

Abstract
The biochemical cascade linking activation of phospholipase C-coupled thyrotropin-releasing hormone (TRH) receptors to rat ERG (r-ERG) channel modulation was studied in situ using perforated-patch clamped adenohypophysial GH3 cells and pharmacological inhibitors. To check the recent suggestion that Rho kinase is involved in the TRH-induced r-ERG current suppression, the hormonal effects were studied in cells pretreated with the Rho kinase inhibitors Y-27632 and HA-1077. The TRH-induced r-ERG inhibition was not significantly modified in the presence of the inhibitors. Surprisingly, the hormonal effects became irreversible in the presence of HA-1077 but not in the presence of the more potent Rho kinase inhibitor Y-27632. Further experiments indicated that the effect of HA-1077 correlated with its ability to inhibit protein kinase C (PKC). The hormonal effects also became irreversible in cells in which PKC activity was selectively impaired with GF109203X, Gö6976 or long-term incubation with phorbol esters. Furthermore, the reversal of the effects of TRH, but not its ability to suppress r-ERG currents, was blocked if diacylglycerol generation was prevented by blocking phospholipase C activity with U-73122. Our results suggest that a pathway involving an as yet unidentified protein kinase is the main cause of r-ERG inhibition in perforated-patch clamped GH3 cells. Furthermore, they demonstrate that although not necessary to trigger the ERG current reductions induced by TRH, an intracellular signal cascade involving phosphatidylinositol-4,5-bisphosphate hydrolysis by phospholipase C, activation of an α/βII conventional PKC and one or more dephosphorylation steps catalysed by protein phosphatase 2A, mediates recovery of ERG currents following TRH withdrawal.
Abundant evidence accumulated over more than a decade indicates that regulation of ether-à-go-go-related (ERG) channel activity by the hypothalamic neuropeptide thyrotropin-releasing hormone (TRH) constitutes an important point of control of electrical activity (and hence of intracellular Ca2+ levels and the secretory process) in anterior pituitary cells (Barros et al. 1994, 1997; Weinsberg et al. 1997; Bauer, 1998; Bauer et al. 1998, 1999). ERG channels have also been recognized as important determinants of action potential characteristics in heart muscle (Keating & Sanguinetti, 2001), neurones (Chiesa et al. 1997) and other cell types (Lecchi et al. 2002; Rosati et al. 2000). Thus inhibition of human ERG (HERG) channel activity by inherited mutations or prescribed drugs causes prolongation of the QT interval on the surface electrocardiogram, associated with an increased risk of cardiac arrhythmia and sudden death (Chiang & Roden, 2000; Keating & Sanguinetti, 2001). ERG channels have also been proposed as important determinants of neuritogenesis and differentiation in neuronal cells (Arcangeli et al. 1993, 1997), ontogeny and potassium homeostasis in glia (Zhou et al. 1998; Emmi et al. 2000), and maintenance of the neoplastic phenotype in cancer cells (Bianchi et al. 1998; Smith et al. 2002).
In spite of the physiological and pathological relevance of ERG channels, the mechanisms of regulation by different physiological agents in neuronal and cardiac cells are largely unknown. In native lactotrophs and clonal GH adenohypophysial cells, ERG currents are suppressed by activation of the G protein-coupled TRH receptor (Bauer et al. 1990, 1994; Barros et al. 1992, 1993; Schäfer et al. 1999; Schledermann et al. 2001). The demonstration that the TRH effect is diminished by loading the cell with an ATP analogue that cannot donate its γ-phosphate in phosphorylation reactions and the irreversibility of the TRH effect caused by the specific protein phosphatase inhibitor okadaic acid strongly suggest that, at least in GH3 cells, one or more phosphorylation/dephosphorylation steps participate in the biochemical cascade linking the TRH receptor to rat-ERG (r-ERG) channel inhibition (Barros et al. 1992, 1993). Whether this phosphorylation takes place on the channel itself, on a regulatory subunit or on a component of the coupling cascade remains to be established. Furthermore, protein phosphatase 2A (PP2A) seems to be involved in the reversal of the inhibitory effects caused by TRH (Barros et al. 1992, 1993), but the identity of the protein kinase(s) leading to r-ERG current reduction is unknown.
The inhibitory modulation of heterologously expressed HERG channels in oocytes by protein kinase C (PKC; Barros et al. 1998) or A (PKA; Kiehn, 2000) has recently been reported. Enhancement of endogenous ERG currents by PKC in guinea-pig ventriculocytes following stimulation of β-adrenergic receptors coupled to cyclic-AMP production and PKA activation (Heath & Terrar, 2000), and opposite effects of cyclic-AMP analogues following activation of PKA or direct binding of the nucleotide to HERG channels expressed in Chinese hamster ovary (CHO) cells (Cui et al. 2000) have also been reported. Unfortunately, these results cannot explain the physiological mechanism of ERG current regulation by TRH in pituitary cells. The TRH receptor is coupled to a G protein of the Gq/11 family resulting in phospholipase C (PLC) activation and the generation of myo-inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) from phosphatidylinositol 4,5-bisphosphate (PIP2). It is also known that TRH is able to activate several PKC isozymes in GH3 cells (Kiley et al. 1991; Akita et al. 1994). However, TRH-induced reduction of ERG current in these cells does not depend on PKC or PKA activation (Bauer et al. 1990, 1994; Barros et al. 1992, 1993; Schäfer et al. 1999; Schledermann et al. 2001). Pharmacological evidence also suggests that activation of mitogen-activated protein (MAP) or tyrosine kinases, arachidonic acid liberation, NO production and actin reorganization do not appear to be involved in the TRH-induced reduction of ERG current in pituitary cells (Schledermann et al. 2001).
An alternative pathway for ERG regulation by TRH involving a G13- and Rho-mediated signalling cascade has recently been described (Storey et al. 2002). Based on the known ability of Rho to act through Ser/Thr protein kinases and the aforementioned requirement of Ser/Thr-directed PP2A for reversal of TRH effects on ERG channels, it has been postulated that Rho kinases are involved in ERG regulation by TRH receptors or other G13- and Rho-coupled receptors.
In this study we used a pharmacological approach to check the possible involvement of Rho kinases (ROCK-I/II or ROK-α/β) in the TRH-induced modification of endogenous ERG current in situ using perforated-patch clamped GH3 cells. Treatment of the cells with the ROCK inhibitors HA-1077 and Y-27632 (Davies et al. 2000; Nagumo et al. 2000) did not modify the TRH-induced current reductions. Surprisingly, the reductions became essentially irreversible in the presence of HA-1077 but not Y-27632 in spite of the considerably higher potency of Y-27632 as a ROCK inhibitor. Further studies indicated that the effect of HA-1077 correlated with its ability to inhibit PKC and that TRH-induced inhibition also became irreversible in response to other treatments that impair the activity of conventional PKC isozymes. Our results demonstrate that although PLC activity is not necessary for TRH-induced ERG current reduction, an intracellular signal cascade involving PIP2 hydrolysis by PLC, activation of a conventional PKC and one or more dephosphorylation steps catalysed by PP2A, participates in the reversal of ERG current inhibition following TRH withdrawal.
METHODS
Cell culture
GH3 rat anterior pituitary cells (ATCC-CCL 82.1) were plated in 35 mm diameter plastic tissue culture dishes containing sterile glass coverslips coated with poly-l-lysine, and grown at 37 °C in a humidified atmosphere of 95 % air and 5 % CO2. The culture medium consisted of Dulbecco's modified Eagle's medium + nutrient mixture F-12 Ham (DME/F12 1:1 mixture, Sigma) supplemented with 100 U ml−1 penicillin, 1.1 mg ml−1 streptomycin and a serum mixture of 15 % horse serum and 2.5 % fetal bovine serum. The coverslips constituted the bottom of a small recording chamber (0.2–0.3 ml) that was continuously perfused with saline at a rate of about 1 ml min−1.
Electrophysiological recordings, solutions and data analysis
Current recordings were performed at room temperature with the perforated-patch variant of the patch-clamp technique as previously described (Barros et al. 1991, 1992, 1994, 1997). Electrodes were fabricated from borosilicate or kimax disposable micropipettes (Boralex, Rochester Scientific, Rochester, NY, USA; Fisherbrand, Fisher Scientific, Pittsburg, PA, USA or Kimble Glass Inc., Vineland, NJ, USA). Electrode resistance was 2–5 MΩ when filled with the pipette solution containing (mm): 65 KCl, 30 K2SO4, 10 NaCl, 1 MgCl2, 50 sucrose and 10 Hepes (pH 7.4 with KOH). The tip of the pipette was initially filled with nystatin-free solution and the remainder of the pipette was back-filled with the same solution supplemented with 250 μg ml−1 nystatin, added from a stock of 50 mg ml−1 nystatin dissolved in dimethylsulphoxide (DMSO). This solution was sonicated just before use. The course of perforation was followed by monitoring the progress of capacitive transients under voltage-clamp mode, setting the pipette voltage to a value of −70 mV. Access resistance, as estimated from the capacitive compensation circuitry on the amplifier, reached 10–30 MΩ within 5–20 min after the seal was made. Solution junction potentials were nulled before seal formation. Once patch permeabilization reached the indicated levels, the extracellular solution was changed as detailed below and the cell was voltage clamped at the desired holding potential. An EPC-7 patch-clamp amplifier (HEKA Elektronic, Lambrecht, Germany) was used to record membrane currents. Stimulation, data acquisition and analysis were carried out using Pulse+PulseFit software (HEKA Elektronic) running on Macintosh computers. Current records were sampled every 1 ms and digitally filtered at 500 Hz. Data are shown without correction for leakage and capacitive transients. Further data processing was performed with PulseFit and Igor-Pro (WaveMetrics, Lake Oswego, OR, USA).
