetic doses of ketamine and other NMDA receptor antagonists may increase the release of endogenous excitatory amino acids, glutamate and aspartate (Bustos et al. 1992; Liu & Moghaddam, 1995; Moghaddam et al. 1997). Therefore, administration of subanaesthetic doses of NMDA receptor antagonists may be able to produce differential modulation of glutamatergic activity, decreasing NMDA neurotransmission and activating AMPA and kainate non-NMDA transmission at the same time.
The aim of the present experiments was to use transcranial magnetic stimulation (TMS) to test the effects of subanaesthetic doses of ketamine on the excitability of cortical networks in the human motor cortex. A number of excitatory and inhibitory circuits were examined with a range of TMS protocols during infusion of incremental doses of ketamine. We evaluated the following TMS parameters: (1) the threshold for EMG responses; (2) recruitment curves of EMG responses; (3) intracortical inhibition and facilitation using the technique of Kujirai et al. (1993); (4) short-latency intracortical facilitation (Tokimura et al. 1996; Ziemann et al. 1998a,b; Di Lazzaro et al. 1999b); and (5) silent period duration. Additionally, we evaluated the amplitude of EMG responses evoked by transcranial electrical stimulation. The results show that the main effect of subanaesthetic doses of ketamine is an increase in cortical excitability to TMS.
METHODS
Subjects
Seven healthy volunteers (mean age ±s.d. 32.1 ± 5.8 years) participated in all experiments including the experiments with electrical stimulation. All the subjects were authors of this study, and gave their written informed consent. The study was performed according to the Declaration of Helsinki and approved by the ethics committee of the Medical Faculty of the Catholic University of Rome.
Magnetic and electric stimulation
Magnetic stimulation was performed using two high-power Magstim 200 magnetic stimulators (Magstim Co., Whitland, UK) connected to the Bistim Module throughout all measurements. A figure-of-eight coil with external loop diameters of 9 cm was held over the right motor cortex at the optimum scalp position to elicit motor responses in the contralateral first dorsal interosseus (FDI). The induced current flowed in a postero-anterior direction. Resting motor threshold (RMT) was defined as the minimum stimulus intensity that produced a liminal motor evoked response (about 50 μV in 50 % of 10 trials) at rest. Active motor threshold (AMT) was defined as the minimum stimulus intensity that produced a liminal motor evoked response (about 200 μV in 50 % of 10 trials) during isometric contraction of the tested muscle at about 20 % maximum. A constant level of voluntary contraction was maintained with reference to an oscilloscope display of EMG in front of the subject. Auditory feedback of the EMG activity was also provided. We evaluated the recruitment curve of EMG responses using magnetic stimuli of increasing intensities. Recruitment curves were recorded with target muscles both at rest and during voluntary contraction at 20 % maximum. At rest, magnetic stimuli were applied at RMT, 10 % and 20 % of maximum stimulator output above RMT and 5 % of maximum stimulator output below RMT. During voluntary contraction, magnetic stimuli were applied at AMT, 110 % AMT, 150 % AMT and 200 % AMT. The four intensity steps of the active and the four intensity steps of resting recruitment curves, were divided into two blocks, each of two different stimulus intensities. Five stimuli were delivered at each intensity. In the first block of both the recruitment curves recorded at rest and during voluntary contraction, magnetic stimuli were randomly intermixed with five electrical stimuli at 110 % electrical resting motor threshold (RMT(E); the minimum stimulus intensity that produced a liminal motor-evoked response of about 50 μV in 50 % of 10 trials at rest). Electrical stimulation of the motor cortex was performed with a Digitimer D180A stimulator, with a time constant of 50 μs. The cathode was located at the vertex and the anode 7 cm laterally (anodal stimulation).
The silent period was elicited whilst subjects held a tonic voluntary contraction of approximately 20 % of maximum voluntary contraction. Five stimuli at 200 % AMT were given. The test after ketamine was performed using cortical magnetic stimuli of the same intensity used in baseline conditions. Moreover, when there was a change in the amplitude of the EMG response, silent period recording was repeated with the intensity of the cortical stimulus adjusted in order to maintain an EMG response of the same amplitude as that recorded in baseline conditions. Silent period duration was measured from the end of the EMG response to the return of sustained post-stimulus EMG activity.
Intracortical inhibition and facilitation
Intracortical inhibition and facilitation were studied using the technique of Kujirai et al. (1993). Using a Bistim module, two magnetic stimuli were given through the same stimulating coil over the motor cortex and the effect of the first (conditioning) stimulus on the second (test) stimulus was investigated. The conditioning stimulus was set at an intensity of 5 % (of stimulator output) below active threshold. The second, test, shock intensity was adjusted to evoke a muscle response in relaxed FDI with an amplitude of approximately 1 mV, peak-to-peak. The timing of the conditioning shock was altered in relation to the test shock. Inhibitory interstimulus intervals (ISIs) of 2 and 3 ms and facilitatory ISIs of 15 and 25 ms were investigated. Five stimuli were delivered at each ISI. For these recordings, muscle relaxation is very important and the subject was given audiovisual feedback at high gain to assist in maintaining complete relaxation.
The amplitude of the conditioned EMG responses was expressed as the percentage of the amplitude of the test EMG responses. The amplitude of the conditioned responses at the two different inhibitory ISIs and the amplitude of the conditioned responses at the two facilitatory ISIs were averaged separately obtaining grand mean amplitudes of the inhibitory and of the facilitatory ISIs.
