

Abstract
Whole-cell patch-clamp recordings were made from neurons in the trigeminal nucleus caudalis and trigeminal ganglion, in vitro, to investigate the cellular actions of the endogenous cannabinoid, anandamide. Anandamide has been shown to act through both the cannabinoid receptor 1 (CB1) and the vanilloid receptor 1 (VR1). Anandamide (30 μm) caused a 54 % increase in the rate of miniature excitatory post-synaptic currents (mEPSCs), without affecting their amplitude. The effect of anandamide was blocked by the VR1 antagonist capsazepine (20 μm), but not by the CB1-specific antagonist AM251 (3 μm). Application of the VR1 receptor agonist capsaicin (300 nm) caused a 4200 % increase in the mEPSC rate. In dissociated trigeminal ganglion neurons, both anandamide and capsaicin caused an outward current in neurons that were voltage clamped at +40 mV. The maximal outward current produced by anandamide (EC50, 10 μm) was 45 % of that produced by capsaicin (10 μm). Co-application of the VR1 antagonist capsazepine (30 μm) completely reversed the effects of both capsaicin and anandamide. The anandamide transport inhibitor, AM404 (30 μm) caused a 40 % increase in mEPSC rate in the slice preparation and an outward current in dissociated neurons. The latter current was reversed by the VR1 antagonist iodoresiniferatoxin (1 μm). The fatty acid amide hydrolase (FAAH) inhibitors phenylmethylsulfonyl fluoride (PMSF) (20 μm) and OL53 (1 μm) did not enhance the effect of anandamide in either the slice or dissociated neuron preparations. These results suggest that within the superficial medullary dorsal horn, anandamide (30 μm) acts presynaptically to enhance the release of glutamate via activation of the VR1 receptor.
Anandamide (arachidonylethanolamide) was originally isolated from the brain as an endogenous ligand for the cannabinoid receptor (Devane et al. 1992) and has been shown to be a partial agonist at cannabinoid type-1 receptors (CB1) but functionally inactive at CB2 receptors (Howlett, 2000). In behavioural studies, anandamide exerts an antinociceptive effect (Calignano, 1998; Di Marzo, 2000). This anandamide-induced antinociception persists in animals in which the gene encoding the CB1 receptor has been knocked out, suggesting that anandamide works through other receptors (Di Marzo, 2000). Furthermore, anandamide has been shown to act at the cloned vanilloid type 1 receptor (VR1) (Caterina et al. 1997) in heterologous systems and at native VR1 receptors in dorsal root and trigeminal ganglion neurons (Zygmunt et al. 1999; Smart et al. 2000; Tognetto et al. 2001; Roberts et al. 2002).
The nucleus caudalis of the spinal trigeminal nucleus in the dorsal part of the caudal medulla receives primary afferent fibres from the head and neck, including unmyelinated C-fibres (Light et al. 1992) on which VR1 is predominantly located (Caterina et al. 1997). We have previously demonstrated an inhibitory action of cannabinoid agonists on inhibitory but not excitatory synaptic transmission in the medullary dorsal horn via CB1 receptors (Jennings et al. 2001).
Since the VR1 is predominantly expressed in small diameter sensory neurons (Caterina et al. 1997) and in view of the reported effects of anandamide on VR1, we examined the effects of anandamide in the medullary dorsal horn and trigeminal ganglia of the rat.
Methods
Medullary dorsal horn slices
Sprague-Dawley rats (12–21 days old) were anaesthetised with halothane, decapitated and horizontal brain slices (250 μm), containing the trigeminal nucleus caudalis were cut (Grudt & Williams, 1994). All procedures reported conformed to the University of Sydney Animal Ethics Committee guidelines. Briefly, a block of brainstem containing the medulla caudal to the obex, was placed in a vibratome bath containing ice cold (< 4 °C) artificial cerebrospinal fluid (ACSF). Two to three slices were taken from near the dorsal surface of the medulla, and hemisected before being transferred to a submerged chamber containing ACSF equilibrated with 95 % O2−5 % CO2, and maintained at 34 °C. The slices were transferred to a superfusion chamber (32 °C) for recording (Jennings et al. 2001). The ACSF contained (mm): NaCl 126, KCl 2.5, NaH2PO4 1.4, MgCl2 1.2, CaCl2 2.4, glucose 11 and NaHCO3 25.
Neurons in the substantia gelatinosa of the trigeminal nucleus caudalis were clearly visible as a translucent band, just medial to the spinal trigeminal tract, which enters the slice 5–6 mm rostral to the nucleus caudalis, and travels along the lateral edge of the slice (Grudt & Williams, 1994). These neurons were visualised using infrared Nomarski optics, and whole-cell patch-clamp recordings (patch electrodes, 3–7 MΩ), under voltage clamp (holding potential, −74 mV), were made using an Axopatch 200B amplifier (Axon Instruments, Union City, CA, USA). Miniature EPSCs (mEPSCs) and electrically evoked EPSCs (eEPSCs) were recorded using a CsCl-based internal solution containing (mm): CsCl 140, EGTA 10, Hepes 5, CaCl2 2 and MgATP 2 (pH 7.3; osmolarity, 275–285 mosmol l−1). Series resistance (< 18 MΩ) was compensated by 80 % and continuously monitored during experiments. A liquid junction potential of −4 mV for the CsCl-based internal solution was corrected for.
