

Abstract
Cerebellar granule cells are inhibited phasically by GABA released synaptically from Golgi cells, but are inhibited more powerfully by tonic activity of high affinity α6 subunit-containing GABAA receptors. During development the tonic activity is generated by the accumulation of GABA released by action potentials, but in the adult the tonic activity is independent of action potentials. Here we show that in adult rats the tonic activation of GABAA receptors is produced by non-vesicular transmitter release and is reduced by the activity of GAT-1 and GAT-3 GABA transporters, demonstrating that alterations of GABA uptake will modulate information flow through granule cells. Acetylcholine (ACh) evokes a large Ca2+-dependent but action potential-independent release of GABA, which activates α6 subunit-containing GABAA receptors. These data show that three separate modes of transmitter release can activate GABAA receptors in adult cerebellar granule cells: action potential-evoked exocytotic GABA release, non-vesicular release, and ACh-evoked Ca2+-dependent release independent of action potentials. The relative magnitudes of the inhibitory charge transfers generated by action potential-evoked release (during high frequency stimulation of the mossy fibres), tonic inhibition and superfused ACh are 1:3:12, indicating that tonic and ACh-mediated inhibition may play a major role in regulating granule cell firing.
At the cerebellar mossy fibre to granule cell synapse, excitatory input to the cerebellar cortex is recoded into the firing of granule cells, which in turn excite the Purkinje cells that modulate motor output. This recoding is regulated by inhibition of the granule cells, which affects the tendency of the granule cells to generate oscillatory firing (Maex & De Schutter, 1998), controls the gain of information transmission between the mossy fibre input and the Purkinje cell output of the cerebellar cortex (Hamann et al. 2002), and increases the capacity of the cerebellum to store motor commands (Marr, 1969; Tyrrell & Willshaw, 1992).
Granule cells are known to experience two sorts of inhibition. First, they receive inhibitory postsynaptic currents (IPSCs) produced by GABA release from Golgi cells (Eccles et al. 1966). These IPSCs contain a fast component due to GABA released from Golgi cells connected directly to the granule cell, but also exhibit a slow component mediated by spillover of GABA released from Golgi cells that are not anatomically presynaptic to the recorded granule cell (Rossi & Hamann, 1998). Secondly, granule cells show a tonic activation of GABAA receptors, which in young animals is generated by the accumulation of GABA released by action potentials, but in adult animals is not blocked by TTX and so is not caused by action potential-dependent release of transmitter (Kaneda et al. 1995; Tia et al. 1996; Brickley et al. 1996; Wall & Usowicz, 1997).
In adult rats the tonic inhibition of granule cells dominates over the inhibition generated by action potential-evoked vesicular release of GABA (Brickley et al. 1996; Wall & Usowicz, 1997; Hamann et al. 2002), making an understanding of its generation and modulation fundamental to understanding information processing in the cerebellum. Surprisingly, however, little is known about the mechanism of transmitter release that generates the tonic inhibition. Since the tonic inhibition is generated by high affinity GABAA receptors containing α6 and probably δ subunits (Laurie et al. 1992; Nusser et al. 1998; Brickley et al. 2001; Hamann et al. 2002), which have a low micromolar EC50 for GABA (0.2 μm for receptors containing the subunit combination α6β2δ and 2 μm for α6β2γ2: Saxena & MacDonald, 1996), one possibility is that there is actually no release mechanism and the receptors are activated simply by the background level of GABA set by transporters. Even in the absence of GABA release, transporters with the stoichiometry of GAT-1 can only lower [GABA]o to around 0.4 μm (Attwell et al. 1993), which is high enough to activate α6 subunit-containing GABAA receptors. Alternatively there may be active release of GABA by an action potential-independent mechanism. If so, then it is important to understand how this release might be modulated, since this will contribute to the behaviour of the cerebellar cortex.
Here we investigate the source of the transmitter activating the tonic inhibition, and whether the inhibition can be modulated. The results show that three distinct modes of transmitter release contribute to inhibiting cerebellar granule cells.
Methods
Preparation of brain slices
Sprague-Dawley rats were killed by cervical dislocation, in accordance with the UK Animals (Scientific Procedures) Act 1986. Patch-clamp recordings from visually identified granule cells in thin (160–220 μm) parasagittal cerebellar vermis slices were performed as previously described (Rossi & Slater, 1993); no attempt was made to restrict recording to particular lobules of the slices (in particular we did not select for cells in the uvula and nodulus, which receive cholinergic innervation from the vestibular nuclei). Slices were from animals of either sex, usually 35–45 days old, although some experiments were on 7- to 8-day-old rats as noted in the text, at 29 ± 3 °C, in extracellular solution containing (mm): NaCl 126, NaHCO3 24, NaH2PO4 1, KCl 2.5, CaCl2 2.5, MgCl2 2, d-glucose 10 (gassed with 95 % O2-5 % CO2), pH 7.4. For experiments with no extracellular Ca2+, CaCl2 was replaced by 2.5 mm MgCl2 and 2 mm EGTA. Kynurenic acid (1 mm) was included in the dissection and incubation solution (to block glutamate receptors, to reduce potential excitotoxic damage) but was omitted from the superfusion solution.
Patch-clamp recording and synaptic stimulation
For recording, slices were placed under flowing solution on the stage of an upright microscope and viewed with a ×40 water immersion objective with Hoffman modulation contrast or differential interference contrast and infrared optics. Whole-cell voltage-clamp recordings were made from the somata of visually identified neurones. Patch pipettes were constructed from thick-walled borosilicate glass capillaries and filled, for voltage-clamp recordings with ECl = 0 mV, with an internal solution containing (mm): CsCl 135, NaCl 4, CaCl2 0.5, Hepes 10, EGTA 5, MgATP 2, Na2GTP 0.5, QX-314 10 (to suppress voltage-gated sodium currents), pH adjusted to 7.2 with CsOH; or in some experiments (which gave indistinguishable results) CsCl 130, NaCl 4, Hepes 10, BAPTA 10, MgATP 4, Na2GTP 0.5, QX-314 10, pH set to 7.2 with CsOH. For experiments with ECl set to −60 mV, the solution contained (mm): caesium gluconate 120, NaCl 4, Hepes 10, BAPTA 10, MgATP 4, Na2GTP 0.5, pH adjusted to 7.2 with CsOH. To stimulate Golgi cells, glass pipettes (resistance ∼1–2 MΩ filled with extracellular medium) were placed in the granular layer. Electrode junction potentials were corrected for.
Application of drugs
Unless otherwise specified, drugs were added to the superfusion solution. In some experiments to investigate the role of exocytotic transmitter release, slices were pre-soaked for at least 2 h in 0.5 μm concanamycin while control slices were soaked in external solution lacking concanamycin (both solutions contained 1 mm kynurenate to block glutamate receptors). Similar pre-soaking was used to load cells with β-alanine and to prevent cell swelling with sucrose, as described in Results. Caffeine (5 mm), which was used to deplete Ca2+ from intracellular stores, reduced the tonic bicuculline-sensitive current reported in this paper, possibly because a caffeine-evoked rise in intracellular calcium inhibits GABAA receptors as reported previously (Desaulles et al. 1991; Akopian et al. 1998). This reduction recovered upon washout of caffeine, and was observed in the presence of a saturating (1 mm) level of exogenous GABA (ruling out the possibility that it reflects a decrease in GABA release). Intracellular Ca2+ stores should not refill after caffeine-induced depletion when the endoplasmic reticulum calcium pump is blocked with thapsigargin, so we analysed the influence of caffeine and thapsigargin on the tonic GABAA receptor current only after the caffeine was washed out.
Data analysis
Data are presented as means ±s.e.m. and significance of changes was assessed with Student's two-tailed t test. Figures showing the tonic activation of GABAA receptors are filtered at 5 Hz for clarity of display and so spontaneous IPSCs are not visible. For analysis and display of spontaneous IPSCs, data were filtered at 5 kHz. When quantifying IPSC occurrence, IPSCs were defined as current deflections that had an amplitude (measured from the mean current) greater than the peak-to-peak amplitude of the current noise, and with a decay time constant at least 10-fold slower than the rise time.
