

Abstract
The cerebellum is important for many aspects of behaviour, from posture maintenance and goal-oriented reaching movements to timing tasks and certain forms of learning. In every case, information flowing through the cerebellum passes through Purkinje neurons, which receive input from the two primary cerebellar afferents and generate continuous streams of action potentials that constitute the sole output from the cerebellar cortex to the deep nuclei. The tonic firing behaviour observed in Purkinje neurons in vivo is maintained in brain slices even when synaptic inputs are blocked, suggesting that Purkinje neuron activity relies to a significant extent on intrinsic conductances. Previous research has suggested that the interplay between Ca2+ currents and Ca2+-activated K+ channels (KCa channels) is important for Purkinje cell activity, but how many different KCa channel types are present and what each channel type contributes to cell behaviour remains unclear. In order to better understand the ionic mechanisms that control the behaviour of these neurons, we investigated the effects of different Ca2+ channel and KCa channel antagonists on Purkinje neurons in acute slices of rat cerebellum. Our data show that Ca2+ entering through P-type voltage-gated Ca2+ channels activates both small-conductance (SK) and large-conductance (BK) KCa channels. SK channels play a role in setting the intrinsic firing frequency, while BK channels regulate action potential shape and may contribute to the unique climbing fibre response.
The cerebellum is highly conserved across vertebrate species and is required for optimal performance of a number of behaviours, including target-directed reaching motions, movements requiring precision timing, balance and posture control, certain forms of motor learning and reflex adaptation, and a number of cognitive tasks that involve the planning and execution of specific action sequences (Dow & Moruzzi, 1958; Ito, 1984; Ivry, 1997; Schmahmann, 1997). A central component of the cerebellar circuitry is the layer of Purkinje neurons within the cerebellar cortex. Information entering the cerebellum through the two principal afferent systems – the mossy fibre-parallel fibre pathway and the olivary climbing fibre pathway – converges in the extensive dendritic trees of the Purkinje neurons, and the GABAergic projection from Purkinje cells to the deep cerebellar nuclei is the only source of output from the cerebellar cortex (Ito, 1984). In keeping with the central position Purkinje neurons occupy in the cerebellar circuitry, insults that disrupt their development or survival generally result in classic cerebellar symptoms such as ataxia of the trunk and limbs, speech disturbances, postural imbalance and nystagmus (Dow & Moruzzi, 1958; Wallesch & Bartels, 1997; Koeppen, 1998).
Previous studies have shown that Purkinje neurons in vivo fire tonically at rates of 10 to 150 Hz and that relevant stimuli such as retinal slip or tactile stimulation modulate the firing rate (Armstrong & Rawson, 1979; Ojakangas & Ebner, 1992; Ebner, 1998). Purkinje cells continue to fire action potentials spontaneously in reduced preparations such as brain slices (Llinas & Sugimori, 1980a; Hausser & Clark, 1997; Williams et al. 2002), and even as dissociated cell bodies from which most of the dendritic tree has been removed (Raman & Bean, 1999). In addition, the spontaneous firing rates observed in these in vitro preparations are quite similar to average firing rates of Purkinje neurons in vivo. Based on these findings, it has been proposed that the tonic activity of Purkinje neurons in vivo is generated by intrinsic conductances, and that synaptic input modulates rather than dictates the firing frequency (Hausser & Clark, 1997).
The main excitatory drive behind the intrinsic spiking behaviour of Purkinje neurons is the presence of resurgent Na+ channels, which favour repetitive firing by briefly reopening during the latter stages of action potential repolarization (Raman & Bean, 1997). Purkinje neurons also express at least four different types of voltage-gated Ca2+ channels, P, L, T and R (Llinas et al. 1989; Regan, 1991; Yokoyama et al. 1995; Chung et al. 2000), but the net effect of inward Ca2+ current near rest is paradoxically to hyperpolarize the membrane potential, presumably by activating one or more types of Ca2+-activated K+ channels (KCa channels) (Crepel & Penit-Soria, 1986; Raman & Bean, 1999; Williams et al. 2002). Accordingly, the removal of extracellular Ca2+ from the bathing solution results in irregular firing behaviour and abnormally high firing rates (Llinas & Sugimori, 1980a). In addition, the interruption of spike firing that follows climbing fibre activation in vivo is thought to be due to KCa channels (Llinas & Sugimori, 1980a,b; Hounsgaard & Midtgaard, 1989), indicating that this class of ion channels is important both for the intrinsic firing pattern and for the modulation of this pattern by synaptic inputs.