Miniature EPSCs were filtered (2 kHz low-pass filter) and sampled at 5 kHz for on-line and later off-line analysis (Axograph 4.6, Axon Instruments), and were recorded in the presence of picrotoxin (100 μm), strychnine (3 μm) and tetrodotoxin (300 nm), to block GABAA, glycine and sodium channels, respectively. Miniature EPSCs above a preset threshold (5.0–5.2 standard deviations above baseline noise) were automatically detected by a sliding template algorithm, and then manually checked off-line. Miniature EPSCs were then counted in 10 s epochs every 15 s to construct rate-time plots. The mEPSC rate and amplitude recorded during a 3 min period in the absence (control) and presence of anandamide were compared.
Electrically evoked EPSCs (eEPSCs) were elicited via bipolar tungsten stimulating electrodes placed in the spinal trigeminal tract about 1 mm rostral to the site of recording. Stimuli were delivered once every 15 s (0.07 Hz) at intensities in the range 3–10 V (duration, 0.06–0.2 ms), and were recorded in the presence of picrotoxin (100 μm) and strychnine (3 μm). For paired-pulse experiments, two stimuli of identical strength were applied with an inter-stimulus interval of 70 ms. Amplitudes were measured to construct time plots of the eEPSC amplitude. For analysis of the paired-pulse experiments, the amplitude of the first eEPSC was normalised to measure the relative amplitude of the second eEPSC (eEPSC2/eEPSC1).
Dissociated trigeminal ganglion neurons
Sprague-Dawley rats (3–4 weeks old) were used for this study. Rats were anaesthetised with halothane (4 %), killed by cervical dislocation and the trigeminal ganglia removed and placed in ice cold ACSF gassed with 95 % O2−5 % CO2. Ganglia were cut up with iridectomy scissors and incubated at 32–34 °C for 30 min in ACSF. The ganglia pieces were then transferred to oxygenated Hepes-buffered saline (HBS) containing 20 units ml−1 papain and incubated at 32–34 °C for 20–25 min. HBS contained (mm): NaCl 154, KCl 2.5, CaCl2 2.5, MgCl2 1.5, Hepes 10 and glucose 10; pH was adjusted to 7.2 with NaOH; osmolarity, 330 ± 5 mosmol l−1. The digestion was terminated by addition of HBS containing 1 mg ml−1 bovine serum albumin (BSA) and 1 mg ml−1 trypsin inhibitor (chicken egg white ovomucoid, Type II-O). Minced ganglia were washed free of enzyme and enzyme inhibitors with HBS at room temperature (22–24 °C). Cells were released by gentle trituration through decreasing bore, silanised Pasteur pipettes with fire-polished tips. The cells were plated onto plastic culture dishes and kept at room temperature in HBS.
Ionic currents from rat trigeminal neurons were recorded in whole-cell configuration of the patch-clamp method (Hamill et al. 1981) at room temperature. Dishes were continually perfused with HBS. The intracellular pipette solution contained (mm): CsCl 120, Hepes 10, EGTA 10, CaCl2 2, MgATP 5, Na2GTP 0.2 and NaCl 5; pH was adjusted to 7.3 with CsOH; osmolarity, 285 ± 5 mosmol l−1. Recordings were made using an Axopatch-1D or 200B amplifier (Axon Instruments) using pClamp acquisition software (v 5.5, Axon Instruments). Currents were sampled at 1 kHz and recorded on hard disk for later analysis. Patch pipettes were pulled from borosilicate glass (AM Systems, Everett, WA, USA). The pipette input resistance was in the range 0.7–1.5 MΩ. The capacitance of individual cells was in the range 8–20 pF, with a series resistance between 1 and 5 MΩ. Series resistance compensation of at least 80 % was used in all experiments. Capacitance transients were compensated by using the manual compensation on the Axopatch-1D.
Currents through VR1 were recorded by voltage clamping the neurons at +40 mV, a potential at which calcium entry through the channel is minimised and desensitisation of the channel to brief exposures of capsaicin is minimal (Piper et al. 1999; Roberts et al. 2002). Drugs were perfused over the cell via a series of pipes positioned 100–200 μm from the cell. Drug solutions were changed by moving the pipes using an electronic micromanipulator; solution exchange between adjacent pipes took less than 1 s. Stock solutions of drugs in ethanol were diluted directly into the HBS. The ethanol concentration did not exceed 0.1 %, a concentration that by itself did not affect the trigeminal neurons. Drugs were applied until the currents they produced reached equilibrium, and the dose-response relationships for anandamide and capsaicin were constructed either cumulatively or by successive perfusion of increasing concentrations of agonist. Concentration-response data was obtained from any one cell for only one drug. Anandamide-induced currents in each cell were expressed as a percentage of the current produced by a maximally effective concentration of capsaicin (10 μm) applied after washout of anandamide.