Results
All data are from adult rats (35–45 days old) unless otherwise stated.
Granule cells show IPSCs produced by exocytotic GABA release from Golgi cells
Stimulating in the granular layer of slices of cerebellum from adult rats, to evoke action potentials in Golgi cells, produces inhibitory postsynaptic currents with a biphasic decay (Fig. 1), comprising a fast decaying component (τ∼30 ms) due to direct synaptic input, followed by a much slower decaying component (τ∼800 ms) produced by spillover of GABA from Golgi cell synapses with other granule cells (for quantification of the two components’ properties see Hamann et al. (2002)). The experiments of Fig. 1 were carried out on granule cells whole-cell voltage-clamped at −60 mV with electrodes containing isotonic Cl− (ECl= 0 mV), so GABAA receptor-mediated currents are inward. Both components of the IPSC were blocked by the GABAA receptor antagonists bicuculline (40 μm: Fig. 1A) and GABAzine (SR95531, 10 μm: Fig. 1B). They were also blocked by TTX (1 μm: Fig. 1C) and by removal of extracellular Ca2+ (replaced with Mg2+ and 2 mm EGTA: Fig. 1D), and so both components of the IPSC reflect conventional action potential-evoked exocytosis driven by voltage-gated Ca2+ entry.
Figure 1. Golgi-granule cell inhibition in adult rat cerebellum mediated by action potential- and calcium-dependent exocytosis of GABA.
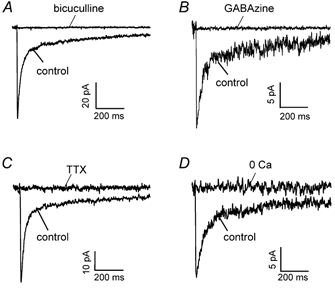
A, IPSC recorded in a granule cell at −60 mV, with ECl set to 0 mV so the current is inward, in the absence (control) and presence of 40 μm bicuculline to block GABAA receptors. B, like A but using GABAzine (SR95531, 10 μm) to block GABAA receptors. C, block of the IPSC by tetrodotoxin (TTX, 1 μm). D, block of the IPSC by removal of external calcium (0 Ca, 2.5 mm CaCl2 replaced by 2.5 mm MgCl2 and 2 mm EGTA).
Tonic inhibition of granule cells is produced by non-vesicular transmitter release
In the absence of stimulation, applying bicuculline (40 μm) generated an outward current shift of 42 ± 4 pA (n = 45; range of values 8–113 pA), corresponding to the suppression of a tonic inward current (when ECl = 0 mV) through a conductance of 700 pS that is generated by GABAA receptor channels (Fig. 2A and F; see also Brickley et al. 1996; Wall & Usowicz, 1997). Similar results were obtained with the GABAA blockers picrotoxin (100 μm, Fig. 2B and F) and GABAzine (10 μm, Fig. 2C and F). Neither TTX (1 μm, see also Kaneda et al. 1995; Wall & Usowicz, 1997) nor removal of external Ca2+ (replaced with Mg2+ and 2 mm EGTA, see also Wall & Usowicz, 1997) affected the bicuculline-sensitive tonic inward current significantly (Fig. 2D–F) showing that it does not depend on action potentials or Ca2+ influx.
Figure 2. Tonic inhibition of granule cells independent of sodium action potentials and calcium.
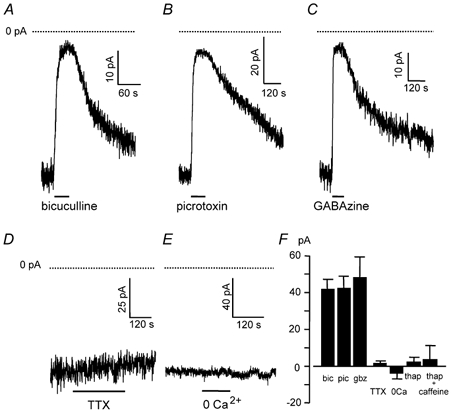
Holding potential −60 mV, and ECl was at 0 mV, so GABA-mediated currents are inward and blocking them gives an outward current shift. Dotted lines show zero current. A-C, suppression of tonic inhibition evoked by blocking GABAA receptors with: A, bicuculline (40 μm); B, picrotoxin (100 μm); C, GABAzine (10 μm). D, the holding current of granule cells was not affected by block of action potentials with tetrodotoxin (TTX, 1 μm), so tonic inhibition is not action potential-dependent. E, the holding current of granule cells was not affected by removal of external calcium (0 Ca2+, 2.5 mm CaCl2 replaced by 2.5 mm MgCl2 and 2 mm EGTA), so tonic inhibition is not activated by transmitter released by exocytosis evoked by Ca2+ entry. F, mean current shifts evoked by bicuculline (bic, 45 cells), picrotoxin (pic, four cells), GABAzine (gbz, nine cells), tetrodotoxin (TTX, four cells), zero calcium solution (0Ca, four cells) and thapsigargin (thap) in zero calcium with or without caffeine pretreatment (three cells each).
To test the possibility that vesicular release might be evoked by the release of calcium from intracellular stores we used thapsigargin, an inhibitor of the endoplasmic reticulum Ca2+ pump, which results in the gradual depletion of intracellular Ca2+ stores (Irving et al. 1992). Application of thapsigargin (1.5 μm) for up to 20 min did not affect the granule cell tonic current (Fig. 2F). To ensure the complete depletion of intracellular stores we also used caffeine (a ryanodine receptor agonist which releases Ca2+ from stores: Kirischuk et al. 1996) during thapsigargin exposure. A transient (2–4 min) application of caffeine (5 mm) in the presence of thapsigargin (1.5 μm, 20 min) did not affect the tonic current recorded in thapsigargin after the caffeine was washed out (Fig. 2F, see Methods). Thus, the mechanism responsible for releasing the GABA that generates the granule cell tonic current is not dependent on Ca2+ influx or release from intracellular stores.
Since exocytosis may be able to occur independently of Ca2+ (Katz & Miledi, 1977; Hille et al. 1999; Zhang & Zhou, 2002), we tested the effect on tonic inhibition of abolishing all exocytotic release of transmitter by soaking slices in the vesicular H+-ATPase inhibitor concanamycin (0.5 μm) prior to recording. By abolishing the proton electrochemical gradient across the vesicle membrane (Manabe et al. 1993; Dröse & Altendorf, 1997) this drug prevents the accumulation of transmitter within vesicles (Zhou et al. 2000). We first checked that concanamycin did indeed block exocytotic release of GABA by recording from young (P7–8) rats, which show easily detectable spontaneous IPSCs (spontaneous IPSCs are harder to quantify accurately in adult rats because of the membrane current noise generated by the tonic activation of GABA receptors). As shown in Fig. 3A–C, soaking in concanamycin abolished spontaneous IPSCs (compared with data recorded from interleaved slices soaked in solution lacking concanamycin). Furthermore, as detailed below (see Fig. 7), concanamycin blocked an increase of IPSC frequency evoked by acetylcholine in granule cells in young rats, and blocked an ACh-evoked Ca2+-dependent increase of GABAA receptor-mediated current in adult rats. Thus, concanamycin does block exocytotic transmitter release both in young and adult rats. However, concanamycin had no effect (P = 0.65) on the tonic inhibitory current present in adult granule cells (Fig. 3D–F). Thus, the tonic current is activated by non-vesicular transmitter release.
Figure 3. The vesicular H+-ATPase blocker concanamycin blocks exocytosis but does not affect the tonic current.