The standard extracellular saline used for perforation and monitoring of intracellular Ca2+ concentration ([Ca2+]i) contained (mm): 137 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose and 10 Hepes (pH 7.4 with NaOH). Recordings of r-ERG currents were performed after changing the extracellular medium to a high-K+, Ca2+-free solution once permeabilization of the patches was complete. This solution contained (mm): 140 KCl, 4 MgCl2, 10 EGTA and 10 Hepes (pH 7.4 with KOH). Inward currents were studied during hyperpolarization pulses to −100 mV from a holding potential of −10 mV. The hyperpolarization pulses were preceded by a 100 ms ramp from 0 to −50 mV, which yields an estimation of the membrane conductance within this voltage range and would tend to potentiate the otherwise voltage-dependent effect of TRH (Bauer et al. 1990; Barros et al. 1992, 1994, 1997). To prevent variations due to differences in deactivation rates from cell to cell, the magnitude of the inward currents was estimated with the PulseFit software as the total inward charge computed between cursors located at 0.5 and 100 % duration of the hyperpolarization pulses. Data are presented as means ±s.e.m. with the number of cells indicated by n and the significance level after Student's t test indicated by P.
Intracellular calcium measurements
Measurements of [Ca2+]i were performed in cells grown as above. In this case the coverslips were transferred to wells containing standard extracellular saline plus 5 μm Fura-2 AM (Molecular Probes) and loaded with the dye for about 60 min at room temperature. Cells were then washed with saline to remove non-hydrolysed Fura-2 AM. Fluorescence measurements were performed using a Zeiss Axiovert 100 microscope equipped with epifluorescence accessories and attached to a photometric set-up (Till Photonics GmbH, Planegg, Germany). Control of the monochromator and fluorescence recordings were performed with the X-Chart program with Fura extension (HEKA Elektronic) running on Macintosh computers. Cells were alternately exposed for 40 ms to excitation light of 360 and 390 nm during the experiment. The fluorescence emission at 510 nm was measured with a photomultiplier tube over the repetitive periods of 100 ms used to integrate and sample the individual data points. Light collection was limited to a spot of diameter equivalent to about the size of a single cell by inserting a pinhole before the photomultiplier in the video port of the microscope. The ratio of the emission intensities (360 nm/390 nm) was used as a measure of changes in intracellular Ca2+ levels. Ca2+ concentration was estimated from the fluorescence ratio by comparison with Fura-2 standards (Barros et al. 1994).
Chemicals
TRH, phorbol ester (4β-phorbol 12-myristate 13-acetate, PMA), U-73122, wortmannin and nystatin were purchased from Sigma. E-4031 was from Alomone Labs. GF109203X, Gö6976 and HA-1077 were purchased from Calbiochem. Fura-2 and Fura-2 AM were from Molecular Probes. Y-27632 was from Tocris. Stock solutions of Fura-2 AM, PMA, GF109203X, U-73122, Gö6976 and wortmannin were prepared in DMSO. The final DMSO concentration never exceeded 0.1 %. All other substances were dissolved in water. Final dilutions were made with external solutions. For long-term treatments, drugs (Y-27632, HA-1077, wortmannin or PMA) were directly added to complete culture medium and preincubation proceeded at 37 °C in the incubator. In these cases, the inhibitors were also present in the extracellular solutions used for electrophysiological recordings.
RESULTS
Specificity of TRH effects on E-4031-sensitive currents under the experimental conditions used to record r-ERG in GH3 cells
The main goal of our study was to investigate the biochemical cascade linking TRH receptor activation to r-ERG channel regulation in rat adenohypophysial GH3 cells. For this purpose we used perforated-patch conditions and a pharmacological approach to maintain the intracellular components of the hormonal response as unaltered as possible and to inhibit individual steps of the cascade. This not only preserves intact the physiological electrical response to hypothalamic neuropeptides (Barros et al. 1991), but also it is necessary to allow observation of the reversibility of the r-ERG current reductions induced by TRH, both of which are minimized by the intracellular dialysis caused by conventional whole-cell conditions (Barros et al. 1991, 1993; Schledermann et al. 2001). Due to the fast inactivation of ERG channels at depolarized potentials and the presence of several outwardly rectifying voltage- and calcium-dependent K+ currents in GH3 cells, currents were studied at negative potentials with established voltage protocols and using high-K+, low-Ca2+ extracellular solutions designed to increase the amplitude of the inwardly rectifying r-ERG currents and to reduce Ca2+ currents and activation of Ca2+-dependent K+ currents (Bauer et al. 1990, 1999; Barros et al. 1992, 1997; Weinsberg et al. 1997; see also Methods). However, even under these circumstances, inward currents at negative potentials are not exclusively carried by ERG channels, but have two different components: one that is fully blocked by micromolar concentrations of the specific ERG channel blocker E-4031 (Weinsberg et al. 1997) and the other which is E-4031 resistant and is independent of ERG operation. This second component is mainly due to deactivation of non-inactivating currents present at depolarized holding potentials (Weinsberg et al. 1997; Schäfer et al. 1999) and to reopening before closing of outwardly rectifying K+ channels inactivated at these potentials (e.g. Kv1.4 currents; Ruppersberg et al. 1991).
As exemplified by the two cells shown in Fig. 1A and B, the relative amount of E-4031-sensitive and -insensitive inward current components greatly varied from cell to cell. Since it has been reported that several K+ currents can be inhibited by TRH in GH3 cells (Dubinsky & Oxford, 1985; Barros et al. 1991; Simasko, 1991), we checked whether under our experimental conditions the hormonal effects were predominantly exerted on the E-4031-sensitive r-ERG current component. As indicated below, this is important to allow quantification of the extent of the TRH-induced reduction of r-ERG current and to ensure that the reversibility of such a reduction is due to the recovery of ERG channel function and not to the reversal of the effects of TRH on other currents. Application of TRH in the presence of 5 μm E-4031 (a concentration that totally blocks the r-ERG current in GH3 cells; see Weinsberg et al. 1997 and Schäfer et al. 1999) left the E-4031-resistant inward currents practically unaltered during the hyperpolarization steps (Fig. 1A and B). This demonstrates that regardless of the presence of additional currents at negative voltages, the TRH-induced reductions were exerted mostly, if not exclusively, on the E-4031-sensitive r-ERG current. It also indicates that a quantification of the hormonal effects on r-ERG can be performed by subtracting the current remaining in the presence of E-4031 from that elicited in control conditions and comparing it with the difference between the TRH- and E-4031-resistant currents.
Figure 1. E-4031-insensitive currents are not a main target for TRH in perforated-patch clamped GH3 cells studied in high-K+, low-Ca2+ extracellular solutions.
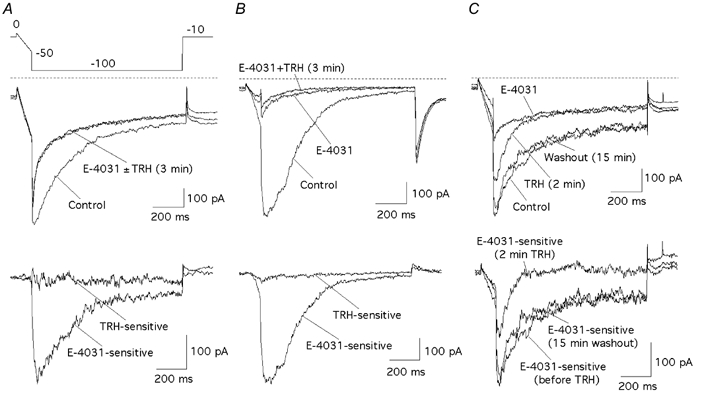
Current recordings from two representative cells showing high and low proportions of E-4031-insensitive inward current components are shown in A and B. The E-4031 sensitivity of currents recovered following TRH-induced inhibition after a prolonged period of TRH washout is illustrated in C. Currents were recorded in response to the voltage protocol shown in A. A and B, upper panel, currents under control conditions, after blockade of r-ERG channels with 5 μm E-4031, and 3 min after adding 100 nm TRH in the continuous presence of the inhibitor. Lower panel, E-4031-sensitive and TRH-sensitive currents obtained by subtracting the traces in the presence of E-4031 or TRH + E-4031 from control currents. C, current traces in the upper panel correspond to those recorded before adding TRH (Control), after 2 min of TRH treatment, following a 15 min period of washout with hormone-free medium, and in the presence of 5 μm E-4031. E-4031-sensitive currents obtained by subtracting the trace in the presence of E-4031 from those without inhibitor are shown in the lower panel. Zero current level is indicated with dashed lines on the non-subtracted current records.
Under perforated-patch conditions, the TRH-induced inhibition of the hyperpolarization-evoked current was reversed following a long period of hormone washout (Fig. 1C; see also below). As an additional control, we also checked the effect of E-4031 following a washout period of 15 min in which the current returned to levels similar to those observed before adding TRH. As shown in Fig. 1C, the recovered current was readily blocked by the inhibitor. This indicates that the increased current is not due to the delayed appearance of a new conductance in response to TRH, but that it is carried by E-4031-sensitive r-ERG channels that have recovered from the TRH-induced inhibition upon hormone washout.