Drugs
Stock solutions of all drugs were diluted to working concentrations using ACSF immediately before use and applied by superfusion unless otherwise noted. Picrotoxin, strychnine and baclofen were obtained from Sigma (Sydney, Australia); CNQX (6-cyano-7-nitroquinoxaline-2, 3-dione), capsaicin, capsazepine, AM251 (N-(4-hydroxyphenyl)arachidonylamide), iodoresiniferatoxin (6,7-deepoxy-6,7-didehydro-5-deoxy-21-dephenyl-21-(phenylmethyl)-daphnetoxin,20-(4-hydroxy-5-iodo-3-methoxybenzeneacetate)) and anandamide (in water soluble emulsion) were from Tocris Cookson (Bristol, UK); anandamide (in ethanol), AM404 (N-(4-hydroxyphenyl) arachidonoylamide) and methanandamide ((r)-(+)-arachidonyl-1′-hydroxy-2′-propylamide) were from Cayman Chemical (Ann Arbor, MI, USA); TTX (tetrodotoxin) was from Alomone Labs Ltd (Jerusalem, Israel); SR141716A (N-piperidino-5-(4-chlorophenyl)-l-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxamide) was donated by Sanofi Recherche; OL53 (compound 53 in the paper by Boger et al. 2000) was donated by Dr Dale Boger (The Scripps Research Institute, San Diego, CA, USA); and CGP56999A was donated by Novartis (Basel, Switzerland). Stock solutions of anandamide, AM404, capsaicin and capsazepine were prepared in ethanol and diluted with ACSF to a final concentration of 0.02–0.1 % ethanol. Similarly, stock solutions of AM251 and SR141716A were prepared in dimethyl sulphoxide and diluted using ACSF to a final concentration of 0.01–0.1 % dimethyl sulphoxide. The superfusion system was dismantled and rinsed with ethanol after each recording. Stock solutions of all other drugs were made in distilled water, or added directly to the ACSF.
All pooled data are expressed as means ±s.e.m. and statistical comparisons were made using Student's t test, paired or unpaired (as appropriate), unless otherwise stated. The sample variances were tested in each unpaired case (F test) and if significantly different, Welch's correction was used.
Results
Anandamide excites glutamatergic nerve terminals in the medullary dorsal horn
Miniature excitatory postsynaptic currents (mEPSCs) were recorded from neurons in the trigeminal nucleus caudalis in the presence of tetrodotoxin (TTX; 300 nm), picrotoxin (100 μm) and strychnine (3 μm). Miniature EPSCs had a mean rate of 1.4 ± 0.4 Hz and mean amplitude of 39 ± 2 pA (n = 14), and were abolished by the AMPA/kainate receptor antagonist CNQX (5 μm). Superfusion of anandamide (30 μm in ethanol) increased the mEPSC rate by 54 ± 6 % (range, 9–82 %; n = 14; P < 0.01), but had no effect on the amplitude. The mean amplitude of mEPSCs was similar in the absence (39 ± 2 pA) or presence of anandamide (30 μm; 38 ± 2 pA; n = 14; P > 0.1). The increase in mEPSC rate following anandamide superfusion was reversed on washout. The vehicle used to dissolve anandamide, ethanol (0.02 %), had no effect on the mEPSC rate (n = 4; P = 0.3). Superfusion of anandamide (30 μm, dissolved in a water-soluble emulsion) caused a 79 ± 16 % increase in mEPSC rate (range, 28–130 %; n = 6; Fig. 1) without affecting the amplitude. The mean mEPSC amplitude was similar in the absence (37 ± 5 pA) and presence of anandamide (44 ± 7 pA; n = 6; P > 0.05; Fig. 1B). Anandamide in water-soluble emulsion caused a similar increase in mEPSC rate to anandamide in ethanol (P > 0.05).
Figure 1. Anandamide increases the mEPSC rate without altering the amplitude.

A, time plot of mEPSC rate showing an increase after application of anandamide (in emulsion; AEA; 30 μm) and then following application of capsaicin (300 nm). Inset shows the same plot on a different scale to demonstrate the magnitude of the capsaicin response. B, traces of raw data showing mEPSCs in control and following superfusion of anandamide, capsaicin or CNQX (5 μm). C, normalised cumulative histograms of mEPSC amplitude in the absence (ctrl; continuous line) and presence of anandamide (AEA; dotted line). Inset shows averaged mEPSCs before (ctrl; continuous line) and during application of anandamide (AEA; dashed line). D, graph showing the mEPSC rate before (control) and during (AEA) application of anandamide. Data in A-C are taken from one neuron.
Superfusion of the VR1 agonist, capsaicin (300 nm) onto some cells, subsequent to either preparation of anandamide, rapidly induced an increase of 4200 ± 2000 % in mEPSC rate (range, 480–24200 %; n = 11; P < 0.0001; Fig. 1A, inset), without significantly changing the amplitude (P > 0.05).
The effect of anandamide is mediated by VR1
In slices incubated with the VR1 antagonist, capsazepine (20 μm, for at least 30 min), mEPSCs had a mean rate of 4.3 ± 2 Hz and mean amplitude of 44 ± 4 pA, in the absence of anandamide. The addition of anandamide (30 μm) had no significant effect on the rate of mEPSCs (increase, 13 ± 6 %; range, −4 to 40 %; n = 7; P > 0.1; Fig. 2). In the presence of capsazepine, the mean amplitude of the mEPSCs was similar in the absence (44 ± 4 pA) and presence of anandamide (44 ± 4 pA; n = 7; P = 0.9; Fig. 2B). The effect of anandamide on mEPSC rate was less in the presence of capsazepine than in its absence (P < 0.05).
Figure 2. The VR1 antagonist capsazepine attenuates the effects of anandamide.