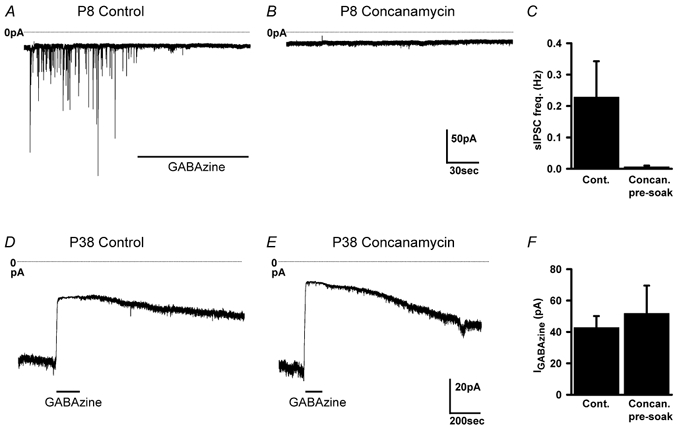
A and B, membrane current in P8 granule cells in slices pre-soaked in solution lacking (A) or containing (B) 0.5 μm concanamycin. Control cells show spontaneous IPSCs that are blocked by GABAzine (10 μm), which are absent after concanamycin soaking. C, mean frequency of spontaneous IPSCs in P7–8 slices pre-soaked in control (seven cells) or concanamycin (six cells) solution. D and E, membrane current in adult granule cells in slices pre-soaked in solution lacking (D) or containing (E) 0.5 μm concanamycin. Control and concanamycin cells show a tonic current that is blocked by GABAzine (10 μm). F, magnitude of tonic current in cells pre-soaked in control (seven cells) or concanamycin (seven cells) solution.
Figure 7. Effect of concanamycin on the current evoked in granule cells by ACh (100 μm).
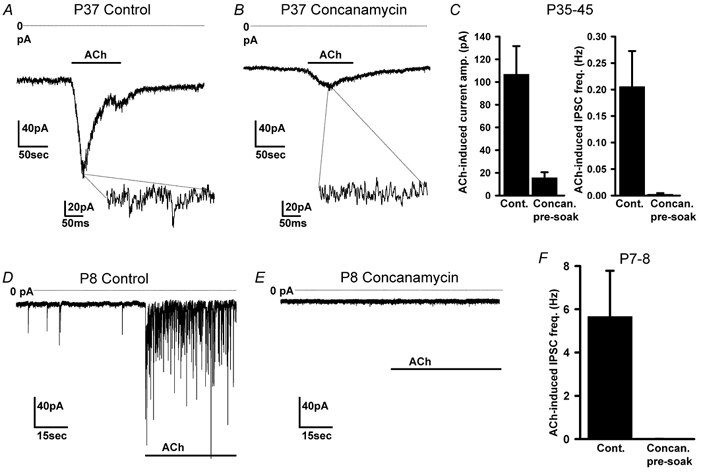
A and B, ACh-evoked current of granule cells in slices from adult (P37) rats, pre-soaked in solution lacking (A) or containing (B) 0.5 μm concanamycin. Insets are expanded sections of the record, showing IPSCs in control solution but not after concanamycin treatment. C, mean ACh-evoked current (left) and IPSC frequency (right) in cells from P35-P45 rats pre-soaked in control solution (seven cells) or in concanamycin (seven cells). D and E, ACh-evoked current of granule cells in slices from P8 rats, pre-soaked in solution lacking (D) or containing (E) 0.5 μm concanamycin. F, mean IPSC frequency in cells from P7-P8 rats pre-soaked in control solution (seven cells) or in concanamycin (six cells).
Tonic inhibition is not via a swelling-activated mechanism
Cell swelling, which might in principle be a consequence of damage caused during the slicing procedure, can release amino acid transmitters in a non-vesicular manner, through a swelling-activated anion channel (Kimelberg et al. 1990; Rutledge & Kimelberg, 1996). To investigate whether this might be the source of the transmitter activating the tonic current, we tried using L-644,711 (300 μm) and NPPB (5-nitro-2-(3-phenylpropylamino) benzoic acid, 100 μm) to block swelling-activated anion channels (Olson & Kimelberg, 1995, Pasantes-Morales et al. 1999), but these agents were found to have non-specific effects: L-644,711 blocked GABAA receptors, reducing the current response to 1 mm exogenous GABA by 85 ± 6 % in five cells, while NPPB appeared to activate the channels directly or increase GABA release, producing a mean current of 36 ± 4 pA at −60 mV in 13 cells which was blocked by bicuculline (data not shown). We therefore attempted to reduce cell swelling by pre-incubating slices in hyperosmotic solution (containing an additional 60 mm sucrose) for 2–4 h and making recordings in this medium (Richerson & Messer, 1995). For granule cells in slices in hyperosmotic solution the mean diameter was reduced by 21 % (from 5.04 ± 0.24 μm (n = 90) in control solution to 4.00 ± 0.20 μm (n = 90) in hyperosmotic solution; significantly different, P = 2.3 × 10−18 by Student's t test), implying a 51 % decrease of volume. The tonic bicuculline-sensitive holding current was still present (40 ± 8 pA in five cells), and was not significantly different from that in control slices (42 ± 4 pA in 45 cells, P = 0.87), suggesting that GABA is not released via swelling-activated channels.
Tonic inhibition is not produced by GABA released by reversed transport, but is modulated by the operation of neuronal and glial GABA transporters
Neurotransmitter transporters can, if the transmembrane ion gradients are appropriate, run backwards and release transmitter in a non-vesicular manner (Attwell et al. 1993). For glutamate, this occurs during pathological conditions that lead to a run-down of ionic gradients (Rossi et al. 2000), and in the case of GABA it may occur under physiological or therapeutic conditions (Schwartz, 1987; Gaspary et al. 1998; Wu et al. 2001). In the cerebellar granule cell layer GABA transport is dominated by the neuronal, and also glial, transporter GAT-1 (also known as GAT-A) and the glial transporter GAT-3 (also known as GAT-B or mouse GAT-4) (Itouji et al. 1996; Morara et al. 1996; Barakat & Bordey, 2002).
To determine whether the tonic current is activated by GABA released through reversed transport on GAT-1, or is modulated by uptake via GAT-1, we tested the effect of SKF-89976A, a specific, non-transported inhibitor of GAT-1 (Borden et al. 1994). SKF-89976A (100 μm) did not block the tonic current but instead produced an inward current of 32 ± 6 pA in 12 cells (Fig. 4A and B), which was unaffected by the presence of 1 μm TTX (four cells, data not shown). This is due to GABA accumulating rapidly in the extracellular space and acting on GABAA receptors, since no inward current was observed when SKF-89976A was applied in the presence of bicuculline (Fig. 4B and C), and the bicuculline-sensitive current was increased by a factor of 2.2 ± 0.3 in the presence of SKF-89976A (relative to control, Fig. 4D). Thus, GAT-1 transporters do not contribute to GABA release but, in fact, are actively taking up GABA from the extracellular space (by contrast Wall & Usowicz (1997) found no effect of SKF-89976A).
Figure 4. Modulation of tonic inhibition of granule cells by GABA transporters.
