

Abstract
In eukaryotic cells, hormones and neurotransmitters that engage the phosphoinositide pathway evoke a biphasic increase in intracellular free Ca2+ concentration: an initial transient release of Ca2+ from intracellular stores is followed by a sustained phase of Ca2+ influx. This influx is generally store-dependent and is required for controlling a host of Ca2+-dependent processes ranging from exocytosis to cell growth and proliferation. In many cell types, store-operated Ca2+ entry is manifest as a non-voltage-gated Ca2+ current called ICRAC (Ca2+ release-activated Ca2+ current). Just how store emptying activates CRAC channels remains unclear, and some of our recent experiments that address this issue will be described. No less important from a physiological perspective is the weak Ca2+ buffer paradox: whereas macroscopic (whole cell) ICRAC can be measured routinely in the presence of strong intracellular Ca2+ buffer, the current is generally not detectable under physiological conditions of weak buffering following store emptying with the second messenger InsP3. In this review, I describe some of our experiments aimed at understanding just why InsP3 is ineffective under these conditions and which lead us to conclude that respiring mitochondria are essential for the activation of ICRAC in weak intracellular Ca2+ buffer. Mitochondrial Ca2+ uptake also increases the dynamic range over which InsP3 functions as the second messenger that controls Ca2+ influx. Finally, we find that Ca2+-dependent slow inactivation of Ca2+ influx, a widespread but poorly understood phenomenon that helps shape the profile of an intracellular Ca2+ signal, is regulated by mitochondrial Ca2+ buffering. Thus, by enabling macroscopic store-operated Ca2+ current to activate and then by controlling its extent and duration, mitochondria play a crucial role in all stages of store-operated Ca2+ influx. Store-operated Ca2+ entry reflects therefore a dynamic interplay between endoplasmic reticulum, mitochondria and plasma membrane.
An increase in intracellular free Ca2+ concentration is a ubiquitous signalling mechanism that regulates a broad spectrum of kinetically disparate processes ranging from exocytosis to cell growth and proliferation (Carafoli, 2002). Two major ways whereby eukaryotic cells can increase their cytosolic Ca2+ concentration are release of the ions from intracellular stores (mainly the endoplasmic/ sarcoplasmic reticulum) or entry of Ca2+ into the cell from the external solution (Berridge, 1993; Pozzan et al. 1994). Because the intracellular stores have a limited capacity, Ca2+ entry into the cell down an enormous electrochemical potential gradient is important for supporting the activities of numerous Ca2+-dependent processes. In excitable tissues, like nerve and muscle, Ca2+ entry is accomplished by the opening of Ca2+-permeable ion channels (voltage- and ligand-gated; Burnashev, 1998; Catterall, 2000). Much is known about the biophysical properties of these proteins, their stoichiometry, and even the identity of amino acids that are crucial for key channel functions like selectivity and gating (Burnashev, 1998; Catterall, 2000; Hille, 2001).
However, until relatively recently, our understanding of how Ca2+ enters non-excitable cells has been weak. Non-excitable cells are cells that do not fire action potentials and include the cells of the immune system, endothelia lining the blood vessels, epithelia that line the respiratory and digestive tracts, and glial cells in the brain. In these cell types, Ca2+ entry is essential for maintaining normal cell function but voltage-operated Ca2+ channels tend not to be expressed and ligand-gated Ca2+-permeable channels are sparse. Instead, a major route for Ca2+ influx in these cells is via store-operated Ca2+ channels (Parekh & Penner, 1997).
Store-operated Ca2+ influx
In an erudite paper published in 1986, James Putney proposed a model for Ca2+ influx in non-excitable cells, which he called capacitative Ca2+ entry (Putney, 1986). The fundamental tenet of this model was that the Ca2+ entry pathway in the plasma membrane was controlled by the Ca2+ content of the intracellular stores. As the store Ca2+ content fell, a signal was somehow sent from the stores which caused the Ca2+ entry pathway to open (now referred to as store-operated Ca2+ entry). Although indirect evidence began to accrete in support of Putney's model, it was not until 1992 that Reinhold Penner directly demonstrated, in an elegant series of experiments, that Ca2+ store depletion resulted in the activation of a Ca2+-selective current which was called ICRAC (Hoth & Penner, 1992). Ca2+ entry through store-operated Ca2+ channels is important for regulating a host of temporally diverse processes from exocytosis and enzymatic activity to gene transcription and cell proliferation (Parekh & Penner, 1997; Putney & McKay, 1999). Aberrant store-operated entry has been reported to underlie debilitating human diseases like certain primary immunodeficiencies (Partiseti et al. 1994) and acute pancreatitis (Raraty et al. 2000; Parekh, 2000). It now seems clear that store-operated Ca2+ channels are not a homogeneous class but instead reflect a family with different biophysical properties. The best characterised store-operated current is ICRAC, which is found in several types of cell including mast cells, lymphocytes, macrophages, megakaryocytes and hepatocytes (Parekh & Penner, 1997; Lewis, 1999).
Although store-operated entry is a widespread Ca2+ influx pathway, it is by no means the only route for Ca2+ entry in non-excitable cells. In addition to a range of second-messenger operated Ca2+-permeable channels (activated by Ca2+, InsP3/InsP4, cyclic nucleotides; reviewed in Clapham, 1995), growing evidence indicates that a Ca2+ influx pathway controlled by arachidonic acid might be prominent under certain conditions (reviewed in Taylor, 2002).
In this Wellcome Prize Lecture review, I shall focus on some of our work over the past several years that has been aimed at understanding the mechanisms that control ICRAC. It is, as befits a prize lecture, a subjective account and is not a general review of the literature. Because of space constraints, certain papers have not been cited and I apologise to those authors for this.
Basic features of ICRAC
CRAC channels are activated by the process of emptying the intracellular Ca2+ stores. It is of little consequence how the stores are actually emptied, the net result is the same (Hoth & Penner, 1992; Parekh & Penner, 1997). Physiologically, stores are emptied following cell-surface receptor stimulation which increases the levels of the Ca2+ releasing messenger InsP3. Experimentally, stores can be emptied by increasing net Ca2+ flux out of the stores following application of receptor agonists, increasing intracellular InsP3 by dialysis with a patch pipette or by exposure to Ca2+ ionophores. Alternatively, suppressing Ca2+ re-uptake into the stores allows the endogenous Ca2+ leak pathway to deplete the stores. Inhibition of store refilling can occur by exposure to thapsigargin, a potent inhibitor of the sarcoplasmic/endoplasmic reticular Ca2+ ATPase (SERCA) pumps on the stores.
The Ca2+ flux through CRAC channels gives rise to the small Ca2+ current ICRAC, which can be measured directly and unambiguously using the whole cell patch clamp technique. With Ca2+ as the charge carrier, ICRAC activates mono-exponentially (time constant of around 20 s; Fig. 1A) and is a non-voltage-gated, inwardly rectifying current with a very positive reversal potential (> +60 mV; Fig. 1B), the latter indicating high selectivity for Ca2+. Although Na+ ions outnumber Ca2+ by 70:1 in the external solution, neither the extent of ICRAC nor the positive reversal potential (still > +60 mV) appear to be altered by substituting external Na+ for organic cations like NMDG+ in the presence of physiological levels of external Ca2+ (1–2 mm) (Fierro & Parekh, 2000). Moreover, removal of external Ca2+ in the continuous presence of external Na+ and Mg2+ abolishes the current completely (Hoth & Penner, 1992; Zweifach & Lewis, 1993; Fierro & Parekh, 2000). Because of the lack of a clear reversal potential for ICRAC with Ca2+ as the charge carrier, the permeability ratio of Ca2+ to Na+ (PCa/PNa) has been hard to establish. Using the ‘added buffer method’, where cells are loaded with sufficient Fura-2 to capture all the incoming Ca2+ through CRAC channels, it has been estimated that CRAC channels are as selective for Ca2+ as their voltage-operated counterparts (Hoth, 1995), the latter exhibiting a PCa/PNa of 1000. However, CRAC channels discriminate between various divalent cations, with Ba2+ and Sr2+ supporting less current than Ca2+ (Hoth & Penner, 1992; Zweifach & Lewis, 1993; Fierro & Parekh, 1999a). The single CRAC channel conductance is low. Stationary noise analysis has estimated it to be well below 1 pS, beyond the current level of detection (Hoth & Penner, 1992; Zweifach & Lewis, 1993). A consequence of this is that there are estimated to be at least 5000 functional CRAC channels in the plasma membrane following stimulation.