The test after ketamine was performed using cortical magnetic stimuli of the same intensity as the test and the conditioning stimuli used in baseline conditions. Additionally, when there was a change in threshold or amplitude, paired stimulation was repeated with the intensity of the cortical stimulus adjusted in order to maintain a control EMG response of the same amplitude as that recorded in baseline conditions and the conditioning stimulus intensity adjusted in order to maintain an intensity of 5 % (of stimulator output) below active threshold. This is important because the intensities of both conditioning and test stimuli have a profound effect on the degree of inhibition /facilitation produced by paired magnetic stimulation (Kujrai et al. 1993; Ziemann et al. 1998a)
Short-latency intracortical facilitation
We also analysed the facilitatory interaction that occurs between pairs of threshold magnetic stimuli given over the motor cortex at short interstimulus intervals (Tokimura et al. 1996; Ziemann et al. 1998a,b; Di Lazzaro et al. 1999b). This is not the same as the facilitation that follows the period of intracortical inhibition described above at ISIs of 15 and 25 ms. Short-latency intracortical facilitation occurs at much shorter intervals and is seen when both stimuli have an intensity that is above the active threshold. It may represent interaction between the mechanisms that produce repetitive I wave firing of pyramidal neurones (Tokimura et al. 1996; Ziemann et al. 1998a,b; Di Lazzaro et al. 1999b). Using a Bistim module, two magnetic stimuli were given through the same stimulating coil over the motor cortex and the effect of the first stimulus on the second stimulus was investigated. The intensity of the first stimulus was set to be 4 % (of stimulator output) above AMT. The second stimulus had an intensity of 2 % (of stimulator output) above AMT. An interstimulus interval of 1.4 ms was investigated. Five stimuli were delivered for each condition. The responses to five single and five paired stimuli were averaged. When both magnetic shocks were applied, the EMG responses evoked by the first stimulus alone were subtracted offline from the responses recorded using paired stimulation in order to evaluate the additional activity evoked by the second stimulus. The amplitude of the EMG responses to both stimuli was expressed as the percentage of the amplitude of the EMG responses to the second stimulus on its own. Because the intensities of both conditioning and test stimuli are critical for the degree of facilitation produced by this short-latency paired magnetic stimulation, short-latency intracortical facilitation during ketamine infusion was repeated with the intensity of the cortical stimulus adjusted in order to maintain an intensity of the conditioning stimulus of 4 % (of stimulator output) above active threshold and of the test stimulus of 2 % (of stimulator output) above active threshold.
Experimental design and data analysis
The effects of graded incremental doses of ketamine were evaluated. Intravenous ketamine infusion (0.01 mg kg−1 min−1) was administered for 20 min and was followed by incremental doses of 0.02 mg kg−1 min−1 for 15 min and then of 0.04 mg kg−1 min−1 for 15 min. Measurements were made immediately before infusion (baseline) and were repeated at each level of infusion, apart from the short-latency intracortical facilitation, which was performed only immediately before infusion and at the highest infusion level.
The first repetition of measurements was started 5 min after the beginning of infusion. An anaesthesiologist administered the drug and continuously monitored the subject. Ketamine effects on vigilance were moderate and did not interfere with subjects' ability to fully comply with the requirements of the experimental protocol.
Sedation/agitation levels, psychiatric symptoms, attention and concentration levels were assessed prior to the start of ketamine infusion and at each different infusion level. Ketamine-induced sedation/agitation was evaluated using the Riker Sedation -Agitation Scale (SAS) (Riker et al. 1999). Drug-induced psychiatric symptoms were assessed by evaluating the psychosis factor score (the sum of the items ‘conceptual disorganisation’, ‘suspiciousness’, ‘hallucinatory behaviour’ and ‘unusual thought content’) of the Brief Psychiatric Rating Scale (BPRS) (Overall & Gorham, 1962). Levels of attention and concentration were evaluated using the attention and concentration score of the Mini-Mental State Examination (MMSE) (Folstein et al. 1975).
Motor cortex excitability parameters were analysed using ANOVA for repeated measures, with the Greenhouse-Geisser correction for lack of sphericity. Planned comparisons vs. baseline were statistically evaluated with Bonferroni's correction (0.05/3 = 0.017). A logarithmic transformation of amplitude data was applied to give a better fit to Gaussian distribution and to reduce the outlier effects. Cognitive and psychiatric scales were analysed by Friedman's non-parametric ANOVA for repeated measures and comparisons vs. baseline by the Wilcoxon signed-rank test. The effects of RMT changes on recruitment curves were evaluated by calculating the best fitting function and coefficients of the recruitment curve obtained in the baseline condition, and of the recruitment curve obtained at the highest ketamine dose with intensity stimulus values in this latter curve shifted relative to the new RMT value for each subject.
In order to check whether the ketamine-induced changes in neurophysiological parameters could depend on psychiatric and cognitive variations, separate ANOVAs for repeated measures with cognitive and psychiatric scales as changing covariates were used. The significance of ketamine dose after removing the effect of behavioural scores on neurophysiological parameters would indicate an independent effect of the drug on cortical excitability.
RESULTS
Neurophysiological results are summarised in Table 1 and in Fig. 1 and Fig. 2, raw data from one subject are shown in Figs 3, 4 and 5.
Table 1.
Effects of ketamine on the measures of motor excitability
| Measure | Baseline | Ketamine 0.01 mg kg−1 min−1 i.v. | Ketamine 0.02 mg kg−1 min−1 i.v. | Ketamine 0.04 mg kg−1 min−1 i.v. | Test | P value |
|---|---|---|---|---|---|---|
| RMT | 49.3 ± 3.3 | 46.6 ± 3.6 | 44.7 ± 3 | 42.6 ± 2.6 | ANOVA | < 0.001 |
| (p = 0.012 post hoc t-test) | (p < 0.001 post hoc t-test) | (p < 0.001 post hoc t-test) | ||||
| AMT | 38 ± 3.3 | 36 ± 5.6 | 34.7 ± 5 | 33 ± 4.4 | ANOVA | < 0.001 |
| (p = 0.012 post hoc t-test) | (p < 0.002 post hoc t-test) | |||||
| ICI | 34 ± 16.7 | 37 ±18.1 | 45 ± 24.5 | 51.2 ± 19.8 | ANOVA | 0.002 |
| (p = 0.007 post hoc t-test) | ||||||
| ICI | — | 40.2 ± 14.5 | 46.3 ± 21.3 | 43.4 ± 20.5 | ANOVA | 0.142 |
| (adjusted stimulus intensities) | ||||||
| ICF | 117.5 ± 20.2 | 118.8 ± 79 | 119.5 ± 30.5 | 124.2 ± 51.1 | ANOVA | 0.982 |
| ICF | — | 136.6 ± 93 | 140 ± 61.3 | 141.8 ± 102 | ANOVA | 0.709 |
| (adjusted stimulus intensities) | ||||||
| SICF | 449.5 ± 325 | — | — | 703.5 ± 1015 | WILCOXON | 1.00 |
| (adjusted stimulus intensities) | ||||||
| SP | 191.4 ± 25 | 201.7 ± 41 | 216.3 ± 36 | 251.7 ± 54 | ANOVA | < 0.001 |
| (5.5 ± 1.7) | (5.9 ± 1.9) | (P = 0.007 post hoc t-test) | (P = 0.004 post hoc t-test) | |||
| (EMG response) | (6.1 ± 2.2) | (6 ± 2.4) | ||||
| SP | — | — | — | 194 ± 30 | Paired t-test | 0.76 |
| (matched EMG response) | (5.6 ± 1.8) | |||||
| Anodal stimulation | ANOVA | |||||
| EMG response active | 2.6 ± 2 | 2.6 ± 2 | 2.6 ± 1.9 | 2.2 ± 1.6 | 0.570 | |
| EMG response rest | 0.66 ± 0.9 | 0.68 ± 1 | 0.7 ± 1 | 0.71 ± 1.3 | 0.771 |
RMT = resting motor threshold; AMT = active motor threshold; ICI = intracortical inhibition; ICF = intracortical facilitation; SICF =short latency intracortical facilitation; SP = silent period. All values are means ± standard deviations. Bold type indicates significant changes from baseline. Post hoc t-test was performed versus baseline.