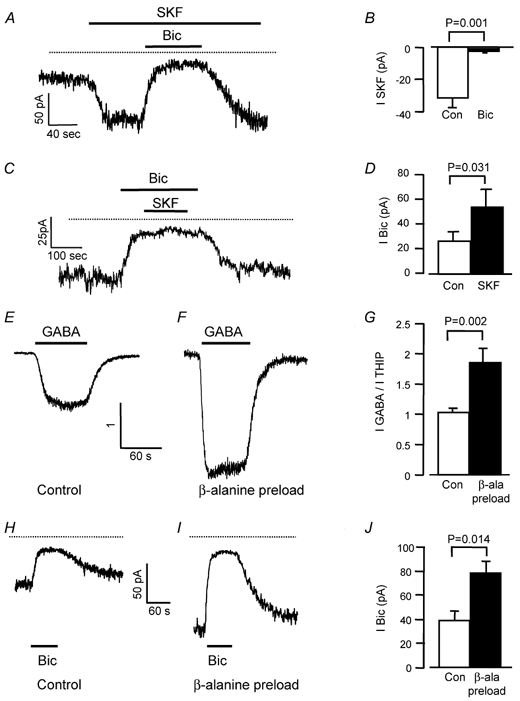
Holding potential −60 mV and ECl was at 0 mV, so GABA-mediated currents are inward, and increasing or blocking them gives an inward or outward current shift, respectively. Dotted lines show zero current. A, inhibiting the GABA transporter GAT-1 with SKF-89976A (SKF, 100 μm) evokes an inward current that is reversed by bicuculline (bic, 40 μm) reflecting increased tonic inhibition. B, mean current evoked by SKF-89976A in the absence (control, 12 cells) and presence (bic, four cells) of bicuculline. C, SKF-89976A (SKF, 100 μm) evokes no current when GABAA receptors are blocked with bicuculline (bic). D, mean bicuculline-evoked current in the absence (control) and presence of SKF-89976A (SKF, four cells). E-G, blocking the GABA transporter GAT-3 by pre-loading with β-alanine inhibits GABA uptake around granule cells. E, specimen response to GABA (5 μm), normalized to the response in the same cell to the non-transported GABAA agonist THIP (10 μm), in a control slice. F, as E, but in a slice pre-loaded with β-alanine. G, mean ratio of currents evoked by 5 μm GABA and 10 μm THIP in nine cells in control slices and seven cells in slices pre-loaded with β-alanine. H, response to bicuculline (40 μm) in a control slice. I, response to bicuculline in a slice pre-loaded with β-alanine. J, mean current change evoked by bicuculline in seven control cells interleaved with seven cells from slices pre-soaked in 1 mmβ-alanine.
To determine whether the tonic current is activated by GABA released by reversed transport on the glial transporter GAT-3, we tested the effect of β-alanine, which is a potent inhibitor of GAT-3 transport (Borden et al. 1994). β-Alanine (200 μm) produced an inward current of 210 ± 24 pA (n = 9), which was blocked by 40 μm bicuculline (by 91 ± 2 %, n = 3; a similar result was obtained by Wall & Usowicz (1997)). However, unlike SKF-89976A, β-alanine is a low affinity transport substrate and a low affinity GABAA receptor agonist (Liu et al. 1993; Wu et al. 1993; Jones et al. 1998), so the current it generates may be due in part to GABA released by heteroexchange on transporters or due to a direct activation of receptors by the β-alanine. We therefore adopted the approach used by Rossi et al. (2000) to block reversed transport of glutamate, i.e. pre-loading cells with the transported inhibitor. We incubated slices for at least 1 h with 1 mm β-alanine, in the presence of 300 μm bicuculline to block GABAA receptor activation (control slices were incubated just with bicuculline), and then washed the β-alanine out of the slice. The aim was to accumulate β-alanine inside cells, where it will occupy the transporter and reduce transport of GABA out of the cells if the transporter is running backwards (or into the cells if the transporter is running forwards). This approach avoids the problems of GABA release by heteroexchange and of β-alanine directly activating receptors, because β-alanine should not be present at a significant concentration extracellularly during the recording period (see below). To check that β-alanine pre-loading succeeded in impairing GABA transport, we compared the granule cell responses to exogenous GABA and THIP (4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol), an agonist of the GABAA receptor that is not transported by GABA transporters (Korn & Dingledine, 1986; Kemp et al. 1986). Under control conditions (Fig. 4E and G) the ratio of the response produced by bath-applied GABA (5 μm) to that produced by THIP (10 μm) was 1.03 ± 0.08. When slices were pre-loaded with β-alanine (Fig. 4F and G) the ratio of the responses to GABA and THIP increased to 1.85 ± 0.23 (significantly different from control conditions, P = 0.002), as expected if GABA transport is reduced and a higher GABA concentration is able to penetrate into the slice. However, β-alanine pre-loading did not reduce the bicuculline-sensitive holding current as would be expected if GAT-3 were releasing GABA, but instead doubled it relative to control slices (Fig. 4H–J).
This increase is unlikely to reflect increased activation of GABAA receptors by a GAT-3-mediated efflux of β-alanine from pre-loaded cells, for the following reasons. Such an efflux is unlikely to be larger than any efflux of GABA that normally occurs on GAT-3 (since GAT-3 has a 120-fold higher apparent affinity for GABA than for β-alanine, from the data of Liu et al. (1993) for mouse GAT-4), and GABAA receptors have an apparent affinity for β-alanine that is 200-fold lower than that for GABA (Wu et al. 1993; Jones et al. 1998), so any replacement of GABA efflux by β-alanine efflux should decrease the tonic activation of GABAA receptors.
These results suggest that, like the neuronal and glial transporter GAT-1, the glial transporter GAT-3 is normally taking up GABA, and pre-loading with β-alanine reduces this uptake. Thus, the tonic current in mature granule cells is not produced by GABA released by reversed transport on either GAT-1 or GAT-3, but the activity of these transporters modulates the strength of tonic inhibition occurring.
Modulation of granule cell inhibition
Tonic inhibition of granule cells by their GABAA receptors seems an energetically wasteful design feature of the cerebellar cortex if the inhibition is constantly present, and a similar functional effect might be achieved by releasing less glutamate at mossy fibre synapses, or expressing more K+ channels (Brickley et al. 2001). We therefore tested whether the tonic GABA conductance could be modulated by agents known to regulate transmitter release by acting on presynaptic receptors, or other neurotransmitters known to be released in the cerebellar cortex. The activators of presynaptic metabotropic glutamate receptors, ACPD (100 μm, Fig. 5A) and l-AP4 (100 μm, Fig. 5B), the GABAB agonist baclofen (20 μm, Fig. 5B), histamine (100 μm, Fig. 5B), noradrenaline (100 μm, Fig. 5C), dopamine (100 μm, Fig. 5C), ATP (100 μm, Fig. 5D) and 5-HT (100 μm, Fig. 5D) had no effect on the resting membrane current of granule cells (mean current changes are shown in Fig. 5F). However, in all 36 cells tested, acetylcholine (ACh, 100 μm) generated a desensitizing inward current of peak amplitude 123 ± 12 pA at −60 mV when ECl was set to zero (Fig. 5E and F), along with superimposed phasic synaptic currents (Fig. 5E, inset), which we show below are both produced by GABA release and which we investigated in more detail. The presence of phasic currents indicates that, as detailed below, only part of the ACh-evoked current is generated by modulation of non-vesicular transmitter release.
Figure 5. Modulation of granule cell holding current by activation of neurotransmitter receptors.
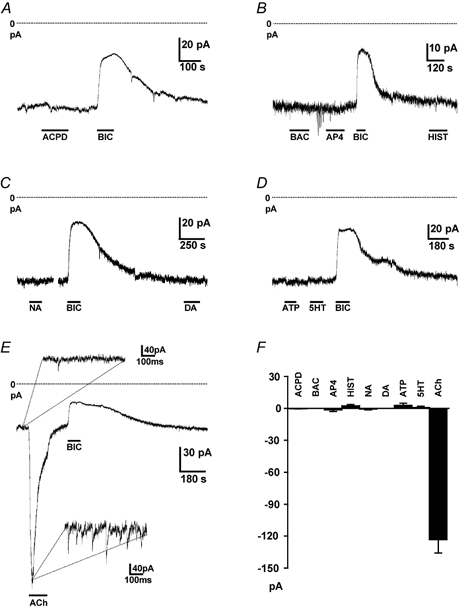
Holding potential −60 mV and ECl was at 0 mV, so GABA-mediated currents are inward, and increasing or blocking them gives an inward or outward current shift, respectively. Dotted lines show zero current. A, effect of the mGluR agonist ACPD (100 μm) on the holding current of a granule cell. Response to bicuculline (40 μm) is shown for comparison. B, effect of the GABAB agonist baclofen (BAC, 40 μm), the mGluR agonist l-AP4 (100 μm), histamine (HIST, 100 μm) and bicuculline. C, effect of noradrenaline (NA, 100 μm), dopamine (DA, 100 μm) and bicuculline. D, effect of ATP (100 μm), 5-HT (100 μm) and bicuculline. E, effect of acetylcholine (ACh, 100 μm) and bicuculline; insets show expanded sections of traces in control solution and in ACh, revealing an increase of IPSC frequency in ACh. F, mean currents evoked by ACPD (four cells), baclofen (four cells), AP4 (eight cells), histamine (seven cells), noradrenaline (four cells), dopamine (four cells), ATP (seven cells), 5-HT (nine cells) and acetylcholine (36 cells).