Figure 1. Development of ICRAC following store depletion.
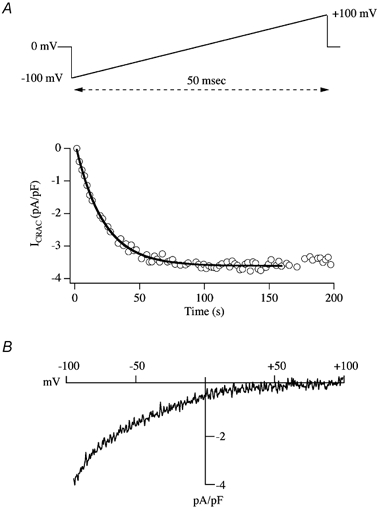
A, the upper panel shows the typical voltage ramp protocol used to monitor ICRAC in mast cells, the related rat basophilic leukaemia (RBL-1) cell line and jurkat T-lymphocytes. The ramp spans −100 to +100 mV in 50 ms, and is applied from a holding potential of 0 mV once every 2 s. The lower panel depicts the time course of activation of ICRAC. The current amplitude, measured at −80 mV from each ramp, has been normalised for cell size by dividing the current by cell capacitance. The thick line shows a mono-exponential fit to the activation time course. B shows the current-voltage relationship, once the current had reached steady state. Note the inward rectification and very positive reversal potential, characteristic of ICRAC. The pipette solution is caesium glutamate based, supplemented with InsP3 and 10 mm EGTA.
Activation mechanism
The unique gating of ICRAC has made it an attractive target of investigation for the identification of new signal transduction pathways. CRAC channels are activated by the emptying of intracellular Ca2+ stores. But just how a reduction in store content is translated into opening of these channels remains unclear. A sensor within the stores presumably detects the fall in Ca2+ content and this then initiates a process resulting in a signal being sent from the stores to the channels in the plasma membrane. Virtually nothing is known about the identity of the sensor, other than it is probably not calreticulin (Fasolato et al. 1998), the major intraluminal Ca2+ binding protein of non-muscle cells. As for the activation signal, three models have been put forward (see Fig. 2; reviewed in Parekh & Penner, 1997; Putney & McKay, 1999): (1) the vesicular fusion hypothesis (in which CRAC channels are inserted into the plasma membrane upon store depletion via an exocytotic mechanism; Yao et al. 1999; Alderton et al. 2000); (2) conformational coupling (where InsP3 receptors on the stores are physically attached to CRAC channels in the plasma membrane; Berridge, 1995; Kiselyov et al. 1999), including the related secretion-like coupling model (where peripheral endoplasmic reticulum (ER) moves to the plasma membrane upon store depletion so that coupling can take place and this movement is regulated by the peripheral cytoskeleton; Patterson et al. 1999; Rosado et al. 2000), and (3) the diffusible messenger model (in which a small mobile factor is released from the stores upon their emptying and which then directly opens CRAC channels; Ramdriamampita & Tsien, 1993; Csutora et al. 1999).
Figure 2. The three contemporary models proposed to account for the activation mechanism of ICRAC.
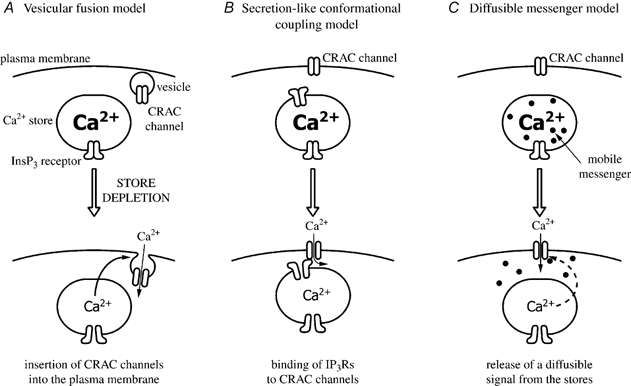
The vesicular fusion model does not provide any insight into the activation signal itself. It postulates instead that this signal results in the insertion of CRAC channels into the plasma membrane. The fusion model therefore could be accommodated within either of the other two contemporary models. However, we found that dialysing the cytosol with small GTP-binding proteins known to be involved in vesicular trafficking (Parekh & Penner, 1996), interfering with GTP-binding protein function (Fierro & Parekh, 1999b) or exposing cells to either clostridial neurotoxins or to recombinant proteins that suppress exocytosis (in collaboration with Professor Burgoyne, Liverpool) all fail to affect the activation of ICRAC, findings that are not readily reconcilable with a fusion mechanism. The secretion-like conformational coupling model predicts that, first, intimate interactions between the ER and plasma membrane are essential for ICRAC to activate, second, InsP3 receptors are required for all stages of store-operated Ca2+ entry, and third, that interfering with the peripheral cytoskeleton should impair the ability of ICRAC to activate. We have tested these predictions in rat basophilic leukaemia (RBL-1) cells (Bakowski et al. 2001). Inflating the cell by applying positive pressure to the patch pipette (cell ballooning) did not interefere with the ability of CRAC channels to activate, in spite of some rather marked changes in the cellular architecture (Bakowski et al. 2001). A variety of InsP3 receptor inhibitors did not affect ICRAC development, in spite of occupying InsP3 receptors, and pharmacological tools directed towards re-shaping the cytoskeleton consistently failed to interfere with the activation of ICRAC (Bakowski et al. 2001). Hence, a secretion-like coupling model is probably not a viable candidate for the activation of CRAC channels in RBL-1 cells. As for the diffusible messenger hypothesis, no convincing candidate has yet been identified. Recently, we have found that inhibition of the lipoxygenase family of enzymes impairs the activation of ICRAC (Glitsch et al. 2002a). The inhibitors are much less effective if administered after CRAC channels have been activated. Such findings raise the intriguing possibility that a lipoxygenase might be involved in the activation mechanism, although, as discussed in Glitsch et al. (2002), such a conclusion is not unequivocal as it is based on a pharmacological approach.
Clearly, the ability to study single CRAC channel currents in an inside-out patch would greatly facilitate identification of key regulatory molecules involved in channel gating, as would the identification of the CRAC channel gene(s). It was recently suggested that the protein TRPV6 comprises all or part of the CRAC channel pore (Yue et al. 2001). But work by us and others suggests that this is probably not the case (Bakowski & Parekh, 2002a,b; Hermosura et al. 2002; Prakriya & Lewis, 2002). Understanding the molecular identity of the CRAC channel as well as the activation signal remain outstanding questions in the field.
The weak buffer paradox
Another major problem concerning ICRAC is the weak Ca2+ buffer paradox. Whereas ICRAC can be activated maximally following whole cell dialysis with a patch pipette containing InsP3 and strong buffer (10 mm EGTA), the current generally fails to develop in the presence of weak Ca2+ buffer (0.1 mm EGTA; Fig. 3, modified from Bakowski & Parekh, 2001; see also Broad et al. 1999; Fierro & Parekh, 2000). Such low concentrations of EGTA are only slightly larger than the levels of endogenous mobile Ca2+ buffers (Zhou & Neher, 1993). InsP3 is able to release Ca2+ from the stores under these conditions (Parekh et al. 1997). Hence the inability of InsP3 to activate ICRAC in weak Ca2+ buffer does not reflect a failure to mobilise Ca2+ from the stores. Clearly, resolution of why InsP3 is so effete in weak buffer is essential if we are to place ICRAC in any kind of physiological context.
Figure 3. The weak buffer paradox.
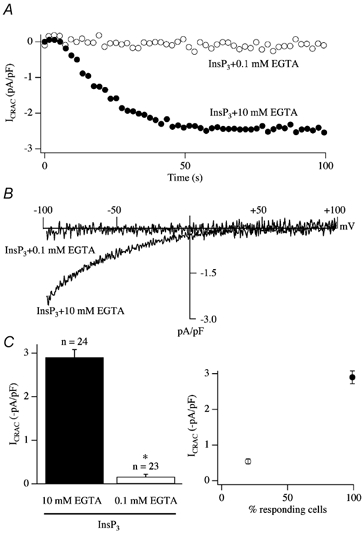
A, InsP3 generally fails to activate ICRAC in weak intracellular Ca2+ buffer (0.1 mm EGTA) whereas it is effective in strong buffer (10 mm EGTA). B, current-voltage relationships for the two recordings from A, taken at steady state. C, the mean amplitude of ICRAC (left-hand panel) and the fraction of cells responding (right-hand panel) for each condition is shown. In this and all subsequent figures, data from RBL-1 cells are shown. *P < 0.01.