Figure 1. Effects of ketamine at increasing doses on resting (RMT) and active (AMT) motor threshold (A), on amplitude of EMG responses evoked by anodal stimulation (B) and recruitment curves obtained with magnetic stimulation at increasing stimulus intensities (C and D).
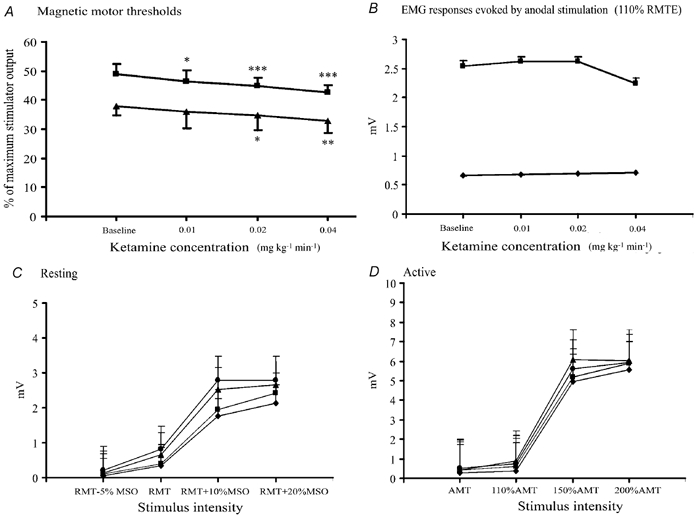
Error bars are s.d. (A) and s.e.m. (B-D). A, administration of ketamine significantly reduced the mean RMT (▪) (F (3, 18) = 22.33, P < 0.001) and AMT (▴) (F(3, 18) = 12.17, P < 0.001). A progressive decrement of RMT was observed with increasing ketamine dose (*P= 0.012 at the lowest dosage; P < 0.001 at the second and third infusion levels). Starting from the second dosage, a significant decrease in AMT was observed (*P= 0.012). The maximal effect of ketamine on AMT occurred at the highest infusion level (P < 0.002). B, the mean amplitude of EMG responses evoked by electrical stimulation at 110 % RMT(E) was not affected by ketamine either at rest (♦) (F (3, 18) = 0.38, P= 0.771) or during voluntary contraction (▪) (F (3, 18) = 0.69, P= 0.570). C, ketamine led to an increase in the amplitude of EMG responses evoked by magnetic stimulation at rest. This increase was a function of ketamine dosage (F (3, 18) = 5.29, P= 0.009). Planned comparisons showed that the only significant increment vs. baseline was reached at the highest dose (***P < 0.001). C and D, ketamine doses: ♦ baseline, ▪ 0.01 mg kg−1 min−1, ▴ 0.02 mg kg−1 min−1 and • 0.04 mg kg−1 min−1. MSO, maximum stimulator output.
Figure 2. Effects of ketamine at increasing doses on cortical silent period duration (A), intracortical inhibition and facilitation (B) and short-latency intracortical facilitation (C).
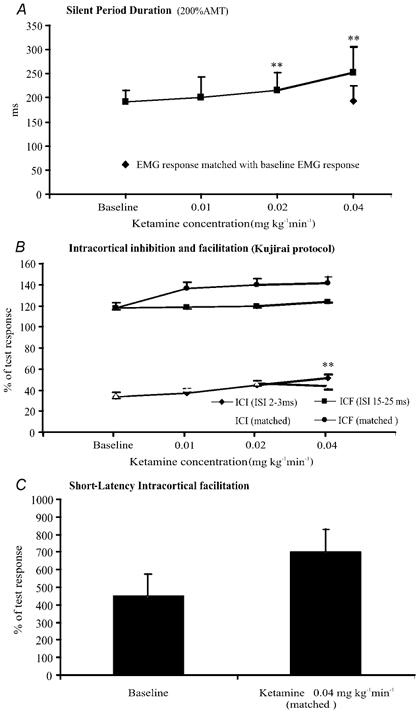
Error bars are s.d. (A) and s.e.m. (B and C). A, the duration of the silent period was increased by ketamine (F (3, 18) = 19.15, P < 0.001). The lengthening reached significance at the second and third steps (**P < 0.01). At the highest dose, the silent period produced by cortical stimuli evoking adjusted EMG responses has the same duration of the silent period recorded before ketamine administration. B, the amount of inhibition was progressively reduced by increasing doses of ketamine (F (3, 18) = 7.11, P= 0.002). The reduction in intracortical inhibition reached statistical significance at the highest dosage (*P < 0.01). With adjusted stimulus intensities and EMG response amplitudes, the amount of inhibition was not significantly modified by ketamine. The amount of facilitation evaluated using the Kujirai protocol was not significantly modified by ketamine (F (3, 18) = 0.06, P= 0.982). The amount of facilitation was not significantly modified with adjusted stimulus intensities and EMG response amplitudes either, but there was a clear trend toward more facilitation. C, short-latency intracortical stimulation (evaluated using adjusted stimulus intensities) was clearly increased by ketamine, but, due to the high variability, the increase did not reach statistical significance.
Figure 3. EMG responses evoked by transcranial electric (A) and magnetic (B) stimulation in first dorsal interosseous muscle in one of the subjects in baseline conditions and after ketamine at the highest dosage.