Purkinje neurons are known to express KCa channels of the BK class, named for their ‘big’ unitary conductance, on their somata and main dendritic trunks (Gahwiler & Llano, 1989; Gruol et al. 1991; Knaus et al. 1996; Jacquin & Gruol, 1999). It was recently demonstrated that they also contain small-conductance (SK) KCa channels (Stocker & Pedarzani, 2000; Cingolani et al. 2002). BK and SK channels are both activated by intracellular Ca2+ and increase the membrane's permeability to K+, driving the membrane potential toward the K+ reversal potential (for reviews, see Latorre et al. 1989; Vergara et al. 1998). They differ from each other, however, in that SK channels are not voltage dependent, responding to Ca2+ equally well at hyperpolarized and depolarized membrane potentials (Blatz & Magleby, 1986; Kohler et al. 1996; Vergara et al. 1998), while BK channels require both membrane depolarization and intracellular Ca2+ for maximal activation under physiological conditions (Latorre, 1994). Consistent with these differing properties, BK and SK channels have been found to be important for different aspects of cell function in studies of other types of neurons. In hippocampal pyramidal neurons, BK channels contribute to action potential repolarization and a fast after-hyperpolarization (Lancaster & Nicoll, 1987; Storm, 1987), and have been proposed to regulate Ca2+ influx during single spikes (Shao et al. 1999) as they do in some presynaptic nerve terminals (Robitaille & Charlton, 1992). BK channels are also important for spike repolarization in hippocampal interneurons (Zhang & McBain, 1995), dorsal vagal neurons (Pedarzani et al. 2000), neurons of the lateral amygdala (Faber & Sah, 2002) and sympathetic ganglia neurons (Marsh & Brown, 1991), suggesting that spike repolarization may be the primary function of somatic BK channels in neurons. By contrast, SK channels contribute to an after-hyperpolarization (AHP) current lasting tens to hundreds of milliseconds following bursts of action potentials, and regulate the firing frequency during such bursts in some cases (Sah, 1996; Stocker et al. 1999; Pedarzani et al. 2000, 2001), but not in others (Faber & Sah, 2002).
To determine how Ca2+ and KCa channels regulate Purkinje cell activity, we examined the effects of different pharmacological agents on Purkinje neurons in acute slices of rat cerebellum. The aim of this study is to answer the following three questions: first, which KCa channels contribute to Purkinje neuron electrical activity; second, what does each channel type contribute to the intrinsic electrical properties of these neurons; and third, which voltage-gated Ca2+ channels are important in KCa channel activation?
Methods
Animal use protocols were approved by the Duke University Institutional Animal Care and Use Committee, and were in accordance with the guidelines set forth in the Animal Welfare Act and the US Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Slice preparation
Slices were prepared according to the method of Finch & Augustine (1998). Parasagittal slices were cut from the cerebellar vermis of 13- to 21-day-old Sprague-Dawley rats (Charles River Laboratories, Inc., Wilmington, MA, USA). Rats were anaesthetized by inhalation of metofane and then decapitated. The cerebellum was rapidly removed and immersed in ice-cold ACSF consisting of (mm): 125 NaCl, 2.5 KCl, 1.3 MgSO4, 1 NaH2PO4, 2.5 CaCl2, 10 dextrose and 26 NaHCO3, bubbled continuously with a mixture of 95 % oxygen and 5 % carbon dioxide. The lateral hemispheres were removed and the vermis was affixed onto a chilled, stainless steel tray (Ted Pella, Inc., Redding, CA, USA) using cyanoacrylate glue (Krazy Glue or Loc-Tite). The tray was filled with ice cold, oxygenated Ringer solution, and slices 200–300 μm thick were cut with a Pelco 101 Vibratome series 1000 (Ted Pella, Inc.). Cut slices were immediately transferred to a submersion chamber containing oxygenated Ringer solution at 37 °C, incubated at this temperature for 45 min to 1 h, then maintained at room temperature until use. Slices generally remained viable for about 4–6 h after the end of the 37 °C incubation period.