The first explanation that we considered was that the inability of InsP3 to activate ICRAC in weak Ca2+ buffer was an idiosyncrasy of our recording conditions. Like most electrophysiologists, we routinely work at room temperature. To see whether InsP3 was effective at physiological temperature, we compared the effects of InsP3 in weak versus strong buffer at 36 °C. However, InsP3 was still ineffective in weak buffer (Fig. 4A and B, modified from Fierro et al. 2000). We also systematically altered the composition of the pipette solution to see whether we could render InsP3 more effective. However, InsP3 was still largely ineffective in weak buffer in either a Cs+- or K+-based pipette solution, when Cl− was the dominant anion or when EGTA was replaced by BAPTA or dimethyl BAPTA (Fierro & Parekh, 2000; Gilabert & Parekh, 2000). Non-metabolisable analogues of InsP3 were also incapable of routinely generating the current, as was receptor stimulation, the latter being used as a physiological route to generate InsP3.
Figure 4. InsP3 still fails to activate ICRAC in weak Ca2+ buffer at physiological temperature.
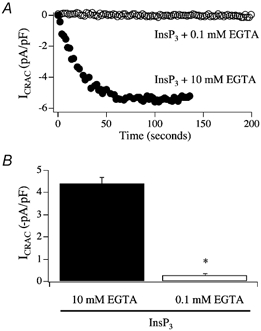
Experiments were carried out at 36 °C. A, whereas InsP3 evoked a large ICRAC at 36 °C in 10 mm EGTA, no current was detectable in 0.1 mm EGTA. B, aggregate data are summarised. *P < 0.01 (Student's t test).
Passive activation of ICRAC
An important clue came from experiments designed to try to understand the mechanism underlying activation of ICRAC following passive depletion of stores (Fierro & Parekh, 1999c). Dialysis with a high concentration of Ca2+ chelator alone (10 mm EGTA or BAPTA) is sufficient to maximally activate ICRAC, a process referred to as passive depletion of stores (Hoth & Penner, 1992). Passive depletion is thought to rely only on the endogenous leak of Ca2+ from the stores. In strong buffer (several millimolar chelator), cytoplasmic Ca2+ is strongly clamped at very low levels, so as Ca2+ leaks out of the stores it is captured by the chelator and cannot therefore be taken back up. Stores will be drained gradually of Ca2+ and hence empty sufficiently for ICRAC to activate (passive activation). Figure 5A–C shows the pattern of development of ICRAC following dialysis with 10 mm of EGTA, BAPTA or dimethyl BAPTA. Superimposed on the EGTA trace in Fig. 5A is a recording from a cell in which InsP3 was also included in the pipette. With InsP3, ICRAC activated rapidly and mono-exponentially. In marked contrast, the pattern of development of the current to passive depletion was much slower than that seen with InsP3. With EGTA (Fig. 5A), BAPTA (Fig. 5B) or dimethyl BAPTA (Fig. 5C), the development of the current was kinetically complex: after a sizeable delay of around 60 s, ICRAC initially activated slowly (first phase of development), followed by a faster developing phase (second phase of development). We analysed the various features of the current (delay, rate of development of the two phases as well as their durations, their relative contributions to total current amplitude) and these were not significantly different between the chelators (Fierro & Parekh, 1999b). These chelators have apparent equilibrium dissociation constants of 150, 225 and 150 nm, respectively, and on-rates of 1.5 × 106m−1 s−1 for EGTA and 5 × 108m−1 s−1 for BAPTA and dimethyl BAPTA at pH 7.2. Hence neither the speed of binding Ca2+ nor the equilibrium affinity of the chelators accounts for the complex kinetic profile.
Figure 5. Passive activation of ICRAC.
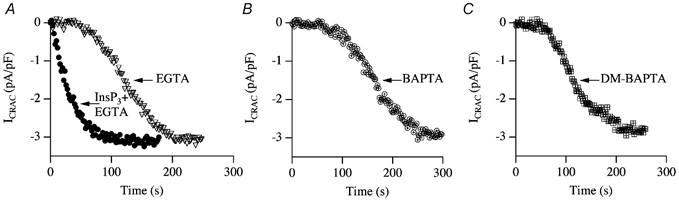
A, comparison of the time course of development of ICRAC following dialysis with either 10 mm EGTA alone or 30 μm InsP3+ EGTA. Note the initial slow development of ICRAC followed by a second, faster phase in response to EGTA alone. B, development of ICRAC following dialysis with 10 mm BAPTA or dimethyl BAPTA (panel C).
Importance of SERCA pumps
We found that SERCA pumps were extremely influential in shaping the pattern of development of ICRAC following passive store depletion (Fierro & Parekh, 1999c). When SERCA pumps were inhibited with either cyclopiazonic acid or thapsigargin, the kinetics of ICRAC changed dramatically compared with the current activated in 10 mm EGTA alone (Fig. 6A). The biphasic nature of the current (initial slow phase followed by a second faster phase), which was always seen with passive depletion using 10 mm EGTA, was not present when SERCA pumps were blocked. Now, the intial slow component was lost and instead the rapid phase developed in isolation. The slow initial activation therefore reflects a dynamic interplay between Ca2+ leakage from the stores and SERCA pump-mediated Ca2+ reuptake. The Ca2+ pumps help shape the profile of ICRAC development, even in 10 mm EGTA.
Figure 6. SERCA pumps shape the pattern of passive activation of ICRAC.
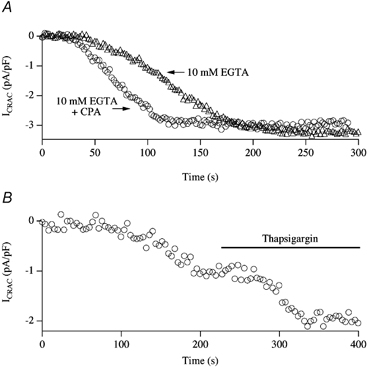
A, inclusion of cyclopiazonic acid (CPA) eliminates the initial slow phase of current development, leaving the second component intact. B, following dialysis with 2.5 mm EGTA, ICRAC activated partially in this cell. Subsequent application of thapsigargin increased the extent of the current, as well as its rate of development.
For lower concentrations of EGTA (1–2.5 mm), the effects of SERCA pumps were even more striking. Following dialysis with EGTA < 2.5 mm, ICRAC activated slowly and mono-phasically to reach a submaximal steady-state amplitude (Fig. 6B). Once the small current had reached steady-state, application of the SERCA pump blocker thapsigargin increased the size of the current further. This demonstrates that the submaximal ICRAC reflected a steady state between Ca2+ efflux from the stores and reuptake by SERCA pump activity (Fierro & Parekh, 1999b). Moreover, even in the presence of a relatively high concentration of EGTA (2.5 mm), SERCA pumps were still active and were able to resequester Ca2+ into the stores.
SERCA and InsP3
Although the preceding results demonstrate that SERCA pumps are very powerful and their activity needs to be overcome in order for ICRAC to activate fully, they do not address the key question as to whether the pumps can sculpt the ability of the natural trigger, namely InsP3, to activate ICRAC under physiological conditions of weak Ca2+ buffering. Because the current cannot be measured when cells are dialysed with InsP3 in weak Ca2+ buffer (Fig. 3), we therefore determined the lowest concentration of EGTA that would still enable us to reliably measure macroscopic ICRAC and, having found this, we examined the effects of SERCA pump block on the features of the current (Glitsch & Parekh, 2000). Figure 7A summarises results from experiments in which cells were dialysed with a maximally effective concentration of InsP3 and different concentrations of EGTA. Concentrations of EGTA less than 0.35 mm generally failed to support any macroscopic activation of ICRAC whereas close to maximal activation was seen with 1 mm EGTA. However, with 0.6 mm EGTA, ICRAC clearly had a sub-maximal amplitude in spite of the cells being continuously exposed to a high concentration of InsP3 via the patch pipette. We reasoned that the stores might nevertheless refill partially in the presence of such a moderate Ca2+ chelator concentration and this would therefore reduce the overall extent of ICRAC activation. If true, then one might expect that the size of ICRAC would increase under these conditions if SERCA pumps were to be blocked since this would suppress store refilling. The results of Fig. 7B show that this was indeed the case. Inclusion of thapsigargin in the recording pipette together with InsP3 and 0.6 mm EGTA significantly increased the size of the current compared with InsP3 and 0.6 mm EGTA alone. Moreover, the delay before the current activated was also reduced by blocking SERCA pumps, although the time to peak was unaffected (Fig. 7C).
