

Abstract
Missense mutations of the human skeletal muscle voltage-gated Na+ channel (hSkM1) cause a variety of neuromuscular disorders. The mutation R1448H results in paramyotonia congenita and causes cold-induced myotonia with subsequent paralysis. The mutation M1360V causes an overlapping syndrome with both K+-induced muscle weakness and cold-induced myotonia. The molecular mechanisms of the temperature dependence of these disorders are not well understood. Therefore we investigated physiological parameters of these Na+ channel mutations at different temperatures. Channel proteins were recombinantly expressed in human embryonic kidney cells and studied electrophysiologically, using the whole-cell patch-clamp technique. We compared the wild-type (WT) channel with both mutants at different temperatures. Both mutations had slower inactivation and faster recovery from inactivation compared to WT channels. This effect was more pronounced at the R1448H mutation, leading to a larger depolarization of the cell membrane causing myotonia and paralysis. The voltage dependence of activation of R1448H was shifted to more negative membrane potentials at lower temperature but not at the M1360V mutation or in the WT. The window current by mutation R1448H was increased at lower temperatures. The results of this study may explain the stronger cold-induced clinical symptoms resulting from the R1448H mutation in contrast to the M1360V mutation.
Voltage-gated Na+ channels are membrane spanning proteins responsible for the initiation and propagation of action potentials in nerve and muscle cells. In response to voltage changes during action potentials, the channels open from their closed (resting) state and inactivate during sustained depolarization. For exact regulation of action potentials and normal excitability of nerve and muscle cells the gating processes of opening and closing of these channels are precisely controlled. Defects of gating and/or conductance of voltage-gated channels cause a number of so called ‘channelopathies’ (Hoffman et al. 1995). Among these genetically transmitted disorders are three neurological syndromes caused by naturally occurring mutations in the human skeletal muscle Na+ channel α-subunit: paramyotonia congenita (PC; Ptacek et al. 1992), hyperkalaemic periodic paralysis (HyperPP; Rojas et al. 1991) and K+-aggravated myotonia (PAM; Lerche et al. 1993). To date, more than 25 different mutations are known (Lehmann-Horn & Jurkat-Rott, 1999). The clinical hallmark of PC is paradoxical myotonia, i.e. muscle stiffness increasing with exercise, which is in contrast to the ‘warm-up’ phenomenon observed at the Cl− channel myotonia Thomsen and Becker (Lehmann-Horn & Jurkat-Rott, 1999). HyperPP is characterized by episodic muscle weakness sometimes accompanied by EMG myotonia (myotonic discharge in needle EMG). Weakness usually appears at rest after exercise or by K+ intake. Temperature sensitivity is not a typical sign of HyperPP (Rojas et al. 1991; Heine et al. 1993). PAM is characterized by muscle stiffness induced or aggravated by K+ and shows no paresis (Lerche et al. 1993; Moran et al. 1999).
Voltage-clamp experiments on native samples of muscle biopsies from patients with Na+ channelopathies revealed incomplete inactivation of Na+ channels (Lehmann-Horn et al. 1987b, 1991). Patch-clamp studies of recombined mutant channels expressed in heterologous systems confirmed these results, but also revealed more detailed features of Na+ channel dysfunction, such as slowed inactivation, shortened recovery from inactivation, shifts in the voltage dependence or steady-state inactivation, uncoupling of activation from inactivation and increase of the window current (Cannon et al. 1991; Yang et al. 1994). All these biophysical phenomena result in an enhanced and prolonged Na+ influx into the muscle cell causing sustained depolarization of the cell membrane. The degree of depolarization is supposed to be decisive if symptoms of increased (myotonia) or decreased excitability (weakness) occur.
The Na+ channel disorders are transmitted with autosomal dominant inheritance and involve the Na+ channel gene, SCN4A. The functionally important α-subunit of the Na+ channel is composed of four homologous domains (D1 to D4), each containing six transmembrane segments (S1 to S6) (Lehmann-Horn & Jurkat-Rott, 1999). For the R1448H mutant causing PC, a positively charged arginine in the S4 segment of domain IV of the skeletal muscle Na+ channel α-subunit is substituted by a histidine, which is predominantly neutral at pH 7.4 (Yang et al. 1994). The segment S4 is proposed to serve as the channel voltage sensor (Stuhmer et al. 1989). The mutation R1448H presents a common PC as a clinical phenotype (Meyer et al. 1994). The M1360V mutation is located in the segment S1 of domain IV (Wagner et al. 1997). Although the exact role of this segment in Na+ channel function is not known, there is evidence that this part of the channel determines inactivation (Stuhmer et al. 1989). The mutation M1360V causes an overlapping syndrome of HyperPP and PC, with episodes of flaccid paresis as well as paradoxical myotonia and cold-induced weakness (Wagner et al. 1997).