Effect of ROCK inhibitors on TRH-induced reduction of r-ERG current
We took advantage of the specific blockade exerted by the class III antiarrhythmics (e.g. E-4031, dofetilide and WAY123, 298; Roden, 1993; Weinsberg et al. 1997) on ERG channels to pharmacologically isolate the E-4031-sensitive r-ERG current present in GH3 cells and to quantify the TRH-induced inhibition. Addition of TRH was immediately followed by a transient increase in holding and voltage-evoked currents that generally lasted for 15–60 s (1–3 pulses at a frequency of 1 pulse delivered to the cell every 15 or 20 s). Such an increase results from transient activation of Ca2+-dependent K+ channels due to massive liberation of Ca2+ from intracellular stores (Bauer et al. 1990; Barros et al. 1992). This initial phase was followed by a strong reduction of the inward r-ERG current in almost every cell tested that reached a maximum within 1–3 min after adding TRH (Fig. 2A and D). To quantify the extent of the TRH-induced r-ERG reduction, the remaining E-4031-resistant current was subtracted from the current obtained without the blocker both before and after adding TRH (see above). This yielded a value of 73.1 ± 6.8 % (n = 7) for the TRH-induced reduction of the E-4031-sensitive current.
Figure 2. TRH-induced r-ERG current suppression is not significantly modified by ROCK inhibitors.
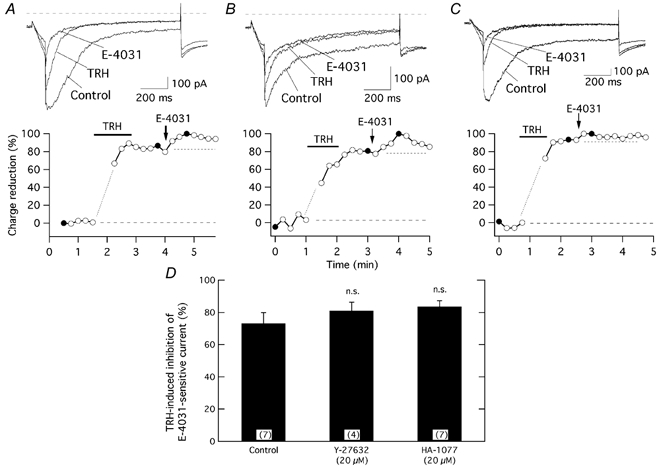
The time course of relative r-ERG current reduction by TRH and E-4031 is shown for three different cells without any previous treatment (A), following preincubation for 1 h with 20 μm Y-27632 (B), or after 1.5 h with HA-1077 (C) at 37 °C. For further details on drug treatments see Methods. Pulse protocols as described in Fig. 1 were used. Current estimations were performed from total inward charge during the hyperpolarization steps at −100 mV as described in Methods. Averaged charge values before any addition and those corresponding to the minimum following addition of E-4031 were considered as 0 and 100 %, respectively. Filled circles correspond to the current traces shown above. The first one or two data points following addition of TRH, when total inward currents became transiently enhanced by activation of Ca2+-dependent K+ channels due to massive liberation of Ca2+ from intracellular stores have been deleted for clarity. Perfusion of 100 nm TRH and addition of 5 μm E-4031 to the recording chamber are indicated. Values for the data in the absence and presence of TRH are indicated by horizontal dashed lines. D, comparison of TRH-induced reduction of E-4031-sensitive r-ERG currents for control cells and cells treated for 1–3 h at 37 °C with Y-27632 or HA-1077. Current reductions were estimated from total inward charge measurements as indicated above. n.s., not significant. Numbers in parentheses indicate the number of experiments.
Previous experiments in GH3 cells indicate that a phosphorylating step seems to be involved in the TRH-induced reduction of r-ERG current (see above). Whereas PKA and PKC do not participate in this effect (Bauer et al. 1990, 1994; Barros et al. 1992, 1993; Schäfer et al. 1999; Schledermann et al. 2001; Storey et al. 2002), it has recently been suggested that a Rho-dependent protein kinase may be involved (Storey et al. 2002). To check this possibility we studied the effect of TRH on the E-4031-sensitive current in cells incubated with the ROCK inhibitors Y-27632 and HA-1077 (Davies et al. 2000; Nagumo et al. 2000). Treatment with the ROCK inhibitors did not significantly modify the extent of the TRH-induced inhibition, which amounted to 81.0 ± 5.3 % (n = 4) and 83.7 ± 3.6 % (n = 7) in the presence of 20 μm Y-27632 and HA-1077, respectively (Fig. 2B–D).
It could be argued that failure to observe any effect of the ROCK inhibitors on the TRH-induced current reduction is due to the inability of the chemicals to reach their targets inside the cell. However, long preincubations at 37 °C and drug concentrations well above the IC50 for ROCK inhibition (0.8 and 1.9 μm for Y-27632 and HA-1077, respectively; Davies et al. 2000) were used. This suggests that under our experimental conditions ROCK is not necessary for TRH-induced inhibition of r-ERG.
Effect of ROCK inhibitors on reversal of the TRH-induced r-ERG current reductions
It is known that the TRH-induced reduction of endogenous GH3 cell r-ERG current becomes irreversible in standard whole-cell recordings, but not in perforated-patch experiments (Barros et al. 1992, 1993; Schledermann et al. 2001). Surprisingly, the TRH-induced inhibition of r-ERG was readily reversed following a 15–20 min washout period both in control cells and in cells incubated with 20 μm Y-27632, but it did not reverse in the presence of 20 μm HA-1077 (Fig. 3). The recovery 15 min after starting TRH washout averaged 74.5 ± 5.3 % (n = 9) and 55 ± 10.9 % (n = 6) in untreated and Y-27632-treated cells, respectively. However, only 9 ± 5.4 % (n = 5) of the inhibition was reversed during the same washout period in HA-1077-treated cells. It is important to note that E-4031 was not always added in these experiments due to the extended periods of time necessary to follow the reversal of hormonal effects before the cell was lost. However, since TRH-induced current reduction is exclusively exerted on r-ERG currents and the recovered currents are carried by E-4031-sensitive r-ERG channels (see above), the time course of the total current recovery provides a good quantification for reversal of the TRH effects on r-ERG. On the other hand, the clear differences in behaviour of the HA-1077-treated cells as compared with untreated and Y-27632-treated cells demonstrates that at least with HA-1077 the inhibitor is acting inside the cell, even though it is not able to impair the current inhibition triggered by TRH.
Figure 3. Effect of ROCK inhibitors on reversal of the TRH-induced r-ERG current reductions.
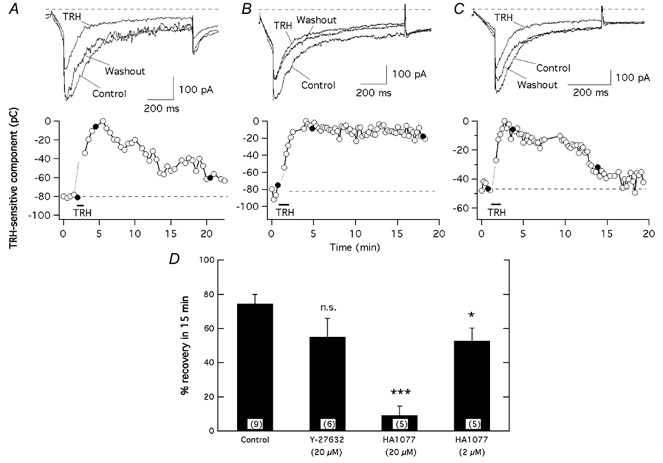
The time course of variations in inward current magnitude is shown for three different cells without any previous treatment (A) or following treatment for 1–3 h at 37 °C with 20 μm HA-1077 (B) or Y-27632 (C). Filled circles correspond to the current traces shown above. Pulse protocols as described in Fig. 1 were used. Current estimations were performed from total inward charge during the hyperpolarization steps at −100 mV. The magnitude of the TRH-sensitive current component was estimated by subtracting the minimum current level reached in the presence of TRH from every data point. Averaged values for the data before adding TRH are indicated by horizontal dashed lines. D, comparison of the magnitude of current recovery upon TRH washout in the absence or presence of ROCK inhibitors. Data are expressed as percentage of TRH-inhibited current recovered after 15 min of hormone washout. The values were derived from the data shown in A–C. Data from cells preincubated with 2 μm HA-1077 are also shown for comparison. ***P = 0.0001, *P = 0.05. Numbers in parentheses indicate the number of experiments.
The greater potency of HA-1077 as compared with Y-27632 in blocking the reversal of the TRH effects contrasts with its lower potency as a ROCK inhibitor (see above). It would also be expected that, since 20 μm is a concentration well above the IC50 values for ROCK inhibition, both HA-1077 and Y-27632 effectively impair cellular ROCK-dependent processes. This raised the possibility that the effect of HA-1077 was not related to its ability to inhibit ROCK but to an action on a different cellular component. Besides ROCK, HA-1077 is also able to inhibit other protein kinases including PKA and PKC (Ki values 1.6 and 3.5 μm, respectively; Nagumo et al. 2000). To check whether one of these kinases was involved in the reversal of the TRH-induced inhibition, cells were incubated with 2 μm HA-1077, a concentration still above the IC50 for ROCK and the Ki for PKA, but almost two times lower than the Ki for PKC. As shown in Fig. 3D, preincubation of the cells in 2 μm HA-1077 increased the extent of current recovery up to 52.8 ± 7.3 % (n = 5) after a 15 min period of hormone washout. This result adds further support to the hypothesis that the effects of HA-1077 are not related to its ability to impair ROCK activity, but to the inhibition of some other cellular component such as PKC. It is also important to note that although Y-27632 concentrations higher than 20 μm were not used to prevent non-specific actions, the small effect of this drug (with Ki values of 0.14, 25 and 26 μm for ROCK, PKA and PKC, respectively), could be also compatible with such a hypothesis.
