

Abstract
The polarized distribution of HCO3− transport was investigated in human nasal epithelial cells from normal and cystic fibrosis (CF) tissues. To test for HCO3− transport via conductive versus electroneutral Cl−/HCO3− exchange (anion exchange, AE) pathways, nasal cells were loaded with the pH probe 2′,7′-bis(carboxyethyl)-5(6)-carboxyfluorescein and mounted in a bilateral perfusion chamber. In normal, but not CF, epithelia, replacing mucosal Cl− with gluconate caused intracellular pH (pHi) to increase, and the initial rates (ΔpH min−1) of this increase were modestly augmented (∼26 %) when normal cells were pretreated with forskolin (10 μm). Recovery from this alkaline shift was dependent on mucosal Cl−, was insensitive to the AE inhibitor 4,4′-diisothiocyanatodihydrostilbene-2,2′-disulfonic acid (H2DIDS; 1.5 mm), but was sensitive to the cystic fibrosis transmembrane conductance regulator (CFTR) channel inhibitor diphenylamine-2-carboxylate (DPC; 100 μm). In contrast, removal of serosal Cl− caused pHi to alkalinize in both normal and CF epithelia. Recovery from this alkaline challenge was dependent on serosal Cl− and blocked by H2DIDS. Additional studies showed that serosally applied Ba2+ (5.0 mm) in normal, but not CF, cells induced influx of HCO3− across the apical membrane that was reversibly blocked by mucosal DPC. In a final series of studies, normal and CF cells acutely alkaline loaded by replacing bilateral Krebs bicarbonate Ringer (KBR) with Hepes-buffered Ringer solution exhibited basolateral, but not apical, recovery from an alkaline challenge that was dependent on Cl−, independent of Na+ and blocked by H2DIDS. We conclude that: (1) normal, but not CF, nasal epithelia have a constitutively active DPC-sensitive HCO3− influx/efflux pathway across the apical membrane of cells, consistent with the movement of HCO3− via CFTR; and (2) both normal and CF nasal epithelia have Na+-independent, H2DIDS-sensitive AE at their basolateral domain.
Studies of airway epithelial salt and water physiology have traditionally centred around measurements of Na+ and Cl− transport, and these studies have provided a large body of information about cellular signalling mechanisms important for regulating these ion transport processes and their protective functions in the respiratory tract. In contrast, fewer studies have focused on identifying the membrane elements important for the transport of H+ and HCO3− (Nord et al. 1988; Willumsen & Boucher, 1992; Lubman et al. 1995; Paradiso, 1997), which are necessary for the maintenance of intracellular pH (pHi) homeostasis. It is predicted that polarized airway epithelia distribute these mechanisms (apical/basolateral membrane) to establish salt and H+ gradients between the cytosol and secreted liquids, important for the regulation of airway surface liquid (ASL) ionic composition and pH.
Since the recognition that the cystic fibrosis transmembrane conductance regulator (CFTR) functions as a Cl− channel (Quinton, 1983), Cl− transport activity by CFTR-expressing tissues has been well studied (Quinton, 1983, 1990; Willumsen et al. 1989; Widdicombe & Wine, 1991; Anderson et al. 1991; Linsdell et al. 1999). However, the mechanisms underlying HCO3− secretion in CFTR-expressing epithelia remain poorly understood, even though HCO3− transport activity is impaired in cystic fibrosis (CF; Kaiser & Drack, 1974; Smith & Welsh, 1992). Of relevance to ASL acid-base homeostasis, a role for CFTR as a HCO3− transporter has been suggested in CF airway models, based on a variety of technical approaches comparing wild-type CFTR-expressing cells with ΔF508 CFTR-expressing CF cells (Smith & Welsh, 1992; Illek et al. 1997) or comparing wild-type mice with CFTR knockout (−/−) mice (Grubb & Gabriel, 1997). These studies suggest the possibility that a defect in HCO3− secretion through CFTR may lead to abnormally acidic ASL pH and thus have the potential to contribute to the pathophysiology of CF airway disease.
In addition to conductive CFTR-mediated HCO3− secretion, it is well known that a major mechanism for HCO3− transport across plasma membranes is Na+-independent, electroneutral Cl−/HCO3− exchange (anion exchange; AE). Both Cl− and HCO3− can be transported via AE and the direction of transport is determined by the gradients of both anions across the plasma membrane. Under normal physiological conditions, the ratio of extracellular-to-intracellular Cl− is greater than the inward-directed HCO3− gradient, and AE exchanges extracellular Cl− for intracellular HCO3−. When pHi is increased, i.e. when cell HCO3− increases, the inward-directed HCO3− gradient is reduced, thus further favouring efflux of HCO3− via electroneutral AE from the cell. Thus, AE functions as an ‘acid-loader’ by removing cell HCO3− during acute cellular alkalosis.
Recent studies have raised the possibility that CFTR and AE may exhibit a complex regulatory interaction when both proteins are co-localized in the same membrane. In this regard, Lee and co-workers (1999b) reported regulation of AE by CFTR expressed in NIH 3T3 and HEK 293 cells, as well as regulation of luminal AE by CFTR in mouse submandibular and pancreatic ducts (Lee et al. 1999a). In contrast to these studies, Mastrocola et al. (1998) reported that the transfection of wt CFTR, ΔF508 CFTR or vector did not influence AE activity in C129 cell lines. Whether such a relationship exists in airway epithelia will, in part, depend on determining whether CFTR and AE indeed co-localize at the apical barrier of airway epithelia. Utilizing RT-PCR, Dudeja and co-workers (1999) reported that the message for the AE2 and brain AE3 isoforms, but not AE1 or cardiac AE3 isoforms, was present in proximal human airways. Currently, however, no definitive data are available as to the apical/basolateral membrane distribution of any AE, or on the relative contributions of AE and CFTR on overall HCO3− fluxes in human normal and CF nasal epithelia.
Consequently, the aim of the present study was to investigate the polarized distribution (apical/basolateral membrane) of HCO3− transport in nasal epithelial cells in the presence and absence of functional CFTR. To this end, polarized normal and CF preparations were loaded with the pH-sensitive probe 2′,7′-bis(carboxyethyl)-5(6)-carboxyfluorescein (BCECF) to measure pHi, mounted in a bilateral perfusion chamber, and the response of pHi to ion substitution and inhibitor protocols employed to determine the relative roles of conductive, i.e. CFTR-mediated, versus AE-mediated HCO3− transport.
Methods
Subjects
Primary human nasal epithelial tissues were obtained, with written consent, from 12 normal subjects (32 ± 5 years old; seven males, five females) undergoing elective surgery for standard medical indications (e.g. sleep apnoea secondary to nasal obstruction) and nine cystic fibrosis patients (17 ± 5 years old; five males, four females) undergoing polypectomy. All procedures were approved by the University of North Carolina Committee for the Rights of Human Subjects and conformed to the Declaration of Helsinki.
Chemicals and solutions
The acetoxymethyl ester of BCECF (BCECF/AM) and 4,4′-diisothiocyanatodihydrostilbene-2,2′-disulfonic acid (H2DIDS) were purchased from Molecular Probes, Inc. (Eugene, OR, USA). Diphenylamine-2-carboxylate (DPC) was obtained from Research Biochemicals International (Natick, MA, USA). All other chemicals were obtained from Sigma Chemical Co. (St Louis, MO, USA).
The standard Krebs bicarbonate Ringer (KBR) solution contained (mm): 125 NaCl, 2.5 K2HPO4, 1.3 CaCl2, 1.3 MgCl2, 25 NaHCO3 and 5 d-glucose (5 % CO2/95 % O2; pH 7.4). For Cl−-free KBR, NaCl and MgCl2 were replaced mole-for-mole by sodium gluconate and MgSO4, respectively, and CaCl2 was replaced with 2 mm CaSO4 to compensate for Ca2+ chelation by gluconate, as previously reported (Clarke & Boucher, 1992). The Hepes-buffered Na+- and Cl−-free Ringer solution contained (mm): 150 N-methyl-d-glucamine (NMG) gluconate, 2.5 K2HPO4, 2.0 CaSO4, 1.3 MgSO4, 5 d-glucose and 10 Hepes (pH 7.4). For the Hepes-buffered Na+-free Ringer solution containing Cl−, the chemicals were identical to Na+- and Cl−-free Ringer solution except that NMG gluconate was replaced mole-for-mole by NMGCl (pH 7.4).
Cell culture and perfusion chamber
Human nasal epithelial (HNE) cells were harvested from polyps by enzymatic digestion (Protease XIV (Sigma) for 24–48 h at 4 °C) as previously described (Wu et al. 1985). HNE cells were plated on porous Transwell Col filters (pore diameter = 0.40 μm; Costar, Corning, Inc., NY, USA) affixed to O-rings and maintained in serum-free Ham's F-12 medium supplemented with insulin (10 μg ml−1), transferrin (5 μg ml−1), triiodothyronine (3 × 10−8m), endothelial cell growth supplement (3.75 μg ml−1), hydrocortisone (5 × 10−9m) and CaCl2 (10−3m). Polarized monolayers were studied 10–12 days after seeding, as previously described (Paradiso et al. 2001).
After achieving confluence, polarized monolayers of HNE were loaded with BCECF (5 μm at 37 °C for 25 min) and mounted in a miniature chamber over an objective (Zeiss LD Achroplan ×40, NA 0.6; working distance 1.8 mm) of a Zeiss Axiovert 35 microscope. The cell chamber and the method for independently perfusing the apical and basolateral membranes of cells have been previously described in detail (Paradiso et al. 2001).