Our experiments were performed to elucidate the molecular mechanisms of cold sensitivity in PC. Therefore, we measured various parameters of Na+ channel function of wild-type (WT) human voltage-dependent skeletal muscle Na+ channels and two mutant channels at different temperatures (15, 25 and 35 °C) using the whole-cell configuration of the patch-clamp technique. R1448H is a typical PC mutation; M1360V can also result in cold-induced symptoms and its physical location in the channel protein differs from the R1448H mutation and all other mutations (Wagner et al. 1997). In the present study we wanted to clarify the molecular mechanisms underlying the cold sensitivity which characterize the clinical picture of Na+ channel mutations R1448H and M1360V.
METHODS
WT and mutant (R1448H and M1360V) α-subunit constructs of human skeletal muscle Na+ channels were assembled in the mammalian expression vector pRC/CMV and transfected into human embryonic kidney cells (HEK 293) by the calcium phosphate precipitation method. Since transient expression was low (< 10 %) stable cell lines were obtained by antibiotic selection (Mitrovic et al. 1995).
Standard whole-cell patch-clamp recordings (Lehmann-Horn et al. 1987a) were performed at 15, 25 and 35 °C (temperature controller II, Luigs & Neumann, Ratingen, Germany) using an EPC-9 patch-clamp amplifier with ‘Pulse’ software (HEKA, Lambrecht, Germany). Cell dishes were placed between two temperature control elements and a temperature sensor was placed within the solution in the cell dish. Temperature could be regulated with a precision of ± 0.2 °C. By an external circulation system the cell dish was surrounded by water with the respective temperature. The desired temperature was reached within around 5 min.
The voltage error due to series resistance was below 5 %. Leakage and capacitive currents were subtracted automatically by a prepulse protocol (−P/4). All data were low-pass filtered at 5 kHz and sampled at 20 kHz. Analysis was based on HEKA and Excel (Microsoft Corporation) software. The solutions were composed as follows (mm, pH 7.4): pipette: 135 CsCl, 5 NaCl, 2 MgCl2, 5 EGTA and 10 Hepes; bath and control solution: 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 4 dextrose and 5 Hepes.
The Student's t test was applied for statistical analysis using SPSS software. Statistical significance was assumed at a P value < 0.05. All data are presented as means ±s.e.m. The amplitude of Na+ currents in non-transfected cells was always below 0.5 nA (0.25 ± 0.05 nA, n = 8). A maximum peak current in transfected cells was up to 20 nA. To minimize both serial resistance and contribution of endogenous Na+ channels, data were recorded only from cells with currents < 5 nA.
RESULTS
Figure 1 shows superimposed whole-cell Na+ current transients of HEK 293 cells expressing either WT or mutant (R1448H or M1360V) voltage-gated skeletal muscle Na+ channels elicited by 40 ms depolarizing test pulses from −100 to 0 mV at 15 (A), 25 (B) and 35 °C (C). The current reached its peak during activation and decayed due to inactivation. The current decay became slower at lower temperatures, indicating slower inactivation. The slower inactivation was significant for R1448H at all temperatures compared to control. Inactivation of M1360V was slightly, but not statistically significantly, slower than inactivation of WT Na+ channel currents (Fig. 1). Inactivation was always complete, i.e. there was no persistent Na+ current.
Figure 1. Superimposed whole-cell current traces.
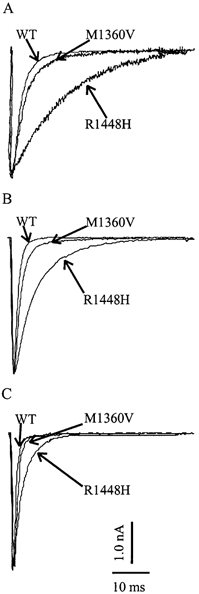
Superimposed representative raw whole-cell current traces for a depolarization from −100 to 0 mV for WT and mutant channels, R1448H and M1360V, at different temperatures: 15 (A), 25 (B) and 35 °C (C).