Effect of PKC inhibition on reversal of the TRH-induced r-ERG current reduction
The TRH receptor is coupled to a Gq/11-type G protein, leading to the activation of PLC and generation of IP3 and DAG, which are able to release Ca2+ from intracellular stores and induce PKC activation. To confirm the hypothesis that the effect of the ROCK inhibitors is related to their ability to block PKC, we studied the influence of specific PKC inhibition on the TRH-induced reduction of r-ERG current and its subsequent recovery upon hormone washout.
Application of the non-toxic PKC-specific inhibitor GF109203X (Palomero et al. 1998) for 5 min before adding TRH resulted in a slight reduction of the hyperpolarization-evoked inward currents in nearly half of the cells, averaging 16.1 ± 4 % (n = 5) of the E-4031-sensitive current. Moreover, GF109203X did not significantly affect the current reduction induced by TRH. Thus the E-4031-sensitive current remaining in the presence of the PKC inhibitor was reduced by 75.7 ± 3.8 % (n = 4) by TRH. However, the TRH-induced current reduction became almost totally irreversible (16.7 ± 2.5 % (n = 6) of recovery after 15 min of TRH washout) in the presence of GF109203X (Fig. 4A and C). This demonstrates that although PKC activity is not required for TRH to reduce r-ERG current levels, the presence of enzymatically active PKC is necessary for subsequent recovery of the current.
Figure 4. Effect of PKC inhibition on reversal of the TRH-induced r-ERG current reductions.
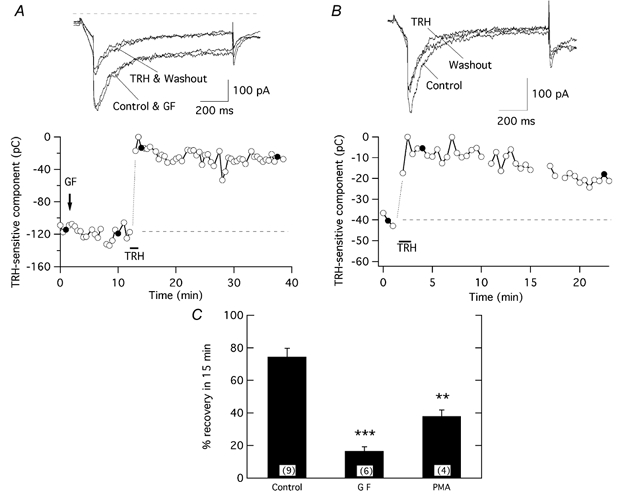
A, time course of variations in inward current magnitude in response to the addition of 1 μm GF109203X (GF) and the subsequent response to a short application of 100 nm TRH in the continuous presence of the inhibitor. B, time course of the TRH effect in a PKC-downregulated cell following a long-term incubation for 24 h with 1 μm PMA. C, comparison of the magnitude of current recovery in control and PKC-inhibited cells. Pulse protocols and data evaluation were performed as described in Fig. 3. ***P = 0.0001, **P = 0.01.
As a further demonstration that PKC participates in current reduction recovery, we investigated the effect of long-term incubation of GH3 cells with a high concentration of phorbol ester (1 μm PMA for at least 24 h). A comparable long incubation with PMA has been reported to cause a 94 % reduction of PKC activity in GH3 cells (Kolesnick, 1989). Although a smaller TRH-induced current reduction was systematically observed in these PKC downregulated cells, the reversion of the hormonal effect was significantly lowered to 38 ± 3.8 % (n = 4; Fig. 4B and C). It is possible that alterations in cellular components during the prolonged exposure to PMA and/or maintenance of a small fraction of highly active PKC after long PMA treatment contributes to the higher reversion levels as compared to those obtained with GF109203X. Nevertheless, these results further support the conclusion that a normally active PKC is necessary for efficient reversal of the TRH effect.
Impairment of PIP2 hydrolysis does not prevent TRH-induced r-ERG current reduction, but blocks the reversion of the TRH effect
It has recently been proposed that consumption of endogenous PIP2 levels following stimulation of PLC by the Gq-coupled α1A-adrenergic receptor regulates HERG channel activity (Bian et al. 2001). To check whether a similar mechanism is involved in the regulation of r-ERG by TRH in pituitary cells, we compared the hormonal effects in the presence and absence of the membrane-permeable PLC inhibitor U-73122 (Broad et al. 2001; Cho et al. 2001). To verify that the drug is effective in inhibiting PLC when applied to GH3 cells, we began by testing whether U-73122 readily antagonizes the initial TRH-induced elevation of [Ca2+]i, a response that requires PLC activation and the generation of IP3. A 3–5 min pretreatment of cells with 10 μm U-73122 has previously been documented to fully and irreversibly prevent PLC activation upon agonist stimulation (Broad et al. 2001). Likewise, a 5 min preincubation of GH3 cells with U-73122 was sufficient to prevent the [Ca2+]i increase triggered by TRH, consistent with a blockade of agonist-activated PLC (Fig. 5). A similar absence of Ca2+ transients was observed in three cells treated with U-73122 and voltage clamped at −60 mV under perforated-patch conditions, as compared with the prominent Ca2+ elevations recorded under the same circumstances but in the absence of the drug (not shown).
Figure 5. Effect of U-73122 on the Ca2+ responses of GH3 cells.
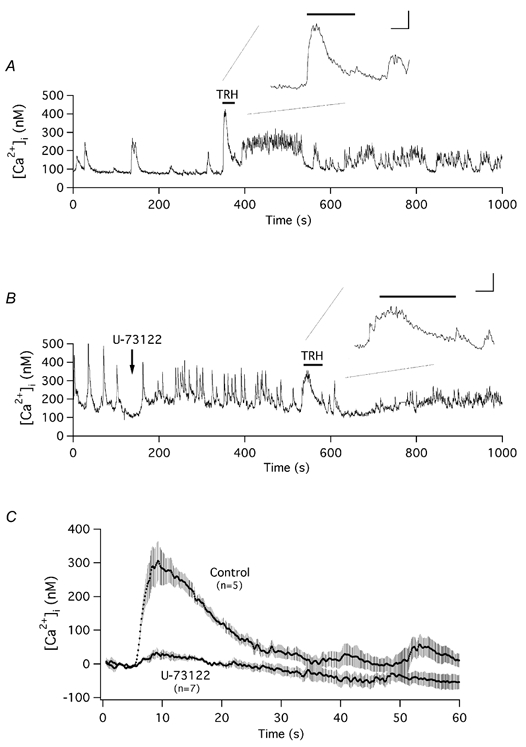
A and B, time course of variation in [Ca2+]i in two representative cells and their response to TRH addition in the absence (A) or the presence (B) of 10 μm U-73122. Measurement of [Ca2+]i in Fura-2-loaded cells was performed as described in Methods. Addition of U-73122 to the recording chamber and the period of perfusion with 100 nm TRH are indicated. Expanded fragments of the [Ca2+]i recordings during perfusion of TRH are shown in the insets. Note the contribution of two precedent spontaneous Ca2+ oscillations to the otherwise quite small Ca2+ elevation induced by TRH entry in B. Inset calibration bars correspond to 100 nm Ca2+ and 10 s. C, averaged recordings of [Ca2+]i showing the initial TRH-induced Ca2+ transients in the absence (Control) or presence of U-73122. Data traces synchronized to the moment of TRH addition were averaged point by point and averaged values ±s.e.m. are shown. Averaged [Ca2+]i values in the first point of the recordings have been subtracted from the data. Numbers in parentheses indicate the number of cells from which averaged data were obtained.
Unlike the effects of U-73122 on the TRH-evoked Ca2+ response, the presence of the inhibitor did not affect the TRH-induced suppression of r-ERG current. Acute application of U-73122 for 5 min resulted in small reductions of the hyperpolarization-evoked currents that averaged 23.4 ± 4.2 % (n = 11) of the total E-4031-sensitive current (Fig. 6). Addition of TRH after U-73122 was still able to drastically suppress the remaining E-4031-sensitive current by 85.3 ± 1.8 % (n = 6). However, the TRH-induced current reductions became almost totally irreversible (10.1 ± 3.4 % (n = 5) of recovery after 15 min of TRH washout) in U-73122-treated cells (Fig. 6B). Provided that PKC activity is required to reverse the TRH-induced current inhibition, these results are completely coherent with the expected absence of DAG production due to PLC inhibition and hence with the subsequent inability to detect such a reversal due to the lack of PKC activation. On the other hand, they also suggest that depletion of PIP2 levels is not a requisite for TRH to suppress r-ERG current in GH3 cells.
Figure 6. Effect of PLC inhibition by U-73122 on the TRH effects.
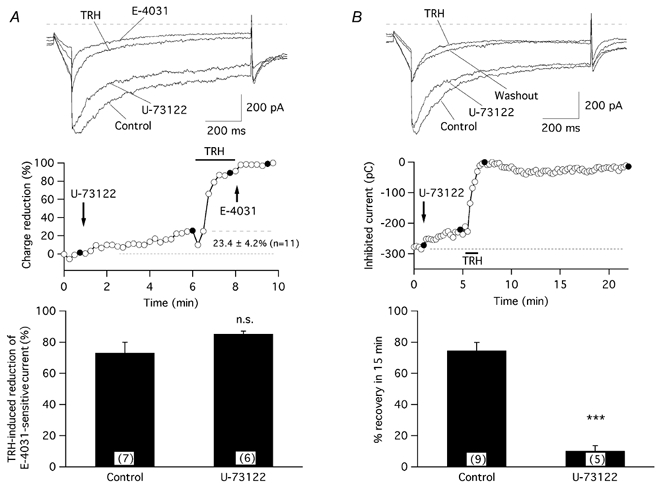
A, TRH-induced r-ERG current suppression is not modified by U-73122. The time course of relative r-ERG current reduction by TRH and E-4031 following a 5 min incubation with 10 μm U-73122 is shown in the middle panel. Filled circles correspond to the current traces shown above. The start of perfusion with U-73122 and the duration of the TRH treatment are marked. Introduction of 5 μm E-4031 into the recording chamber and the averaged magnitude of the current reduction at the end of the 5 min treatment with U-73122 before adding TRH are also indicated on the graph. A comparison of TRH-induced reduction of E-4031-sensitive r-ERG currents for control cells and cells treated with U-73122 is shown in the lower panel. B, the r-ERG current reductions caused by TRH became irreversible in the presence of U-73122. The time course of variations in inward current magnitude in response to an addition of 10 μm U-73122 and the subsequent response to a short application of 100 nm TRH in the continuous presence of the inhibitor are shown in the middle panel. Filled circles correspond to the current traces shown above. A comparison of the magnitude of current recovery upon TRH washout in the absence and presence of U-73122 is shown in the lower panel. Pulse protocols and data evaluation were performed as described in Figs 2 and 3. ***P = 0.0001. Numbers in parentheses indicate the number of experiments.