Figure 7. SERCA pumps function in the presence of InsP3.
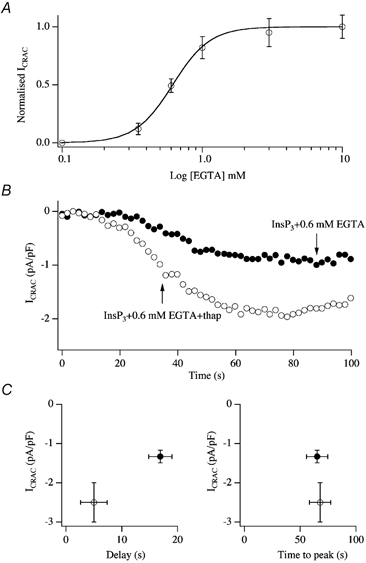
A, the relationship between intrapipette EGTA concentration and amplitude of ICRAC (evoked by dialysis with 30 μm InsP3) is plotted. Note that ICRAC was clearly submaximal in the presence of 0.6 mm EGTA. Each point is the mean ±s.e.m. of at least 5 cells. B, recordings are shown for a cell dialysed with 30 μm InsP3 and 0.6 mm EGTA (•) and for one dialysed with this solution but supplemented with 2 μm thapsigargin, a SERCA blocker (○). Note the increase in the size of ICRAC following inhibition of the pumps. C, the amplitudes, delays and times to peak are summarised for experiments as in B. Filled circles denote InsP3+ 0.6 mm EGTA whereas open cirlces represent InsP3+ 0.6 mm EGTA + thapsigargin.
Collectively, these results demonstrate that SERCA pumps can refill stores, even in the continuous presence of InsP3, and to the extent that ICRAC activates only partially.
InsP3 activates ICRAC in physiological buffer provided SERCA pumps are blocked
The preceding results provide an explanation as to why InsP3 is largely ineffective in activating ICRAC under physiological conditions of weak Ca2+ buffering. The SERCA pumps are so effective that they can prevent InsP3 from depleting the stores sufficiently or long enough for ICRAC to activate. If true, then one would expect InsP3 together with a SERCA pump blocker to activate ICRAC in weak buffer. Figure 8 shows experiments designed to test this (Fierro & Parekh, 2000). Whereas InsP3 was ineffective in weak Ca2+ buffer, the combination of InsP3 and thapsigargin evoked a large current (Fig. 8A). The effects of thapsigargin were mimicked by the different SERCA pump blockers cyclopiazonic acid (Fig. 8B) and thapsigargicin (which is almost as potent as thapsigargin but less hydrophobic). Inhibition of SERCA pumps alone, in the absence of InsP3, also activated ICRAC (Fig. 8B; Fierro & Parekh, 2000). This occurred at a much slower rate than that seen in the presence of InsP3 because store depletion to thapsigargin alone occurs via the background leak pathway.
Figure 8. InsP3 activates ICRAC in weak buffer provided SERCA pump activity is compromised.
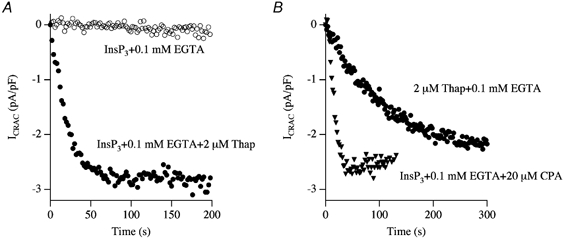
A, InsP3 together with thapsigargin activates ICRAC in weak buffer whereas InsP3 alone is ineffective. B, InsP3 with cyclopiazonic acid, a structurally distinct SERCA pump blocker, is also effective. SERCA pump block alone (2 μm Thap + 0.1 mm EGTA) can also activate ICRAC, albeit at a much slower rate.
In fact, provided SERCA pumps were blocked, ICRAC could activate even in the absence of any exogenous Ca2+ chelator (Fierro & Parekh, 2000). Figure 9 shows the development of ICRAC following dialysis with InsP3, thapsigargin, 100 μm Ca2+ and no added Ca2+ chelator. Ca2+ in the pipette solution was weakly buffered by ATP (2 mm) and glutamate (main anion in the patch pipette). The current activated at the normal rate (trace marked InsP3+ Thap + Ca2+ in Fig. 9A). Dialysis with thapsigargin and 100 μm Ca2+ (in the absence of InsP3) also activated ICRAC, with the typical slow kinetics expected for thapsigargin alone (trace marked Thap + Ca2+). Aggregate data are summarised in Fig. 9B. Although the free Ca2+ concentration in the cells is not known precisely under these conditions (due to exogenous buffering/removal), in experiments with thapsigargin alone the slow development of ICRAC would ensure that reasonable dialysis with micromolar Ca2+ concentrations would have occurred as ICRAC activated. ICRAC can therefore activate when cytosolic Ca2+ is in the micromolar range. Moreover, exogenous Ca2+ chelators are not required for ICRAC to be activated.
Figure 9. SERCA pump block is sufficient to activate ICRAC even in high intracellular Ca2+.
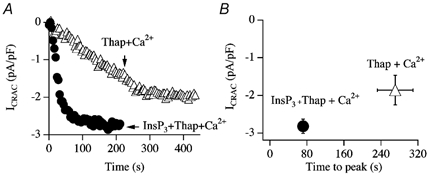
A, InsP3 and thapsigargin can activate ICRAC in weak buffer even in the presence of high intracellular Ca2+ (100 μm total CaCl2 in the pipette solution, free Ca2+ concentration estimated to be in the micromolar range). Thapsigargin alone was also able to evoke ICRAC under these conditions, but after a delay and at a slower rate. B, aggregate data are summarised.
SERCA and secretion
Upon stimulation, mast cells and basophils degranulate, thereby releasing a host of factors into the extracellular medium which are important in shaping a local inflammatory response. We measured secretion from individual RBL-1 cells using the capacitance method to track vesicular fusion (Artalejo et al. 1998). We found that exocytosis required both an increase in Ca2+ and a GTP-dependent step, at least in the whole cell configuration of the patch clamp technique. Strikingly, dialysis with InsP3 in weak Ca2+ buffer failed to evoke any clear secretory response unless thapsigargin was present (Fig. 10). No doubt, part of the effects of thapsigargin are likely to involve reduced Ca2+ removal by SERCA pumps. But it seems likely that Ca2+ entry following the development of ICRAC, as a consequence of exposure to the combination of InsP3 and thapsigargin, will also promote a larger secretory response.
Figure 10. The combination of InsP3 and thapsigargin evokes a robust secretory response in weak buffer, whereas InsP3 alone is largely ineffective.
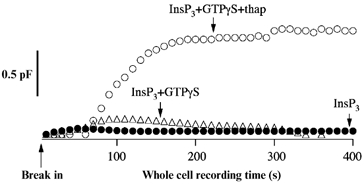
InsP3 alone (•) or in combination with GTPγS (▵) did not trigger a detectable increase in membrane capacitance, whereas InsP3 together with thapsigargin and GTPγS produced a prominent secretory response (○). In these experiments, cells were clamped at −60 mV.