Fluorimeter and measurements of pHi
Measurements of pHi in polarized HNE cells were obtained using a RatioMaster fluorimeter (Photon Technology International, Brunswick, NJ, USA) attached via fibre optics to the microscope. BCECF fluorescence from 30–40 cells (spot diameter ∼65 μm) was acquired alternately at 440 and 490 nm (emission ≥ 520 nm). At a given excitation wavelength (440 or 490 nm), background light levels were measured in non-loaded cells and subtracted from the corresponding signal measured in BCECF-loaded cells prior to taking the ratio (490/440). The corrected ratio was converted to pHi as previously described (Paradiso, 1997).
Data analysis
Means ±s.e.m. were calculated from the total number of measurements for a given experimental condition. Statistical significance was determined using the Student's paired t test, with P < 0.05 being considered significant.
Results
Basal pHi in normal and CF HNE cells bilaterally perfused with nominally CO2/HCO3−-free Ringer solution or KBR
Employing H+-sensitive microelectrodes, an earlier study by Willumsen & Boucher (1992) reported that basal pHi was 7.1–7.15 in normal and CF HNE cells bilaterally perfused with KBR (5 % CO2/25 mm HCO3−, pH 7.4). They also reported that basal steady-state pHi was not affected by the removal of HCO3− from the bathing medium, suggesting that HCO3− may not be important for the maintenance of steady-state basal pHi.
To reassess the role of a CO2/HCO3− buffer system on basal pHi, we compared the effects of Hepes-buffered NaCl Ringer solution and KBR on basal pHi in normal and CF HNE cells. As shown in Fig. 1, when normal (Fig. 1A, n = 10, four individuals) and CF (Fig. 1B, n = 10, four individuals) HNE cells were bilaterally exposed to Hepes-buffered NaCl Ringer (pH 7.4) solution, basal pHi was ∼7.1 in both cell preparations. When normal and CF cells were subsequently exposed to symmetrical KBR (Fig. 1A and B), pHi first rapidly decreased (due to the hydration of CO2) and then regulated back to a new steady-state basal pHi, which was lower than the values measured in a Hepes-buffered Ringer solution, in both cell preparations. On average, in Hepes-buffered NaCl Ringer solution, basal pHi was 7.16 ± 0.03 (n = 17; four individuals) and 7.13 ± 0.02 (n = 17; four individuals) in normal and CF HNE cells, respectively, consistent with our previous measurements of basal pHi in Hepes-buffered Ringer solution (also refer to Table 1 of Paradiso, 1997). In contrast, in the presence of bilateral KBR, basal pHi was 6.95 ± 0.02 (n = 138; twelve individuals) and 6.94 ± 0.01 (n = 102; nine individuals) in normal and CF HNE cells, respectively.
Figure 1. Basal intracellular pH (pHi) in polarized monolayers of normal and cystic fibrosis (CF) human nasal epithelial (HNE) cells bilaterally perfused with nominally CO2/HCO3−-free Ringer solution or Krebs bicarbonate Ringer (KBR).
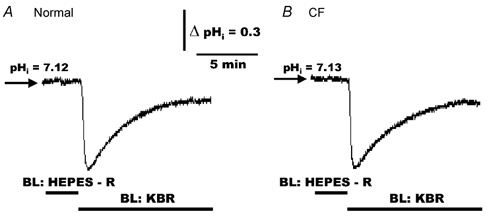
For both normal (A) and CF (B) cell preparations, HNE cells were initially bilaterally (BL) perfused with Hepes-buffered NaCl Ringer solution (pH 7.4) before changing to bilateral KBR (pH 7.4) at the times indicated in the tracings. Each trace is representative of ten separate experiments (four different individuals).
Our results demonstrate that steady-state basal pHi in normal and CF HNE cells is ∼0.15 pH units more acidic in KBR compared to Hepes-buffered NaCl Ringer solution, clearly suggesting a role for a CO2/HCO3− buffer system for the maintenance of cell pH. This issue is discussed further below.
Effects of amiloride on pHi in response to an CO2/HCO3− challenge in normal and CF HNE cells
In nominally CO2/HCO3−-free NaCl Ringer solution, we have previously identified an amiloride-sensitive Na+/H+ exchanger that was restricted to the basolateral membrane in both normal and CF HNE cells (Paradiso, 1997). To extend these observations, we examined whether the Na+/H+ exchanger was the major transporter that could account for the recovery of pHi during an acid challenge in a CO2/HCO3− buffer system (see Fig. 1). As shown in Fig. 2, when normal (Fig. 2A, n = 7, three individuals) and CF (Fig. 2B, n = 7, three individuals) HNE cells were symmetrically perfused with Hepes-buffered NaCl Ringer solutions, the addition of amiloride (500 μm) to the serosal compartment elicited no change in basal pHi. However, in the presence of serosal amiloride, when the perfusate was subsequently changed to bilateral KBR, pHi rapidly decreased to ∼6.6 and remained acidic in both normal and CF cell preparations; pHi recovered towards a new steady-state basal level in both normal and CF HNE cells only when amiloride was removed from the serosal perfusate (Fig. 2).
Figure 2. Effects of amiloride on pHi in response to a CO2/HCO3− challenge in polarized monolayers of normal and CF HNE cells.
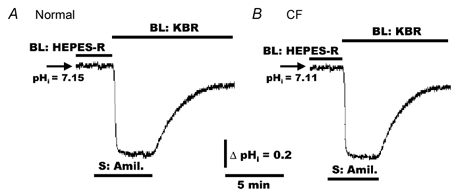
For both normal (A) and CF (B) cell preparations, HNE cells were initially bilaterally perfused with Hepes-buffered NaCl Ringer solution (pH 7.4) before adding amiloride (Amil., 500 μm) to the serosal (S) perfusate. In the presence of amiloride, the perfusate was subsequently changed to bilateral KBR (pH 7.4) and amiloride removed from the serosal compartment at the times indicated in the tracings. Each trace is representative of seven separate experiments (three different individuals).
The data thus far strongly suggest that the major membrane mechanism for the recovery of pHi in response to an acid challenge in the presence (Fig. 2) and absence (Paradiso, 1997) of a CO2/HCO3−-buffered solution is a basolateral Na+/H+ exchanger in normal and CF HNE cells. Because amiloride completely and reversibly blocked the recovery phase of the acid-load, a role for a Na+-HCO3− cotransporter, i.e. the influx of Na+ and HCO3− across the apical/basolateral membrane, is unlikely. Finally, the observation that amiloride added to the serosal perfusate induced no change of pHi in nominally CO2/HCO3−-free (Hepes-buffered) Ringer solution (Fig. 2) indicates that the basolateral Na+/H+ exchanger is inactive, i.e. there is no net efflux of H+ coupled to influx of Na+ via the basolateral Na+/H+ exchanger at a basal pHi ≥ 7.1, as previously reported in normal and CF HNE cells (Paradiso, 1997).
Acute effects of H2DIDS and amiloride on basal pHi
To identify the membrane transporters that acutely modulate pHi under basal conditions in a CO2/HCO3− buffer system, we tested the effects of mucosal and serosal H2DIDS (to block a putative AE) and amiloride (to inhibit the Na+/H+ exchanger) in normal and CF HNE cells bilaterally perfused with KBR.
As depicted in Fig. 3, exposing normal (Fig. 3A) or CF (Fig. 3B) cells to H2DIDS (1.5 mm) added to the mucosal perfusate caused no changes in basal pHi. In contrast, serosally applied H2DIDS elicited an alkalinization in both normal (Fig. 3A) and CF (Fig. 3B) HNE cells. On average, the magnitude of the increase (peak-basal value) of pHi in response to serosal H2DIDS was 0.10 ± 0.01 (n = 84, nine individuals) and 0.11 ± 0.01 (n = 68, nine individuals) in normal and CF HNE cells, respectively.
Figure 3. Response of pHi to mucosal and serosal additions of H2DIDS and amiloride in polarized monolayers of normal and CF HNE cells.
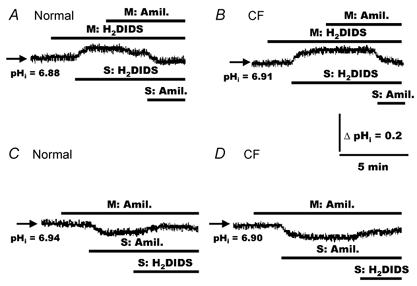
For both normal (A and C) and CF (B and D) cell preparations, HNE cells were bilaterally perfused with KBR before adding 4,4′-diisothiocyanatodihydrostilbene-2,2′-disulfonic acid (H2DIDS; 1.5 mm) and amiloride (500 μm) to the mucosal (M) and serosal (S) compartments as indicated in the tracings. Each trace is representative of seven separate experiments (three different individuals).
The increase in pHi in response to H2DIDS could potentially result from two sources: (i) the activity of a Na+/H+ exchanger during inhibition of AE and/or (ii) the cellular accumulation of HCO3− during the inhibition of a basolateral AE. To test whether Na+/H+ exchange contributed to the alkalinization of cells in response to H2DIDS added to the serosal perfusate, we examined the response of pHi to amiloride after H2DIDS treatment. As shown in Fig. 3, the addition of amiloride (500 μm) to the mucosal perfusate failed to alter pHi in either H2DIDS-treated normal (Fig. 3A) or CF (Fig. 3B) HNE cells. In contrast to mucosal amiloride addition, serosally applied amiloride (500 μm) following H2DIDS-elicited alkalinization induced a re-acidification of cell pH back to basal values in both normal (Fig. 3A) and CF (Fig. 3B) cell preparations, consistent with the inhibition of basolateral Na+/H+ exchange. Furthermore, under basal conditions, mucosal addition of amiloride again failed to alter pHi, whereas the addition of amiloride to the serosal perfusate caused cell pH to decrease in both normal (Fig. 3C) and CF (Fig. 3D) HNE cells. As noted above, these data are consistent with our previous report that the Na+/H+ exchanger is restricted to the basolateral membrane in both normal and CF HNE cells (Paradiso, 1997). On average, the magnitude of the decrease of pHi in response to serosal amiloride was 0.08 ± 0.02 (n = 7, three individuals) and 0.09 ± 0.01 (n = 7, three individuals) in normal and CF HNE cells, respectively.