A, time plot of mEPSC rate in slices pre-treated with capsazepine (20 μm). The addition of anandamide does not significantly change the mEPSC rate. Note that the capsaicin-induced increase in rate is significantly reduced by capsazepine. B, cumulative plot analysis of mEPSCs in the same neuron showing the peak amplitude; inset shows averaged mEPSCs before (control; continuous line) and during application of anandamide (AEA; dotted line). C, pooled results demonstrating that anandamide has no effect on the mEPSC rate in cells pre-treated with capsazepine.
Pre-incubation with capsazepine (20 μm) greatly attenuated the response to capsaicin (300 nm). Capsaicin (300 nm) caused a small significant increase in the mEPSC rate of 32 ± 16 % under these conditions (n = 3; P < 0.05; Fig. 2A), without changing the amplitude (P > 0.1). The capsaicin-mediated increase in mEPSC rate was significantly less in the presence of capsazepine than in its absence (P < 0.05).
In the presence of the CB1 antagonist, SR141716 (3 μm), anandamide (30 μm) induced a non-significant increase of 28 ± 15 % in the mEPSC rate (range; −7 to 93 %; n = 6; P > 0.1) without altering the mEPSC amplitude (n = 6; P > 0.5). In the presence of the CB1 antagonist AM251 (3 μm) anandamide (30 μm) induced a 36 ± 9 % increase in the mEPSC rate (range, 10–64 %; n = 6; P < 0.05 compared to control; Fig. 3A), without significantly altering the amplitude of mEPSCs (n = 6; P > 0.1). The mean anandamide-induced increase in mEPSC rate was similar (P > 0.1; unpaired t test) in the absence (54 ± 6 %; n = 14) and presence of AM251 (36 ± 9 %; n = 6). The addition of AM251 to the perfusate did not significantly increase the baseline mEPSC rate (P = 0.98; unpaired Student's t test). The anandamide-induced increase in mEPSC rate in the presence of capsazepine was not significantly different to that in the presence of SR141716A (P = 0.35).
Figure 3. Effects of agents on the action of anandamide on mEPSC frequency.

Pooled data showing the increase in mEPSC frequency in the presence of anandamide (30 μm) and the CB1 antagonist AM251 (3 μm) (A), methanandamide (30 μm) (B), anandamide (mAEA; 30 μm) and the FAAH inhibitor PMSF (20 μm) (C) and the anandamide transport inhibitor AM404 (30 μm) (D). There was no mean mEPSC rate increase in the presence of anandamide and the FAAH inhibitor PMSF.
Methanandamide and AM404 also increase the mEPSC rate
The stable anandamide analogue R1-methanandamide (30 μm) caused a 60 ± 13 % increase in mEPSC rate (range, 16–132 %; n = 9; P < 0.05; Fig. 3B), without altering the amplitude. The mean mEPSC amplitude was similar in the absence (36 ± 3 pA) and presence of methanandamide (37 ± 3 pA; n = 9; P > 0.01). The increase in mEPSC rate induced by methanandamide was similar to that caused by superfusion of anandamide when compared to basal rates.
Superfusion of the anandamide transporter inhibitor, AM404 (30 μm), caused an increase of 40 ± 10 % in the frequency (range, 4–67 %; n = 6; P < 0.05; Fig. 3D), but not the amplitude of mEPSCs. The mean amplitude of mEPSCs was similar in the absence (33 ± 5 pA) or presence of AM404 (33 ± 5 pA; n = 6; P > 0.5).
Effects of anandamide in the presence of FAAH inhibitors
In neurons from slices pre-treated and maintained in the presence of the serine protease and FAAH inhibitor PMSF (20 μm), anandamide (30 μm) had no effect on the mEPSC rate (increase, 23 ± 15 %; range, −23 to 118 %; n = 10; P > 0.1; Fig. 3C). The mean amplitude of mEPSCs was similar in the presence of PMSF alone (34.9 ± 3.5 pA) or the presence of PMSF and anandamide (30 μm; 34 ± 3 pA; n = 10; P > 0.5).
In slices incubated in the presence of the novel FAAH inhibitor OL53 (1 μm, for at least 30 mins), the mean mEPSC rate was 3.0 ± 0.7 Hz (n = 7). There was no change in the mEPSC amplitude following incubation with OL53 (38 ± 2 pA without OL53 treatment and 37 ± 6 pA following incubation; P > 0.5). Superfusion of anandamide (30 μm) in the presence of OL53 had no effect on the mEPSC rate (increase, 8 ± 12 %; n = 7; P > 0.5 compared to control; range, −17 to 57 %). There was no significant change in the amplitude in the absence (38 ± 6 pA) and presence of anandamide (40 ± 7 pA; n = 7; P > 0.1).
The basal mEPSC rate in the absence of PMSF or OL53 (1.7 ± 0.2 Hz; n = 45) was less than in the presence of PMSF (5.0 ± 2.0 Hz; n = 10; P < 0.05) or OL53 (3.0 ± 0.7 Hz; n = 7; P < 0.05). Preincubation of slices with the VR1 antagonist iodoresiniferatoxin (1 μm) did not prevent the increase in mEPSC rate produced by OL53 (4.1 ± 0.8 Hz; n = 8; P = 0.5), suggesting that the increase in mEPSC rate seen in the presence of OL53 is not mediated by VR1.