Time course of inactivation
To determine the time course of inactivation in dependence of temperature, cells were depolarized from −100 mV in a cyclic pulse program with 40 ms test pulses in 15 mV steps. The current decay was best described by a sum of two exponentials yielding two time constants of inactivation, τh1 and τh2, with a relative weight of the faster time constant, τh1, of more than 90 % on the whole current amplitude. Figure 2 shows τh1 (left column) and τh2 (right column) at 15, 25 and 35 °C versus the respective test potential for WT (A), M1360V (B) and R1448H (C). Especially at lower test potentials (e.g. to −45 mV) the cold-induced slowing of inactivation, i.e. the increase of τh1, was significant (at 15°C, τh1= 3.8 ± 0.1 and 3.2 ± 0.3 ms for R1448H and M1360V, respectively), when compared to WT channels (at 15 °C, τh1= 2.5 ± 0.4 ms; P < 0.05). When the cells were depolarized to 0 mV at 15 °C (Fig. 1) R1448H channels inactivated significantly more slowly (τh1= 3.9 ms) compared to M1360V (τh1= 0.8 ms) and WT (τh1= 0.6 ms) (see also Table 1). As can be seen from Fig. 2, M1360V and WT channels showed a monotonic voltage dependence of inactivation at the temperatures tested, whereas it was non-monotonic for R1448H channels. The slower time constant of inactivation (τh2) showed no temperature dependence for WT, M1360V and R1448H. Additionally there was no significant difference of τh2 between WT and M1360V channels; however, τh2 of R1448H was always slower. When the cells were depolarized to 0 mV at 25°C, R1448H channels inactivated significantly slower (τh2= 14.7 ± 2.4 ms) compared to M1360V (τh2= 3.2 ± 0.5 ms) and WT (τh2= 2.9 ± 0.2 ms) (P < 0.05).
Figure 2. The temperature dependency of the time course of inactivation.
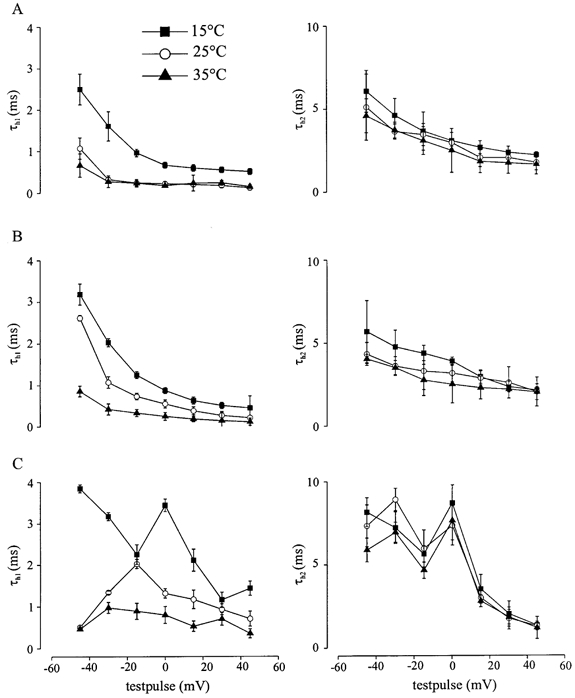
Cells were depolarized from −100 mV in a cyclic pulse program with 40 ms test pulses in 15 mV steps. The current decay was best described by a sum of two exponentials yielding two time constants of inactivation, τh1 and τh2, with a relative weight of the faster time constant, τh1, of more than 90 %. τh1 (left column) and τh2 (right column) at 15, 25 and 35 °C in WT (A), M1360V (B) and R1448H (C).
Table 1.