Previous experiments in the neuroblastoma cell line SH-SY5Y indicated that blockade of PIP2 resynthesis with concentrations of wortmannin high enough to inhibit phosphatidylinositol 4-kinase (Nakanishi et al. 1995) leads to a fast depletion of the phospholipid associated with a failure in receptor-mediated phosphoinositide and Ca2+ signalling (Willars et al. 1998). Neither a 5–10 min treatment of GH3 cells with 10 μm wortmannin nor a longer preincubation (up to 2 h at 37 °C) with the drug modified the TRH-induced reduction of the E-4031-sensitive r-ERG current, which averaged 72 ± 3.6 % (n = 6) in the presence of wortmannin. Interestingly, in five out of eight cells in which the effect was studied over a prolonged period of time, an apparently normal reversal of the hormonal effect took place during the first 5 min of TRH washout. However, such a reversal was followed by an additional phase in which the current became suppressed again, reaching a reduced level similar to that observed during the initial addition of the agonist (Fig. 7). In two additional cells a partial reversion to around 50 % of the initial current magnitude was obtained after 15 min of washout. As discussed below, it is possible that although depletion of PIP2 is not involved in the fast and initial current suppression, it could determine the delayed inhibition if resynthesis of PIP2 is prevented.
Figure 7. Effect of wortmannin on the TRH-induced current reductions.
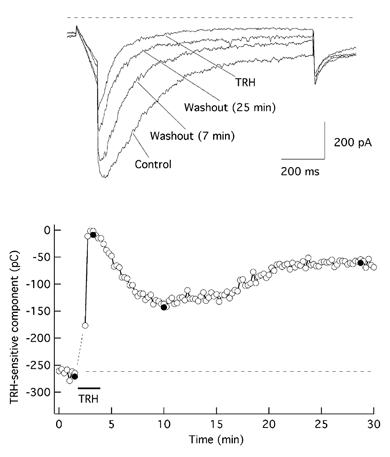
Data from a cell preincubated for 2 h with 10 μm wortmannin at 37 °C are shown. The inhibitor was also maintained in the chamber at the same concentration during the full time course of the experiment. Filled circles correspond to the current traces shown above. Similar results were obtained in five of the eight cells treated with wortmannin.
Identification of the PKC isozyme involved in the reversal of the TRH effect as a conventional PKC of the α/β type
In GH3 cells expression of five different PKC isozymes has been detected: cPKCα, cPKCβII, nPKCδ, nPKCε and aPKCζ (Kiley et al. 1991; Akita et al. 1994). The fact that aPKCζ does not need DAG production for activation and that this isozyme is not inhibited by GF109203X (Way et al. 2000) excludes it as the isoform involved in the reversal of TRH-triggered r-ERG inhibition. To distinguish whether a conventional (α and βII) or a novel (δ and ε) PKC is acting on r-ERG current recovery, we tested the TRH effects in the presence of Gö6976, an agent known to potently inhibit cPKC isozymes (IC50 values of 2.3 and 6.2 nm for PKCα and PKCβI, respectively; Hofmann, 1997; Way et al. 2000) that does not affect the kinase activity of nPKCs up to micromolar concentrations.
Preincubation of GH3 cells with 0.5 μm Gö6976 for 5 min did not alter the r-ERG current levels of the perforated-patch clamped GH3 cells (Fig. 8). Neither did the presence of the inhibitor modify the extent of current inhibition induced by TRH, which amounted to 70.3 ± 5.1 % (n = 5). However, the TRH-induced current reductions became irreversible in the presence of Gö6976. Thus, in cells treated with Gö6976 the current recovery after 15 min of TRH washout averaged only 16.8 ± 9.4 % (n = 5). These results again confirm the need for a normally active PKC for efficient reversal of the TRH effect. Furthermore, since cPKCγ has not been identified in GH3 cells, they identify a conventional PKC of the α or βII subtype as the more likely candidate to participate in recovery of the TRH effect.
Figure 8. Effect of the PKC inhibitor Gö6976 on the TRH effects.
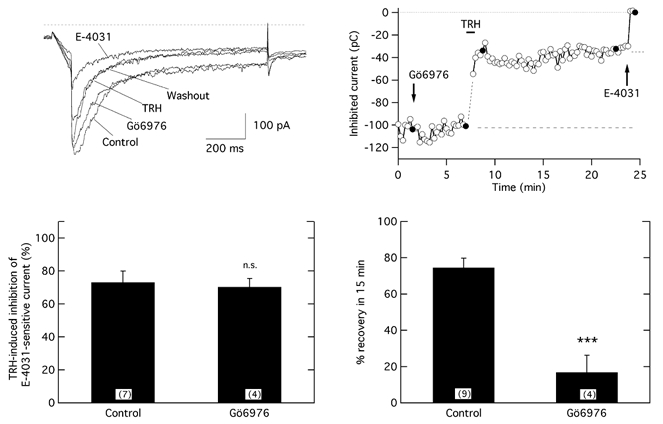
The time course of relative r-ERG current reduction by TRH and E-4031 following a 5 min treatment with 0.5 μm Gö6976 is shown in the upper right panel. Filled circles correspond to the current traces shown on the left. Note the almost complete absence of reversal of the TRH-induced inhibition after more than 20 min of hormone washout. A comparison of the TRH-induced reductions of the E-4031-sensitive r-ERG currents for control cells and cells treated with Gö6976 is shown in the lower left panel. Averaged values of the magnitude of current recovery upon TRH washout in the absence or the presence of Gö6976 is shown in the lower right panel. ***P = 0.0001.
DISCUSSION
We have studied the biochemical cascade linking activation of PLC-coupled TRH receptors to the regulation of endogenous r-ERG channels in situ using perforated-patch clamped GH3 cells. Comparison of the results obtained here with previous results in oocytes co-expressing TRH receptors and HERG channels (Barros et al. 1998) or in other cells such as guinea-pig ventriculocytes studied with perforated-patch techniques (Heath & Terrar, 2000), or CHO cells heterologously expressing HERG channels studied under conventional whole-cell conditions (Cui et al. 2000), indicates that the choice of cellular background and/or the technical environment can influence the results and the mechanisms involved in receptor-mediated modulation of the currents. Thus in oocytes the effects of both TRH and PMA on HERG are antagonized by the specific PKC inhibitor GF109203X (Barros et al. 1998), but the TRH-induced suppression of endogenous r-ERG current in GH3 cells remains unaltered in the presence of the drug. The strong modulatory effects of cyclic AMP analogues and PKA in CHO cells (Kiehn, 2000) are not directly applicable to the pituitary cells either, since endogenous GH3 cell ERG currents are not modified in the presence of cyclic AMP or permeant cyclic AMP analogues (Barros et al. 1992, 1994), and PKA and PKC do not participate in the TRH-induced inhibition of r-ERG (Bauer et al. 1990, 1994; Barros et al. 1992, 1993; Schäfer et al. 1999; Schledermann et al. 2001; Storey et al. 2002). Interestingly, we have not been able to reproduce the effects of cyclic AMP analogues in a HEK293 cell line permanently expressing HERG channels and TRH receptors showing a strong TRH effect, and in which the intracellular presence of the nucleotide was demonstrated by its ability to activate an expressed olfactory cyclic nucleotide-gated channel (T. Giráldez & F. Barros, unpublished data). Some discrepancies between different cell systems have also been observed for the current parameters modified by the agonist. Whereas in oocytes the shifts in the inflection potentials of the I–V curves and the accelerations in deactivation are not accompanied by significant decreases in the maximal tail current magnitudes, prominent current reductions are observed in GH3 cells. Given the high degree of identity between the r-ERG and HERG proteins, it is unlikely that differences in the channel structure are the main cause of these discrepancies. Furthermore, reductions equivalent to those shown here are induced by TRH on HERG channels when GH3 cells injected with HERG-coding cDNA are studied (Schledermann et al. 2001). The possibility remains that the presence of heteromultimers erg1/erg2 as the molecular basis of r-ERG channels in GH3 cells (Wimmers et al. 2001) or the formation of macromolecular complexes between ERG channels and specific regulatory components in native cells may contribute to differences in regulation between pituitary cells and heterologous expression systems. Altogether, this highlights the danger of extrapolating results obtained with a pharmacological tool in a specific cellular background to another cell type and/or physiological regulator.