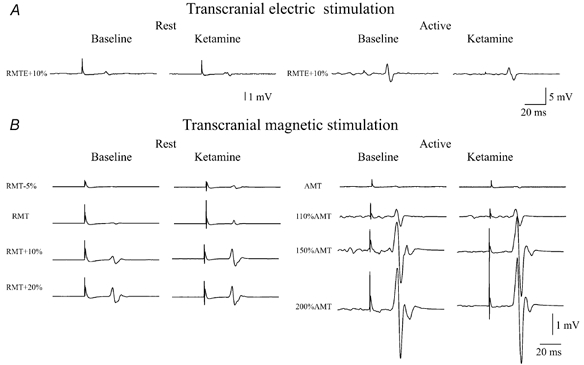
A, the amplitude of EMG responses evoked by electric stimulation was not affected by ketamine, either at rest or during voluntary contraction. B, the amplitude of EMG responses evoked by magnetic stimulation was increased by ketamine and this effect was more pronounced at rest. Ketamine also determined a reduction in resting motor threshold (RMT); after ketamine, a clear EMG response was recorded even at a stimulus intensity of 5 % of maximum stimulator output below the baseline RMT value.
Figure 4. Intracortical inhibition (2 and 3 ms ISIs) and facilitation (15 and 25 ms ISIs), as evaluated using the Kujirai protocol and short-latency intracortical facilitation (1.4 ms ISI) in baseline conditions and after ketamine at the highest dosage in one of the subjects.
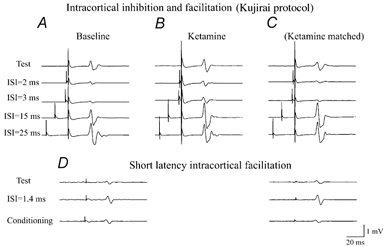
A, the average (of 5 trials each) of EMG responses evoked in the FDI by cortical stimulation alone and by cortical stimulation conditioned by a cortical stimulus subthreshold for motor responses (5 % of maximum magnetic stimulator output below active motor threshold) given 2–3 ms and 15–25 ms earlier. In baseline conditions, the conditioning cortical stimulus suppressed the EMG response when the ISI was 2 or 3 ms and increased the EMG response when the ISI was 15 or 25 ms ISIs. B, the average (of 5 trials each) of EMG responses evoked in the FDI by cortical stimulation alone and by cortical stimulation conditioned by a cortical stimulus subthreshold for motor responses performed using the same cortical stimulus intensities used in baseline conditions. The amplitude of the EMG response evoked by the test stimulus was increased, the inhibition observed when the ISI was 2 or 3 ms was slightly reduced and the facilitation observed when the ISI was 15 or 25 ms was slightly increased. C, intracortical inhibition and facilitation after ketamine repeated with the intensity of the test cortical stimulus adjusted in order to maintain a control EMG response of the same amplitude as that recorded in baseline conditions and a conditioning stimulus intensity adjusted in order to maintain an intensity of 5 % (of stimulator output) below active threshold. Intracortical inhibition (2 and 3 ms ISIs) was unaffected, whilst intracortical facilitation (15 and 25 ms ISIs) was increased. D, short-latency intracortical facilitation in baseline conditions (left) and after ketamine (right). EMG responses evoked by a conditioning magnetic stimulus alone, by test magnetic stimulus alone and by both conditioning and test stimuli with an ISI of 1.4 ms are shown. The EMG responses evoked by the first stimulus, alone, were subtracted offline from the responses recorded using paired stimulation in order to evaluate the additional activity evoked by the second stimulus. The test after ketamine was performed with the intensity of the test and conditioning cortical stimuli adjusted in order to obtain EMG responses of the same amplitude as those recorded in baseline conditions. After ketamine, short-latency intracortical facilitation was more pronounced.
Figure 5. Silent periods evoked in baseline conditions and at the highest ketamine dosage in one of the subjects.
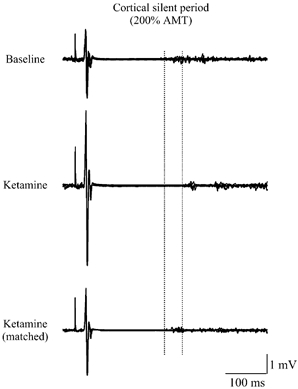
Five traces are superimposed. The amplitude of EMG responses was increased and the duration of silent periods was prolonged after ketamine. When the test after ketamine was performed with the intensity of the cortical stimulus adjusted in order to obtain EMG responses of the same amplitude as those recorded in baseline conditions, the duration of the silent period was unaffected.
Magnetic stimulation
Administration of ketamine significantly reduced the mean RMT (F (3, 18) = 22.33, P < 0.001) and AMT (F (3, 18) = 12.17, P < 0.001).
A linear decrement in RMT was observed with increasing ketamine dose (linearity was tested after logarithmic transformation of doses; F (1, 6) = 43.58, P= 0.001), being significant starting at the lowest dosage (P= 0.012). Changes were also significant at the second (P < 0.001) and third infusion levels (P < 0.001).
A linear decrement in AMT was also observed with increasing ketamine dose (linearity was tested after logarithmic transformation of doses; F (1, 6) = 36.00, P= 0.001). The lowest ketamine dosage did not statistically modify AMT (P= 0.128). Starting from the second dosage, a significant decrease in AMT was observed (P= 0.012). The maximal effect of ketamine on AMT occurred at the highest infusion level (P < 0.002).
When measured at rest, the amplitude of EMG responses, besides the well-known dependence on stimulation intensity (F (1.2, 7.2) = 109.64, P < 0.001), was a function of ketamine dosage (F (3, 18) = 5.29, P= 0.009) without any significant interaction (F (2.9, 17.4) = 1.13, P= 0.361), indicating a similar ketamine effect at each stimulus intensity. Planned comparisons showed that the only significant increment vs. baseline was reached at the highest dose (P < 0.001, significant after Bonferroni's correction). There was no substantial change in the sigmoidal shape of recruitment curves. Also, when the recruitment curve obtained in baseline conditions and that obtained at the highest ketamine dose with shifted intensity stimulus values (relative to the new RMT value for each subject) were compared, the sigmoidal equation was the best fitting function for both baseline (R2= 0.667) and high dose ketamine curves (R2= 0.665). The coefficients were not statistically different (baseline: 0.848, 95 % confidence interval = 0.809–0.887; high dose ketamine: 0.869, 95 % confidence interval = 0.836–0.903; P > 0.05).
When measured during voluntary contraction, the amplitude of EMG responses, besides the well-known dependence on stimulation intensity (F (1.3, 7.6) = 65.69, P < 0.001), was not significantly modified by ketamine infusion (F (3, 18) = 2.40, P= 0.101), without any significant interaction (F (2.5, 14.9) = 1.86, P= 0.185). However, a clear trend toward an increase in EMG response amplitude was also evident during voluntary contraction (Fig. 1).