Central role for mitochondria in development of ICRAC
SERCA pump activity therefore appears to be one of the main determinants of whether ICRAC activates or not under physiological conditions. Only when SERCA pump activity is reduced does ICRAC develop. How might SERCA pump activity be reduced under physiological conditions of weak Ca2+ buffering? We reasoned that an increase in the rate of Ca2+ removal from the cytosol by another clearance mechanism might be particularly effective because this would compete effectively with SERCA pumps and hence reduce the rate and extent of store refilling. Stores would therefore empty more and ICRAC should activate. Moreover, enhanced Ca2+ clearance away from the endoplasmic reticulum might also reduce Ca2+-dependent inactivation of InsP3 receptors (Taylor & Traynor, 1995). Combined, this would enable InsP3 to deplete stores sufficiently for ICRAC to activate. We focused on mitochondria for three reasons. First, much recent evidence has established that they can and do take up significant amounts of Ca2+ following cytosolic Ca2+ increases under physiological conditions (Rizzuto et al. 1993, 1998; Jouavaille et al. 1995; Babcock et al. 1997; Hajnoczky et al. 1999; Tinel et al. 1999; Montero et al. 1999; Park et al. 2001; Arnaudeau et al. 2001) and this Ca2+ uptake, which occurs via a relatively low affinity ruthenium red-sensitive uniporter, is driven by the large inner mitochondrial membrane potential (Duchen, 2000). Second, mitochondria are often physically very close to the endoplasmic reticulum (Rizzuto et al. 1998; Park et al. 2001; Csordas & Hajnoczky, 2001), an arrangement that would greatly facilitate interaction between the two Ca2+-handling organelles. Third, mitochondria can take up Ca2+ that has entered via the store-operated pathway (Lawrie et al. 1996; Hoth et al. 1997; Hartmann & Verkhratsky, 1999).
To examine the effects of mitochondria on the ability of InsP3 to activate ICRAC in weak Ca2+ buffer, we dialysed cells with a pipette solution containing weak buffer and no thapsigargin but supplemented with a cocktail (malate, pyruvate, NaH2PO4, cAMP and GTP) known to be important for sustaining respiring mitochondria in the whole cell recording configuration (Gunter & Pfeiffer, 1990; Herrington et al. 1996). The results were dramatic (Fig. 11). InsP3 in the absence of cocktail usually failed to activate any detectable ICRAC (Fig. 11A–C) and, in the small fraction of cells that responded (Fig. 11D), the current was very small. Inclusion of the cocktail had striking effects (Gilabert & Parekh, 2000). A robust ICRAC could now be activated (Fig. 11A–C) and the vast majority of cells responded (Fig. 11D), although the size of the current was smaller than that seen in strong buffer. The effects of the cocktail were prevented by pre-treatment with the combination of antimycin A and oligomycin or by including ruthenium red in the pipette together with InsP3 and cocktail (Fig. 11C). Oligomycin alone was without effect (Glitsch et al. 2002b). Hence mitochondrial Ca2+ uptake is required for InsP3 to activate ICRAC under physiological conditions.
Figure 11. InsP3 activates ICRAC in weak buffer provided mitochondria are energised.
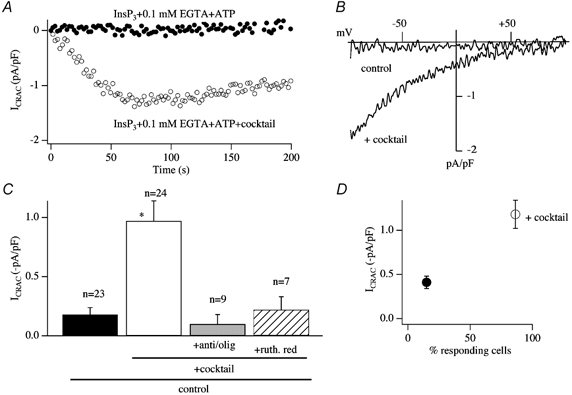
A, time course of activation of ICRAC in the presence of the mitochondrial cocktail (○). ICRAC does not activate in weak Ca2+ buffer in response to InsP3 alone (•). B, steady-state I-V relationships are shown for the cells in A. C, the effects of cocktail are suppressed by preventing mitochondrial Ca2+ uptake by either depolarising mitochondria (pre-treatment with antimycin A (5 μg ml−1) and oligomycin (0.5 μg ml−1)) or inhibiting the uniporter with ruthenium red. D, the presence of cocktail dramatically increases the fraction of cells that respond to InsP3 in weak Ca2+ buffer. *P < 0.01.
The effects of the mitochondrial cocktail solution were Ca2+-dependent, because no enhancing effect was seen in strong Ca2+ buffer (Fig. 12A). Strong Ca2+ buffer prevents a rise in intracellular Ca2+ by chelating Ca2+ both released from the stores and entering the cell, and hence a requirement for mitochondrial Ca2+ uptake would be obviated. Importantly, neither antimycin A and oligomycin nor ruthenium red impaired the ability of ICRAC to activate in strong buffer, arguing against a direct channel blocking action of these drugs (Glitsch et al. 2002b).
Figure 12. Physiological antagonism between SERCA pumps and mitochondria.
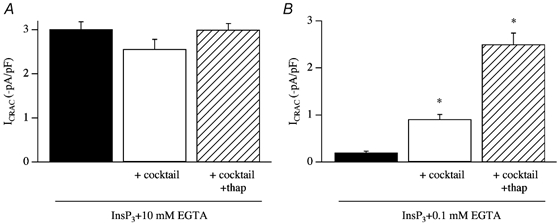
A, the potentiation of ICRAC by cocktail is suppressed by strong intracellular Ca2+ buffer (10 mm EGTA), indicating that the effect is Ca2+-dependent. B, the extent of ICRAC to InsP3 in weak buffer alone, in mitochondrial cocktail and cocktail plus thapsigargin is compared. *P < 0.01.
SERCA pumps compete with mitochondria for removal of cytosolic Ca2+
In the presence of energised mitochondria, the extent of ICRAC following dialysis with InsP3 is only around 35 % the size of that seen in strong buffer. This smaller current in weak buffer could reflect some Ca2+-dependent store refilling, which would imply that not all the Ca2+ that has been released from the stores has been taken up by mitochondria but instead that some has been resequestrated into the stores by the SERCA pumps. To examine this, we measured the size of ICRAC evoked by dialysis with InsP3 in weak buffer in the absence and presence of thapsigargin in the cocktail (Gilabert et al. 2001). Results are summarised in Fig. 12B. Inclusion of thapsigargin resulted in an almost threefold increase in the size of the current, and the amplitude now was similar to that seen in strong buffer. No such potentiating effects were seen when these experiments were repeated in strong buffer (Fig. 12A). The results are consistent with the notion that mitochondria and SERCA pumps compete for removing Ca2+ and this form of physiological antagonism between two major Ca2+ clearance mechanisms, which is particularly acute under physiological conditions of weak intracellular Ca2+ buffering, impacts upon the extent of store depletion and subsequent activation of ICRAC. In RBL cells, there is good morphological evidence documenting a very close apposition between mitochondria and SERCA pumps on the endoplasmic reticulum (Csordas & Hajnoczky, 2001), which would greatly promote competition for Ca2+ removal between these two organelles.
Respiring mitochondria reduce the threshold for activation of ICRAC by InsP3
The previous experiments have all involved supra-maximal concentrations of InsP3. To investigate whether mitochondrial Ca2+ uptake potentiates ICRAC to more moderate levels of InsP3, we compared the size of ICRAC over a range of InsP3 concentrations in the presence and absence of mitochondrial cocktail (Gilabert et al. 2001). The results are summarised in Fig. 13. Whereas 5 μm InsP3 generally failed to activate ICRAC in weak Ca2+ buffer (Fig. 13A and B), it evoked a sizeable current in the presence of the cocktail (Fig. 13A and B). Aggregate data are depicted in Fig. 13C. Comparing the results in the absence and presence of mitochondrial cocktail revealed several striking differences. First, the size of ICRAC was potentiated in cells exposed to mitochondrial cocktail over a range of concentrations of InsP3 (5–30 μm; Fig. 13C). Second, the fraction of cells that responded over this concentration range increased substantially in the presence of energised mitochondria (Fig. 13D). Third, whereas 3 μm InsP3 consistently failed to evoke any current and is therefore a subthreshold concentration in the absence of the cocktail, around half the cells responded to this dose when mitochondria were energised (Fig. 13C and D). Mitochondrial Ca2+ uptake therefore increases the sensitivity of store-operated Ca2+ influx to InsP3, and lowers the threshold concentration of InsP3 that is required to activate ICRAC. Mitochondrial Ca2+ buffering increases the dynamic range of concentrations over which InsP3 is able to function as the physiological messenger that triggers activation of store-operated Ca2+ influx (Gilabert et al. 2001).