The absolute change of pHi in normal and CF cells in response to serosally applied amiloride was not significantly different from the increase in cell pH induced by the serosal addition of H2DIDS, measured in both cell preparations. Furthermore, serosally applied H2DIDS following serosal amiloride treatment caused only a small or no change of pHi in both normal (Fig. 3C) and CF (Fig. 3D) HNE cells, consistent with little accumulation of HCO3− within cells under basal conditions during the short time period employed in these studies. Taken together, these results indicate that constitutively active basolateral, but not apical, H2DIDS-sensitive AE and Na+/H+ exchange contribute to the acute regulation of pHi in normal and CF airway epithelia under basal conditions in the presence of a CO2/HCO3− buffer system.
Effects of mucosal Cl− substitution on pHi in normal and CF HNE cells
The results thus far indicate that normal and CF airway epithelia lack a H2DIDS-sensitive pathway for modulating pHi at their apical membrane (see Fig. 3), suggesting the absence of an apically located AE. To test this notion more rigorously, we investigated whether changes in the luminal Cl− concentration could alter pHi in normal and CF HNE cells. Our expectation was that if AE were present at the apical membrane of cells, then removal of luminal Cl−, i.e. establishing a cell-to-lumen Cl− gradient, should increase pHi via a lumen-to-cell influx of HCO3−. For these studies, normal and CF HNE cells were perfused initially with bilateral KBR and effects of unilateral replacement of mucosal Cl− with gluconate (constant 5 %PCO2/25 mm HCO3−; pH 7.4) examined. As depicted in Fig. 4, removal of luminal Cl− (serosal bath constant KBR, pH 7.4) induced a transient alkalinization that returned to baseline in normal HNE cells (Fig. 4A). The mean maximum change (peak-basal value) of pHi elicited by this mucosal Cl−-free treatment was 0.18 ± 0.02 (n = 7, four individuals). In contrast, this manoeuvre failed to alter pHi in CF airway epithelia (Fig. 4B; n = 8, three individuals).
Figure 4. Effects of mucosal Cl−-free Ringer solution on pHi in polarized monolayers of normal and CF HNE cells.
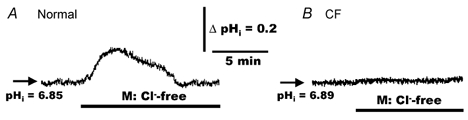
For both normal (A) and CF (B) cell preparations, HNE cells were initially bilaterally perfused with KBR before changing to mucosal (M) Cl−-free (gluconate replacing Cl−; constant 5 % PCO2/25 mm HCO3−, pH 7.4) Ringer solution at the times indicated in the tracings. The serosal perfusate remained KBR throughout these studies. A, representative example of seven separate experiments (four different individuals). B, representative example of eight separate experiments (three different individuals).
Because HNE cells may have at their basolateral border an H2DIDS-sensitive AE (see Fig. 3), it is possible that any Cl− gradient imposed across the apical membrane was ultimately also imposed across the basolateral membrane by lowering intracellular Cl−, resulting in an increase in the serosal-to-cell Cl− gradient across the basolateral membrane. For example, a change in pHi due to an outward-directed (cell-to-lumen) Cl− gradient coupled to an inward-directed (lumen-to-cell) HCO3− influx pathway may be reduced (shunted) if a dominant basolateral AE removes HCO3− entering the cell across the apical membrane. Therefore, to ‘isolate’ the influx of HCO3− across the apical membrane from the efflux of HCO3− across the basolateral membrane in response to changes in luminal Cl− concentration, we added H2DIDS to the serosal perfusate to block any compensatory effects of the putative basolateral AE on HCO3−-dependent changes of pHi. As shown in Fig. 5, in the presence of serosal H2DIDS, replacing luminal Cl− with gluconate (imposing a cell-to-lumen Cl− gradient) alkalinized normal cells, consistent with significant lumen-to-cell HCO3− uptake in normal HNE cells (Fig. 5A). The maximum change of pHi from steady-state level (induced by H2DIDS) to peak value (elicited by Cl− removal) was 0.38 ± 0.02 (n = 11, four individuals), which was significantly greater (P < 0.01) than the change in pHi induced by luminal Cl− removal in non-H2DIDS-treated normal cells (i.e. 0.18 ± 0.02 pH units; see above). Furthermore, returning Cl− to the luminal perfusate (increasing the lumen-to-cell Cl− gradient) stimulated cells to re-acidify back to basal levels, consistent with cell-to-lumen HCO3− movement in normal airway epithelia (see Fig. 5A). In contrast, removal of luminal Cl− in the presence of serosal H2DIDS induced no detectable change of pHi in CF airway cells (Fig. 5B; n = 8, three individuals). It should be noted that we have previously shown that these Cl−-linked changes of pHi in normal (or CF HNE) cells are absent in a nominally CO2/HCO3−-free environment (see Fig. 3 of Paradiso, 1992), indicating that the responses of pHi to altered luminal Cl− concentrations reflect movement of HCO3− across the apical membrane in normal nasal cells.
Figure 5. Effects of mucosal Cl− substitution on pHi in polarized monolayers of normal and CF HNE cells exposed to serosal and/or mucosal H2DIDS.
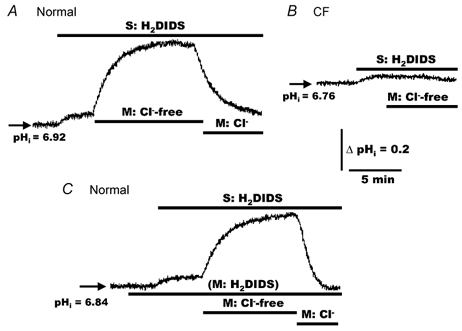
For both normal (A and C) and CF (B) HNE cell preparations, cells were initially perfused bilaterally with KBR and H2DIDS (1.5 mm) added to the serosal and/or mucosal compartment before removing Cl− (gluconate replacing Cl−; constant 5 % PCO2/25 mm HCO3−, pH 7.4) or re-adding Cl− to the mucosal perfusate, as shown in the tracings. The serosal perfusate remained KBR throughout the study. A and C, representative examples of eleven separate experiments (four different individuals). B, is representative of eight separate experiments (three different individuals).
Although the changes in cell pH shown in Fig. 5A in response to altering mucosal Cl− could be interpreted as being consistent with the activity of AE at the apical membrane in normal HNE cells, Fig. 5C shows that these Cl−-dependent changes of pHi were not blocked by mucosal H2DIDS (1.5 mm). On average, in the presence of mucosal H2DIDS, the magnitude of change of pHi from steady-state level (induced by serosal H2DIDS) to peak value obtained in luminal Cl−-free was 0.36 ± 0.03 (n = 11, four individuals), which was not significantly different from the mean value determined in the absence of mucosal H2DIDS.
Effects of forskolin and DPC on pHi in response to mucosal Cl- substitution in normal HNE cells
Three observations reported here and by others suggest that the change in pHi in response to the removal/re-addition of luminal Cl− may be mediated by the movement of HCO3− across the apical membrane via CFTR, which appears to be constitutively active in normal HNE cells. First, unilaterally replacing mucosal Cl− with gluconate induced an alkalinization of pHi under basal conditions in normal (Fig. 5A), but not CF (Fig. 5B), HNE cells. Second, removal of luminal Cl− under basal conditions causes a major depolarization of the apical membrane electrical potential difference (Va) in normal, but not CF, HNE cells (Willumsen et al. 1989). Depolarization of Va would favour conductive entry of HCO3− into the cell, perhaps via CFTR (Illek et al. 1997), which would lead to cell alkalinization. Finally, recent studies by Huang et al. (2001) reported substantial CFTR channel activity in Calu-3 cells, a human airway cell line, under basal conditions that was approximately half the activity measured after forskolin addition, suggesting that CFTR Cl− channels were half-maximally activated under basal conditions. Moreover, they showed that the constitutive activation of the CFTR Cl− channel reflected release of 5′-adenosine triphosphate (ATP) into the lumen that was metabolically degraded into adenosine. Adenosine activated the apically located adenosine receptors (A2B-subtype) and adenylyl cyclase present in the apical membrane by means of a G protein (i.e. Gs). Sufficient adenosine 3′,5′-cyclic monophosphate (cAMP) was generated to activate protein kinase A, and hence CFTR, in a diffusionally restricted apical microdomain, without increasing cAMP in other cellular compartments. Similar constitutive release of ATP and generation of adenosine have been reported both in vivo (Donaldson et al. 2000) and in vitro (Watt et al. 1998) in normal and CF HNE cells, and constitutive activation of CFTR (∼half-maximal) has been reported in normal nasal epithelium in vivo (Knowles et al. 1995) under basal conditions.
To investigate the effects of maximal CFTR activation on pHi, we examined the effects of forskolin on changes in mucosal Cl− concentration in normal HNE cells. As shown in Fig. 6, in the presence of serosal H2DIDS the addition of forskolin (10 μm) to the mucosal/serosal perfusate and subsequently H2DIDS to the mucosal compartment elicited no change in pHi in normal HNE cells perfused bilaterally with KBR. In the presence of forskolin and bilateral H2DIDS, replacing luminal Cl− with gluconate (imposing a cell-to-lumen Cl− gradient) again alkalinized cells, consistent with significant lumen-to-cell HCO3− uptake in normal HNE cells (Fig. 6A). The maximum change of pHi from steady-state level (induced by H2DIDS) to peak value (elicited by Cl− removal) was 0.37 ± 0.02 (n = 7, three individuals), which was not significantly different from the change in pHi induced by luminal Cl− removal in non-stimulated normal cells treated with bilateral H2DIDS (i.e. 0.36 ± 0.03 pH units; also see Fig. 5C). Furthermore, returning Cl− to the luminal perfusate (increasing the lumen-to-cell Cl− gradient) stimulated cells to re-acidify back to basal levels, again consistent with cell-to-lumen HCO3− movement in normal airway epithelia (see Fig. 6A).