The duration of the silent period was increased by ketamine (F (3, 18) = 19.15, P < 0.001), although the lowest ketamine dosage did not statistically modify this (P= 0.255). Starting from the second dosage, a significant prolongation of silent period duration was observed (P= 0.007, significant after Bonferroni's correction) and the maximal effect of ketamine on the silent period occurred at the highest infusion level (P= 0.004, significant after Bonferroni's correction). The amplitude of EMG responses evoked at 200 % AMT during ketamine infusion was larger than that of EMG responses recorded at the same stimulus intensity before ketamine administration. Therefore, at the highest infusion level of ketamine, which produced the most pronounced effect on silent period duration, silent period recording was repeated with the intensity of the cortical stimulus adjusted (lowering the intensity of cortical stimulus) in order to maintain an EMG response of the same amplitude to that recorded in baseline conditions. When the stimulus intensity was reduced so that the EMG response amplitudes were matched (Student's t test (d.f., 6) = 0.550, P= 0.602 when compared with baseline values), there was no significant effect of ketamine on silent period duration (Student's t test (d.f., 6) = 0.32, P= 0.760 when compared with baseline values).
Electrical anodal stimulation
The mean amplitude of EMG responses evoked at 110 % RMT(E) was not affected by ketamine, either at rest (F (3, 18) = 0.38, P= 0.771) or during voluntary contraction (F (3, 18) = 0.69, P= 0.570).
Intracortical inhibition and facilitation
Intracortical inhibition and facilitation were studied according to the Kujirai protocol (Kujirai et al. 1993; Di Lazzaro et al. 1998b). When two stimuli were delivered, the EMG responses to the test stimulus were dramatically suppressed at interstimulus intervals of 2 and 3 ms. Before ketamine, the mean amplitude of the conditioned response over these intervals was 34 ± 16.7 % of control. The amount of inhibition over ISIs of 2–3 ms was progressively reduced by increasing doses of ketamine (F (3, 18) = 7.11, P= 0.002). However, the first two ketamine dosages did not statistically modify the amount of inhibition (P= 0.470 at the lowest dosage and P= 0.044 (not significant after Bonferroni's correction) at the intermediate dosage). The maximal effect of ketamine on the amount of inhibition occurred at the highest infusion level (P= 0.007, significant after Bonferroni correction).
Because AMT was decreased and EMG response amplitude was increased by ketamine, we repeated the experiments after adjusting the stimulus intensities to compensate for the changes in threshold and EMG response amplitude after ketamine. With adjusted stimulus intensities and EMG response amplitudes, the amount of inhibition over ISIs of 2–3 ms was not modified by ketamine (F (3, 18) =2.06, P= 0.142).
When two stimuli were delivered, the EMG responses to the test stimulus were increased at interstimulus intervals of 15 and 25 ms. Before ketamine, the mean amplitude of the conditioned response over these intervals was 117.5 ± 20.2 % of control. The amount of facilitation was not significantly modified by ketamine (F (3, 18) = 0.06, P= 0.982).
Because AMT was decreased and EMG response amplitude was increased by ketamine, we repeated the experiments after adjusting the stimulus intensities to compensate for the changes in threshold and EMG response amplitude after ketamine. With adjusted stimulus intensities, the amount of facilitation was increased by ketamine. However, because of the high variability, the observed ketamine-induced increment of facilitation was not statistically significant (F (3, 18) = 0.47, P= 0.709).
Short-latency intracortical facilitation
When two suprathreshold stimuli were delivered, the EMG responses to the test stimulus were increased at an interstimulus interval of 1.4 ms. Before ketamine treatment, the mean amplitude of the conditioned response was 449.5 ± 325.2 % control.
Because the AMT was decreased by ketamine, we performed short-latency intracortical facilitation after adjusting the stimulus intensities to compensate for the change in threshold. The amount of facilitation was increased by ketamine. However, because of the high variability, the observed ketamine-induced increment of facilitation was not statistically significant (Wilcoxon Z= 0.00, P= 1.00).
Ketamine did not induce anaesthesia at any infusion level or in any subject, but only produced a slight sedation at the highest infusion level. However, the slight changes that occurred in SAS scores were not significant (Friedman's test, χ2(3) = 6.8, P= 0.079). With regard to other clinical scales, ketamine induced significant changes in BPRS (Friedman's test, χ2(3) = 15.9, P= 0.001) and in MMSE (Friedman's test, χ2(3) = 15.6, P= 0.001) (Fig. 6). Comparisons vs. baseline indicated that significant changes in BPRS and MMSE occurred only at the highest dosage (Wilcoxon's test, Z= 2.232, P= 0.026 and Z= 2.226, P= 0.026, respectively). In order to verify whether the observed significant changes in neurophysiological parameters could depend on psychiatric and cognitive variations, RMT, AMT and log amplitude at the highest TMS intensity were analysed by ANOVA for repeated measures, with BPRS and MMSE as changing covariates. The effect of ketamine remained significant after controlling for concomitant mental status on RMT (F (3, 15) = 5.19, P= 0.012), on AMT (F (3, 15) = 3.76, P= 0.034) and on log amplitude (F (3, 15) = 4.64, P= 0.017). This suggests that neurophysiological results and mental status are independently changed by ketamine administration.
Figure 6. Sedation/agitation levels, psychiatric symptoms, attention and concentration levels during ketamine infusion.
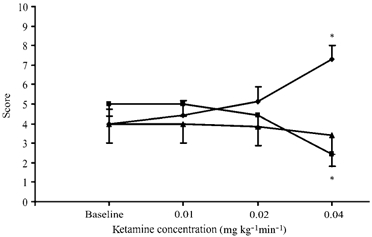
Sedation/ agitation levels were assessed according to the Riker Sedation-Agitation Scale (▴; SAS), psychiatric symptoms using the psychosis factor score of the Brief Psychiatric Rating Scale (♦; BPRS) and attention and concentration levels according to the Mini-Mental State Examination (▪; MMSE). Ketamine induced significant changes in BPRS (Friedman's test, χ2 (3) = 15.9, P= 0.001) and in MMSE (Friedman's test, χ2 (3) = 15.6, P= 0.001). Comparisons vs. baseline indicated that significant changes in BPRS and MMSE occurred at the highest dosage (*P < 0.05).
DISCUSSION
The present results provide the first evidence that the excitability of motor cortical networks as tested by transcranial magnetic stimulation in humans can be increased by subanaesthetic doses of ketamine.