Figure 13. Energised mitochondria increase the ability of InsP3 to activate ICRAC in weak Ca2+ buffer.
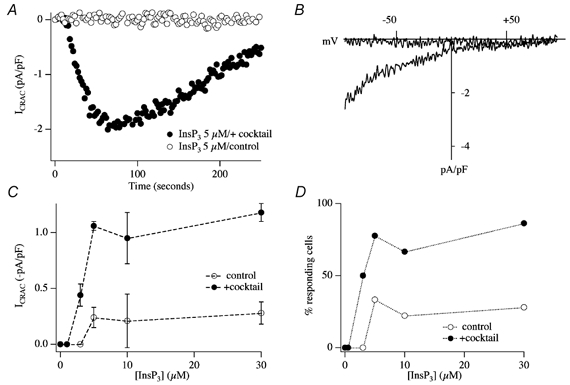
A, whereas 5 μm InsP3 is ineffective in weak buffer (○), it evokes a prominent ICRAC in the presence of energised mitochondria (•). B, I-V relationships for the cells shown in A, taken at steady state. C, concentration-response curve to InsP3 in the absence (○) and presence (•) of cocktail. D, the fraction of cells responding versus concentration of InsP3 is plotted in the absence (○) and presence (•) of energised mitochondria.
Ca2+-dependent inactivation of CRAC channels is reduced by mitochondria
We have identified three mechanisms whereby a rise in cytosolic Ca2+ can inhibit CRAC channel activity. Ca2+-dependent fast inactivation occurs when permeating Ca2+ ions feed back to partially inactivate the channel through which they permeated (Fierro & Parekh, 1999a). Inactivation occurs with time constants of 10 and 100 ms, is largely independent of the macroscopic current (consistent with local feedback) and is more effectively reduced by BAPTA than EGTA, suggesting that the intracellular Ca2+ binding site is within a few nanometres of the pore (Fierro & Parekh, 1999a). This is very similar to fast inactivation first described in mast cells and jurkat T-cells (Hoth & Penner, 1993; Zweifach & Lewis, 1995a). Importantly, fast inactivation is unaffected by either energising mitochondria (Gilabert & Parekh, 2000) or depolarising them (Glitsch et al. 2002b), indicating that mitochondrial Ca2+ uptake does not seem to interfere with this form of Ca2+-dependent inactivation. Two slower Ca2+-dependent regulatory pathways also exist, operating over a time frame of several tens of seconds: Ca2+-dependent store refilling (Bakowski et al. 2001) and Ca2+ entry-dependent but store-independent inactivation (Parekh, 1998). These pathways can be separated by the use of thapsigargin, as was originally described in jurkat T cells (Zweifach & Lewis, 1995b). By inhibiting store refilling, thapsigargin eliminates the store-dependent component and enables the slow Ca2+-dependent inactivation mechanism to be studied in relative isolation, although the latter's contribution is probably exaggerated somewhat by thapsigargin because a major Ca2+ removal mechanism has been inhibited. Figure 14A shows an experiment in which a cell was dialysed with a pipette solution containing InsP3 and thaspigargin in weak Ca2+ buffer. ICRAC activated but then gradually decayed with a half-time of around 100 s, as Ca2+-dependent slow inactivation developed (• in Fig. 14A). Strikingly, if mitochondria were maintained in an energised state by including the cocktail, the extent of this slow inactivation was reduced significantly and the rate at which it developed was slowed (○ in Fig. 14A). Pooled data are summarised in Fig. 14B and C. Blocking mitochondrial Ca2+ uptake with ruthenium red prevented the cocktail from reducing the extent of slow inactivation (Fig. 14B and C). Hence mitochondrial Ca2+ uptake can prolong the time course of store-operated Ca2+ influx by reducing Ca2+-dependent slow inactivation (Gilabert & Parekh, 2000). A similar process has recently been reported in jurkat T cells (Hoth et al. 2000).
Figure 14. Mitochondrial Ca2+ uptake reduces the rate and extent of Ca2+-dependent slow inactivation.
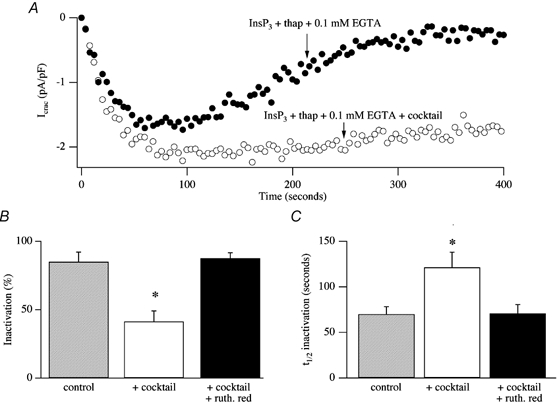
A, dialysis with InsP3 and thapsigargin in weak buffer (control) activates ICRAC but then the current inactivates slowly (•). The extent of inactivation is reduced if mitochondria are energised (○). B and C summarise aggregate data comparing the extent (B) and rate (C) of inactivation of ICRAC in control cells, in those in which cocktail was added to the solution and in cells exposed to cocktail and ruthenium red, to inhibit mitochondrial Ca2+ uptake. *P < 0.01. t1/2, time at which ICRAC had inactivated by 50 %.
Dual actions of mitochondria on ICRAC
It is particularly striking that the same mechanism, Ca2+ uptake by mitochondria, should have such pronounced effects on both the activation and inactivation of ICRAC. By facilitating the ability of InsP3 to activate ICRAC (by promoting more extensive store emptying) and, at the same time, reducing Ca2+-dependent slow inactivation, mitochondria will both increase the extent of ICRAC as well as its duration to a much greater extent than through an action on activation or inactivation alone. A diagram summary depicting this dual action is shown in Fig. 15.
Figure 15. A diagrammatic scheme for the role of mitochondria in controlling ICRAC under physiological conditions of weak intracellular Ca2+ buffering.
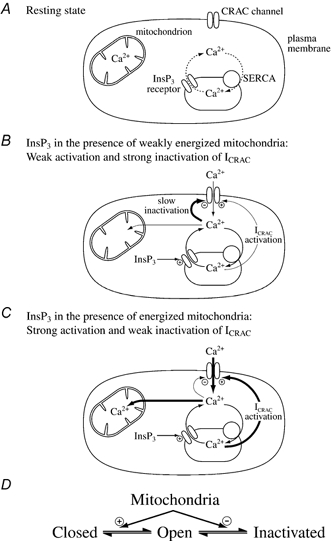
A, the resting state, where ICRAC is not functioning, is shown. Stores are largely full and any Ca2+ that leaks from the stores is taken back up by the SERCA pumps. B, increasing InsP3 levels in the absence of active mitochondrial Ca2+ uptake, releases Ca2+ from the stores. However, the SERCA pumps are able to resequester sufficient Ca2+ so only a very small fraction of CRAC channels are activated (undetectable in whole cell mode). Furthermore, the rise in cytosolic Ca2+ results in Ca2+-dependent slow inactivation of CRAC channels, and possibly InsP3 receptors as well. C, in the presence of respiring mitochondria, InsP3 activates macroscopic ICRAC. Ca2+ released from the stores by InsP3 is taken up by mitochondria. This reduces the amount of Ca2+ available to the SERCA pumps and in the vicinity of open InsP3 receptors such that the stores are depleted sufficiently for macroscopic ICRAC to activate (less refilling by SERCA pumps and less inactivation of InsP3 receptors). Some refilling does occur because inclusion of thapsigargin enhances the size of the current. Furthermore, mitochondrial Ca2+ buffering reduces the rate and extent of Ca2+-dependent slow inactivation, thereby increasing the size and duration of the current. D, a simplified gating scheme for CRAC channels summarising the role of mitochondrial Ca2+ buffering. Mitochondria facilitate opening (Closed to Open transition) whilst, simultaneously, reducing inactivation (Open to Inactivated transition). In this way, mitochondria have a much larger impact on ICRAC than through either transition alone.
Under physiological conditions therefore, mitochondria play a pivotal role in all stages of store-operated Ca2+ entry, determining whether the whole cell current develops or not, to what extent and for how long it stays operational. We are now trying to understand how mitochondrial Ca2+ buffering impacts upon physiologically relevant responses like exocytosis.