The midpoint of steady-state of fast inactivation (Inactivation V0.5(mV))and steady-state activation curves (Activation V0.5(mV)), time course of fast inactivation (τh1(ms)) and recovery from inactivation (τrec (ms))
| 35°C | 25°C | 15°C | |
|---|---|---|---|
| Inactivation V0.5 (mV) | |||
| WT | −51.4 ± 0.6 | −51.5 ± 0.4 | −50.7 ± 1.5 |
| MV | −61.8 ± 3.0 | −63.6 ± 3.4 | −61.8 ± 1.3 |
| RH | −64.5 ± 2.9 | −62.3 ± 5.9 | −62.4 ± 1.2 |
| Activation V0.5 (mV) | |||
| WT | −15.0 ± 1.8 | −13.9 ± 0.6 | −15.1 ± 2.8 |
| MV | −14.8 ± 1.9 | −13.8 ± 0.5 | −15.0 ± 3.4 |
| RH | −18.2 ± 1.5 | −21.7 ± 0.9 | −36.3 ± 0.7 |
| τh1 (ms) | |||
| WT | 0.17 ± 0.01 | 0.22 ± 0.01 | 0.66 ± 0.03 |
| MV | 0.25 ± 0.03 | 0.55 ± 0.01 | 0.87 ± 0.05 |
| RH | 0.8 ± 0.2 | 1.32 ± 0.11 | 3.44 ± 1.15 |
| τrec (ms) | |||
| WT | 1.06 ± 0.53 | 1.6 ± 0.59 | 6.45 ± 0.53 |
| MV | 0.72 ± 0.26 | 1.50 ± 0.33 | 3.45 ± 0.46 |
| RH | 0.57 ± 0.3 | 1.19 ± 0.2 | 2.91 ± 0.3 |
The data are means ±s.e.m. for all three channel types and at three temperatures (15,25 and 35 °C).
Steady-state inactivation
To characterize steady-state inactivation, which determines the potential dependence of the probability of Na+ channels being available for activation, a variable 40 ms prepulse ranging from −150 to −5 mV in 5 mV steps was applied prior to a 40 ms test pulse to 0 mV. The steadystate inactivation curve was fitted by the Boltzmann equation (I/Imax= (1 + exp[(V−V0.5)/k])−1). The midpoint of the inactivation curve (V0.5) was shifted for both mutant channels to lower potentials (P < 0.05) compared to WT at all temperatures (Table 1). The temperature, however, had no influence on V0.5 or the slope of the curve at all three channels tested, i.e. the steady-state inactivation curve was temperature-independent (Fig. 3, Table 1). In this study, we investigated the properties of fast inactivation; however the fraction of excitable channel could vary slowly over time due to slow inactivation.
Figure 3. Steady state of fast inactivation.
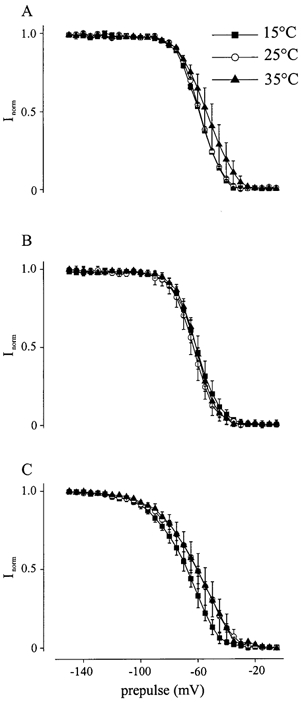
Steady-state of fast inactivation curves of WT (A), M1360V (B) and R1448H (C) at different temperatures (15, 25, and 35 °C). The steady-state inactivation curve was fitted by the Boltzmann equation (I/Imax= (1 + exp[(V−V0.5)/k])−1). The midpoint of the inactivation curve (V0.5) was shifted for both mutant channels to lower potentials (P < 0.05) compared to WT at all temperatures (Table 1). The temperature, however, had no influence on V0.5 or the slope of the curve at all three channels tested.
Steady-state activation
Steady-state activation is a measure for the fraction of channels activated by a depolarization. It was determined by using a series of depolarizing test pulses lasting 15 ms starting at −50 mV and increasing in 5 mV steps after a 30 ms conditioning pulse to −150 mV, from a holding potential of −100 mV. In these experiments the current amplitudes were normalized to the maximal current amplitude of the respective experiment. Mathematical transformation and fitting data points to a standard Boltzmann equation resulted in the steady-state activation curve (conductance G versus test pulse potential). As shown in Fig. 4 and Table 1, the steady-state activation curves and the midpoint of the activation curve (V0.5) were nearly superimposed at 35 °C (Fig. 3C). At 25 °C (Fig. 3B) the activation curve of R1448H (V0.5=−21.68 ± 0.92 mV) was shifted to more negative potentials (P < 0.05), whereas WT (V0.5=−13.88 ± 0.55 mV) and M1360V (V0.5=−13.77 ± 0.51 mV) remained unchanged. This shift of steady-state activation curve for R1448H was more pronounced at 15 °C (V0.5=−36.30 ± 0.68 mV; P < 0.05) while lower temperatures had nearly no effect on WT (V0.5=−14.96 ± 1.80 mV) and M1360V Na+ channel currents (V0.5=−14.77 ± 1.85 mV). There was no temperature dependence on the slopes of the steady-state activation curves of any of the channels tested.