Figure 6. Effects of forskolin and diphenylamine-2-carboxylate (DPC) on pHi in response to mucosal Cl− substitution in polarized monolayers of normal HNE cells.
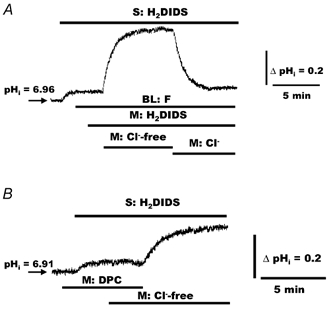
A, normal HNE cells were initially perfused with bilateral (BL) KBR. Serosal H2DIDS (1.5 mm) and bilateral forskolin (F; 10 μm) were added to the perfusate at the times indicated in the tracing, before changing the mucosal perfusate from KBR to Cl−-free (gluconate replacing Cl−; constant 5 % PCO2/25 mm HCO3−, pH 7.4) KBR. Cl− was subsequently re-added to the mucosal compartment at the time indicated in the tracing. B, normal HNE cells were bilaterally perfused with KBR and mucosal DPC (100 μm) and serosal H2DIDS (1.5 mm) added to the perfusate at the times indicated in the tracing. In the presence of DPC, changing the mucosal perfusate from KBR to Cl−-free KBR elicited no change in pHi, whereas removal of DPC induced an alkalinization of the cells. For both traces, the serosal perfusate remained KBR throughout the study. A, representative example of seven separate experiments (three different individuals). B, representative example of seven separate experiments (four different individuals).
In the absence (see Fig. 5C) and presence (see Fig. 6A) of forskolin, the initial rate of alkalinization (ΔpHi min−1) in normal HNE cells, measured over the pHi range 7.05 to 7.20 in response to mucosal Cl− removal, was 0.14 ± 0.02 (n = 11; four individuals) and 0.19 ± 0.02 (n = 7; four individuals), respectively. In the presence of forskolin, the initial rate in the change in pHi (i.e. ΔpHi min−1 = 0.19) induced by luminal Cl− removal in normal cells was marginally greater (P < 0.05) than the change in pHi (i.e. ΔpHi min−1= 0.14) induced by luminal Cl− removal in non-stimulated normal HNE cells. This difference represents an increase in the rate of alkalinization in forskolin-treated normal cells over non-stimulated cells by ∼26 %. Furthermore, in the presence of bilateral H2DIDS, bilaterally applied forskolin (10 μm) had no effect on steady-state pHi in the presence or absence of luminal Cl− in CF HNE cells (n = 7; three individuals; data not shown).
Although there are no available blockers that selectively inhibit the CFTR Cl− channel activity without potentially affecting other membrane channels/transporters, DPC has been reported (Stutts et al. 1990; Zhang et al. 2000) to decrease CFTR anion conductance in several cell types, including airway epithelial cells. We utilized DPC to investigate whether the Cl−-induced changes in pHi in response to unilaterally replacing Cl− with gluconate in the mucosal bath were mediated via CFTR. As depicted in Fig. 6B, in normal HNE cells bilaterally perfused with KBR, the addition of DPC (100 μm) to the mucosal perfusate elicited little or no change in basal pHi, whereas the subsequent administration of serosal H2DIDS again caused pHi to increase. However, mucosal DPC attenuated the increase of pHi in response to mucosal Cl−-free Ringer solution (compare Fig. 6B with Fig. 5A). Notably, when DPC was removed from the mucosal compartment, the inhibitory effect of DPC was reversed, and the cells alkalinized. The maximum average change of pHi from steady-state level (induced by serosal H2DIDS) to peak value following luminal DPC removal was 0.18 ± 0.02 (n = 7, four individuals).
Effects of mucosal DPC on pHi in response to serosal Ba2+ in normal and CF HNE cells
Previous studies on HNE cells have reported that the Va also reflects, in part, the basolateral K+ conductance, i.e. cell depolarization in response to the blockade of a basolateral K+ conductance will be reflected in a depolarization of Va (Willumsen et al. 1989). Based on this information, we designed protocols to test whether CFTR transports HCO3− in response to altered Va by exposing polarized HNE cells to serosal Ba2+, a known K+ channel blocker (Devor & Frizzell, 1998).
For these studies, we first tested whether Ba2+ could alter pHi in cells perfused with a nominally CO2/HCO3− buffer solution. As illustrated in Fig. 7A, when normal HNE cells were bilaterally perfused with Hepes-buffered NaCl Ringer solution, the addition of H2DIDS (1.5 mm) and subsequently Ba2+ (5.0 mm) to the serosal compartment induced no change in basal pHi. However, when normal (Fig. 7B and C) and CF (Fig. 7D) HNE cells were perfused with bilateral KBR, the addition of H2DIDS (1.5 mm) to the serosal perfusate again caused pHi to increase. Moreover, in the presence of bilateral KBR, the subsequent addition of serosally applied Ba2+ (5.0 mm) to normal airway epithelia induced a substantial and further increase in pHi that was completely reversible with the removal of Ba2+ from the serosal compartment (Fig. 7B). The magnitude of the change of pHi from the H2DIDS-treated to peak values elicited by Ba2+ was 0.19 ± 0.03 (n = 6, three individuals). Furthermore, in normal HNE cells, the mucosal addition of DPC (100 μm) reversibly blocked changes of pHi in response to serosal Ba2+ (Fig. 7C). Finally, neither mucosal DPC (100 μm) nor serosal Ba2+ (5.0 mm) altered pHi in CF airway cells (Fig. 7D). Collectively, these data support a role for CFTR in HCO3− transport across the apical membrane in normal HNE cells.
Figure 7. Effects of mucosal DPC on pHi in response to serosal Ba2+ in polarized monolayers of normal and CF HNE cells.
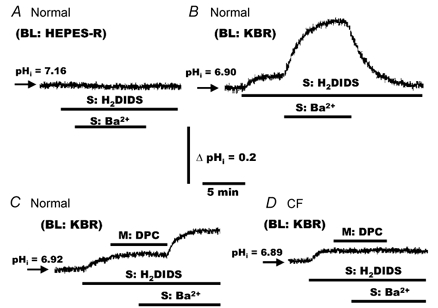
A, normal HNE cells were bilaterally perfused with Hepes-buffered NaCl Ringer solution before adding H2DIDS (1.5 mm) and Ba2+ (5.0 mm) to the serosal compartment at the times shown in the trace. For both normal (B and C) and CF (D) HNE cell preparations, cells were perfused bilaterally with KBR and H2DIDS (1.5 mm) administrated to the serosal perfusate. Following H2DIDS treatment, DPC (100 μm) and/or Ba2+ (5.0 mm) were added to and/or removed from the serosal and/or mucosal compartment in both normal (B and C) and CF (D) cell preparations at the times shown in the tracings. Each trace is representative of six separate experiments (three different individuals).
Effects of serosal Cl− substitution on pHi in normal and CF HNE cells
For these studies, normal and CF HNE cells were initially perfused in bilateral KBR with H2DIDS (1.5 mm) added to the serosal perfusate to block the putative basolateral AE. As shown in Fig. 8, in the presence of serosal H2DIDS, changing the serosal perfusate to Cl−-free (gluconate replacement) KBR (mucosal bath constant KBR, pH 7.4) caused no change in pHi in either normal (Fig. 8A) or CF (Fig. 8B) airway cells. However, with serosal Cl−-free Ringer solution, i.e. in the presence of a cell-to-serosal Cl− gradient, cell pH increased in both normal and CF HNE cells when H2DIDS was removed from the serosal compartment. The initial rate of alkalinization (ΔpHi min−1) measured over the pHi range 7.05–7.25 in response to serosal Cl− substitution was 0.16 ± 0.04 (n = 10; four individuals) and 0.18 ± 0.03 (n = 10; four individuals) in normal and CF HNE cells, respectively. These initial rates were not significantly different. It should be noted that the maximum absolute change of pHi in response to serosal Cl− substitution was slightly smaller in normal (ΔpHi= 0.48 ± 0.02) compared to CF (ΔpHi= 0.56 ± 0.01) HNE cells. Moreover, unlike CF HNE cells, there was a spontaneous secondary slow re-acidification of pHi following the peak alkalinization in normal airway cells (Fig. 6A). These two observations for normal and CF HNE cells are addressed later in the study. Furthermore, as shown in Fig. 8, the re-addition of serosal Cl− (establishing a serosal-to-cell Cl− gradient) caused both normal (Fig. 8A) and CF (Fig. 8B) HNE cells to re-acidify rapidly back toward basal pHi values. Taken together, these results are consistent with the activity of a H2DIDS-sensitive AE at the basolateral membrane in both normal and CF cell preparations.
Figure 8. Effects of H2DIDS and serosal Cl− substitution on pHi in polarized monolayers of normal and CF HNE cells.
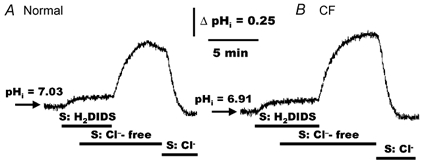
For both normal (A) and CF (B) HNE cell preparations, cells were initially perfused bilaterally with KBR and H2DIDS (1.5 mm) added to the serosal compartment before removing Cl− (gluconate replacing Cl−; constant 5 % PCO2/25 mm HCO3−, pH 7.4) from the serosal perfusate, as shown in the tracings. In the presence of serosal Cl−-free KBR, removal of serosal H2DIDS caused pHi to alkalinize, whereas the re-addition of serosal Cl− caused cell pH to re-acidify back to basal level. The mucosal perfusate remained KBR throughout the study. Each tracing is representative of ten separate experiments (four different individuals).