Responses to single magnetic stimuli
Ketamine reduced active and resting motor threshold. The effect of ketamine on threshold was dose dependent in the dose range studied, with larger reductions in threshold produced by higher doses.
The reduction in motor threshold was accompanied by an increase in the amplitude of EMG responses evoked by magnetic stimulation at rest; this was particularly evident at the highest dose. The amplitude of EMG responses evoked by magnetic stimulation during voluntary contraction showed a trend toward an increase particularly at suprathreshold intensities, but this effect was less evident than for the amplitude of EMG responses evoked at rest and did not reach significance. Because voluntary contraction increases the excitability of corticospinal cells, increasing the number and size of I-waves (Di Lazzaro et al. 1998c), it can be hypothesised that voluntary contraction shifts the excitability close to the maximum level. This limits the further increase that can be obtained with ketamine. Previous studies in normal subjects during ischaemic anaesthesia and in amputees have reported similar findings, demonstrating changes in recruitment curves that were more pronounced in resting than in active muscles (Ridding & Rothwell, 1997).
In contrast with EMG responses evoked by TMS, the responses evoked by electrical stimulation both at rest and during voluntary contraction were unaffected by administration of ketamine. Transcranial electrical stimulation tends to activate the axons of corticospinal neurones in the white matter, whereas magnetic stimulation activates the same fibres trans-synaptically (Di Lazzaro et al. 1998a,c,1999a,b). Thus, electrically evoked responses are not as sensitive to changes in cortical excitability as those evoked by magnetic stimulation. Therefore, a differential effect of ketamine on EMG responses evoked by magnetic and electric stimulation demonstrates that the increase in excitability takes place at the level of circuits within the motor cortex.
The increase in cortical excitability observed after ketamine treatment that produces a depression of glutamatergic transmission through a blockade of NMDA receptors might appear a paradox. However, it has been recently demonstrated that NMDA receptor antagonists may increase the release of the endogenous excitatory amino acids glutamate and aspartate (Bustos et al. 1992; Liu & Moghaddam, 1995). Therefore, administration of NMDA receptor antagonists may activate glutamatergic neurotransmission at AMPA and kainate non-NMDA receptors. The question is why the activation of non-NMDA glutamatergic transmission should result in an increase of excitability in response to TMS. The increase in excitability of the motor cortex to transcranial magnetic stimulation may be explained if one considers the different kinetics of the ionotropic glutamatergic receptors AMPA and NMDA and the physiological basis of TMS. The AMPA receptor opens quickly (< 1 ms) and briefly, whilst the NMDA receptor takes longer to open (> 2 ms) and remains open for longer (see Conti & Wienberg, 1999 for a review). Due to their kinetics, NMDA channels participate selectively in low-frequency transmission (Collingridge et al. 1988a; Priesol et al. 2000) and may act as a low-pass filter for high frequencies due to their slow depolarising potential, whilst AMPA channels are more involved in high-frequency transmission. Since transcranial magnetic stimulation of the human brain produces a high-frequency repetitive discharge of corticospinal cells with a periodicity of about 700 Hz (Di Lazzaro et al. 1998a,c), it is conceivable that TMS depends mostly on short-lasting AMPA transmission that is characterised by shorter EPSPs and shorter refractory periods. Therefore, the activation of glutamatergic neurotransmission at non-NMDA receptors induced by ketamine may result in an increase of excitability of the motor cortex to TMS. Moreover, because a prolonged opening of ionic channels as induced by NMDA receptor activation may limit the repetitive discharge of pyramidal cells induced by TMS, the blocking of NMDA receptors might further facilitate the repetitive discharge of pyramidal cells, determining larger EMG responses and lower thresholds. A similar finding has been reported in an experimental study that evaluated the synaptic response evoked by 100 Hz stimulation of Schaffer collateral-commissural fibres after NMDA blockade with the antagonist d-2-amino-5-phosphovalerate (APV) (Collingridge et al. 1988b). In this study the authors observed that the amplitude of the fast EPSPs evoked at 100 Hz tended to be increased by APV and suggested that this was due to the increased driving force consequent on the reduction of the slow depolarising potentials (Collingridge et al. 1988b).
Unlike cortical pyramidal neurones, spinal motoneurones have well-developed, post-spike after-hyperpolarising potentials of long duration, which act as a powerful brake on sustained firing frequency (Kernell, 1965; Calvin & Schwindt, 1972). This electrophysiological property of spinal motor neurone prevents high-frequency repetitive discharge of spinal motoneurones after activation from corticospinal projections and might explain why there was no effect of ketamine on EMG responses evoked by electrical stimulation.
The duration of the silent period was increased by ketamine. The silent period represents an inhibitory phenomenon that appears to be related to GABAB mechanisms (Sanger et al. 2001). It is known that GABAB-mediated slow inhibition is activated through glutamatergic receptors, both NMDA and non-NMDA, providing an important means to offset excessive excitation (Benardo, 1995). Because the main effect we observed after ketamine is an increase in cortical excitability, probably due to an enhancement of AMPA-mediated fast excitatory intracortical transmission leading to an increase of repetitive pyramidal cell discharge, it can be hypothesised that when fast glutamatergic transmission is increased by ketamine, GABAB-mediated slow inhibition is increased consequently and this results in silent period prolongation. In agreement with this is the fact that silent period duration evaluated for EMG responses of matched amplitude was not modified after ketamine treatment.
At the highest infusion level, ketamine produced psychosis-like effects and reduced attention and concentration. We checked whether the ketamine-induced changes in RMT, AMT and EMG response amplitudes could depend on these behavioural and psychological effects. Statistical analysis showed that the effect of ketamine on RMT, AMT and EMG response amplitudes remained significant after controlling for concomitant mental status. Therefore, our data suggest that neurophysiological results and mental status are independently modified by ketamine administration. However, we cannot exclude the possibility that cognitive changes less pronounced than those measured could have influenced the results.