Our present study looking at the TRH-induced modification of endogenous ERG current in situ using perforated-patch clamped GH3 cells does not provide a definitive clue about the biochemical pathway(s) linking the activation of the TRH receptor to r-ERG inhibition. However, the results firmly support the participation of the PKC branch of the PLC signalling cascade in the reversal of the hormonal effect. Evidence in favour of such an interpretation includes: (a) the correlation in the potency of the putative ROCK inhibitors Y-27632 and HA-1077 to act as PKC inhibitors (but not to inhibit ROCK), and their ability to render the TRH effect irreversible; (b) the irreversibility obtained in the presence of the specific PKC inhibitors GF109203X and Gö6976; (c) the significantly smaller reversal of the hormonal effect observed in PKC-downregulated cells following long-term incubation in a high concentration of PMA; and (d) the demonstration that the TRH effect becomes irreversible in the absence of DAG production due to PLC inhibition with U-73122.
Based in their structure and substrate requirements, three groups of PKC isoforms have been established: (1) conventional or classical PKCs (cPKCs), including isoforms α, β and γ, which are Ca2+ dependent and activated by phosphatidylserine and DAG; (2) novel PKCs (nPKCs: δ, ε, η and θ), which are also regulated by DAG and phosphatidylserine but are Ca2+ independent; and (3) atypical PKCs (aPKCs: ζ and λ), which are Ca2+ independent and do not require DAG for activation, but are regulated by phosphatidylserine (Hofmann, 1997; Way et al. 2000). Expression of five different PKC isozymes has been detected in GH3 cells: cPKCα, cPKCβII, nPKCδ, nPKCε and aPKCζ (Kiley et al. 1991; Akita et al. 1994). The impairment of current reversion in the absence of DAG production and the insensitivity of aPKCζ to GF109203X and Gö6976 (Way et al. 2000) exclude the involvement of this isoform in the reversal of TRH-triggered r-ERG inhibition. The insensitivity of nPKCs to Gö6976 also excludes the participation of δ and ε isozymes in the current reversion. Since cPKCγ has not been identified in GH3 cells, our results identify a cPKC of the α or βII subtype as the more likely candidate to participate in the reversal of the TRH effect. Further work based in selective knock-out or specific inhibition of each isoform in situ will be necessary to confirm the identity of the isoform that mediates the recovery.
The exact mechanism by which PKC activation leads to reversal of the TRH-induced inhibition is unclear. The reported irreversibility of the TRH effect in the presence of specific protein phosphatase inhibitors indicates that besides PKC a protein phosphatase (most probably PP2A; Barros et al. 1992, 1993) is necessary for such a reversal. In addition to its known ability to generate the second messengers IP3 and DAG, activation of Gq/PLC-coupled receptors (e.g. TRH receptors) may also cause consumption of endogenous PIP2 due to PLC activation (Hilgemann et al. 2001). It has recently been suggested that normal activity of HERG channels depends on the presence of PIP2 and that depletion of this essential lipid phosphate triggered by activation of α1A-adrenergic receptors coexpressed with HERG in HEK293 cells leads to HERG current reduction (Bian et al. 2001). It is possible that the smaller reductions observed by Bian et al. (2000) as compared to those reported here are related to the use of a heterologous expression system instead of our GH3 cells endogenously expressing channels and hormone receptors. This PI-polyphosphate hypothesis could also be consistent with the observed irreversibility of the hormonal effect in the presence of the PP2A inhibitor okadaic acid (Barros et al. 1992), since it has been reported that a PKC-enhanced PP2A activity is required to activate phosphatidylinositol 4-phosphate 5-kinase, the last enzyme in the PIP2 synthetic pathway (Park et al. 2001).
Nevertheless, our results also indicate that depletion of PIP2 is not the main reason for the TRH-induced inhibition of r-ERG. The TRH-induced suppression of the current remains unaltered even when PIP2 consumption is blocked by PLC inhibition with U-73122. Furthermore, in other systems in which a decreased PIP2 content directly inhibits ion channels only the recovery from receptor-mediated inhibition, not the inhibitory influence of the agonist, is impaired by non-hydrolysable ATP analogues (Xie et al. 1999; Suh & Hille, 2002). However, in GH3 cells the TRH-induced inhibition is minimized by loading the cell with ADP-NH-P (Barros et al. 1993). Interestingly, a delayed inhibitory phase following a partial recovery of the initial inhibition was systematically detected when PIP2 replenishment was blocked with high concentrations of wortmannin (Nakanishi et al. 1995). It seems unlikely that a long-term reduction in DAG levels by depletion of PLC substrates and the subsequent lowering of PKC activation could explain this delay in current recovery, since only the initial DAG accumulation due to TRH-induced hydrolysis of PIP2 participates in GH3 cell PKC activation (Martin et al. 1990). As shown in other cells, it is possible that an exhaustive depletion of PIP2 is induced by the agonist only after the recycling of the lipid is blocked with wortmannin (Nakanishi et al. 1995; Broad et al. 2001). This raises the possibility that depletion of PIP2 could determine the delayed inhibition observed in wortmannin-treated cells in which resynthesis of PIP2 is prevented.
Together with previous data (Barros et al. 1992, 1993), our present results further suggest that a pathway involving a protein kinase is the main cause of r-ERG inhibition in perforated-patch clamped GH3 cells. The kinase identity remains to be established, but it seems to be different from PKC, PKA or ROCK. Attempts to prevent the TRH effect under perforated-patch conditions with inhibitors of tyrosine kinases (tyrphostin A23 and genistein), Ca2+-calmodulin kinase II (KN-62) or MAP kinase pathways (PD98059 and SB202190) proved unsuccessful (Schledermann et al. 2001; D. Gómez-Varela & F. Barros, unpublished observations). On the other hand, the results presented here indicate that a mechanism exists for reversal of TRH-induced r-ERG inhibition in which a PP2A acting downstream of PKC is directly involved in phosphate removal either from the channel itself or from a regulatory component associated with it.
In summary, as depicted in the simplified scheme shown in Fig. 9, coupling of TRH receptors to PLC activation through a Gq/11-type G protein promotes PIP2 hydrolysis. Subsequent generation of IP3 releases Ca2+ from intracellular stores leading to an initial and transient elevation of [Ca2+]i that mediates a peak of secretion associated with a transient hyperpolarization of the cell membrane due to the activation of Ca2+-dependent K+ channels (phase 1 of hormone action). By a different pathway, activation of the receptor is also able to suppress the activity of r-ERG channels, triggering an increase in resting potential and a delayed enhancement in electrical activity. The concomitant increase in intracellular Ca2+ oscillations at the single cell level, manifested in a cell population as a plateau that follows the initial peak of [Ca2+]i, mediates phase 2 of the secretory response. It has been shown that r-ERG channel inhibition is enhanced by short-term treatment with cholera toxin and is abolished when the toxin treatment is prolonged (Barros et al. 1993, 1994; Bauer et al. 1994). On the other hand, TRH-induced r-ERG current reductions are decreased if G13 and/or Rho operation is blunted (Storey et al. 2002). This has been interpreted as an indication that either a Gs-like G protein or a G13- and Rho-based pathway couples the TRH receptor to r-ERG inhibition. As discussed above, it also appears that a phosphorylation step plays the main role in such inhibition. However, the identity of the kinase catalysing this step and the exact molecular target of the phosphorylation process remain to be established.
Figure 9. Schematic representation of the biochemical pathways controlling hormone secretion in GH3 cells (A) and the steps blocked by different pharmacological agents as shown in this report (B).
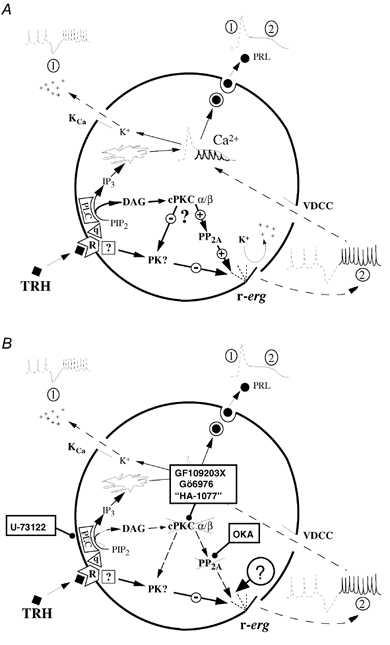
The first phase (marked 1 on the scheme) corresponds to the transient release of stored Ca2+ into the cytosol after production of IP3 in response to PIP2 hydrolysis catalysed by PLC, with concomitant hyperpolarization due to activation of Ca2+-dependent K+ channels. The second phase (marked 2) is the result of the increase in Ca2+-dependent action potential frequency due to reductions in r-ERG conductance, leading to an increase in intracellular Ca2+ oscillations and manifested in a cell population as a plateau of increased Ca2+ and secretion. Participation of a protein kinase-catalysed step is marked PK on the scheme. The minus sign indicates development of the TRH-induced r-ERG current inhibition. The plus sign stands for reversal of the inhibitory effect. The proposed mechanism for reversal of the hormonal inhibition by means of DAG elevation, activation of an α or βII subtype of cPKC, and one or more dephosphorylation steps catalysed by PP2A is depicted in the centre of the cell. Whether phosphorylation and/or phosphate removal are exerted on the channel itself or on a regulatory component associated with it remains to be established. The alternative possibilities that PKC activation either increases PP2A activity or acts by inactivating the kinase involved in r-ERG inhibition are also illustrated. For more explanation see text. TRH, thyrotropin-releasing hormone; R, TRH receptor; q, G protein of the Gq/11 type; PLC, phospholipase C; PIP2, phosphatidylinositol 4,5-bisphosphate; IP3, inositol 1,4,5-trisphosphate; DAG, diacylglycerol; cPKC α/β, conventional protein kinase C of the α or β type; PP2A, protein phosphatase 2A; PRL, prolactin; OKA, okadaic acid; VDCC, voltage-dependent calcium channel; KCa, calcium-dependent potassium channel.