Acetylcholine-evoked inhibition of granule cells
When ECl was set to −60 mV, ACh evoked an outward current at −30 mV and an inward current at −90 mV (Fig. 6A). The mean reversal potential of the ACh-evoked current (derived by subtracting the current response to voltage ramps, applied in the presence and absence of ACh, from a holding potential of −70 mV) was −55 ± 1 mV in five cells, close to ECl. These data are not expected for ACh activating postsynaptic nicotinic receptors linked to cation channels with a reversal potential near 0 mV, but are consistent with activation of a GABA receptor-gated Cl− current. Responses to ACh ran down with time so, to analyse the effect of GABA and ACh receptor blocking drugs on this current, we compared the responses to two sequential applications of ACh, either both in control solution, or the first in control solution and the second in the blocker (Fig. 6B–G). The ACh-evoked current (including the superimposed phasic synaptic currents) was abolished by the nicotinic blocker mecamylamine (25 μm, four cells, P = 0.0005 comparing the second responses to ACh in control solution and in mecamylamine: Fig. 6C and H), but also by the GABAA blockers GABAzine (10 μm, eight cells, P = 6 × 10−6, Fig. 6D and H), picrotoxin (100 μm, three cells, data not shown) and bicuculline (20 μm, two cells, data not shown). The use of GABAzine is an important control for these experiments since bicuculline, unlike GABAzine (Zhang & Feltz, 1991), blocks central nicotinic as well as GABAA receptors. These data suggest that ACh produces a current indirectly, by acting on presynaptic nicotinic receptors (presumably on Golgi cells) and promoting GABA release, as reported previously for inhibitory interneurones in the hippocampus and thalamus (Lena & Changeux, 1997; Jones & Yakel, 1997). The peak inhibitory conductance activated by ACh was 3.99 ± 0.97-fold larger than the tonic GABAzine/bicuculline-sensitive conductance (six cells, Fig. 5E).
Figure 6. Cholinergic control of activation of GABA receptors on granule cells by action potential-independent calcium-dependent GABA release.
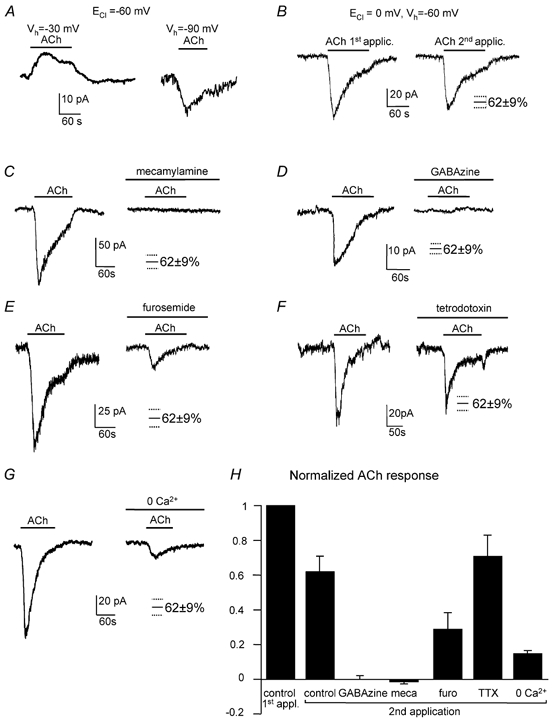
A, with ECl set to −61 mV, ACh (100 μm) evokes an outward current at −30 mV and an inward current at −90 mV, implying a reversal potential for the ACh-evoked current near ECl. B, with ECl = 0 mV, the inward current evoked by ACh decreased slightly with repeated applications of ACh. Mean size of the second control response to ACh (62 ± 9 %) is shown here and in subsequent panels (which also had ECl = 0 mV) to compare with responses in drugs. C, sequential responses to ACh in the absence and presence of the nicotinic blocker mecamylamine (25 μm). D, sequential responses to ACh in the absence and presence of the GABAA blocker GABAzine (10 μm). E, sequential responses to ACh in the absence and presence of furosemide (100 μm). F, sequential responses to ACh in the absence and presence of tetrodotoxin (TTX, 1 μm). G, sequential responses to ACh in normal solution and in zero calcium solution (with 2 mm EGTA and 2.5 mm MgCl2 added). H, summary of responses to two sequential applications of ACh (normalised to the current produced by the first application), with the second application being in normal solution (five cells), GABAzine (eight cells), mecamylamine (meca, four cells), furosemide (furo, five cells), TTX (1 μm, eight cells), or zero calcium solution (five cells).
Furosemide (100 μm), which specifically blocks α6 subunit-containing GABAA receptors (Korpi et al. 1995), approximately halves the bicuculline-sensitive tonic current seen in the absence of ACh and the spillover component of the IPSC generated by Golgi cells (Hamann et al. 2002). Similarly furosemide approximately halved the ACh-evoked current (Fig. 6E and H): a second application of ACh in control solution produced a response which was 62 ± 9 % of the first response in five cells, while applying furosemide during the second ACh application resulted in a response that was 30 ± 11 % of the initial response in five cells (P = 0.05 compared with the second control response). Thus, the GABA released by ACh also acts largely on α6 subunit-containing receptors.
Tetrodotoxin (TTX, 1 μm) had no effect on the ACh-evoked current (Fig. 6F and H, second response in TTX was 71 ± 13 % of the first control response in eight cells, P = 0.61 compared with a second control response of 62 ± 9 %), so Na+-dependent action potentials are not needed to release the GABA (although in vivo they may be needed to release the ACh which here we applied exogenously). This implies that the nicotinic receptors are located close to GABA release sites. Zero calcium solution reduced the ACh-evoked current by about 74 % (Fig. 6G and H: the second response to ACh in zero Ca2+ was 16 ± 2 % of the first control response in five cells, P = 0.001 compared with a second control response of 62 ± 9 %). This suggests that, as in other regions (Lena & Changeux, 1997), much of the ACh-evoked release is produced by Ca2+ entry (either directly through nicotinic channels or through voltage-gated Ca2+ channels activated by nicotinic depolarization), which activates exocytosis from presynaptic, presumably Golgi cell, terminals. The ACh-evoked GABAA current remaining in zero calcium (26 % of the second response in control solution) is unlikely to result from incomplete Ca2+ removal, since trace Ca2+ was chelated with 2 mm EGTA, which completely abolished stimulus-evoked IPSCs (Fig. 1D). Consistent with these results, pre-soaking with 0.5 μm concanamycin to block vesicular release reduced the ACh-evoked current by 85 % in adult rat granule cells (from 107 ± 25 pA in seven control cells to 16 ± 5 pA in seven cells pre-soaked in concanamycin) and abolished the superimposed phasic synaptic currents (Fig. 7A–C). These and the zero-Ca2+ data suggest that, although most of the ACh-evoked current in the adult is generated by vesicular release of GABA, approximately 15–26 % is generated by non-vesicular GABA release. For granule cells in slices from young rats, where the ACh-evoked current appeared to be composed entirely of phasic synaptic currents, pre-soaking with concanamycin completely blocked the response to ACh (Fig. 7D–F).