Figure 4. Steady state of activation.
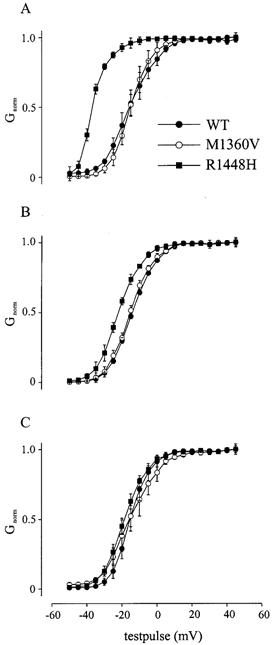
Steady-state of activation curves of WT, M1360V and R1448H at different temperatures: 15 (A), 25 (B) and 35 °C (C). These curves were gained by mathematical transformation of the current-voltage curves to conductance-voltage curves.
Window current
The current flowing in the voltage range where Na+ channels are not inactivated by the mechanism of steady-state inactivation but depolarization is sufficient to open the channels, is determined as the window current. The window current is illustrated by steady-state inactivation and activation diagrams. In Fig. 5, the overlapping of the steady-state inactivation and activation curves was depicted for each of the channel clones. Since steady-state of fast inactivation was temperature-independent for each of the Na+ channels investigated (see above), inactivation curves at all temperatures were averaged to obtain a single curve and depicted with the activation curves at the different temperatures of the different Na+ channels. The window current was temperature-independent for WT (Fig. 5A) and M1360V (Fig. 5B). However, because the activation curve of R1448H Na+ channel currents was shifted to the left at lower temperatures the window current increased (Fig. 5C), resulting in an additional persistent Na+ current.
Figure 5. Window current.
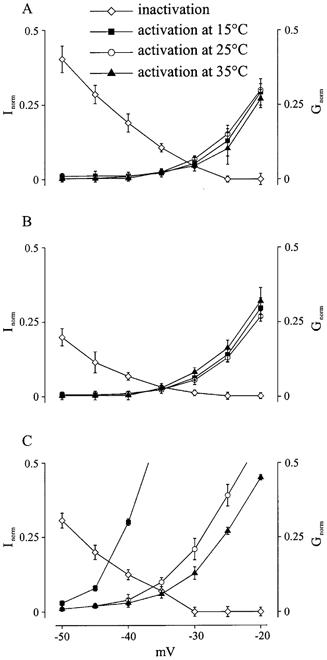
This figure demonstrates the window current for WT (A), M1360V (B) and R1448H (C) at different temperatures (15, 25 and 35 °C). Four curves are plotted in each panel. The only one going downwards is the average curve of steady-state of fast inactivation at the three studied temperatures. It shows the percentage of the normalized current (Inorm) achieved during a depolarization to 0 mV following a previous depolarization from resting potential to variable test potentials plotted on the x-axis. This determines the probability of Na+ channels being non-inactivated and available for activation. The other three curves going upwards are the curves of steady-state of activation at the three studied temperatures. They give the fraction of channels (Gnorm) that are activated by a depolarization with a certain test potential plotted on the x-axis. The overlapping area of these curves gives a range of potential at which there are certain channels being non-inactivated and available for activation. The Na+ channels activated in this way cause a current that is named window current.
Recovery from inactivation
Recovery from inactivation determines the time which is necessary for an inactivated Na+ channel to reach the active state. It therefore limits the maximal firing rate of nerve and muscle cells. Recovery from inactivation was investigated by double pulse experiments. Na+ channels were depolarized by a 36 ms test pulse from −100 to 0 mV followed by a second test pulse with an increasing interval between pulses (steps of 0.5 ms). The time course of recovery from inactivation could be fitted with a single exponential (τrec), which was significantly faster in both mutations (R1448H > M1360V) than in WT at all temperatures (P < 0.05; Table 1). However, the difference between WT and mutations was more distinct at 15 °C (Fig. 6A). At this temperature τrec of R1448H channel currents was 2.91 ± 0.3 ms, τrec of M1360V channel currents was 3.45 ± 0.46 ms and that of WT channel currents 6.45 ± 0.53 ms.