Effects of mucosal and serosal Cl− on pHi in alkalinized normal and CF HNE cells
Because AE can be broadly divided into Na+-dependent AE, i.e. requiring external Na+ for the cycling of extracellular Cl− for intracellular HCO3−, or Na+-independent AE (Boron, 1986), we next tested for Na+-dependent and Na+-independent AE activity at the apical and basolateral domains of normal and CF HNE cells. For Na+-dependent AE, HNE cell preparations were initially perfused bilaterally with KBR, and the perfusate subsequently changed to bilateral Hepes-buffered Cl−-free (sodium gluconate; pH 7.4 (nominally CO2- and HCO3−-free)) Ringer solution. This manoeuvre alkalinized cells due to rapid loss of cell CO2, thereby establishing a cell-to-extracellular HCO3− gradient across the apical/basolateral domain of airway cells. Following cell alkalinization, the Cl− dependency of recovery from an alkaline/HCO3− load in the presence of extracellular Na+ was tested by exposing cells to mucosal or serosal Cl− (Hepes-buffered NaCl Ringer solution).
As shown in Fig. 9A, the pattern in the change of pHi was markedly different between normal and CF HNE cells when the perfusate was changed from symmetrical KBR to symmetrical Hepes-buffered Cl−-free Ringer solution. In normal cells, the initial increase of pHi following bilateral Cl−-free treatment was followed by a spontaneous secondary re-acidification back towards the basal value. In contrast to normal cells, CF HNE cells rapidly alkalinized and no secondary re-acidification was noted. Moreover, the magnitude of the increase of pHi following bilateral Cl−-free treatment was significantly (P < 0.01) reduced in normal (ΔpHi= 0.54 ± 0.04, n = 9; three individuals) compared to CF (ΔpHi= 0.79 ± 0.03, n = 9; three individuals) HNE cells. In the presence of bilateral Na+, the addition of Cl− to the mucosal bath (a condition that established a lumen-to-cell gradient for Cl− and a cell-to-lumen gradient for HCO3−) had little effect on the rate of the secondary re-acidification in normal HNE cells and elicited no change in pHi in CF cells (Fig. 9A). However, when Cl− was added to the serosal perfusate, pHi rapidly returned towards baseline in both cell preparations (Fig. 9A).
Figure 9. Effects of mucosal and serosal Cl−(± Na+) on pHi in polarized monolayers of alkalinized normal and CF HNE cells.
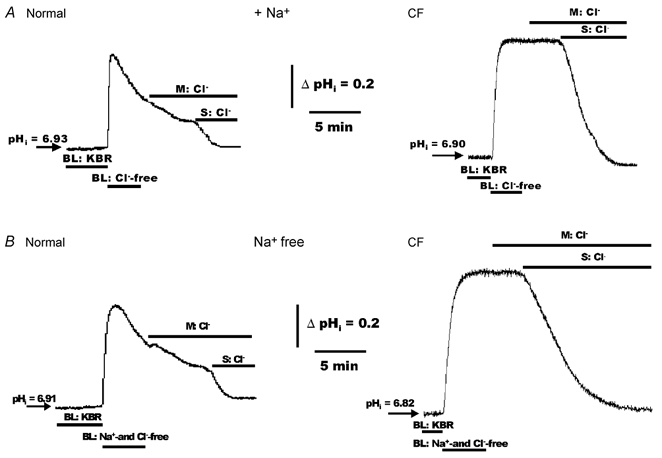
For both normal and CF HNE cell preparations, cells were initially perfused bilaterally with KBR. In one set of experiments shown in panel A, HNE (normal and CF) cells were exposed to bilateral Hepes-buffered Cl−-free (sodium gluconate; no CO2 or HCO3−, pH 7.4) Ringer solution to alkalinize the cells (due to the rapid loss of cell CO2). In a second set of experiments, shown in panel B, HNE (normal and CF) cells were exposed to bilateral Hepes-buffered Na+- and Cl−-free (N-methyl-d-glucamine gluconate replacing Na+ and Cl−; no CO2 or HCO3−, pH 7.4) Ringer solution. Following alkalinization of the cells, Cl− was re-added to the mucosal or serosal perfusate at the times depicted in the tracings. Each trace is representative of nine separate experiments (three different individuals).
To test for the Na+ dependency of AE activity, HNE cell preparations were initially perfused bilaterally with KBR and the perfusate subsequently changed to bilateral Hepes-buffered Na+- and Cl−-free (NMG gluconate; pH 7.4 (nominally CO2- and HCO3−-free)) Ringer solution to alkalinize cells (Fig. 9B). Following cell alkalinization, the Cl− dependency of recovery from an alkaline load was again tested by exposing cells to mucosal or serosal Cl− (Hepes-buffered NMGCl Ringer solution). As shown in Fig. 9B, in normal HNE cells, the initial increase of pHi following bilateral Na+- and Cl−-free treatment was followed by a spontaneous secondary re-acidification back towards baseline. Again in contrast to normal cells, CF HNE cells rapidly alkalinized and no secondary re-acidification was detected. Moreover, the maximum absolute increase of pHi following bilateral Na+- and Cl−-free treatment was again significantly (P < 0.01) reduced in normal (ΔpHi= 0.59 ± 0.03, n = 9; three individuals) compared to CF (ΔpHi= 0.83 ± 0.02, n = 9; three individuals) HNE cells. The addition of Cl− to the mucosal bath (again, a condition that established a lumen-to-cell gradient for Cl− and a cell-to-lumen gradient for HCO3−) slightly decreased or had no effect on the rate of the secondary re-acidification in normal HNE cells and elicited no change in pHi in CF cells (Fig. 9B). In contrast to the mucosal addition of Cl−, when Cl− was added to the serosal perfusate, pHi rapidly accelerated towards baseline in both airway cell preparations (Fig. 9B). The observation that re-acidification of pHi in response to serosal addition of Cl− in alkalinized cells was similar in normal and CF epithelia with (see Fig. 9A) or without Na+ (see Fig. 9B) suggests that the predominant AE pathway is Na+-independent.
Effects of serosal H2DIDS on pHi in alkalinized normal and CF HNE cells
We next investigated whether re-acidification of pHi in response to serosal Cl− could be blocked by H2DIDS in airway cells challenged with an alkaline/HCO3− load (Fig. 10). In normal (Fig. 10A) and CF (Fig. 10B) cells bilaterally perfused with KBR, the addition of serosal H2DIDS (1.5 mm) caused a small increase in pHi. When the serosal and mucosal compartments were subsequently changed to Hepes-buffered Na+- and Cl−-free (NMG gluconate; nominally CO2- and HCO3−-free) Ringer solution to alkalinize cells, no recovery of pHi was detected when Cl− was added to the serosal compartment in the presence of H2DIDS. However, removal of H2DIDS from the serosal compartment in the continual presence of Cl− caused an increase in the rate of pHi recovery from the alkaline challenge in both normal (Fig. 10A) and CF (Fig. 10B) HNE cell preparations.
Figure 10. Effects of serosal H2DIDS (± Cl−) on changes of pHi in polarized monolayers of alkalinized normal and CF HNE cells.
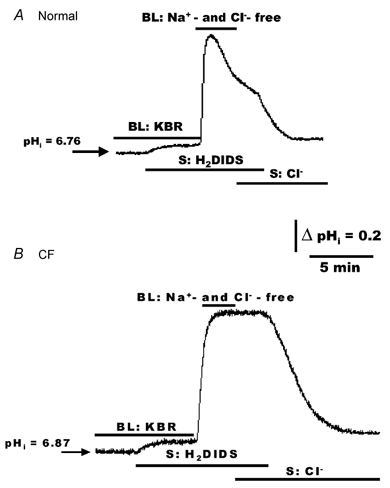
For both normal (A) and CF (B) HNE cell preparations, cells were initially perfused bilaterally (BL) with KBR. In the presence of bilateral KBR, H2DIDS (1.5 mm) was added to the serosal compartment (A and B) at the times shown in the tracings, before changing from bilateral KBR to bilateral Hepes-buffered Na+- and Cl−-free (N-methyl-d-glucamine gluconate replacing Na+ and Cl−; no CO2 or HCO3−, pH 7.4) Ringer solution. Following alkalinization of the cells, serosal H2DIDS blocked the re-acidification of pHi when Cl− was re-added to the serosal perfusate in both normal (A) and CF (B) HNE cells, as shown in the tracings. Note that in the presence of serosal Cl−, removal of H2DIDS from the serosal compartment resulted in a rapid re-acidification of pHi in both normal and CF HNE cells (A and B). Each trace is representative of seven separate experiments (three different individuals).
Spontaneous acidification and effects of mucosal DPC on pHi in alkalinized normal HNE cells
The spontaneous secondary acidification following an alkaline shift in normal HNE cells could potentially result from: (i) the reversal of a Na+/H+ exchanger (i.e. a cell-to-extracellular gradient of Na+ linked to an extracellular-to-cell gradient of H+), which is known to be present on the basolateral, but not apical, membrane of human nasal cells (Paradiso, 1997); (ii) the reversal of a Na+-HCO3− cotransporter (i.e. loss of both Na+ and HCO3− from cells); and/or (iii) loss of cell HCO3− via CFTR.