Recording conditions
Slices were positioned in a custom-designed perfusion chamber and visualized with an upright microscope using a × 60 water-immersion objective lens (Olympus America, Inc., Melville, NY, USA). At this magnification, Purkinje neurons were easily identified based on their location, size, shape and large dendritic arbor. Neurons were excluded from study if they did not show a stable baseline activity pattern (for spontaneously active neurons), if the resting membrane potential was more positive than −45 mV (for neurons in TTX), or if the access resistance exceeded 20 MΩ. For Ca2+ spike experiments, the bath solution consisted of ACSF supplemented with 0.3 μm tetrodotoxin. For experiments examining spontaneous Na+ spikes, the bath solution consisted either of ACSF alone or of ACSF supplemented with 100 μm picrotoxin and 1 mm kynurenic acid to block ionotropic GABA and glutamate receptors, respectively. The presence of picrotoxin and kynurenic acid had no detectable effect on any of the cell properties we measured in these experiments, hence the two groups of cells were combined for analysis. The bath was continuously bubbled with a mixture of 95 % O2-5 % CO2, maintained at 33 ± 2 °C using an inline solution heater (Warner Instruments, Inc., Hamden, CT, USA) and slices were perfused at 1–2 ml min−1.
Pipettes and pipette solution
Whole-cell electrodes were pulled from borosilicate glass (Corning no. 7056, Warner Instruments, Inc.), lightly fire-polished, and coated with Sylgard (Dow Corning, Midland, MI, USA) to reduce electrode capacitance. The pipette solution for most whole-cell recordings consisted of (mm): 130 potassium gluconate, 2 NaCl, 4 MgCl2, 4 Na2-ATP, 0.4 Na-GTP, 15 Mops, pH 7.3, and 0.5 EGTA. For BAPTA experiments, the EGTA was replaced with 10 mm BAPTA. Recordings were obtained from three additional Purkinje cells using pipette solution with 50 μm EGTA rather than 0.5 mm EGTA. No difference between these two EGTA concentrations could be detected, either on the baseline properties of the cells or on their responses to apamin. Electrodes had tip resistances of 3–5 MΩ.
Channel blockers
Iberiotoxin, ω-Aga-IVA and SNX-482 were obtained from Peptides International, Inc. (Louisville, KY, USA). They were prepared as 100 μm stock solutions in buffer consisting of 100 mm NaCl, 10 mm Tris HCl, 1 mm EDTA, 1 mg ml−1 BSA, pH 7.5, and stored at −80 °C in single-use aliquots. Nifedipine, Bay K8644, apamin and TTX were obtained from Sigma-Aldritch Corp. (St Louis, MO, USA). Paxilline was obtained from Alamone Labs (Jerusalem, Israel). Cadmium chloride and 4-aminopyridine (4-AP) were prepared as 100 mm stock solutions in distilled water. Paxilline, nifedipine and Bay K8644 were prepared as 1000 × stock solutions in DMSO. Bath solutions containing peptide blockers were supplemented with 0.1 mg ml−1 BSA to minimize non-specific binding.
Data acquisition and analysis
Recordings from most Purkinje neurons (53 of 63 cells) were made using an Axopatch-200B amplifier (Axon Instruments Inc., Union City, CA, USA) in the fast current clamp mode. To verify that these recordings were not distorted by the imperfect current clamp of the patch clamp amplifier (see Magistretti et al. 1996), we obtained recordings from 10 additional Purkinje neurons under identical conditions using an Axoclamp-2A amplifier in bridge mode. There was no detectable difference between the two amplifiers in terms of the action potential initial voltage, peak voltage, spike amplitude or AHP amplitude, nor were there any qualitative differences in the appearance of the spikes. From this we conclude that the fast current clamp of the Axopatch-200B is suitable for recording action potentials from Purkinje neurons in cerebellar slices. Series resistance was monitored throughout the experiment and compensated using the built-in amplifier circuitry. Traces were filtered at 5 KHz, digitized with an Axon Digidata 1200 A/D converter and stored on a PC using pCLAMP version 6 (Axon Instruments). Data were analysed and plotted using pCLAMP and Origin version 6 (OriginLab Corp., Northampton, MA, USA).
Quantitative analysis of spike shape
For measurements of AHP amplitude and spike amplitude, we used the membrane potential just before the spike as a reference point. For Na+ spikes, this initial voltage was measured as the potential at which the slope of the trace exceeded 4 V s−1, an empirically determined value that was found to reliably represent the beginning of action potentials under all conditions used. Variability in the initial voltage measurement did not account for the effects of any of the blockers on the AHP (ANCOVA interaction term, P ≈ 0.97). The Na+ spike amplitude was measured as the peak voltage minus the initial voltage. The width of Na+ spikes was measured at 25 % of the amplitude. For Ca2+ spikes, the membrane potential 3 ms before the peak of each Ca2+ spike was used as the reference point. The Ca2+ spike initial voltage measured in this manner was not significantly affected by any of the blockers used in this study (ANOVA, P ≈ 0.78) and did not account for the effects of the blockers on the AHP (ANCOVA, P ≈ 0.4).