Anandamide does not alter evoked EPSCs
Glutamatergic EPSCs were electrically evoked by stimulating the trigeminal tract. Recordings were made in the medullary dorsal horn in the presence of strychnine (3 μm) and picrotoxin (100 μm). Evoked EPSCs (eEPSCs) had a mean amplitude of 156 ± 40 pA (n = 7) and were abolished by CNQX (5 μm). Superfusion of anandamide (30 μm) had no effect on the amplitude of the eEPSCs (decrease, 5 ± 3 %; range, −5 to 18 %; n = 10; P > 0.1; Fig. 4). As a control, baclofen was also used on three of the cells reducing the eEPSCs by 89 ± 6 %. Under control conditions, the mean ratio of the paired eEPSCs (inter-stimulus interval, 70 ms) was 0.8 ± 0.1 (eEPSC2/eEPSC1; range, 0.6–0.9; n = 8; Fig. 4). Superfusion of anandamide produced no significant change in the mean ratio of eEPSC2/eEPSC1 (110 ± 9 % of control; P > 0.1). Baclofen (10 μm) facilitated the second eEPSC relative to the first, by 117 and 148 % in two of the cells (Fig. 4C). In the third cell, baclofen abolished both the first and second eEPSC.
Figure 4. Anandamide does not alter the evoked EPSC amplitude.
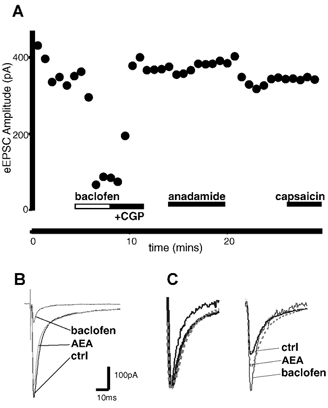
A, time plot of the evoked EPSC amplitude before and during application of anandamide (30 μm; AEA). Neither anandamide nor capsaicin (300 nm) altered the eEPSC. Baclofen (10 μm) causes a decrease in amplitude. B, averaged eEPSC before (control; thick line) and during application of anandamide (AEA; grey line), and after application of baclofen (10 μm; thin line). C, normalised averaged responses to identical paired stimuli (inter-stimulus interval, 70 ms), with the eEPSC1 normalised (left) to demonstrate the lack of change in eEPSC2 relative to eEPSC1, following application of anandamide (AEA). Baclofen caused a facilitation of the eEPSC2 relative to eEPSC1.
Anandamide and AM404 activate VR1 in dissociated trigeminal neurons
Superfusion of anandamide (1–100 μm) or capsaicin (100 nm−30 μm) produced an outward current in 28 out of 30 small rat trigeminal neurons that were voltage clamped at + 40 mV (Fig. 5). The increases in outward current were concentration-dependent, with an EC50 for capsaicin of 500 nm, and for anandamide of 10 μm. The maximum increase in outward current produced by anandamide was 45 % of that produced by a maximally effective concentration of capsaicin (10 μm) in the same cells. The effects of anandamide (100 μm, n = 7; Fig. 5) and capsaicin (1 μm−10 μm, n = 6) were completely reversed by co-superfusion of the VR1 antagonist capsazepine (30 μm).
Figure 5. Anandamide activates VR1 in dissociated trigeminal ganglion neurons.

A, time plot of the VR1-mediated current at +40 mV illustrating the effects of anandamide (AEA; 10 μm and 100 μm) and the reversal of the 100 μm anandamide-induced current following the addition of capsazepine (30 μm). Also illustrated is the response of the cell to capsaicin (10 μm). B, concentration-response relationship for capsaicin (EC50, 500 nm) and anandamide (EC50, 10 μm) in trigeminal ganglion neurons. C, histogram showing that the outward current induced by AM404 (30 μm) can be reversed by the addition of the VR1 receptor antagonist iodoresiniferatoxin (1 μm). The fatty acid amide hydrolase (FAAH) inhibitor, OL53 (300 nm) has no significant effect on the anandamide-induced outward current, and causes only a small outward current when applied alone.
Superfusion of the anandamide transport inhibitor AM404 (30 μm) produced a mean outward current of 28 ± 6 % of that produced by 10 μm capsaicin in the same cells. This outward current was largely reversed (to 3 ± 1 % of the capsaicin-dependent (10 μm) current; n = 4; P < 0.05; Fig. 5C) by the co-application of the VR1-specific antagonist iodoresiniferatoxin (1 μm).
Superfusion of anandamide (3 μm) induced an outward current of 24 ± 2 % of the current produced by 10 μm capsaicin in the same cells. After washout, application of the FAAH inhibitor OL53 (300 nm) produced no current (0.8 ± 1 %), and subsequent re-addition of anandamide (3 μm) resulted in an outward current of 21 ± 4 %, which was not significantly different from the application of anandamide alone (n = 6; P > 0.5; Fig. 5C).
Discussion
In the present study we have demonstrated a role for anandamide as a vanilloid receptor agonist in the medullary dorsal horn. Anandamide mimicked the effect of capsaicin, causing an outward current in dissociated trigeminal ganglion neurons, which was abolished by the VR1 antagonist capsazepine. In addition, anandamide caused an increase in the mEPSC rate, without changing the amplitude, which was also blocked by capsazepine. These findings suggest that anandamide has presynaptic VR1-mediated actions on primary afferent terminals at the level of the medullary dorsal horn and that the actions of anandamide are peripherally mediated. However, an effect via excitatory interneuron terminals cannot be excluded. Recent studies in CB1 receptor knockout animals have suggested a non-CB1-mediated effect of anandamide (Di Marzo, 2000), and it has been suggested that this non-CB1 receptor-mediated effect is sensitive to vanilloid antagonists (Hajos & Freund, 2002). It is likely therefore that part of the anandamide acitivity described in this study is mediated by this vanilloid-sensitive non-CB1 receptor.