Effects of ketamine on cortical inhibition and facilitation
Intracortical inhibition was substantially reduced by ketamine. However, when the test was performed adjusting the stimulus intensities to compensate for the change in threshold and EMG response amplitudes the reduction in intracortical inhibition was less pronounced and did not reach significance. The amount of suppression produced by paired magnetic stimulation depends on the intensity of both the test and conditioning shock (Kujirai et al. 1993; Schafer et al. 1997), and, in particular, larger conditioning shocks may result in less suppression. It is well known that maximum inhibition occurs with a conditioning intensity of 0.8 RMT (Kujirai et al. 1993; Schafer et al. 1997), whilst higher intensities result in less suppression. Therefore, most of the reduction of intracortical inhibition was probably due to the ketamine-induced lowering of threshold to TMS. The pronounced change in threshold produced by ketamine that made the intensity of the conditioning shock clearly above the level for optimal inhibition is conceivably the cause of the change in intracortical inhibition. The fact that intracortical inhibition evaluated by adjusting the stimulus intensities to compensate for the changes in threshold and EMG response amplitude was only slightly modified after ketamine treatment supports this hypothesis. However, there is a possible explanation for the slight reduction in intracortical inhibition that persisted when the stimulus intensities were adjusted. Intracortical inhibition, as tested with paired magnetic stimulation, has been suggested to be related to GABAA mechanisms (Ziemann et al. 1996a,b; Di Lazzaro et al. 2000). It has recently been reported that kainate receptor stimulation can decrease GABA release at the level of connections between fast-spiking inhibitory interneurones and layer V pyramidal cells in rat motor cortex involving GABAA receptors (Ali et al. 2001). Therefore, the activation of neurotransmission at kainate receptors produced by the ketamine-induced release of endogenous glutamate may be responsible for the slight reduction in intracortical inhibition that persisted when the stimulus intensities were adjusted.
Ketamine apparently did not modify either intracortical facilitation as evaluated using the Kujrai protocol or short-latency intracortical facilitation. However, when the tests were performed adjusting the stimulus intensities to compensate for the change in threshold and EMG response amplitude, a trend toward an increase in intracortical facilitation was observed with both protocols. The increase in intracortical facilitation was more pronounced for the protocol testing short-latency intracortical facilitation. However, because of high variability, the change was not statistically significant. It should also be considered that ketamine conceivably shifts the excitability close to the maximum after single magnetic stimulation and only a minor further increase can be obtained with paired magnetic stimulation.
In conclusion, our results demonstrate that low doses of ketamine can increase motor cortex excitability to transcranial magnetic stimulation. Because ketamine produces a differential modulation of glutamatergic activity decreasing NMDA neurotransmission and activating non-NMDA transmission, the observed increase in motor cortex excitability is consistent with the hypothesis that TMS parameters involving intracortical excitatory phenomena (RMT, AMT, recruitment curves and intracortical facilitation after paired stimulation) are dependent on non-NMDA glutamatergic neurotransmission.
Acknowledgments
The authors thank Ulf Ziemann for his useful comments.
REFERENCES
- Ali AB, Rossier J, Staiger JF, Audinat E. Kainate receptors regulate unitary IPSCs elicited in pyramidal cells by fast-spiking interneurons in the neocortex. J Neurosci. 2001;21:2992–2999. doi: 10.1523/JNEUROSCI.21-09-02992.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anis NA, Berry SC, Burton NR, Lodge D. The dissociative anaesthetics, ketamine and phenyclidine, selectively reduce excitation of central mammalian neurones by N-methyl aspartate. B J Pharmacol. 1983;79:565–575. doi: 10.1111/j.1476-5381.1983.tb11031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benardo LS. N-Methyl-d-aspartate transmission modulates GABAB-mediated inhibition of rat hippocampal pyramidal neurons in vitro. Neuroscience. 1995;68:637–643. doi: 10.1016/0306-4522(95)00164-e. [DOI] [PubMed] [Google Scholar]
- Bowyer JF, Albertson TE, Winters WD, Baselt RC. Ketamine-induced changes in kindled amygdaloid seizures. Neuropharmacology. 1983;22:887–894. doi: 10.1016/0028-3908(83)90136-3. [DOI] [PubMed] [Google Scholar]
- Bustos G, Abarca J, Forray MI, Gysling K, Bradberry CW, Roth RH. Regulation of excitatory amino acid release by N-methyl-d-aspartate receptors in rat striatum: in vivo microdialysis studies. Brain Res. 1992;585:105–115. doi: 10.1016/0006-8993(92)91195-k. [DOI] [PubMed] [Google Scholar]
- Calvin WH, Schwindt PC. Steps in the production of motoneuron spikes during rhythmic firing. J Neurophysiol. 1972;35:297–310. doi: 10.1152/jn.1972.35.3.297. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Herron CE, Lester RA. Frequency-dependent N-methyl-d-aspartate receptor-mediated synaptic transmission in rat hippocampus. J Physiol. 1988a;399:301–312. doi: 10.1113/jphysiol.1988.sp017081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Herron CE, Lester RA. Synaptic activation of N-methyl-d-aspartate receptors in the Schaffer collateral-commissural pathway of rat hippocampus. J Physiol. 1988b;399:283–300. doi: 10.1113/jphysiol.1988.sp017080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti F, Weinberg RJ. Shaping excitation at glutamatergic synapses. Trends Neurosci. 1999;22:451–458. doi: 10.1016/s0166-2236(99)01445-9. [DOI] [PubMed] [Google Scholar]
- Di Lazzaro V, Oliviero A, Meglio M, Cioni B, Tamburrini G, Tonali P, Rothwell JC. Direct demonstration of the effect of lorazepam on the excitability of the human motor cortex. Clin Neurophysiol. 2000;111:794–799. doi: 10.1016/s1388-2457(99)00314-4. [DOI] [PubMed] [Google Scholar]
- Di Lazzaro V, Oliviero A, Profice P, Insola A, Mazzone P, Tonali P, Rothwell JC. Effects of voluntary contraction on descending volleys evoked by transcranial electrical stimulation over the motor cortex hand area in conscious humans. Exp Brain Res. 1999a;124:525–528. doi: 10.1007/s002210050649. [DOI] [PubMed] [Google Scholar]