Our present data indicate that the DAG/PKC branch of the PLC-mediated signalling pathway is involved in the recovery of the TRH-induced inhibition of r-ERG. Thus it is possible that by activating a conventional PKC of the α or βII subtype, DAG and initial Ca2+ elevations produced in response to the PIP2 hydrolysis mediate activation of PP2A. Concomitant phosphate removal either from the channel itself or from a regulatory component associated with it determines the reversal of TRH-induced inhibition of r-ERG. As shown in Fig. 9B, blockade of PP2A activity with okadaic acid (Barros et al. 1992, 1993), but also impairment of the biochemical steps upstream of the phosphatase (i.e. inhibiting PKC with GF109203X, Gö6976 and adequate concentrations of HA-1077, or blocking PLC activity with U-73122), prevent progress through this feedback cascade, making the hormonal inhibition of the channels irreversible. Albeit indirectly, recent data demonstrating a direct physical association of PP2A and PKCα in mammalian cells (Boudreau et al. 2002) and PKC-induced phosphorylation of PP2A on its regulatory B subunit (Ricciarelli & Azzi, 1998), are in agreement with this model. However, whereas functional regulation of PKCα by PP2A-catalysed dephosphorylation has been repeatedly reported (Hansra et al. 1996; Ricciarelli & Azzi, 1998; Boudreau et al. 2002), PKCα activation does not affect PP2A activity or its membrane association (Hansra et al. 1996; Ricciarelli & Azzi, 1998). As depicted in Fig. 9, this raises an alternative possibility that activation of PKC indirectly mediates the dephosphorylation event involved in r-ERG recovery, by phosphorylating the kinase involved in r-ERG inhibition and returning it to its inactive state. This scheme parallels another previously proposed for c-Jun dephosphorylation by PKC (Goode et al. 1992) and would be compatible with a constitutively active and permanently membrane-associated PP2A. Thus, the actual molecular target of PKC remains to be established. It would also be interesting to know whether mechanisms similar to those proposed here are operative in other cell types in which ERG channel operation and activation of Gq/11-coupled receptors (e.g. α1A-adrenergic, endothelin or angiotensin II receptors in cardiac cells) bear important physiological and pathological consequences such as action potential repolarization, long-QT syndrome and cardiac hypertrophy.
Acknowledgments
We thank Drs B. Kaupp and W. Bönigk for kindly providing the plasmid containing the olfactory cyclic nucleotide-gated channel CNCα3 and Drs P. Domínguez and J. R. Schwarz for comments on the manuscript. We also thank Noelia S. Durán for technical assistance. This work was supported by grant PM99-0152 from DGICYT of Spain. D.G.-V. was supported by the Fundación Inocente, Madrid, Spain, and presently holds a predoctoral fellowship from Spanish Ministerio de Ciencia y Tecnolog'a (ref. no. FP2000-5736). D.G.-M. is a predoctoral fellow from Spanish Ministerio de Ciencia y Tecnolog'a (ref. no. AP2000-4363). S.G.D. is presently supported by Fundación Inocente, Madrid, Spain. T.G. was supported by a predoctoral fellowship from F.I.S. of Spain (ref. no. 99/9298).
REFERENCES
- Akita Y, Ohno S, Yajima Y, Konno Y, Saido TC, Mizuno K, Chida K, Osada S, Kuroki T, Kawashima S, Suzuki K. J Biol Chem. 1994;269:4653–4660. [PubMed] [Google Scholar]
- Arcangeli A, Rosati B, Cherubini A, Crociani O, Fontana L, Ziller C, Wanke E, Olivotto M. HERG- and IRK-like inward rectifier currents are sequentially expressed during neuronal development of neural crest cells and their derivatives. Eur J Neurosci. 1997;9:2596–2604. doi: 10.1111/j.1460-9568.1997.tb01689.x. [DOI] [PubMed] [Google Scholar]
- Barros F, Del Camino D, Pardo LA, Palomero T, Giráldez T, de la Peña P. Demonstration of an inwardly rectifying K+ current component modulated by thyrotropin-releasing hormone and caffeine in GH3 rat anterior pituitary cells. Pflugers Arch. 1997;435:119–129. doi: 10.1007/s004240050491. [DOI] [PubMed] [Google Scholar]
- Barros F, Delgado LM, Del Camino D, de la Peña P. Characteristics and modulation by thyrotropin-releasing hormone of an inwardly rectifying K+ current in patch-perforated GH3 anterior pituitary cells. Pflugers Arch. 1992;422:31–39. doi: 10.1007/BF00381510. [DOI] [PubMed] [Google Scholar]
- Barros F, Delgado LM, Maciá C, de la Peña P. Effects of hypothalamic peptides on electrical activity and membrane currents of ‘patch perforated’ clamped GH3 anterior pituitary cells. FEBS Lett. 1991;279:33–37. doi: 10.1016/0014-5793(91)80243-v. [DOI] [PubMed] [Google Scholar]
- Barros F, Gómez-Varela D, Viloria CG, Palomero T, Giráldez T, de la Peña P. Modulation of human erg K+ channel gating by activation of a G protein-coupled receptor and protein kinase C. J Physiol. 1998;511:333–346. doi: 10.1111/j.1469-7793.1998.333bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros F, Mieskes G, Del Camino D, de la Peña P. Protein phosphatase 2A reverses inhibition of inward rectifying K+ currents by thyrotropin-releasing hormone in GH3 pituitary cells. FEBS Lett. 1993;336:433–439. doi: 10.1016/0014-5793(93)80851-k. [DOI] [PubMed] [Google Scholar]
- Barros F, Villalobos C, García-Sancho J, Del Camino D, de la Peña P. The role of the inwardly rectifying K+ current in resting potential and thyrotropin-releasing hormone-induced changes in cell excitability of GH3 rat anterior pituitary cells. Pflugers Arch. 1994;426:221–230. doi: 10.1007/BF00374775. [DOI] [PubMed] [Google Scholar]
- Bauer CK. The erg inwardly rectifying K+ current and its modulation by thyrotropin-releasing hormone in giant clonal rat anterior pituitary cells. J Physiol. 1998;510:63–70. doi: 10.1111/j.1469-7793.1998.063bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CK, Davison I, Kubasov I, Schwarz JR, Mason WT. Different G proteins are involved in the biphasic response of clonal rat pituitary cells to thyrotropin-releasing hormone. Pflugers Arch. 1994;428:17–25. doi: 10.1007/BF00374747. [DOI] [PubMed] [Google Scholar]
- Bauer CK, Engeland B, Wulfsen I, Ludwig J, Pongs O, Schwarz JR. RERG is a molecular correlate of the inward-rectifying K current in clonal rat pituitary cells. Receptors Channels. 1998;6:19–29. [PubMed] [Google Scholar]
- Bauer CK, Meyerhof W, Schwarz JR. An inward-rectifying K+ current in clonal rat pituitary cells and its modulation by thyrotropin-releasing hormone. J Physiol. 1990;429:169–189. doi: 10.1113/jphysiol.1990.sp018250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CK, Schäfer R, Schiemann D, Reid G, Hanganu I, Schwarz JR. A functional role of the erg-like inward-rectifying K+ current in prolactin secretion from rat lactotrophs. Mol Cell Endocrinol. 1999;148:37–45. doi: 10.1016/s0303-7207(98)00241-x. [DOI] [PubMed] [Google Scholar]
- Bian J, Cui J, McDonald TV. HERG K+ channel activity is regulated by changes in phosphatidyl inositol 4,5-bisphosphate. Circ Res. 2001;89:1168–1176. doi: 10.1161/hh2401.101375. [DOI] [PubMed] [Google Scholar]
- Bianchi L, Wible B, Arcangeli A, Taglialatela M, Morra F, Castaldo P, Crociani O, Rosati B, Faravelli L, Olivotto M, Wanke E. Herg encodes a K+ current highly conserved in tumors of different histogenesis: A selective advantage for cancer cells? Cancer Res. 1998;58:815–822. [PubMed] [Google Scholar]
- Boudreau RTM, Garduno R, Lin T-J. Protein phosphatase 2A and protein kinase Cα are physically associated and are involved in Pseudomonas aeruginosa-induced interleukin 6 production by mast cells. J Biol Chem. 2002;277:5322–5329. doi: 10.1074/jbc.M108623200. [DOI] [PubMed] [Google Scholar]
- Broad LM, Braun F-J, Lievremont J-P, Bird G, St J, Kurosaki T, Putney JW., Jr Role of the phospholipase C-inositol 1,4,5-trisphosphate pathway in calcium release-activated calcium current and capacitative calcium entry. J Biol Chem. 2001;276:15945–15952. doi: 10.1074/jbc.M011571200. [DOI] [PubMed] [Google Scholar]
- Chiang C-E, Roden DM. The long QT syndromes: genetic basis and clinical implications. J Am Coll Cardiol. 2000;36:1–12. doi: 10.1016/s0735-1097(00)00716-6. [DOI] [PubMed] [Google Scholar]