The presynaptic effect of anandamide was likely to be mediated by VR1 receptors and not cannabinoid CB1 receptors. Thus, the effect of anandamide on mEPSC rate was blocked by the VR1 antagonist capsazepine, but not by the CB1 receptor antagonist AM251. However, another CB1 receptor antagonist, SR141716A (3 μm) prevented the anandamide-induced increase in mEPSC rate. It is likely that SR141716A prevented the actions of anandamide on VR1 because it has also been reported to act as a VR1 antagonist at concentrations greater than 1 μm, attenuating the VR1-mediated responses of both anandamide and capsaicin, in a similar fashion to capsazepine (De Petrocellis et al. 2001). Capsaicin also produced an increase in mEPSC frequency, as previously reported in the spinal dorsal horn (Urban & Dray, 1992; Yang et al. 1998, 1999), which was blocked by capsazepine.
While anandamide produced a VR1-mediated increase in mEPSC in trigeminal dorsal horn, the maximal response was less than 1 % of that produced by capsaicin. This may have been due to a number of factors. Firstly, anandamide is a partial agonist at VR1, unlike capsaicin. In dissociated trigeminal ganglion neurons the maximal VR1-mediated response to anandamide was 45 % of that produced by capsaicin (Roberts et al. 2002). Secondly, the choice of vehicle appears to influence the diffusion of anandamide to its site of action, since the preparation of anandamide in water-soluble emulsion caused a larger increase in the mEPSC rate than anandamide in ethanol vehicle. If a reduced concentration of anandamide reaches synapses throughout the slice preparation, the concentration of drug may be well below the EC50. Thirdly, anandamide may be metabolised in slices before reaching its target. The effect of R1-methanandamide was greater than that of anandamide suggesting that anandamide is rapidly metabolised, particularly since anandamide is reported to be 10 times more potent than methanandamide at VR1 in isolated cells (Zygmunt et al. 1999; Smart et al. 2000; Roberts et al. 2002). In an attempt to limit the intracellular metabolism thought to be responsible for hydrolysis of anandamide, slices were pre-treated with either PMSF or OL53, because both agents have been reported to inhibit FAAH (Boger et al. 2000; De Petrocellis et al. 2001). It is surprising that this treatment significantly increased the basal mEPSC rate and prevented the actions of anandamide. It is possible therefore that the pre-treatment with FAAH inhibitors increased endogenous anandamide levels and caused VR1 activation. This would mask the effects of exogenously applied anandamide. However, the failure of iodoresiniferatoxin to reverse the mEPSC rate increase, indicates that FAAH inhibitors increase mEPSC rate via a mechanism independent of the anandamide binding site on VR1.
Blockade of FAAH increases the efficacy of anandamide in whole tissue preparations, presumably by preventing metabolism (Boger et al. 2000; De Petrocellis et al. 2001). However, it is also possible that both PMSF and OL53 have non-specific actions in slice preparations or that metabolites of anandamide are the active constituents. In the guinea-pig bronchus, PMSF increased the VR1-mediated contractile effect of anandamide in the presence of the non-steroidal anti-inflammatory drug, indomethacin (Craib et al. 2001), suggesting that the blockade of cyclo-oxygenase is a necessary prerequisite for anandamide-induced contraction of the guinea-pig bronchus. They further suggested that lipoxygenase metabolites contribute to the anandamide-induced effect by acting directly on VR1 (Craib et al. 2001). It is possible therefore that the metabolites of anandamide activate the VR1.
The activity of anandamide at VR1 is thought to require facilitated transport into the cell (De Petrocellis et al. 2001). We sought to modulate the efficacy of the anandamide interaction with VR1 by blocking the anandamide transporter with the drug AM404. Unfortunately, AM404 alone increased the mEPSC rate. In the present study AM404 activated VR1 in isolated trigeminal ganglion neurons. AM404 has also been shown to act as an agonist at VR1 in heterologous systems and mouse trigeminal neurons (Zygmunt et al. 2000; De Petrocellis et al. 2001; Ralevic et al. 2001; Roberts et al. 2002). Indeed it was reported that the concentration of AM404 required to activate VR1 is 400-fold less than that required to inhibit the anandamide membrane transporter (De Petrocellis et al. 2000). This direct effect of AM404 on the vanilloid receptors negates its usefulness in blocking the anandamide transporter when examining VR1-mediated effects.