Figure 6. Recovery from inactivation.
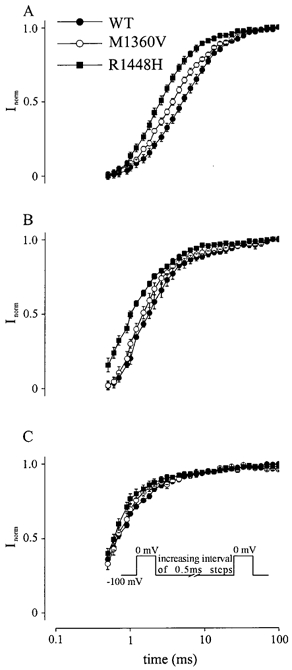
Na+ current amplitudes were measured with double pulse depolarization (see inset) to 0 mV with a variable latency after the first depolarization. The normalized currents Inorm are plotted against the respective latency (time) for WT, M1360V and R1448H at 15 (A), 25 (B) and 35 °C (C).
DISCUSSION
In the present study we have examined the temperature dependence of electrophysiological properties of two mutations of the α-subunit of the human muscle Na+ channel responsible for different channelopathies, and WT Na+ channels. The mutant R1448H Na+ channel causes PC with cold-induced myotonia and subsequent weakness (Meyer et al. 1994), and the mutant M1360V (Stuhmer et al. 1989), an overlapping syndrome of HyperPP and PC with weakness periods after exercise and less marked cold-induced myotonia or weakness (Wagner et al. 1997).
The electrophysiological properties of the mutant M1360V channels have been previously studied at room temperature (Wagner et al. 1997). The time course of inactivation of these mutant channels was slower than that of WT channels, the steady-state inactivation curve was shifted in the hyperpolarizing direction and the time course of recovery from inactivation was faster. There was no difference between M1360V and WT Na+ channels for steady-state activation at room temperature.
Previous investigations of electrophysiological properties of the mutant R1448H channels showed that the time course of inactivation was slower than that of WT channels at all temperatures investigated (Chahine et al. 1994). The steady-state inactivation curve was shifted in the hyperpolarizing direction, the steady-state activation curve showed a slight shift to the hyperpolarizing direction and the time course of recovery from inactivation was faster for the mutant R1448H channels compared to the WT channels at room temperature.
Our studies with WT channels showed no change of the voltage dependencies for activation and fast inactivation. Previous studies on myoballs cultured from biopsies of adult human skeletal muscle (Pröbstle et al. 1988) and rat skeletal muscle Na+ channels (Ruff, 1999) showed no temperature dependence of the voltage dependencies for activation and fast inactivation. In contrast fast inactivation of juvenile muscle Na+ channel, shifted in the hyperpolarizing direction at reduced temperatures, without alteration of the voltage dependence of activation (Pröbstle et al. 1988). In the present study we showed that the steady-state curve of fast inactivation for R1448H and M1360V channels was shifted in the hyperpolarizing direction. This shift was more pronounced for R1448H channels (Table 1). The steady-state properties of inactivation will determine the number of Na+ channels available to open during a depolarization. The left shift of the steady-state inactivation curves of the mutant channels may result in hypoexcitability of the fibre membrane leading to muscle weakness. The data of this study represent the properties of fast inactivation. The slow inactivation impact on the fraction of excitable Na+ channels must also be mentioned. Other studies have shown that the mutation L689I leading to HyperPP causes an impaired slow inactivation (Bendahhou et al. 2002), and that both mutants R1441P and R1441C leading to PC cause slowed deactivation (Featherstone et al. 1998). These biophysical properties are outside the scope of this study, but they might also be differentially affected by changes in temperature.
The time course of inactivation (τh1) of both mutant channels was slower than that of WT channels at all temperatures investigated (Fig. 1 and 2, Table 1). This was more pronounced for R1448H channels. We demonstrated that the time course of recovery from inactivation got slower with cooling at each channel type. The recovery from inactivation was faster for R1448H channels at every temperature (Fig. 6, Table 1). The decreased rate of inactivation and an increased rate of recovery from inactivation would increase excitability during and following action potentials.