With regard to Na+/H+ exchange, it is possible that during an alkaline shift in pHi, Na+ could exit the cell in exchange for extracellular H+ via the Na+/H+ exchanger, and consequently, cells would acidify. However, this possibility seems unlikely, for three reasons. First, the pattern and rate of the spontaneous secondary re-acidification were similar in the presence and absence of extracellular Na+ (see Fig. 9). Second, we have repeated these experiments in the absence of extracellular Na+, pretreating cells with serosal amiloride (500 μm; n = 6, three different individuals) to block the activity of the basolateral Na+/H+ exchanger (Paradiso, 1997). This manoeuvre did not prevent the secondary re-acidification in normal HNE cells (data not shown). Finally, we have previously reported (Paradiso, 1997) that CF HNE cells also have Na+/H+ exchanger activity at their basolateral border that is pharmacologically and kinetically identical to that of the Na+/H+ exchanger in normal nasal tissue, yet no spontaneous secondary re-acidification was detected in CF tissue following the initial alkaline shift in pHi (see Fig. 9).
Although we do not have functional evidence for the presence of a Na+-HCO3− cotransporter at the apical/basolateral aspect of human nasal cells, Devor and co-workers (1999) have functionally identified a basolateral Na+-HCO− cotransporter in Calu-3 cells. Thus, it is possible that the spontaneous re-acidification in response to an alkaline shift of pHi in normal HNE cells resulted from the reversal of the cotransporter. However, like the argument against the involvement of Na+/H+ exchange in the re-acidification in response to an alkaline shift in pHi in normal HNE cells, it again seems unlikely that a cell-to-extracellular movement of Na+ and HCO3− via Na+-HCO− cotransport could account for the spontaneous recovery following an alkaline challenge, since, as noted above, the pattern of re-acidification was identical in Na+-containing (see Fig. 9A) and Na+-free (see Fig. 9B) Ringer solutions. Moreover, as noted above in the discussion of Fig. 2, a role for a Na+-HCO3− cotransporter in the regulation of pHi in surface nasal epithelial cells is not suggested by this study, since basolaterally applied amiloride could completely and reversibly inhibit recovery of pHi from an acid challenge when airway cells were perfused with KBR.
To elucidate whether the spontaneous secondary re-acidification towards basal values in normal cells challenged by CO2 removal was mediated via CFTR, we tested whether mucosally applied DPC blocked this process. As shown in Fig. 11A, when normal HNE cells were initially perfused with symmetrical KBR, the addition of mucosal DPC (100 μm) elicited no change in basal pHi. However, changing the mucosal/serosal perfusate to symmetrical Hepes-buffered Na+- and Cl−-free Ringer solution resulted in a larger alkalinization in cell pH and markedly reduced the secondary re-acidification in DPC-treated compared to non-DPC-treated normal cells (see Fig. 10A). On average, the maximum absolute increase of pHi following bilateral Na+- and Cl−-free treatment was significantly (P < 0.01) increased in DPC-treated cells (ΔpHi= 0.78 ± 0.02, n = 7; three different individuals) compared to non-treated (i.e. ΔpHi of 0.59 ± 0.03, see Fig. 10A) normal HNE cells. Moreover, the subsequent removal of DPC from the mucosal bath caused pHi to return rapidly to baseline (Fig. 11A). In contrast to normal cells, the mucosal addition or removal of DPC (100 μm) had no effect on the magnitude change of pHi in CF HNE cells when the perfusate was changed from symmetrical KBR to symmetrical Hepes-buffered Na+- and Cl−-free Ringer solution (Fig. 11B). On average, the maximum absolute increase of pHi following bilateral Na+- and Cl−-free treatment was not significantly different in CF cells treated with mucosal DPC (ΔpHi= 0.85 ± 0.03, n = 7; three individuals) compared to non-DPC-treated CF cells (ΔpHi= 0.83 ± 0.02; see Fig. 10B).
Figure 11. Effects of mucosal DPC on changes of pHi in polarized monolayers of alkalinized normal and CF HNE cells.
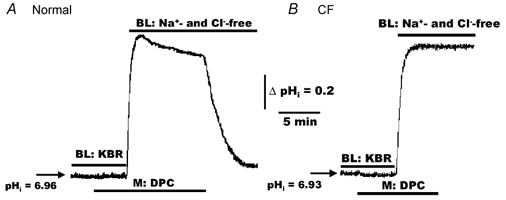
For both normal (A) and CF (B) HNE cell preparations, cells were initially perfused bilaterally (BL) with KBR before adding DPC (100 μm) to the mucosal perfusate at the times shown in each trace. In the presence of DPC, changing from bilateral KBR to bilateral Hepes-buffered Na+- and Cl−-free (N-methyl-d-glucamine gluconate replacing Na+ and Cl−; no CO2 or HCO3−, pH 7.4) Ringer solution, resulted in a larger alkalinization of pHi in normal DPC-treated cells (A) compared to normal cells not treated with DPC (see trace A of Fig. 10), and that removal of DPC elicited a rapid re-acidification of pHi back to base level in normal (A), but not CF (B), HNE cells. Each trace is representative of seven separate experiments (three different individuals).
Discussion
Regulation of basal pHi in normal and CF HNE cells
In a previous study (Paradiso, 1997) using polarized human nasal epithelial cells in primary culture, we reported that basal pHi was ∼7.1 in both normal and CF cells bilaterally perfused with nominally CO2/HCO3−-free Hepes-buffered NaCl Ringer solution. Furthermore, we showed in this earlier study that both normal and CF HNE cells expressed at their basolateral membrane a Na+/H+ exchanger that was inactive at basal pHi≥ 7.1 (Paradiso, 1997).
In the present study, when polarized normal and CF HNE cells were bilaterally perfused in CO2-containing KBR, basal pHi was 6.94 and 6.95, respectively (also see Fig. 1). We speculate that the lower basal pHi in normal and CF HNE cells bilaterally perfused with KBR resulted from the inclusion of CO2 in the perfusate, which lowered pHi and led to the activation of both a basolateral H2DIDS-sensitive AE and a Na+/H+ exchanger. This speculation is based on the following observations generated by this study. First, serosally applied H2DIDS elicited an alkalinization of pHi in both normal and CF cell preparations (see Fig. 3A and B), a response predicted for the inhibition of an active basolateral AE. Second, serosally applied amiloride induced an acidification of basal pHi in both normal and CF cell preparations, a response consistent with the inhibition of an active basolateral Na+/H+ exchanger (see Fig. 3C and D). Third, the parallel activities of AE and a Na+/H+ exchanger were revealed by the observation that serosally applied amiloride acidified pHi following post-H2DIDS-induced alkalinization of cell pH (Fig. 3A and B), and that the addition of H2DIDS subsequent to amiloride caused a small increase in pHi (Fig. 3C and D). Taken together, our data strongly suggest that basal pHi in airway epithelia exposed to physiological PCO2 is maintained via the coordinated activities of a Na+/H+ exchanger and AE at the basolateral membrane in normal and CF HNE cells. The coordinated activities of both exchangers may play an important compensatory function in maintaining cell pH within narrow limits during dynamic shifts in CO2 tension in the airway lumen during the respiratory cycle, i.e. CO2 falls (∼0 Torr) during inspiration and rises (∼40 Torr) during expiration.
HCO3− transport across the apical membrane in HNE cells
In the present study, ion gradients were manipulated across the apical membrane to investigate whether HCO3− influx/efflux pathways were linked to luminal perfusate Cl− concentrations in normal and CF airway cells. In the presence of serosal H2DIDS (to block HCO3− efflux across the basolateral membrane; see Fig. 4 and Fig. 5) and symmetrical HCO3− (constant 5 %PCO2), removal of luminal Cl− resulted in cytoplasmic alkalinization in normal HNE cells due to the influx of HCO3− across the apical membrane (see Fig. 5A; also see Paradiso, 1992). Conversely, when alkalinized normal HNE cells were subsequently exposed to luminal Cl−, cells rapidly re-acidified towards basal levels (see Fig. 5A), consistent with the cellular efflux of HCO3− across the apical membrane. Furthermore, as discussed above, these Cl−-dependent changes of pHi are largely independent of exogenously applied forskolin in normal HNE cells (see Fig. 6A), suggesting that the membrane mechanism responsible for the transport of HCO3− across the apical membrane is constitutively active under basal conditions.
Although these Cl−-dependent changes of pHi, in response to changes in luminal Cl− appeared to be consistent with an apically located AE in normal HNE cells, we argue that the mechanism mediating these changes in pHi is the constitutively active CFTR Cl−/HCO3− conductance in the apical membrane in normal airway epithelia, based on a series of observations (also see discussion above relevant to Fig. 6A). Previous microelectrode studies (Willumsen et al. 1989) showed that removal of luminal Cl− caused Va to depolarize in normal (but not CF) cells by ∼30 mV, whereas the re-addition of luminal Cl− repolarized Va. As reported above, normal and CF HNE cells exhibit an average pHi of ∼6.95 when cells are exposed to bilateral KBR. From this mean pHi value, cell HCO3− concentrations, calculated from the Henderson-Hasselbach equation (Roos & Boron, 1981), is ∼8 mm. Accordingly, in the presence of luminal KBR, the ratio of HCO3− concentration across the apical membrane ([25 mm]out/[∼8 mm]in) is approximately equal to and opposite from the electrical gradient (Va∼30 mV; Willumsen et al. 1989) across this barrier. Therefore, we predict no net movement of HCO3− via CFTR under basal conditions. However, removal of luminal Cl− induces a depolarization of ∼30 mV across the apical membrane, i.e. Va approaches 0 mV, which now favours HCO3− influx driven by the lumen-to-cell electrochemical gradient for this anion, and, as seen in Fig. 5A, pHi increases from ∼6.97 to ∼7.4. The concentration of cellular HCO3− at pHi of ∼7.4 will be close to 25 mm, and [HCO3−]in=[HCO3−]out, consistent with the absence of an electrical driving potential across the apical membrane. When Cl− is restored to the luminal bath (repolarizing Va by ∼30 mV), HCO3− now moves from cell-to-lumen, again driven by the electrochemical gradient across the apical barrier, and the cell re-acidifies.