- Chiesa N, Rosati B, Arcangeli A, Olivotto M, Wanke E. A novel role for HERG K+ channels: spike-frequency adaptation. J Physiol. 1997;501:313–318. doi: 10.1111/j.1469-7793.1997.313bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Youm JB, Ryu SY, Earm YE, Ho W-K. Inhibition of acetylcholine-activated K+ currents by U73122 is mediated by the inhibition of PIP2-channel interaction. Br J Pharmacol. 2001;134:1066–1072. doi: 10.1038/sj.bjp.0704347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Melman Y, Palma E, Fishman GI, McDonald TV. Cyclic AMP regulates the HERG K+ channel by dual pathways. Curr Biol. 2000;10:671–674. doi: 10.1016/s0960-9822(00)00516-9. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubinsky JM, Oxford GS. Dual modulation of K channels by thyrotropin-releasing hormone in clonal pituitary cells. Proc Natl Acad Sci U S A. 1985;82:4282–4286. doi: 10.1073/pnas.82.12.4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmi A, Wenzel HJ, Schwatzkroin PA, Taglialatela M, Castaldo P, Bianchi L, Nerbonne J, Robertson GA, Janigro D. Do glia have heart? Expression and functional role for Ether-a-go-go currents in hippocampal astrocytes. J Neurosci. 2000;20:3915–3925. doi: 10.1523/JNEUROSCI.20-10-03915.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode N, Hughes K, Woodgett JR, Parker PJ. Differential regulation of glycogen synthase kinase-3 beta by protein kinase C isotypes. J Biol Chem. 1992;267:16878–16882. [PubMed] [Google Scholar]
- Hansra G, Bornancin F, Whelan R, Hemmings BA, Parker PJ. 12-O-Tetradecanoylphorbol-13-acetate-induced dephosphorylation of protein kinase Cα correlates with the presence of a membrane-associated protein phosphatase 2A heterotrimer. J Biol Chem. 1996;271:32785–32788. doi: 10.1074/jbc.271.51.32785. [DOI] [PubMed] [Google Scholar]
- Heath BM, Terrar DA. Protein kinase C enhances the rapidly activating delayed rectifier potassium current, IKr, through a reduction in C-type inactivation in guinea-pig ventricular myocytes. J Physiol. 2000;522:391–402. doi: 10.1111/j.1469-7793.2000.t01-2-00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann DW, Feng S, Nasuhoglu C. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE. 2001 doi: 10.1126/stke.2001.111.re19. http://wwwstke.org/cgi/content/full/OC-sigtrans;2001/111/re19. [DOI] [PubMed] [Google Scholar]
- Hofmann J. The potential for isoenzyme-selective modulation of protein kinase C. FASEB J. 1997;11:649–669. doi: 10.1096/fasebj.11.8.9240967. [DOI] [PubMed] [Google Scholar]
- Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–580. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- Kiehn J. Regulation of the cardiac repolarizing HERG potassium channel by protein kinase A. Trends Cardiovasc Med. 2000;10:205–209. doi: 10.1016/s1050-1738(00)00071-2. [DOI] [PubMed] [Google Scholar]
- Kiley SC, Parker PJ, Fabbro D, Jaken S. Differential regulation of protein kinase C isozymes by thyrotropin-releasing hormone in GH4C1 cells. J Biol Chem. 1991;266:23761–23768. [PubMed] [Google Scholar]
- Kolesnick RN. Thyrotropin-releasing hormone and phorbol esters stimulate sphingomyelin synthesis in GH3 pituitary cells. J Biol Chem. 1989;264:11688–11692. [PubMed] [Google Scholar]
- Lecchi M, Redaelli E, Rosati B, Gurrola G, Florio T, Crociani O, Curia G, Cassulini RR, Masi A, Arcangeli A, Olivotto M, Schettini G, Possani LD, Wanke E. Isolation of a long-lasting eag-related gene-type K+ current and its accommodating role during slow firing and prolactin release. J Neurosci. 2000;22:3414–3425. doi: 10.1523/JNEUROSCI.22-09-03414.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TFJ, Hsieh K-P, Porter BW. The sustained second phase of hormone-stimulated diacylglycerol accumulation does not activate protein kinase C in GH3 cells. J Biol Chem. 1990;265:7623–7631. [PubMed] [Google Scholar]
- Nagumo H, Sasaki Y, Ono Y, Okamoto H, Seto M, Takuwa Y. Rho kinase inhibitor HA-1077 prevents Rho-mediated myosin phosphatase inhibition in smooth muscle cells. Am J Physiol Cell Physiol. 2000;278:C57–65. doi: 10.1152/ajpcell.2000.278.1.C57. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, Catt KJ, Balla T. A wortmannin-sensitive phosphatidylinositol 4-kinase that regulates hormone-sensitive pools of inositolphospholipids. Proc Natl Acad Sci U S A. 1995;92:5317–5321. doi: 10.1073/pnas.92.12.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomero T, Barros F, Del Camino D, Viloria CG, de la Peña P. A G protein βγ dimer-mediated pathway contributes to mitogen-activated protein kinase activation by thyrotropin-releasing hormone receptors in transfected COS-7 cells. Mol Pharmacol. 1998;53:613–622. doi: 10.1124/mol.53.4.613. [DOI] [PubMed] [Google Scholar]
- Park SJ, Itoh T, Takenawa T. Phosphatidylinositol 4-phosphate 5-kinase type I is regulated through phosphorylation response by extracellular stimuli. J Biol Chem. 2001;276:4781–4787. doi: 10.1074/jbc.M010177200. [DOI] [PubMed] [Google Scholar]
- Ricciarelli R, Azzi A. Regulation of recombinant PKC alpha activity by protein phosphatase 1 and protein phosphatase 2A. Arch Biochem Biophys. 1998;355:197–200. doi: 10.1006/abbi.1998.0732. [DOI] [PubMed] [Google Scholar]
- Roden DM. Current status of class III antiarrhythmic drug therapy. Am J Cardiol. 1993;72:44–49B. doi: 10.1016/0002-9149(93)90040-j. [DOI] [PubMed] [Google Scholar]
- Rosati B, Marchetti P, Crociani O, Lecchi M, Lupi R, Arcangeli A, Olivotto M, Wanke E. Glucose- and arginine-induced insulin secretion by human pancreatic β-cells: the role of HERG K+ channels in firing and release. FASEB J. 2000;14:2601–2610. doi: 10.1096/fj.00-0077com. [DOI] [PubMed] [Google Scholar]
- Ruppersberg JP, Frank R, Pongs O, Stocker M. Cloned neuronal IK (A) channels reopen during recovery from inactivation. Nature. 1991;353:657–660. doi: 10.1038/353657a0. [DOI] [PubMed] [Google Scholar]
- Sch, äfer R, Wulfsen I, Behrens S, Weinsberg F, Bauer CK, Schwarz JR. The erg-like current in rat lactotrophs. J Physiol. 1999;518:401–416. doi: 10.1111/j.1469-7793.1999.0401p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schledermann W, Wulfsen I, Schwarz JR, Bauer CK. Modulation of rat erg1, erg2, erg3 and HERG K+ currents by thyrotropin-releasing hormone in anterior pituitary cells via the native signal cascade. J Physiol. 2001;532:143–163. doi: 10.1111/j.1469-7793.2001.0143g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simasko SM. Reevaluation of the electrophysiological actions of thyrotropin-releasing hormone in a rat pituitary cell line (GH3) Endocrinology. 1991;128:2015–2026. doi: 10.1210/endo-128-4-2015. [DOI] [PubMed] [Google Scholar]
- Smith GAM, Tsui H-W, Newell EW, Jiang X, Zhu X-P, Tsui FWL, Schlichter LC. Functional up-regulation of HERG K+ channels in neoplastic hematopoietic cells. J Biol Chem. 2002;277:18528–18534. doi: 10.1074/jbc.M200592200. [DOI] [PubMed] [Google Scholar]
- Storey NM, O'Brian JP, Armstrong DL. Rac and Rho mediate opposing hormonal regulation of the ether-a-go-go-related potassium channel. Curr Biol. 2002;12:27–33. doi: 10.1016/s0960-9822(01)00625-x. [DOI] [PubMed] [Google Scholar]
- Suh B-C, Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4, 5-bisphosphate synthesis. Neuron. 2002;35:507–520. doi: 10.1016/s0896-6273(02)00790-0. [DOI] [PubMed] [Google Scholar]
- Way KJ, Chou E, King GL. Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol Sci. 2000;21:181–187. doi: 10.1016/s0165-6147(00)01468-1. [DOI] [PubMed] [Google Scholar]
- Weinsberg F, Bauer CK, Schwarz JR. The class III antiarrhythmic agent E-4031 selectively blocks the inactivating inward-rectifying potassium current in rat anterior pituitary tumor cells (GH3/B6 cells) Pflugers Arch. 1997;434:1–10. doi: 10.1007/s004240050356. [DOI] [PubMed] [Google Scholar]
- Willars GB, Nahorski SR, Challis RAJ. Differential regulation of muscarinic acetylcholine receptor-sensitive polyphosphoinositide pools and consequences for signaling in human neuroblastoma cells. J Biol Chem. 1998;273:5037–5046. doi: 10.1074/jbc.273.9.5037. [DOI] [PubMed] [Google Scholar]
- Wimmers S, Wulfsen I, Bauer CK, Schwarz JR. Erg1, erg2 and erg3 K channel subunits are able to form heteromultimers. Pflugers Arch. 2001;441:450–455. doi: 10.1007/s004240000467. [DOI] [PubMed] [Google Scholar]
- Xie L-H, Horie M, Takano M. Phospholipase C-linked receptors regulate the ATP-sensitive potassium channel by means of phosphatidylinositol 4, 5-bisphosphate metabolism. Proc Natl Acad Sci U S A. 1999;96:15292–15297. doi: 10.1073/pnas.96.26.15292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Cayabyab FS, Pennefather PS, Schlichter LC, De Coursey TE. HERG-like K+ channels in microglia. J Gen Physiol. 1998;111:781–794. doi: 10.1085/jgp.111.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcangeli A, Becchetti A, Mannini A, Mugnai G, De Filipi P, Tarone G, Del Bene MR, Barletta E, Wanke E, Olivotto M. Integrin-mediated neurite outgrowth in neuroblastoma cells depends on the activation of potassium channels. J Cell Biol. 1993;12:1131–1143. doi: 10.1083/jcb.122.5.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]