- Di Lazzaro V, Oliviero A, Profice P, Saturno E, Pilato F, Insola A, Mazzone P, Tonali P, Rothwell JC. Comparison of descending volleys evoked by transcranial magnetic and electric stimulation in conscious humans. Electroenceph Clin Neurophysiol. 1998a;109:397–401. doi: 10.1016/s0924-980x(98)00038-1. [DOI] [PubMed] [Google Scholar]
- Di Lazzaro V, Restuccia D, Oliviero A, Profice P, Ferrara L, Insola A, Mazzone P, Tonali P, Rothwell JC. Magnetic transcranial stimulation at intensities below active motor threshold activates intracortical inhibitory circuits. Exp Brain Res. 1998b;119:265–268. doi: 10.1007/s002210050341. [DOI] [PubMed] [Google Scholar]
- Di Lazzaro V, Restuccia D, Oliviero A, Profice P, Ferrara L, Insola A, Mazzone P, Tonali P, Rothwell JC. Effects of voluntary contraction on descending volleys evoked by transcranial stimulation in conscious humans. J Physiol. 1998c;508:625–633. doi: 10.1111/j.1469-7793.1998.625bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lazzaro V, Rothwell JC, Oliviero A, Profice P, Insola A, Mazzone P, Tonali P. Intracortical origin of the short latency facilitation produced by pairs of threshold magnetic stimuli applied to human motor cortex. Exp Brain Res. 1999b;129:494–499. doi: 10.1007/s002210050919. [DOI] [PubMed] [Google Scholar]
- Ferrer-Allado T, Brechner VL, Dymond A, Cozen H, Crandall P. Ketamine-induced electroconvulsive phenomena in the human limbic and thalamic regions. Anesthesiology; 1973;38:333–344. doi: 10.1097/00000542-197304000-00006. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, MacHugh PR. ‘Mini-mental state’. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Ghoneim MM, Hinrichs JV, Mewaldt SP, Petersen RC. Ketamine: behavioral effects of subanesthetic doses. J Clin Psychopharmacol. 1985;5:70–77. [PubMed] [Google Scholar]
- Harrison NL, Simmonds MA. Quantitative studies on some antagonists of N-methyl-D-aspartate in slices of rat cerebral cortex. J Pharmacol. 1985;84:381–391. doi: 10.1111/j.1476-5381.1985.tb12922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irifune M, Shimizu T, Nomoto M, Fukuda T. Ketamine-induced anesthesia involves N-methyl-d-aspartate receptor-channel complex in mice. Brain Res. 1992;596:1–9. doi: 10.1016/0006-8993(92)91525-j. [DOI] [PubMed] [Google Scholar]
- Kernell D. The limits of firing frequency in cat lumbosacral motoneurones possessing different time course of afterhyperpolarization. Acta Physiol Scand. 1965;65:87–100. [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- Kujirai T, Caramia MD, Rothwell JC, Day BL, Thompson PD, Ferbert A, Wroe S, Asselman P, Marsden CD. Corticocortical inhibition in human motor cortex. J Physiol. 1993;471:501–519. doi: 10.1113/jphysiol.1993.sp019912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Moghaddam B. Regulation of glutamate efflux by excitatory amino acid receptors: evidence for tonic inhibitory and phasic excitatory regulation. J Pharmacol Exp Ther. 1995;274:1209–1215. [PubMed] [Google Scholar]
- MacDonald JF, Miljkovic Z, Pennefather P. Use-dependent block of excitatory amino acid currents in cultured neurons by ketamine. J Neurophysiol. 1987;58:251–266. doi: 10.1152/jn.1987.58.2.251. [DOI] [PubMed] [Google Scholar]
- Miller RD. Non-barbiturate intravenous anesthetics. In: Miller RD, Reves JG, Roizen MF, Savarese JJ, Cucchiara RF, Ross A, editors. Anesthesia. 5. Philadelphia: Churchill Livingstone; 2000. pp. 228–272. chap. 9. [Google Scholar]
- Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overall JE, Gorham DR. Brief psychiatric rating scale. Psychol Rep. 1962;10:799–812. [Google Scholar]
- Priesol AJ, Jones GE, Tomlinson RD, Broussard DM. Frequency-dependent effects of glutamate antagonists on the vestibulo-ocular reflex of the cat. Brain Res. 2000;857:252–264. doi: 10.1016/s0006-8993(99)02441-5. [DOI] [PubMed] [Google Scholar]
- Ridding MC, Rothwell JC. Stimulus/response curves as a method of measuring motor cortical excitability in man. Electroenceph Clin Neurophysiol. 1997;105:340–344. doi: 10.1016/s0924-980x(97)00041-6. [DOI] [PubMed] [Google Scholar]
- Riker RR, Picard JT, Fraser GL. Prospective evaluation of the Sedation-Agitation Scale for adult critically ill patients. Crit Care Med. 1999;27:1325–1329. doi: 10.1097/00003246-199907000-00022. [DOI] [PubMed] [Google Scholar]
- Sanger TD, Garg RR, Chen R. Interaction between two different inhibitory systems in the human motor cortex. J Physiol. 2001;530:307–317. doi: 10.1111/j.1469-7793.2001.0307l.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer M, Biesecker JC, Schulze-Bonhage A, Ferbert A. Transcranial magnetic double stimulation: influence of the intensity of the conditioning stimulus. Electroenceph Clin Neurophysiol. 1997;105:462–469. doi: 10.1016/s0924-980x(97)00054-4. [DOI] [PubMed] [Google Scholar]
- Thomson AM, West DC, Lodge D. An N-methyl-aspartate receptor-mediated synapse in rat cerebral cortex: a site of action of ketamine? Nature. 1985;313:479–481. doi: 10.1038/313479a0. [DOI] [PubMed] [Google Scholar]
- Tokimura H, Ridding MC, Tokimura Y, Amassian V, Rothwell JC. Short latency facilitation between pairs of threshold magnetic stimuli applied to human motor cortex. Electroenceph Clin Neurophysiol. 1996;101:263–272. doi: 10.1016/0924-980x(96)95664-7. [DOI] [PubMed] [Google Scholar]
- Ziemann U, Lonnecker S, Steinhof BJ, Paulus W. The effect of lorazepam on the motor cortical excitability in man. Exp Brain Res. 1996a;109:127–135. doi: 10.1007/BF00228633. [DOI] [PubMed] [Google Scholar]
- Ziemann U, Lonnecker S, Steinhoff BJ, Paulus W. Effects of antiepileptic drugs on motor cortex excitability in humans: a transcranial magnetic stimulation study. Ann Neurol. 1996b;40:367–378. doi: 10.1002/ana.410400306. [DOI] [PubMed] [Google Scholar]
- Ziemann U, Tergau F, Wassermann EM, Wischer S, Hildebrandt J, Paulus W. Demonstration of facilitatory I-wave interaction in the human motor cortex by paired transcranial magnetic stimulation. J Physiol. 1998a;511:181–190. doi: 10.1111/j.1469-7793.1998.181bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemann U, Tergau F, Wischer S, Hildebrandt J, Paulus W. Pharmacological control of facilitatory I-wave interaction in the human motor cortex. A paired transcranial magnetic stimulation study. Electroenceph Clin Neurophysiol. 1998b;109:321–330. doi: 10.1016/s0924-980x(98)00023-x. [DOI] [PubMed] [Google Scholar]