Several other lines of evidence support the notion of electrogenic HCO3− transport via CFTR across the apical membrane in normal HNE cells, rather than electroneutral exchange for Cl− via an AE. First, removal of luminal Cl− failed to induce changes of pHi in CF HNE cells (Fig. 5B), clearly suggesting the requirement for functional CFTR to mediate Cl−-associated changes of pHi in normal airway cells. Second, mucosal H2DIDS, a known inhibitor of AE (Mastrocola et al. 1998), but not CFTR (Paradiso et al. 2001), failed to block Cl−-linked changes of pHi in normal airway cells in the absence (Fig. 5C) and presence (Fig. 6A) of forskolin. Third, mucosal DPC, an inhibitor of CFTR, reversibly blocked the increase of pHi in normal airway epithelia in response to mucosal Cl−-free Ringer solution (see Fig. 6B). Fourth, both normal and CF cells exposed to bilateral Hepes-buffered Cl−- and HCO3−-free Ringer solution acutely alkalinized, whereas only the normal nasal preparations exhibited a secondary re-acidification (see Fig. 9 and Fig. 10). We propose that this re-acidification resulted from loss of cell HCO3− via CFTR, since it was absent in CF airway epithelia (see Fig. 9 and Fig. 10) and was reversibly inhibited by mucosal DPC in normal airway cells (see Fig. 11). Finally, in normal (but not CF) nasal cells bilaterally perfused with KBR (but not Hepes-buffered Ringer solution), serosally applied Ba2+ induced an increase in pHi that was reversibly blocked by mucosal DPC (see Fig. 7), strongly arguing for the conductive movement of HCO3− via CFTR.
Further support for the concept that CFTR mediates apical membrane HCO3− translocation is derived from patch clamp studies by Poulsen et al. (1994), who reported CFTR-mediated HCO3− conductance in NIH 3T3 cells recombinantly expressing wild-type CFTR. Similar findings have been reported by Linsdell et al. (1999), Illek et al. (1997) and Hogan et al. (1997) in a variety of epithelial cell types.
We would note here that some of our findings differ from those of others. Unlike the finding in mouse pancreatic and submandibular ducts, which have at their apical membrane a CFTR-regulated AE system (Lee et al. 1999a), our findings strongly indicate that CFTR, and not AE, is the primary HCO3− transport route in normal airway cells and that AE is restricted to the basolateral membrane in both normal and CF HNE cells. Moreover, recent studies by Wheat and co-workers (2000) reported that CFTR induces the mRNA expression of ‘downregulated in adenoma’ (DRA), and that DRA is itself an AE at the apical membrane in a tracheal epithelial cell line (CFT-1) derived from a CF patient. They suggested that the tracheal HCO3− secretion defect in patients with CF is partly a result of the downregulation of the apically located AE activity mediated by DRA with loss of CFTR function. The apparent discrepancy between their data and ours may be in the selection of tissues used for the studies, i.e. a transformed cell line versus epithelial cells in primary culture.
The concept that CFTR mediates HCO3− translocation across the apical membrane has implications for regulation of ASL pH as well as pHi. For example, HCO3− secretion via CFTR should have an impact on altering pH and HCO3− content of ASL. Consistent with this notion, there is strong evidence that lack of HCO3− secretion renders the pH of liquids lining epithelial surfaces abnormally acidic in CF patients. For example, Kaplan and co-workers reported that CF ejaculate is acidic (pH ∼6.6) relative to normal semen (pH > 8.0), consistent with the absence of HCO3− in seminal fluid (Kaplan et al. 1968). Furthermore, studies by Durie (1989) reported that reduced HCO3− and Cl− transport within pancreatic ducts could account for deficient fluid secretion in the pancreas of CF subjects. In addition, Kaiser & Drack (1974) reported diminished excretion of HCO3− from single sweat glands of patients with CF. With relevance to human airways, studies by Smith & Welsh (1992) have reported that cAMP stimulates HCO3− secretion across normal but not CF nasal epithelia. Importantly, recent studies (Coakley et al. 2000) on well differentiated bronchial epithelial cells have shown that ASL pH is more acidic and exhibits a lower HCO3− concentration in CF compared to normal airway cultures.
HCO3− transport across the basolateral border in HNE cells
Utilizing two experimental approaches to assess for AE, our data demonstrate that normal and CF HNE cells express an AE that is restricted to the basolateral domain of cells. In the first approach, changes in pHi were detected in response to imposed Cl− gradients across the basolateral membrane, and the changes of pHi were reversibly blocked by serosal H2DIDS in both normal and CF HNE cells (see Fig. 8). In the second approach, recovery from an alkaline challenge was independent of extracellular Na+, accelerated by serosal, but not mucosal Cl−, and reversibly inhibited by serosal H2DIDS in normal HNE cells (see Fig. 9 and Fig. 10). Like normal airway cells, the recovery from an alkaline load in CF cells was again independent of extracellular Na+, absolutely dependent on serosal, but not mucosal addition of Cl−, and reversibly blocked by serosal H2DIDS (see Fig. 9 and Fig. 10).
The biological role of the basolateral AE provides several important functions that are interrelated. For example, this exchanger serves as an important mechanism for Cl− entry and HCO3− exit from the cell and thus functions as an acid-loader during acute cellular alkalosis. In addition to the metabolic role for AE, AE coupled to a Na+/H+ exchanger will have important influences on the distribution of ions and water between the extracellular space and the cell during volume regulation (Grinstein et al. 1985). Finally, for Cl−-driven secretion in airways, the basolateral Na+/H+ exchanger and Cl−/HCO3− exchanger, operating in parallel, are functionally equivalent to the basolateral Na+-K+-2Cl− cotransporter and thus can provide an alternative mechanism for the serosal-to-cell uptake of Cl−.
Conclusion
We have identified pathways for HCO3− translocation across the apical and basolateral domains of polarized airway epithelia. Across the apical membrane of normal airway epithelia, HCO3− appears to translocate via a CFTR-mediated electrodiffusive pathway, with no evidence for AE. In contrast, a H2DIDS-sensitive AE pathway exists for HCO3− translocation across the basolateral barrier. The basolateral AE, in concert with the basolateral Na+/H+ exchanger, appears to control responses to local environmental changes (e.g. PCO2) that can acutely alter pHi. We speculate that in normal airway tissues, the transport of HCO3− via CFTR is central to maintaining ASL pH within physiological levels. Moreover, the secretion of HCO3− into ASL appears to be balanced by the secretion of H+ via a recently reported (Paradiso et al. 2000) non-gastric form of the H+,K+-ATPase, which is localized at the apical membrane in both normal and CF airway epithelia. Furthermore, it is reasonable to suggest that it is the activity of the H+,K+-ATPase that, in part, maintains the electrochemical driving force for HCO3− secretion via CFTR in normal airway cells by lowering ASL pH. In addition, in CF airways, ASL pH is predicted to be more acidic than normal airway epithelia with loss of CFTR-mediated HCO3− secretion. In support of this later notion, our recent in vitro studies (Coakley et al. 2000) showing that ASL pH is lower (pH 6.13) in CF airway epithelial cells relative to normal cells (pH 6.44) was linked to a reduction in ASL HCO3− content in CF compared to normal airway cells. We speculate that the lack of CFTR-mediated HCO3− secretion, coupled to a constitutively active H+,K+-ATPase, rather than the absence of a regulatory interaction between apical CFTR and AE, accounts for this difference in luminal pH between CF and normal airway epithelia.
Acknowledgments
We thank L. Brown for editorial assistance, and Drs J. R. Yankaskas and S. Randell for providing normal and CF human nasal tissues required for the study. We especially thank Dr E. H. Larsen and his laboratory (Zoophysiological Laboratory A, August Krogh Institute, University of Copenhagen, Denmark) for constructing the miniature perfusion chamber that made these studies possible. This work was supported by NIH grant NHLBI R01 44173, and a grant from the American Cystic Fibrosis Foundation to A.M.P.