Acknowledgments
I am very grateful to the Physiological Society for conferring the Wellcome Prize on me, and to the Wellcome Trust (Career Development Fellowship) and the Lister Institute (Senior Research Fellowship) for their generous support over the years. I would also like to thank members of my group whose work I have presented: Daniel Bakowski, Leonardo Fierro, Juan-Antonio Gilabert and Maike Glitsch. Finally, I would like to express my deep gratitude to Professors Alison Brading (Oxford), Clive Ellory (Oxford), Reinhold Penner (Hawaii), Ole H. Petersen (Liverpool) and Walter Stuehmer (Goettingen) for profoundly shaping my scientific development.
REFERENCES
- Alderton JM, Ahmed SA, Smith LA, Steinhardt RA. Evidence for a vesicle-mediated maintenance of store-operated calcium channels in a human embryonic kidney cell line. Cell Calcium. 2000;28:161–169. doi: 10.1054/ceca.2000.0144. [DOI] [PubMed] [Google Scholar]
- Arnaudeau S, Kelley WL, Walsh JV, Demaurex N. Mitochondria recycle Ca2+ to the endoplasmic reticulum and prevent the depletion of neighbouring endoplasmic reticulum regions. J Biol Chem. 2001;276:29430–29439. doi: 10.1074/jbc.M103274200. [DOI] [PubMed] [Google Scholar]
- Artalejo AR, Ellory JC, Parekh AB. Ca2+ -dependent capacitance increases in rat basophilic leukemia cells following activation of store-operated Ca2+ entry and dialysis with high-Ca2+-containing intracellular solution. Pflugers Arch. 1998;436:934–939. doi: 10.1007/pl00008088. [DOI] [PubMed] [Google Scholar]
- Babcock DF, Herrington J, Goodwin PC, Park YB, Hille B. Mitochondrial participation in the intracellular Ca2+ network. J Cell Biol. 1997;136:833–844. doi: 10.1083/jcb.136.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakowski D, Glitsch MD, Parekh AB. An examination of the secretion-like coupling model for the activation of the Ca2+ release-activated Ca2+ current ICRAC in RBl-1 cells. J Physiol. 2001;532:55–71. doi: 10.1111/j.1469-7793.2001.0055g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakowski D, Parekh AB. Sarcoplasmic/endoplasmic-reticulum-Ca2+-ATPase-mediated Ca2+ reuptake, and not Ins(1,4,5)P3 receptor inactivation, prevents the activation of macroscopic Ca2+ release-activated Ca2+ current in the presence of physiological Ca2+ buffer in rat basophilic leukaemia-1 cells. Biochem J. 2001;353:561–567. doi: 10.1042/0264-6021:3530561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakowski D, Parekh AB. Monovalent cation permeability and Ca2+ block of the store-operated calcium current ICRAC in rat basophilic leukaemia cells. Pflugers Arch. 2002a;443:892–902. doi: 10.1007/s00424-001-0775-8. [DOI] [PubMed] [Google Scholar]
- Bakowski D, Parekh AB. Permeation through store-operated CRAC channels in divalent-free solution: potential problems and implications for putative CRAC channel genes. Cell Calcium. 2002b;32:379–391. doi: 10.1016/s0143416002001914. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Capacitative calcium entry. Biochem J. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad LM, Armstrong DL, Putney JW. Role of the inositol 1,4,5-trisphosphate receptor in Ca2+ feedback inhibition of calcium release-activated calcium current ICRAC. J Biol Chem. 1999;274:32881–32888. doi: 10.1074/jbc.274.46.32881. [DOI] [PubMed] [Google Scholar]
- Burnashev N. Calcium permeability of ligand-gated channels. Cell Calcium. 1998;24:325–332. doi: 10.1016/s0143-4160(98)90056-2. [DOI] [PubMed] [Google Scholar]
- Carafoli E. Calcium signalling: a tale for all seasons. Proc Natl Acad Sci U S A. 2002;99:1115–1122. doi: 10.1073/pnas.032427999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Ann Rev Cell and Developmental Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Calcium signalling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- Csordas G, Hajnoczky G. Sorting of calcium signals at the junctions of endoplasmic reticulum and mitochondria. Cell Calcium. 2001;29:249–262. doi: 10.1054/ceca.2000.0191. [DOI] [PubMed] [Google Scholar]
- Csutora P, Su Z, Kim HY, Bugrim A, Cunningham KW, Nuccitelli R, Keizer JE, Hanley MR, Blalock JE, Marchase RB. Calcium influx factor is synthesized by yeast and mammalian cells depleted of organellar calcium stores. Proc Natl Acad Sci U S A. 1999;96:121–126. doi: 10.1073/pnas.96.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium. 2000;28:339–348. doi: 10.1054/ceca.2000.0170. [DOI] [PubMed] [Google Scholar]
- Fasolato C, Pizzo P, Pozzan T. Delayed activation of the store-operated calcium current induced by calreticulin overexpression in RBL-1 cells. Mol Biol Cell. 1998;9:1513–1522. doi: 10.1091/mbc.9.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro L, Lund P-E, Parekh AB. Comparison of the activation of the Ca2+ release-activated Ca2+ current ICRAC to InsP3 in Jurkat T-lymphocytes, pulmonary artery endothelia and RBL-1 cells. Pflugers Arch. 2000;440:580–587. doi: 10.1007/s004240000336. [DOI] [PubMed] [Google Scholar]
- Fierro L, Parekh AB. Fast calcium-dependent inactivation of calcium release-activated calcium current (CRAC) in RBL-1 cells. J Memb Biol. 1999a;168:9–17. doi: 10.1007/s002329900493. [DOI] [PubMed] [Google Scholar]
- Fierro L, Parekh AB. The effects of interfering with GTP-binding proteins on the activation mechanism of calcium release-activated calcium current. Pflugers Arch. 1999b;437:547–552. doi: 10.1007/s004240050816. [DOI] [PubMed] [Google Scholar]
- Fierro L, Parekh AB. On the characterisation of the mechanism underlying passive activation of the Ca2+ release-activated Ca2+ current ICRAC in rat basophilic leukaemia cells. J Physiol. 1999c;520:407–416. doi: 10.1111/j.1469-7793.1999.00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro L, Parekh AB. Substantial depletion of the intracellular Ca2+ stores is required for macroscopic activation of the Ca2+ release-activated Ca2+ current in rat basophilic leukaemia cells. J Physiol. 2000;522:247–257. doi: 10.1111/j.1469-7793.2000.t01-1-00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilabert J-A, Bakowski D, Parekh AB. Energized mitochondria increase the dynamic range over which inositol 1,4,5-trisphosphate activates store-operated calcium influx. EMBO J. 2001;20:2672–2679. doi: 10.1093/emboj/20.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilabert J-A, Parekh AB. Respiring mitochondria determine the pattern of activation and inactivation of the store-operated Ca2+ current ICRAC. EMBO J. 2000;19:6401–6407. doi: 10.1093/emboj/19.23.6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glitsch MD, Bakowski D, Parekh AB. Activation of the store-operated Ca2+ current ICRAC is compromised by inhibitors of the lipoxygenase family of enzymes. J Physiol. 2002a;539:93–106. doi: 10.1113/jphysiol.2001.012826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glitsch MD, Bakowski D, Parekh AB. Store-operated Ca2+ entry depends on mitochondrial Ca2+ uptake. EMBO J. 2002b;21:6744–6754. doi: 10.1093/emboj/cdf675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glitsch MD, Parekh AB. Ca2+ store dynamics determines the pattern of activation and inactivation of the store-operated Ca2+ current ICRAC in response to InsP3 in rat basophilic leukaemia cells. J Physiol. 2000;523:283–290. doi: 10.1111/j.1469-7793.2000.t01-2-00283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Hager R, Thomas AP. Mitochondria suppress local feedback activation of inositol 1, 4, 5-trisphosphate receptors by Ca2+ J Biol Chem. 1999;274:14157–14162. doi: 10.1074/jbc.274.20.14157. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Verkhratsky A. Relations between intracellular Ca2+ stores and store-operated Ca2+ entry in primary cultured human glioblastoma cells. J Physiol. 1999;513:411–424. doi: 10.1111/j.1469-7793.1998.411bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermosura M, Monteilh-Zoller MK, Scharenberg AM, Penner R, Fleig A. Dissociation of the store-operated calcium current ICRAC and the Mg-nucleotide-regulated metal ion current MagNuM. J Physiol. 2002;539:445–458. doi: 10.1113/jphysiol.2001.013361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington J, Park YB, Babcock D, Hille B. Dominant role of mitochondria in clearance of large calcium loads from rat adrenal chromaffin cells. Neuron. 1996;16:219–228. doi: 10.1016/s0896-6273(00)80038-0. [DOI] [PubMed] [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. 3. Sunderland, MA, USA: Sinauer Associates, Inc.; 2001. [Google Scholar]