We showed that the activation of mutant M1360V and WT channels was temperature-independent. However, the steady-state activation curve of mutant R1448H channels was significantly shifted to hyperpolarized potentials at low temperature (Fig. 4, Table 1). Previous studies demonstrated (Stuhmer et al. 1989) that a positive shift of the activation curve is expected from neutralization of positive charges near the intracellular surface of the membrane, whereas a negative shift is expected from neutralization of positive charges near the extracellular surface. The mutant R1448H is placed on domain IV segment S4 (Lehmann-Horn & Jurkat-Rott, 1999). This causes the exchange of a positively charged residue at the voltage sensor on domain IV near the extracellular surface of the membrane by the neutral amino acid histidine. We demonstrated that this neutralization on the S4 segment caused a cold-induced shift of the activation to more negative membrane potentials.
It is also supposed that the shift of the activation curve can be the result of slower inactivation and not a real change of activation (Chahine et al. 1994). In our experiments the inactivation was slower at lower temperatures for all three channel types, but only the mutation R1448H showed a cold-induced left shift of the activation curve. Therefore we propose that the change of activation of R1448H Na+ channels is not only the result of slower inactivation at low temperature.
The cold-induced left shift of the activation curve of R1448H channels also resulted in an increase of the window current at lower temperatures (Fig. 5). The increase of the window current results in an increased inward Na+ current, causing an increase of the excitability and a subsequent paralysis.
In the current study we demonstrated for the first time the effects of lower temperature on mutant M1360V channels and also investigated the mutant R1448H channels. Summarizing our results, compared with WT Na+ channels both mutants showed slower time course of inactivation (τh1) and faster recovery from inactivation which resulted in a larger depolarization of the cell membrane leading to myotonia. Additionally the mutant R1448H channels showed a cold-induced shift of the activation to more negative membrane potentials and an increase of window current resulting in an increase of the excitability and a subsequent paralysis. This additional property of the mutant R1448H channels was missing in mutant M1360V channels.
The shift of the steady-state activation curve to hyperpolarized potentials at lower temperatures, the resulting increase of the window current, slower inactivation and faster recovery from inactivation may explain the pronounced clinical symptoms of R1448H muscle in a cold environment in contrast to M1360V.
Acknowledgments
This study was supported by grants of the Deutsche Forschungsgemeinschaft (Bu 938/2–3) and of the Medical School Hannover.
REFERENCES
- Bendahhou S, Cummins TR, Kula RW, Fu YH, Ptacek LJ. Impairment of slow inactivation as a common mechanism for periodic paralysis in DIIS4-S5. Neurology. 2002;58:1266–1272. doi: 10.1212/wnl.58.8.1266. [DOI] [PubMed] [Google Scholar]
- Cannon SC, Brown RH, Corey DP. A sodium channel defect in hyperkalemic periodic paralysis: potassium-induced failure of inactivation. Neuron. 1991;6:619–626. doi: 10.1016/0896-6273(91)90064-7. [DOI] [PubMed] [Google Scholar]
- Chahine M, George AL, Zhou M, Ji S, Sun W, Barchi RL, Horn R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Featherstone DE, Fujimoto E, Ruben PC. A defect in skeletal muscle sodium channel deactivation exacerbates hyperexcitability in human paramyotonia congenita. J Physiol. 1998;506:627–638. doi: 10.1111/j.1469-7793.1998.627bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine R, Pika U, Lehmann-Horn F. A novel SCN4A mutation causing myotonia aggravated by cold and potassium. Hum Mol Genet. 1993;2:1349–1353. doi: 10.1093/hmg/2.9.1349. [DOI] [PubMed] [Google Scholar]