The low potency and efficacy of anandamide in the present study suggests that it is unlikely that it reaches sufficient concentrations in vivo to stimulate VR1 under basal conditions. In a study using in vivo micro-dialysis, the basal level of anandamide in the peri-aqueductal grey of 3 fm was recorded, and electrical stimulation identical to that which elicits a stimulus produced antinociception in behavioural experiments, and increased the extracellular anandamide concentration by 140 %. This stimulus-produced antinociception could be markedly attenuated following administration of the CB1 antagonist SR141716A (Walker et al. 1999). Anandamide levels of 20–30 pmol g−1 were recorded using liquid chromatography-mass spectrometry analysis of brain tissue samples in the mouse (Di Marzo, 2000). In another study, Yang et al. (1999) failed to detect anandamide in any central nervous system region tested, suggesting that the concentration is below 0.1 pmol (mg protein)−1, the detection threshold of their system. In the same study, they were able to measure levels of an anandamide precursor, N-arachidonoylphosphatidylethanolamide, the highest levels of which are found in the spinal cord. Although the basal anandamide concentration in primary afferent terminals is unknown, it would appear that both basal and depolarisation-induced levels are several orders of magnitude less than those applied exogenously in this study and many others. Considering that the highest concentration of anandamide precursor is found in the spinal cord (Yang et al. 1999), it remains possible that sufficient anandamide to activate VR1 might be released following extreme noxious stimulation. If this were the case, anandamide could either be considered to be an agent of hyperalgesia and participate in chronic pain or it may act to desensitise VR1 (Smart et al. 2000) so serving a neuro-protective role. A further consideration is that anandamide might exert VR1-mediated effects predominantly in the periphery where endogenous anandamide levels are reported to be 10 times higher than those in brain or plasma (Calignano, 1998).
Anandamide has been shown to act at both VR1 and CB1 receptors. In relation to pain modulation, it has been shown that both VR1 and CB1 receptors are expressed in both dorsal root ganglion (DRG) and trigeminal ganglion neurons. VR1 mRNA is found in small to medium neurons in both the trigeminal and dorsal root ganglia (Caterina et al. 1997; Helliwell et al. 1998), but in the trigeminal ganglia CB1 mRNA is found almost exclusively in medium to large neurons, and there is little co-localisation with markers for peptides or VR1 (Price et al. 2001). In contrast, both CB1 and VR1 are expressed on small diameter neurons in DRG and there is significant co-localisation (Ahluwalia et al. 2000). This suggests that modulation of nociceptive information by CB1 receptors in the spinal and medullary dorsal horn may differ. Indeed, studies examining the cellular effects of the cannabinoid agonist WIN 55,212 on cells in the superficial medullary or spinal dorsal horn have produced different results. In the spinal dorsal horn, WIN 55,212 presynaptically inhibited excitatory transmission (Morisset & Urban, 2001); whilst in the medullary dorsal horn, WIN 55,212 has no effect on excitatory transmission but inhibited inhibitory transmission (Jennings et al. 2001). In the spinal dorsal horn, the VR1-mediated effects of both capsaicin (Yang et al. 1998) and preliminary data for anandamide (Morisset et al. 2001) are similar to those reported here for the medullary dorsal horn. This difference could be exploited for delivering specific therapies to control pain with an origin in the head and neck region.
In conclusion, anandamide acts presynaptically to increase excitatory neurotransmission in the medullary dorsal horn, and this effect is mediated by VR1. This effect mimics that of capsaicin on the same receptor. Whether the concentration of anandamide used in this study is ‘physiologically relevant’, and what role it plays in VR1-mediated pain modulation has yet to be ascertained.
Acknowledgments
E.A.J., C.W.V. and M.J.C. were supported by the NH & MRC. M.J.C. is also supported by The Medical Foundation of The University of Sydney. The authors thank Dr Mark Connor for his helpful comments on the manuscript, and for providing the preliminary trigeminal ganglion data. The authors gratefully acknowledge the donation of SR141716A from Dr Madeline Mossé, Sanofi Recherche, Montpellier, France; the donation of OL53 by Dr Dale Boger, The Scripps Research Institute, La Jolla, California, and the donation of sumatriptan by Glaxo Smith Kline (UK).
REFERENCES
- Ahluwalia J, Urban L, Capogna M, Bevan S, Nagy I. Cannabinoid 1 receptors are expressed in nociceptive primary sensory neurons. Neuroscience. 2000;100:685–688. doi: 10.1016/s0306-4522(00)00389-4. [DOI] [PubMed] [Google Scholar]