- Hoth M. Calcium and barium permeation through calcium release-activated (CRAC) channels. Pflugers Arch. 1995;430:315–322. doi: 10.1007/BF00373905. [DOI] [PubMed] [Google Scholar]
- Hoth M, Button D, Lewis RS. Mitochondrial control of calcium-channel gating: a mechanism for sustained signaling and transcriptional activation in T lymphocytes. Proc Natl Acad Sci U S A. 2000;97:10607–10612. doi: 10.1073/pnas.180143997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, Fanger C, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J Cell Biol. 1997;137:633–648. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. J Physiol. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouaville LS, Ichas F, Holmuhamedov EL, Camacho P, Lechleiter JD. Synchronization of calcium waves by mitochondrial substrates in Xenopus laevis oocytes. Nature. 1995;377:438–411. doi: 10.1038/377438a0. [DOI] [PubMed] [Google Scholar]
- Kiselyov K, Xu X, Mozhayeva G, Kuo T, Pessah I, Mignery G, Zhu X, Birnbaumer L, Muallem S. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature. 1999;396:478–482. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- Lawrie AM, Rizzuto R, Pozzan T, Simpson AW. A role for calcium influx in the regulation of mitochondrial calcium in endothelial cells. J Biol Chem. 1996;271:10753–10759. doi: 10.1074/jbc.271.18.10753. [DOI] [PubMed] [Google Scholar]
- Lewis RS. Store-operated calcium channels. Adv Second Messenger Phosphoprotein Res. 1999;33:279–307. doi: 10.1016/s1040-7952(99)80014-7. [DOI] [PubMed] [Google Scholar]
- Mogami H, Gardner J, Gerasimenko OV, Camello P, Petersen OH, Tepikin AV. Calcium binding capacity of the cytosol and endoplasmic reticulum of mouse pancreatic acinar cells. J Physiol. 1999;518:463–467. doi: 10.1111/j.1469-7793.1999.0463p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogami H, Tepikin AV, Petersen OH. Termination of cytosolic Ca2+ signals: Ca2+ reuptake into intracellular stores is regulated by the free Ca2+ concentration in the store lumen. EMBO J. 1998;17:435–442. doi: 10.1093/emboj/17.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero M, Alonso MT, Carnicero E, Cuchillo-Ibanez I, Albillos A, Garcia AG, Garcia-Sancho J, Alvarez J. Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nature Cell Biol. 1999;2:57–61. doi: 10.1038/35000001. [DOI] [PubMed] [Google Scholar]
- Neher E. The use of Fura 2 for estimating Ca2+ buffers and Ca2+ fluxes. Neuropharm. 1995;34:1423–1442. doi: 10.1016/0028-3908(95)00144-u. [DOI] [PubMed] [Google Scholar]
- Parekh AB. Slow feedback inhibition of calcium release-activated calcium current by calcium entry. J Biol Chem. 1998;273:14925–14932. doi: 10.1074/jbc.273.24.14925. [DOI] [PubMed] [Google Scholar]
- Parekh AB. Calcium signalling and acute pancreatitis: specific response to a promiscuous messenger. Proc Natl Acad Sci U S A. 2000;97:12933–12934. doi: 10.1073/pnas.97.24.12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Fleig A, Penner R. The store-operated calcium current ICRAC: nonlinear activation by InsP3 and dissociation from calcium release. Cell. 1997;89:973–980. doi: 10.1016/s0092-8674(00)80282-2. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Regulation of the store-operated calcium influx in mast cells in organellar ion channels and transporters. In: Clapham D, Ehrlich B, editors. J Gen Physiol. supplement. New York: The Rockerfeller University Press; 1996. [Google Scholar]
- Parekh AB, Penner R. Store depletion and calcium influx. Physiol Revs. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Park MA, Ashby MC, Eddemli G, Petersen OH, Tepikin AV. Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J. 2001;19:5729–5739. doi: 10.1093/emboj/20.8.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partiseti M, Ledeist F, Hivroz C, Fischer A, Korn H, Choquet D. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J Biol Chem. 1994;269:32327–32335. [PubMed] [Google Scholar]
- Patterson RL, Van Rossum DB, Gill DL. Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell. 1999;98:487–499. doi: 10.1016/s0092-8674(00)81977-7. [DOI] [PubMed] [Google Scholar]
- Pozzan T, Rizzuto R, Volpe P, Meldolesi J. Molecular and cellular physiology of intracellular calcium stores. Physiol Revs. 1994;74:595–636. doi: 10.1152/physrev.1994.74.3.595. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Lewis RS. Separation and characterization of currents through store-operated CRAC channels and Mg(2+)-inhibited cation (MIC) channels. J Gen Physiol. 2002;119:487–507. doi: 10.1085/jgp.20028551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney JW. A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Putney JW, McKay RR. Capacitative calcium entry channels. Bioessays. 1999;21:38–46. doi: 10.1002/(SICI)1521-1878(199901)21:1<38::AID-BIES5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Randriamampita C, Tsien RY. Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx (1993) Nature. 1993;364:809–814. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]
- Raraty M, Ward J, Erdemli G, Vaillant C, Neoptolemos JP, Sutton R, Petersen OH. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc Natl Acad Sci U S A. 2000;97:13126–13131. doi: 10.1073/pnas.97.24.13126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LS, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Rosado J, Jenner S, Sage SO. A role for the actin cytoskeleton in the initiation and maintenance of store-mediated calcium entry in human platelets. Evidence for conformational coupling. J Biol Chem. 2000;275:7527–7533. doi: 10.1074/jbc.275.11.7527. [DOI] [PubMed] [Google Scholar]
- Taylor CW. Controlling calcium entry. Cell. 2002;111:767–769. doi: 10.1016/s0092-8674(02)01197-2. [DOI] [PubMed] [Google Scholar]
- Taylor CW, Traynor D. Calcium and inositol trisphosphate receptors. J Memb Biol. 1995;145:109–118. doi: 10.1007/BF00237369. [DOI] [PubMed] [Google Scholar]
- Tinel H, Cancela JM, Mogami H, Gerasimenko JV, Gerasimenko OV, Tepikin AV, Petersen OH. Active mitochondria surrounding the pancreatic acinar granule region prevent spreading of inositol trisphosphate-evoked local cytosolic Ca2+ signals. EMBO J. 1999;18:4999–5008. doi: 10.1093/emboj/18.18.4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Ferrer-Montiel AV, Montal M, Tsien RY. Activation of store-operated Ca2+ current in Xenopus oocytes requires SNAP-25 but not a diffusible messenger. Cell. 1999;98:475–485. doi: 10.1016/s0092-8674(00)81976-5. [DOI] [PubMed] [Google Scholar]
- Yue L, Peng JB, Hediger MA, Clapham DE. CaT1 manifests the pore properties of the calcium-release-activated calcium channel. Nature. 2001;410:705–709. doi: 10.1038/35070596. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Neher E. Mobile and immobile calcium buffers in bovine adrenal chromaffin cells. J Physiol. 1993;469:245–273. doi: 10.1113/jphysiol.1993.sp019813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci U S A. 1993;90:6295–6269. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Rapid inactivation of depletion-activated calcium current (ICRAC) due to local calcium feedback. J Gen Physiol. 1995a;105:209–226. doi: 10.1085/jgp.105.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Slow calcium-dependent inactivation of depletion-activated calcium current. Store-dependent and -independent mechanisms. J Biol Chem. 1995b;270:14445–14551. doi: 10.1074/jbc.270.24.14445. [DOI] [PubMed] [Google Scholar]