- Hoffman EP, Lehmann-Horn F, Rüdel R. Overexcited or inactive: ion channels in muscle diseases. Cell. 1995;80:681–686. doi: 10.1016/0092-8674(95)90345-3. [DOI] [PubMed] [Google Scholar]
- Lehmann-Horn F, Iaizzo PA, Hatt H, Franke C. Altered gating and conductance of Na+ channels in hyperkalemic periodic paralysis. Pflugers Arch. 1991;418:297–299. doi: 10.1007/BF00370530. [DOI] [PubMed] [Google Scholar]
- Lehmann-Horn F, Jurkat-Rott K. Voltage-gated ion channels and hereditary disease. Physiol Rev. 1999;79:1317–1371. doi: 10.1152/physrev.1999.79.4.1317. [DOI] [PubMed] [Google Scholar]
- Lehmann-Horn F, Kuther G, Ricker K, Grafe P, Ballanyi K, Rudel R. Adynamia episodica hereditaria with myotonia: a non-inactivating sodium current and the effect of extracellular pH. Muscle Nerve. 1987a;10:363–374. doi: 10.1002/mus.880100414. [DOI] [PubMed] [Google Scholar]
- Lehmann-Horn F, Rudel R, Ricker K. Membrane defects in paramyotonia congenita (Eulenburg) Muscle Nerve. 1987b;10:633–641. doi: 10.1002/mus.880100709. [DOI] [PubMed] [Google Scholar]
- Lerche H, Heine R, Pika U, George AL, Mitrovic N, Browatzki M, Weiss T, Bastide-Rivet M, Franke C, Lomonaco M, Ricker K, Lehmann-Horn F. Human sodium channel myotonia: slowed channel inactivation due to substitutions for a glycine within the III/IV linker. J Physiol. 1993;470:13–22. doi: 10.1113/jphysiol.1993.sp019843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KC, Otto M, Zoll B, Koch MC. Molecular and genetic characterisation of German families with paramyotonia congenita and demonstration of founder effect in the Ravensberg families. Hum Genet. 1994;93:707–710. doi: 10.1007/BF00201577. [DOI] [PubMed] [Google Scholar]
- Mitrovic N, George AL, Lerche H, Wagner S, Fahlke C, Lehmann-Horn F. Different effects on gating of three myotonia-causing mutations in the inactivation gate of the human muscle sodium channel. J Physiol. 1995;487:107–114. doi: 10.1113/jphysiol.1995.sp020864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran O, Nizzari M, Conti F. Myopathic mutations affect differently the inactivation of the two gating modes of sodium channels. J Bioenerg Biomembr. 1999;31:591–608. doi: 10.1023/a:1005473129183. [DOI] [PubMed] [Google Scholar]
- Probstle T, Rudel R, Ruppersberg JP. Hodgkin-Huxley parameters of the sodium channels in human myoballs. Pflugers Arch. 1988;412:264–269. doi: 10.1007/BF00582507. [DOI] [PubMed] [Google Scholar]
- Ptacek LJ, George AL, Jr, Barchi RL, Griggs RC, Riggs JE, Robertson M, Leppert MF. Mutations in an S4 segment of the adult skeletal muscle sodium channel cause paramyotonia congenita. Neuron. 1992;8:891–897. doi: 10.1016/0896-6273(92)90203-p. [DOI] [PubMed] [Google Scholar]
- Rojas CV, Wang J, Schwartz L, Hoffman EP, Powell BR, Brown RH., Jr A Met-to-Val mutation in the skeletal muscle sodium channel α-subunit in hyperkalemic periodic paralysis. Nature. 1991;354:387–389. doi: 10.1038/354387a0. [DOI] [PubMed] [Google Scholar]
- Ruff RL. Effects of temperature on slow and fast inactivation of rat skeletal muscle Na(+) channels. Am J Physiol. 1999;277:C937–947. doi: 10.1152/ajpcell.1999.277.5.C937. [DOI] [PubMed] [Google Scholar]
- Stuhmer W, Conti F, Suzuki H, Wang X, Noda M, Yahagi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- Wagner S, Lerche H, Mitrovic N, Heine R, George AL, Lehmann-Horn F. A novel sodium channel mutation causing a hyperkalemic paralytic and paramyotonic syndrome with variable clinical expressivity. Neurology. 1997;49:1018–1025. doi: 10.1212/wnl.49.4.1018. [DOI] [PubMed] [Google Scholar]
- Yang N, Ji S, Zhou M, Ptacek LJ, Barchi RL, Horn R, George AL., Jr Sodium channel mutations in paramyotonia congenita exhibit similar biophysical phenotypes in vitro. Proc Natl Acad Sci U S A. 1994;91:12785–12789. doi: 10.1073/pnas.91.26.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]