Mecamylamine alone (in the absence of exogenous ACh) did not alter the membrane current of granule cells (four cells, data not shown) suggesting that in the slice preparation, with afferents cut, there is no spontaneous ACh release regulating GABAergic inhibition. Similarly, the acetylcholinesterase inhibitor eserine (100 μm) did not affect the current (in five cells, data not shown).
Discussion
We have shown that GABAA receptors in adult cerebellar granule cells are activated by transmitter released by three distinct mechanisms: (i) conventional action potential-evoked calcium-dependent exocytosis from Golgi cells, (ii) a non-vesicular transmitter release mechanism independent of calcium and action potentials that activates GABAA receptors tonically, and (iii) acetylcholine-evoked release via a mechanism that is independent of action potentials but is largely (∼80 %) by calcium-dependent exocytosis. The tonic inhibitory conductance is 3-fold larger than the mean conductance generated by high frequency stimulation of exocytotic GABA release from Golgi cells (Hamann et al. 2002), and the peak extra conductance activated by ACh is 4-fold larger than the tonic conductance present in the absence of ACh (Fig. 5E) and hence 12-fold larger than the conductance produced by exocytosis of GABA from Golgi cells. Thus, all three transmitter release mechanisms have the potential to contribute significantly to the control of granule cell firing.
Previous work has shown that the tonic inhibition of adult granule cells is not mediated by action potential-evoked GABA release, e.g. from the Golgi cells which release GABA synaptically onto granule cells (Kaneda et al. 1995; Wall & Usowicz, 1997). We have confirmed this result and shown that the release is not by calcium-dependent or calcium-independent exocytosis, nor via a swelling-activated channel, nor by reversed operation of the GAT-1 or GAT-3 GABA transporters. The possibility that there is actually no active release of GABA, and that high affinity α6 subunit-containing GABAA receptors are activated by the extracellular GABA concentration produced when GABA transporters are at equilibrium (mediating no net GABA flux), the value of which is determined by the stoichiometry of the transporters (see Introduction), or that the receptors are spontaneously active without binding GABA (Neelands et al. 1999), is unlikely for the following reasons. Blocking GAT-1 or GAT-3 increased the tonic inhibition, consistent with these transporters constantly removing GABA (or a related activating substance), which is constantly being released into the extracellular space. Furthermore Wall (2002) has shown that enzymatically degrading extracellular GABA reduces the magnitude of the tonic inhibition.
By exclusion, we suggest that GABA is released (from Golgi cell synaptic terminals or perhaps from astrocytes), by a non-vesicular calcium-independent mechanism, as occurs for glutamate and GABA in the hippocampus (Jabaudon et al. 1999; Liu et al. 2000). We cannot rule out the possibility that, in part, it is not GABA, but taurine or β-alanine that activates the receptors generating the tonic current, although the enzymatic degradation experiment of Wall (2002) shows that at least 30 % of the current is activated by GABA itself. Unfortunately guanidinoethanesulphonic acid (GES), which blocks transporters for β-alanine and taurine, activates GABAA receptors (Mellor et al. 2000), preventing its use as a tool to alter the extracellular concentration of β-alanine and taurine and thus test whether they activate part of the tonic current.
The glomerular structure that makes up the mossy fibre to granule cell synapses is increasingly wrapped by glia as rats become adult (Hamori & Somogyi, 1983). If the GABA (or related substance) which activates the tonic current is released within this structure, for example from Golgi cell synaptic terminals, then the likely restriction to diffusion away imposed by this anatomical structure may help to keep the basal extracellular concentration of transmitter high and contribute to production of the tonic current (Wall & Usowicz, 1997). This would add to the list of unusual information processing features conferred by this structure, including long-lasting Golgi-granule cell IPSCs and mossy fibre-granule cell EPSCs (Rossi & Hamann, 1998; DiGregorio et al. 2002), and spillover-mediated heterosynaptic modulation (Mitchell & Silver, 2000). Interestingly, although the transmitter release evoked by acetylcholine is mainly vesicular, the resulting current evoked in granule cells is sustained, with only a small component of discrete IPSCs (Fig. 5E), suggesting that it is mainly produced by an accumulation within the glomerulus of GABA released exocytotically at release sites that do not form synapses with the recorded cell. This suggests that in adult animals in vivo, where the Golgi cell firing rate may be higher than in our slices, there could be an extra component of sustained inhibition (on top of the tonic inhibition generated by non-vesicular transmitter release) produced by the accumulation of GABA released by Golgi cell action potentials.
If the tonic activation of granule cell GABAA receptors were constant, it would not be obvious why evolution has favoured limiting the activity of granule cells in this way, rather than by decreasing either glutamate release or the postsynaptic response to glutamate at the mossy fibre synapses. We have shown that alterations in the rate of GABA uptake by neurones and glia (GAT-1) or glia (GAT-3), for example by phosphorylation (Corey et al. 1994), will alter the strength of tonic inhibition. Furthermore, our demonstration that ∼20 % of the current evoked by acetylcholine (i.e. the 15–26 % remaining in concanamycin or zero Ca2+) apparently is due to an activation of GABAA receptors by non-vesicular transmitter release suggests another mechanism for modulation of the tonic inhibition. Conceivably, in addition to ACh inducing Ca2+-dependent GABA release (see below), the depolarization produced by ACh may modulate tonic Ca2+-independent release of GABA, perhaps by modulating the release mechanism that generates the tonic conductance (cf. Parnas et al. 2000) or by inhibiting or reversing the operation of GAT-1 (Schwartz, 1987; Attwell et al. 1993; Gaspary et al. 1998).
The increase of vesicular GABA release evoked by ACh is presumably similar to that in other brain regions, where calcium entry through nicotinic receptors, or through voltage-gated calcium channels activated by nicotinic receptor-mediated depolarization, generates increased exocytotic GABA release from presynaptic terminals (Lena & Changeux, 1997). In hippocampus and neocortex, cholinergic modulation may be involved in memory formation (Hasselmo, 1999). In the cerebellar cortex, nicotinic receptor subunits have been reported on Purkinje cells, granule cells and Golgi cells (Dominguez del Toro et al. 1994; Nakayama et al. 1997), of which the receptors on Golgi cells are the most likely substrate for modulating the GABAA receptor-mediated current in granule cells. The predominant cholinergic innervation of the rat cerebellum (Jaarsma et al. 1997) consists of mossy fibres from the vestibular nuclei to the uvula and nodulus of the vermis and of more diffusely terminating fibres from the pedunculopontine tegmental and lateral paragigantocellular nuclei (although some Golgi cells may be cholinergic too: Illing, 1990; De Lacalle et al. 1993). Our data suggest that these inputs may, by increasing tonic inhibition of granule cells, decrease the fraction of granule cells that are excited by incoming mossy fibre signals.
Phasic and tonic activation of granule cell GABAA receptors are predicted by modelling work to have different effects on the pattern of firing of granule and Golgi cells, with phasic activation (due to synaptic GABA release from Golgi cells) producing more synchronous rhythmic firing, and tonic inhibition reducing synchronicity and rhythmicity (Maex & De Schutter, 1998; De Schutter, 2002). The effect on firing pattern of the ACh-evoked GABA release will depend critically on the timing and duration of the physiological release of ACh in the cerebellum, about which we know little at present. All three transmitter release modes will decrease the number of granule cells that are simultaneously active, and may thus increase the number of motor programmes that can be stored in the cerebellum (Marr, 1969; Tyrrell & Willshaw, 1992).
Acknowledgments
This work was supported by the Wellcome Trust, a Wolfson- Royal Society award (D.A.), and a Burroughs-Wellcome fellowship (D.J.R.). We thank Harold Kimelberg for providing L-644,711 and Céline Auger, Daniela Billups, Alasdair Gibb, Mike Häusser, Païkan Marcaggi and Angus Silver for comments on an earlier version of the manuscript.