- Boger DL, Sato H, Lerner AE, Hedrick MP, Fecik RA, Miyauchi H, Wilkie GD, Austin BJ, Patricelli MP, Cravatt BF. Exceptionally potent inhibitors of fatty acid amide hydrolase: the enzyme responsible for degradation of endogenous oleamide and anandamide. Proc Natl Acad Sci U S A. 2000;97:5044–5049. doi: 10.1073/pnas.97.10.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calignano A. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor – a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Craib SJ, Ellington HC, Pertwee RG, Ross RA. A possible role of lipoxygenase in the activation of vanilloid receptors by anandamide in the guinea-pig bronchus. Br J Pharmacol. 2001;134:30–37. doi: 10.1038/sj.bjp.0704223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Davis JB, Pertwee RG, Di Marzo V. Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett. 2000;483:52–56. doi: 10.1016/s0014-5793(00)02082-2. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Maccarrone M, Davis JB, Finazzi-Agro A, Di Marzo V. The activity of anandamide at vanilloid VR1 receptors requires facilitated transport across the cell membrane and is limited by intracellular metabolism. J Biol Chem. 2001;276:12856–12863. doi: 10.1074/jbc.M008555200. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. Levels, metabolism, and pharmacological activity of anandamide in CB(1) cannabinoid receptor knockout mice: evidence for non-CB(1), non-CB(2) receptor-mediated actions of anandamide in mouse brain. J Neurochem. 2000;75:2434–2444. doi: 10.1046/j.1471-4159.2000.0752434.x. [DOI] [PubMed] [Google Scholar]
- Grudt TJ, Williams JT. mu-Opioid agonists inhibit spinal trigeminal substantia gelatinosa neurons in guinea pig and rat. J Neurosci. 1994;14:1646–1654. doi: 10.1523/JNEUROSCI.14-03-01646.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajos N, Freund TF. Pharmacological separation of cannabinoid sensitive receptors on hippocampal excitatory and inhibitory fibers. Neuropharmacology. 2002;43:503–510. doi: 10.1016/s0028-3908(02)00157-0. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Helliwell RJ, McLatchie LM, Clarke M, Winter J, Bevan S, McIntyre P. Capsaicin sensitivity is associated with the expression of the vanilloid (capsaicin) receptor (VR1) mRNA in adult rat sensory ganglia. Neurosci Lett. 1998;250:177–180. doi: 10.1016/s0304-3940(98)00475-3. [DOI] [PubMed] [Google Scholar]
- Howlett AC. Cellular signal transduction by anandamide and 2-arachidonoylglycerol. Chem Phys Lipids. 2000;108:53–70. doi: 10.1016/s0009-3084(00)00187-0. [DOI] [PubMed] [Google Scholar]
- Jennings EA, Vaughan CW, Christie MJ. Cannabinoid actions on rat superficial medullary dorsal horn neurons in vitro. J Physiol. 2001;534:805–812. doi: 10.1111/j.1469-7793.2001.00805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light AR, Shults RC, Jones SL. The Initial Processing of Pain and its Descending Control: Spinal and Trigeminal Systems. Basel: Blackwell Science Inc; 1992. [Google Scholar]
- Morisset V, Ahluwalia J, Nagy I, Urban L. Possible mechanisms of cannabinoid-induced antinociception in the spinal cord. Eur J Pharmacol. 2001;429:93–100. doi: 10.1016/s0014-2999(01)01309-7. [DOI] [PubMed] [Google Scholar]
- Morisset V, Urban L. Cannabinoid-induced presynaptic inhibition of glutamatergic EPSCs in substantia gelatinosa neurons of the rat spinal cord. J Neurophysiol. 2001;86:40–48. doi: 10.1152/jn.2001.86.1.40. [DOI] [PubMed] [Google Scholar]
- Piper AS, Yeats JC, Bevan S, Docherty RJ. A study of the voltage dependence of capsaicin-activated membrane currents in rat sensory neurones before and after acute desensitization. J Physiol. 1999;518:721–733. doi: 10.1111/j.1469-7793.1999.0721p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Dussor GD, Hargreaves RJ, Flores CM. Expression and distribution of cannabinoid receptor subtypes in trigeminal ganglion: colocalization with nociceptive markers. Abstr Soc Neurosci. 2001;31:716. [Google Scholar]
- Ralevic V, Kendall DA, Jerman JC, Middlemiss DN, Smart D. Cannabinoid activation of recombinant and endogenous vanilloid receptors. Eur J Pharmacol. 2001;424:211–219. doi: 10.1016/s0014-2999(01)01153-0. [DOI] [PubMed] [Google Scholar]
- Roberts LA, Christie MJ, Connor M. Anandamide is a partial agonist at native vanilloid receptors in acutely isolated mouse trigeminal sensory neurons. Br J Pharmacol. 2002;137:421–428. doi: 10.1038/sj.bjp.0704904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart D, Gunthorpe MJ, Jerman JC, Nasir S, Gray J, Muir AI, Chambers JK, Randall AD, Davis JB. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1) Br J Pharmacol. 2000;129:227–230. doi: 10.1038/sj.bjp.0703050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognetto M, Amadesi S, Harrison S, Creminon C, Trevisani M, Carreras M, Matera M, Geppetti P, Bianchi A. Anandamide excites central terminals of dorsal root ganglion neurons via vanilloid receptor-1 activation. J Neurosci. 2001;21:1104–1109. doi: 10.1523/JNEUROSCI.21-04-01104.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban L, Dray A. Synaptic activation of dorsal horn neurons by selective C-fibre excitation with capsaicin in the mouse spinal cord in vitro. Neuroscience. 1992;47:693–702. doi: 10.1016/0306-4522(92)90177-4. [DOI] [PubMed] [Google Scholar]
- Walker JM, Huang SM, Strangman NM, Tsou K, Sanudo-Pena MC. Pain modulation by release of the endogenous cannabinoid anandamide. Proc Natl Acad Sci U S A. 1999;96:12198–12203. doi: 10.1073/pnas.96.21.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HYT, Karoum F, Felder C, Badger H, Wang TCL, Markey SP. GC/MS analysis of anandamide and quantification of N-arachidonoylphosphatidylethanolamides in various brain regions, spinal cord, testis, and spleen of the rat. J Neurochem. 1999;72:1959–1968. doi: 10.1046/j.1471-4159.1999.0721959.x. [DOI] [PubMed] [Google Scholar]
- Yang K, Kumamoto E, Furue H, Yoshimura M. Capsaicin facilitates excitatory but not inhibitory synaptic transmission in substantia gelatinosa of the rat spinal cord. Neurosci Lett. 1998;255:135–138. doi: 10.1016/s0304-3940(98)00730-7. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Chuang HH, Movahed P, Julius D, Hogestatt ED. The anandamide transport inhibitor AM404 activates vanilloid receptors. Eur J Pharmacol. 2000;396:39–42. doi: 10.1016/s0014-2999(00)00207-7. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang HH, Sorgard M, Di Marzo V, Julius D, Hogestatt ED. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]