REFERENCES
- Anderson MP, Gregory RJ, Thompson S, Souza DW, Paul S, Mulligan RC, Smith AE, Welsh MJ. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991;253:202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- Boron WF. Intracellular pH regulation in epithelial cells. Annu Rev Physiol. 1986;48:377–388. doi: 10.1146/annurev.ph.48.030186.002113. [DOI] [PubMed] [Google Scholar]
- Clarke LL, Boucher RC. Chloride secretory response to extracellular ATP in normal and cystic fibrosis nasal epithelia. Am J Physiol. 1992;263:C348–356. doi: 10.1152/ajpcell.1992.263.2.C348. [DOI] [PubMed] [Google Scholar]
- Coakley RD, Paradiso AM, Grubb BR, Gatzy JT, Chadburn JL, Boucher RC. Abnormal airway surface liquid pH (pHASL) regulation in cultured CF bronchial epithelium. Pediatr Pulmonol Suppl. 2000;14:194. [Google Scholar]
- Devor DC, Frizzell RA. Modulation of K+ channels by arachidonic acid in T84 cells. II. Activation of a Ca2+-independent K+ channel. Am J Physiol. 1998;274:C149–160. doi: 10.1152/ajpcell.1998.274.1.C149. [DOI] [PubMed] [Google Scholar]
- Devor DC, Singh AK, Lambert LC, Deluca A, Frizzell RA, Bridges RJ. Bicarbonate and chloride secretion in Calu-3 human airway epithelial cells. J Gen Physiol. 1999;113:743–760. doi: 10.1085/jgp.113.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson SH, Lazarowski ER, Picher M, Knowles MR, Stutts MJ, Boucher RC. Basal nucleotide levels, release, and metabolism in normal and cystic fibrosis airways. Mol Med. 2000;6:969–982. [PMC free article] [PubMed] [Google Scholar]
- Dudeja PK, Hafez N, Tyagi S, Gailey CA, Toofanfard M, Alrefai WA, Nazir TM, Ramaswamy K, Al-Bazzaz FJ. Expression of the Na+/H+ and Cl−/HCO3− exchanger isoforms in proximal and distal human airways. Am J Physiol. 1999;276:L971–978. doi: 10.1152/ajplung.1999.276.6.L971. [DOI] [PubMed] [Google Scholar]
- Durie PR. The pathophysiology of the pancreatic defect in cystic fibrosis. Acta Paediatr Scand Suppl. 1989;363:41–44. doi: 10.1111/apa.1989.78.s363.41. [DOI] [PubMed] [Google Scholar]
- Grinstein S, Rothstein A, Cohen S. Mechanism of osmotic activation of Na+/H+ exchange in rat thymic lymphocytes. J Gen Physiol. 1985;85:765–787. doi: 10.1085/jgp.85.5.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb BR, Gabriel SE. Intestinal physiology and pathology in gene-targeted mouse models of cystic fibrosis. Am J Physiol. 1997;273:G258–266. doi: 10.1152/ajpgi.1997.273.2.G258. [DOI] [PubMed] [Google Scholar]
- Hogan DL, Crombie DL, Isenberg JI, Svendsen P, Schaffalitzky De Muckadell OB, Ainsworth MA. CFTR mediates cAMP- and Ca2+-activated duodenal epithelial HCO3− secretion. Am J Physiol. 1997;272:G872–878. doi: 10.1152/ajpgi.1997.272.4.G872. [DOI] [PubMed] [Google Scholar]
- Huang P, Lazarowski ER, Tarran R, Milgram SL, Boucher RC, Stutts MJ. Compartmentalized autocrine signaling to cystic fibrosis transmembrane conductance regulator at the apical membrane of airway epithelial cells. Proc Natl Acad Sci U S A. 2001;98:14120–14125. doi: 10.1073/pnas.241318498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illek B, Yankaskas JR, Machen TE. cAMP and genistein stimulate HCO3− conductance through CFTR in human airway epithelia. Am J Physiol. 1997;272:L752–761. doi: 10.1152/ajplung.1997.272.4.L752. [DOI] [PubMed] [Google Scholar]
- Kaiser D, Drack E. Diminished excretion of bicarbonate from the single sweat gland of patients with cystic fibrosis of the pancreas. Eur J Clin Invest. 1974;4:261–265. doi: 10.1111/j.1365-2362.1974.tb00402.x. [DOI] [PubMed] [Google Scholar]
- Kaplan E, Shwachman H, Perlmutter AD, Rule A, Khaw KT, Holsclaw DS. Reproductive failure in males with cystic fibrosis. N Engl J Med. 1968;279:65–69. doi: 10.1056/NEJM196807112790203. [DOI] [PubMed] [Google Scholar]
- Knowles MR, Paradiso AM, Boucher RC. In vivo nasal potential difference: techniques and protocols for assessing efficacy of gene transfer in cystic fibrosis. Hum Gene Ther. 1995;6:447–457. doi: 10.1089/hum.1995.6.4-445. [DOI] [PubMed] [Google Scholar]
- Lee MG, Choi JY, Luo X, Strickland E, Thomas PJ, Muallem S. Cystic fibrosis transmembrane conductance regulator regulates luminal Cl−/HCO3− exchange in mouse submandibular and pancreatic ducts. J Biol Chem. 1999a;274:14670–14677. doi: 10.1074/jbc.274.21.14670. [DOI] [PubMed] [Google Scholar]
- Lee MG, Wigley WC, Zeng W, Noel LE, Marino CR, Thomas PJ, Muallem S. Regulation of Cl−/HCO3− exchange by cystic fibrosis transmembrane conductance regulator expressed in NIH 3T3 and HEK 293 cells. J Biol Chem. 1999b;274:3414–3421. doi: 10.1074/jbc.274.6.3414. [DOI] [PubMed] [Google Scholar]
- Linsdell P, Tabcharani JA, Rommens JM, Hou YX, Chang XB, Tsui LC, Riordan JR, Hanrahan JW. Permeability of wild-type and mutant cystic fibrosis transmembrane conductance regulator chloride channels to polyatomic anions. J Gen Physiol. 1999;110:355–364. doi: 10.1085/jgp.110.4.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubman RL, Danto SI, Chao DC, Fricks CE, Crandall ED. Cl−-HCO3− exchanger isoform AE2 is restricted to the basolateral surface of alveolar epithelial cell monolayers. Am J Respir Cell Mol Biol. 1995;12:211–219. doi: 10.1165/ajrcmb.12.2.7865219. [DOI] [PubMed] [Google Scholar]
- Mastrocola T, Porcelli AM, Rugolo M. Role of CFTR and anion exchanger in bicarbonate fluxes in C127 cell lines. FEBS Lett. 1998;440:268–272. doi: 10.1016/s0014-5793(98)01468-9. [DOI] [PubMed] [Google Scholar]
- Nord EP, Brown SES, Crandall ED. Cl−/HCO3− exchange modulates intracellular pH in rat type II alveolar epithelial cells. J Biol Chem. 1988;263:5599–5606. [PubMed] [Google Scholar]
- Paradiso AM. Identification of Na+/H+ exchange in human normal and cystic fibrosis ciliated airway epithelium. Am J Physiol. 1992;262:757–764. doi: 10.1152/ajplung.1992.262.6.L757. [DOI] [PubMed] [Google Scholar]
- Paradiso AM. ATP-activated basolateral Na+/H+ exchange in human normal and cystic fibrosis airway epithelium. Am J Physiol. 1997;273:L148–158. doi: 10.1152/ajplung.1997.273.1.L148. [DOI] [PubMed] [Google Scholar]
- Paradiso AM, Coakley RD, Winders A, Ribeiro CM, Kreda SM, Rochelle LG, Burch LH, Boucher RC. Functional identification and tissue distribution of two forms of H+,K+-ATPase in proximal human airway. Pediatr Pulmonol Suppl. 2000;20:206. [Google Scholar]
- Paradiso AM, Ribeiro CMP, Boucher RC. Polarized signaling via purinoceptors in normal and cystic fibrosis airway epithelia. J Gen Physiol. 2001;117:53–68. doi: 10.1085/jgp.117.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen JH, Fischer H, Illek B, Machen TE. Bicarbonate conductance and pH regulatory capability of cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci U S A. 1994;91:5340–5344. doi: 10.1073/pnas.91.12.5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinton PM. Chloride impermeability in cystic fibrosis. Nature. 1983;301:421–422. doi: 10.1038/301421a0. [DOI] [PubMed] [Google Scholar]
- Quinton PM. Cystic fibrosis: a disease in electrolyte transport. FASEB J. 1990;4:2709–2717. doi: 10.1096/fasebj.4.10.2197151. [DOI] [PubMed] [Google Scholar]
- Roos A, Boron WF. Intracellular pH. Physiol Rev. 1981;61:296–434. doi: 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- Smith JJ, Welsh MJ. cAMP stimulates bicarbonate secretion across normal, but not cystic fibrosis airway epithelia. J Clin Invest. 1992;89:1148–1153. doi: 10.1172/JCI115696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutts MJ, Henke DC, Boucher RC. Diphenylamine-2-carboxylate (DPC) inhibits both Cl− conductance and cyclooxygenase of canine tracheal epithelium. Pflugers Arch. 1990;415:611–616. doi: 10.1007/BF02583514. [DOI] [PubMed] [Google Scholar]
- Watt WC, Lazarowski ER, Boucher RC. Cystic fibrosis transmembrane regulator-independent release of ATP. Its implications for the regulation of P2Y2 receptors in airway epithelia. J Biol Chem. 1998;273:14053–14058. doi: 10.1074/jbc.273.22.14053. [DOI] [PubMed] [Google Scholar]
- Wheat VJ, Shumaker H, Burnham C, Shull GE, Yankaskas JR, Soleimani M. CFTR induces the expression of DRA along with Cl−/HCO3− exchange activity in tracheal epithelial cells. Am J Physiol Cell Physiol. 2000;279:C62–71. doi: 10.1152/ajpcell.2000.279.1.C62. [DOI] [PubMed] [Google Scholar]
- Widdicombe JH, Wine JJ. The basic defect in cystic fibrosis. Trends Biochem Sci. 1991;16:474–477. doi: 10.1016/0968-0004(91)90183-v. [DOI] [PubMed] [Google Scholar]
- Willumsen NJ, Boucher RC. Intracellular pH and its relationship to regulation of ion transport in normal and cystic fibrosis human nasal epithelia. J Physiol. 1992;455:247–269. doi: 10.1113/jphysiol.1992.sp019300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willumsen NJ, Davis CW, Boucher RC. Cellular Cl− transport in cultured cystic fibrosis airway epithelium. Am J Physiol. 1989;256:C1045–1053. doi: 10.1152/ajpcell.1989.256.5.C1045. [DOI] [PubMed] [Google Scholar]
- Wu R, Yankaskas J, Cheng E, Knowles MR, Boucher R. Growth and differentiation of human nasal epithelial cells in culture. Serum-free, hormone-supplemented medium and proteoglycan synthesis. Am Rev Respir Dis. 1985;132:311–320. doi: 10.1164/arrd.1985.132.2.311. [DOI] [PubMed] [Google Scholar]
- Zhang ZR, Zeltwanger S, McCarty NA. Direct comparison of NPPB and DPC as probes of CFTR expressed in Xenopus oocytes. J Membr Biol. 2000;175:35–52. doi: 10.1007/s002320001053. [DOI] [PubMed] [Google Scholar]