References
- Akopian A, Gabriel R, Witkovsky P. Calcium released from intracellular stores inhibits GABAA-mediated currents in ganglion cells of the turtle retina. J Neurophysiol. 1998;80:1105–1115. doi: 10.1152/jn.1998.80.3.1105. [DOI] [PubMed] [Google Scholar]
- Attwell D, Barbour B, Szatkowski M. Nonvesicular release of neurotransmitter. Neuron. 1993;11:401–407. doi: 10.1016/0896-6273(93)90145-h. [DOI] [PubMed] [Google Scholar]
- Barakat L, Bordey A. GAT-1 and reversible GABA transport in Bergmann glia in slices. J Neurophysiol. 2002;88:1407–1419. doi: 10.1152/jn.2002.88.3.1407. [DOI] [PubMed] [Google Scholar]
- Borden LA, Murali Dhar TG, Smith KE, Weinshank RL, Branchek TA, Gluchowski C. Tiagabine, SK&F 89976-A, CI-966, and NNC-711 are selective for the cloned GABA transporter GAT-1. Eur J Pharmacol. 1994;269:219–224. doi: 10.1016/0922-4106(94)90089-2. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Cull-Candy SG, Farrant M. Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J Physiol. 1996;497:753–759. doi: 10.1113/jphysiol.1996.sp021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley SG, Revilla V, Cull-Candy SG, Wisden W, Farrant M. Adaptive regulation of neuronal excitability by a voltage-independent potassium conductance. Nature. 2001;409:88–92. doi: 10.1038/35051086. [DOI] [PubMed] [Google Scholar]
- Corey JL, Davidson N, Lester HA, Brecha N, Quick MW. Protein kinase C modulates the activity of a cloned GABA transporter expressed in Xenopus oocytes via regulated subcellular redistribution of the transporter. J Biol Chem. 1994;269:14 759–14 767. [PubMed] [Google Scholar]
- Desaulles E, Boux O, Feltz P. Caffeine induced Ca2+ release inhibits GABAA responsiveness in rat identified native primary afferents. Eur J Pharmacol. 1991;203:137–140. doi: 10.1016/0014-2999(91)90803-x. [DOI] [PubMed] [Google Scholar]
- De Lacalle S, Hersh LB, Saper CB. Cholinergic innervation of the human cerebellum. J Comp Neurol. 1993;328:364–376. doi: 10.1002/cne.903280304. [DOI] [PubMed] [Google Scholar]
- De Schutter E. Cerebellar cortex: computation by extrasynaptic inhibition? Curr Biol. 2002;12:R363–365. doi: 10.1016/s0960-9822(02)00861-8. [DOI] [PubMed] [Google Scholar]
- Dominguez del Toro E, Juiz JM, Peng X, Lindstrom J, Criado M. Immunocytochemical localization of the α7 subunit of the nicotinic acetylcholine receptor in the rat central nervous system. J Comp Neurol. 1994;349:325–342. doi: 10.1002/cne.903490302. [DOI] [PubMed] [Google Scholar]
- Dröse S, Altendorf K. Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases. J Exp Biol. 1997;200:1–8. doi: 10.1242/jeb.200.1.1. [DOI] [PubMed] [Google Scholar]
- Eccles JC, Llinas R, Sasaki K. The inhibitory interneurones within the cerebellar cortex. Exp Brain Res. 1966;1:1–16. doi: 10.1007/BF00235206. [DOI] [PubMed] [Google Scholar]
- Gaspary HL, Wang W, Richerson GB. Carrier-mediated GABA release activates GABA receptors on hippocampal neurons. J Neurophysiol. 1998;80:270–281. doi: 10.1152/jn.1998.80.1.270. [DOI] [PubMed] [Google Scholar]
- Hamann M, Rossi D, Attwell D. Tonic and spillover inhibition control information flow through cerebellar cortex. Neuron. 2002;33:625–633. doi: 10.1016/s0896-6273(02)00593-7. [DOI] [PubMed] [Google Scholar]
- Hamori J, Somogyi J. Differentiation of cerebellar mossy fiber synapses in the rat: a quantitative electron microscope study. J Comp Neurol. 1983;220:365–377. doi: 10.1002/cne.902200402. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME. Neuromodulation: acetylcholine and memory consolidation. Trends Cogn Sci. 1999;3:353–359. doi: 10.1016/s1364-6613(99)01365-0. [DOI] [PubMed] [Google Scholar]
- Hille B, Billiard J, Babcock DF, Nguyen T, Koh DS. Stimulation of exocytosis without a calcium signal. J Physiol. 1999;520:23–31. doi: 10.1111/j.1469-7793.1999.00023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illing RB. A subtype of cerebellar Golgi cells may be cholinergic. Brain Res. 1990;522:267–274. doi: 10.1016/0006-8993(90)91471-r. [DOI] [PubMed] [Google Scholar]
- Irving AJ, Collingridge GL, Schofield JG. Interactions between Ca2+ mobilizing mechanisms in cultured rat cerebellar granule cells. J Physiol. 1992;456:667–680. doi: 10.1113/jphysiol.1992.sp019360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itouji A, Sakai N, Tanaka C, Saito N. Neuronal and glial localization of two GABA transporters (GAT1 and GAT3) in the rat cerebellum. Brain Res Mol Brain Res. 1996;37:309–316. doi: 10.1016/0169-328x(95)00342-p. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Ruigrok TJ, Caffe R, Cozzari C, Levey AI, Mugnaini E, Voogd J. Cholinergic innervation and receptors in the cerebellum. Prog Brain Res. 1997;114:67–96. doi: 10.1016/s0079-6123(08)63359-2. [DOI] [PubMed] [Google Scholar]
- Jabaudon D, Shimamoto K, Yasuda-Kamatani Y, Scanziani M, Gähwiler BH, Gerber U. Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc Natl Acad Sci USA. 1999;96:8733–8738. doi: 10.1073/pnas.96.15.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Sahara Y, Dzubay JA, Westbrook GL. Defining affinity with the GABAA receptor. J Neurosci. 1998;18:8590–8604. doi: 10.1523/JNEUROSCI.18-21-08590.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Yakel JL. Functional nicotinic ACh receptors on interneurones in the rat hippocampus. J Physiol. 1997;504:603–610. doi: 10.1111/j.1469-7793.1997.603bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M, Farrant M, Cull-Candy SG. Whole-cell and single-channel currents activated by GABA and glycine in granule cells of the rat cerebellum. J Physiol. 1995;485:419–435. doi: 10.1113/jphysiol.1995.sp020739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R. Transmitter leakage from motor nerve endings. Proc Roy Soc Lond B. 1977;196:59–72. doi: 10.1098/rspb.1977.0029. [DOI] [PubMed] [Google Scholar]
- Kemp JA, Marshall GR, Woodruff GN. Quantitative evaluation of the potencies of GABA-receptor agonists and antagonists using the rat hippocampal slice preparation. Br J Pharmacol. 1986;87:677–684. doi: 10.1111/j.1476-5381.1986.tb14585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelberg HK, Goderie SK, Higman S, Pang S, Waniewski RA. Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J Neurosci. 1990;10:1583–1591. doi: 10.1523/JNEUROSCI.10-05-01583.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirischuk S, Voitenko N, Kostyuk P, Verkhratsky A. Calcium signalling in granule neurones studied in cerebellar slices. Cell Calcium. 1996;19:59–71. doi: 10.1016/s0143-4160(96)90013-5. [DOI] [PubMed] [Google Scholar]
- Korn SJ, Dingledine R. Inhibition of GABA uptake in the rat hippocampal slice. Brain Res. 1986;368:247–255. doi: 10.1016/0006-8993(86)90568-8. [DOI] [PubMed] [Google Scholar]
- Korpi ER, Kuner T, Seeburg PH, Lüddens H. Selective antagonist for the cerebellar granule cell-specific γ-aminobutyric acid type A receptor. Mol Pharmacol. 1995;47:283–289. [PubMed] [Google Scholar]
- Laurie DJ, Seeburg PH, Wisden W. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain II: olfactory bulb and cerebellum. J Neurosci. 1992;12:1063–1076. doi: 10.1523/JNEUROSCI.12-03-01063.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lena C, Changeux JP. Role of Ca2+ ions in nicotinic facilitation of GABA release in mouse thalamus. J Neurosci. 1997;17:576–585. doi: 10.1523/JNEUROSCI.17-02-00576.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QR, Lopez-Corcuera B, Mandiyan S, Nelson H, Nelson N. Molecular characterization of four pharmacologically distinct gamma-aminobutyric acid transporters in mouse brain. J Biol Chem. 1993;268:2106–2112. [PubMed] [Google Scholar]
- Liu QY, Schaffner AE, Chang YH, Maric D, Barker JL. Persistent activation of GABAA receptor/Cl− channels by astrocyte-derived GABA in cultured embryonic rat hippocampal neurons. J Neurophysiol. 2000;84:1392–1403. doi: 10.1152/jn.2000.84.3.1392. [DOI] [PubMed] [Google Scholar]
- Maex R, De Schutter E. Synchronization of Golgi and granule cell firing in a detailed network model of the cerebellar granule cell layer. J Neurophysiol. 1998;80:2521–2537. doi: 10.1152/jn.1998.80.5.2521. [DOI] [PubMed] [Google Scholar]
- Manabe T, Yoshimori T, Henomatsu N, Tashiro Y. Inhibitors of vacuolar-type H+-ATPase suppress proliferation of cultured cells. J Cell Physiol. 1993;157:445–452. doi: 10.1002/jcp.1041570303. [DOI] [PubMed] [Google Scholar]
- Marr D. A theory of cerebellar cortex. J Physiol. 1969;202:437–470. doi: 10.1113/jphysiol.1969.sp008820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor JR, Gunthorpe MJ, Randall AD. The taurine uptake inhibitor guanidinoethyl sulphonate is an agonist at gamma-aminobutyric acid(A) receptors in cultured murine cerebellar granule cells. Neurosci Lett. 2000;286:25–28. doi: 10.1016/s0304-3940(00)01082-x. [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. Glutamate spillover suppresses inhibition by activating presynaptic mGluRs. Nature. 2000;404:498–502. doi: 10.1038/35006649. [DOI] [PubMed] [Google Scholar]
- Morara S, Brecha NC, Marcotti W, Provini L, Rosina A. Neuronal and glial localization of the GABA transporter GAT-1 in the cerebellar cortex. Neuroreport. 1996;7:2993–2996. doi: 10.1097/00001756-199611250-00039. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Shioda S, Nakajo S, Ueno S, Nakashima T, Nakai Y. Immunocytochemical localization of nicotinic acetylcholine receptor in the rat cerebellar cortex. Neurosci Res. 1997;29:233–239. doi: 10.1016/s0168-0102(97)00100-4. [DOI] [PubMed] [Google Scholar]
- Neelands TR, Fisher JL, Bianchi M, MacDonald RL. Spontaneous and gamma-aminobutyric acid (GABA)-activated GABAA receptor channels formed by epsilon subunit-containing isoforms. Mol Pharmacol. 1999;55:168–178. doi: 10.1124/mol.55.1.168. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Sieghart W, Somogyi P. Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J Neurosci. 1998;18:1693–1703. doi: 10.1523/JNEUROSCI.18-05-01693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JE, Kimelberg HK. Hypoosmotic volume regulation and osmolyte transport in astrocytes is blocked by an anion transport inhibitor, L-644,711. Brain Res. 1995;682:197–202. doi: 10.1016/0006-8993(95)00368-z. [DOI] [PubMed] [Google Scholar]
- Parnas H, Segel L, Dudel J, Parnas I. Autoreceptors, membrane potential and the regulation of transmitter release. Trends Neurosci. 2000;23:60–68. doi: 10.1016/s0166-2236(99)01498-8. [DOI] [PubMed] [Google Scholar]
- Pasantes-Morales H, Ochoa de la Paz LD, Sepulveda J, Quesada O. Amino acids as osmolytes in the retina. Neurochem Res. 1999;24:1339–1346. doi: 10.1023/a:1022568203717. [DOI] [PubMed] [Google Scholar]
- Richerson GB, Messer C. Effect of composition of experimental solutions on neuronal survival during rat brain slicing. Exp Neurol. 1995;131:133–143. doi: 10.1016/0014-4886(95)90015-2. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Hamann M. Spillover-mediated transmission at inhibitory synapses promoted by high affinity α6 subunit GABAA receptors and glomerular affinity. Neuron. 1998;20:783–795. doi: 10.1016/s0896-6273(00)81016-8. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403:316–321. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Slater NT. The developmental onset of NMDA receptor-channel activity during neuronal migration. Neuropharmacol. 1993;32:1239–1248. doi: 10.1016/0028-3908(93)90018-x. [DOI] [PubMed] [Google Scholar]
- Rutledge EM, Kimelberg HK. Release of [3H]-d-aspartate from primary astrocyte cultures in response to raised external potassium. J Neurosci. 1996;16:7803–7811. doi: 10.1523/JNEUROSCI.16-24-07803.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena NC, MacDonald RL. Properties of putative cerebellar γ-aminobutyric acidA receptor isoforms. Mol Pharmacol. 1996;49:567–579. [PubMed] [Google Scholar]
- Schwartz EA. Depolarization without calcium can release gamma-aminobutyric acid from a retinal neuron. Science. 1987;238:350–355. doi: 10.1126/science.2443977. [DOI] [PubMed] [Google Scholar]
- Tia S, Wang JF, Kotchabhakdi N, Vicini S. Developmental changes of inhibitory synaptic currents in cerebellar granule neurons: role of GABAA receptor α6 subunit. J Neurosci. 1996;16:3630–3640. doi: 10.1523/JNEUROSCI.16-11-03630.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrrell T, Willshaw D. Cerebellar cortex: its simulation and the relevance of Marr's theory. Phil Trans Roy Soc Lond B Biol Sci. 1992;336:239–257. doi: 10.1098/rstb.1992.0059. [DOI] [PubMed] [Google Scholar]
- Wall MJ. Furosemide reveals heterogeneous GABAA receptor expression at adult rat Golgi cell to granule cell synapses. Neuropharmacol. 2002;43:737–749. doi: 10.1016/s0028-3908(02)00085-0. [DOI] [PubMed] [Google Scholar]
- Wall MJ, Usowicz MM. Development of action potential-dependent and independent spontaneous GABAA receptor-mediated currents in granule cells of postnatal rat cerebellum. Eur J Neurosci. 1997;9:533–548. doi: 10.1111/j.1460-9568.1997.tb01630.x. [DOI] [PubMed] [Google Scholar]
- Wu FS, Gibbs TT, Farb DH. Dual activation of GABAA and glycine receptors by beta-alanine: inverse modulation by progesterone and 5-alpha-pregnan-3-alpha-ol-20-one. Eur J Pharmacol. 1993;246:239–246. doi: 10.1016/0922-4106(93)90037-a. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wang W, Richerson GB. GABA transaminase inhibition induces spontaneous and enhances depolarization-evoked GABA efflux via reversal of the GABA transporter. J Neurosci. 2001;21:2630–2639. doi: 10.1523/JNEUROSCI.21-08-02630.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Zhou Z. Ca2+-independent but voltage-dependent secretion in mammalian dorsal root ganglion cells. Nat Neurosci. 2002;5:425–430. doi: 10.1038/nn845. [DOI] [PubMed] [Google Scholar]
- Zhang ZW, Feltz P. Bicuculline blocks nicotinic acetylcholine response in isolated intermediate lobe cells of the pig. Br J Pharmacol. 1991;102:19–22. doi: 10.1111/j.1476-5381.1991.tb12125.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Petersen CCH, Nicoll RA. Effects of reduced vesicular filling on synaptic transmission in rat hippocampal neurones. J Physiol. 2000;525:195–206. doi: 10.1111/j.1469-7793.2000.t01-1-00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]