Statistics
Statistical comparisons were made using Statview version 5.0 (SAS Institute, Inc., Cary, NC, USA). Treatment effects were first tested for significance by ANOVA, and pair-wise comparisons between groups were then carried out using Fisher's protected least significant difference (PLSD) test, significance level 0.05. Reported P values refer to Fisher's PLSD test comparisons unless otherwise specified.
Results
We recorded the electrical activity of Purkinje cells in acute cerebellar slices at 33 ± 2 °C. Except where TTX was present in the bath, all of the neurons included in this study were spontaneously active under control conditions, firing Na+ spikes at rates ranging from 4 to 115 Hz (mean = 34 Hz, median = 30 Hz). The great majority of cells (88 %) exhibited tonic firing behaviour during the baseline period, with the remaining 12 % showing rhythmic oscillations in their spontaneous activity. These results differ from studies of adult guinea-pig Purkinje neurons (Llinas & Sugimori, 1980a), but are similar to a previous study of rat Purkinje neurons in slices, in which less than 5 % of the Purkinje neurons studied using extracellular and cell-attached recordings showed bursting patterns (Hausser & Clark, 1997). Ca2+ spikes, which normally occur only in response to climbing fibre activation (Eccles et al. 1966; Llinas & Sugimori, 1980b), could be evoked in most Purkinje neurons by depolarizing current injections. This enabled us to study the ionic basis of the Ca2+ spike AHP in the absence of the large synaptic currents that normally accompany climbing fibre activity.
Figure 1 shows examples of Na+ spikes and Ca2+ spikes in the cerebellar slice preparation. In normal ACSF, injection of depolarizing current evoked both Na+ spikes (arrows, Fig. 1A) and Ca2+ spikes (▵, Fig. 1A). The addition of TTX to the bath eliminated the Na+ spikes and revealed the presence of Ca2+ spikes, each followed by a large AHP (Fig. 1B). It is this large AHP that is believed to cause the interruption in spike firing that follows climbing fibre activation in vivo (McDevitt et al. 1982). Only Na+ spikes were observed when the bath solution contained the non-specific Ca2+ channel blocker cadmium instead of TTX (Fig. 1C, different cell). Under these conditions, cells fired a few Na+ spikes and then settled at a depolarized plateau potential of about −30 mV for the remainder of the current step, possibly due to Na+ channel inactivation.
Figure 1. Current clamp recordings from Purkinje neurons in cerebellar slices demonstrate both Na+ and Ca2+ action potentials.

Right-hand traces are at expanded time scales. A, under control conditions, injection of 0.5 nA depolarizing current through the recording pipette evoked bursts of Na+ spikes (arrows) interrupted by single Ca2+ spikes (▵). B, when TTX was added to the bath solution, the same cell fired only Ca2+ spikes. Each spike was followed by a deep after-hyperpolarization (AHP). C, recordings from a different cell demonstrate Na+ spikes in the absence of Ca2+ spikes due to the block of Ca2+ channels with 100 μm bath-applied cadmium chloride.
Ca2+ dependence of Purkinje neuron firing properties
When Ca2+-dependent conductances were inhibited by blocking Ca2+ influx with cadmium, the electrical activity of Purkinje neurons changed considerably. The spontaneous activity pattern of a Purkinje neuron under control conditions (ACSF + picrotoxin and kynurenic acid) is shown in the left-hand trace of Fig. 2A. The firing frequency was very stable, as can be seen from the inter-spike interval distribution for this trace (Fig. 2B, top). In contrast, when Ca2+ channels were blocked by the addition of 100 μm cadmium chloride to the bath solution (Fig. 2A, middle), the neuron switched to an irregular firing pattern in which it fired at a higher frequency (Fig. 2B, middle) and then settled at a depolarized plateau potential of approximately −33 mV. After 48 s at the plateau level, the cell abruptly hyperpolarized to −60 mV, slowly depolarized to −45 mV over the course of 10 s, and spontaneously began another burst of spikes (not shown). This type of activity was always observed in 100 μm cadmium, with depolarized plateau periods lasting 11 ± 5 s and hyperpolarized quiescent periods also lasting 11 ± 5 s. Provided that the cadmium application lasted only a few minutes, the effects could be reversed by extensive perfusion (Fig. 2A, right). Longer-lasting cadmium exposures resulted in irreversible depolarization of the resting membrane potential and loss of spike firing.
Figure 2. The spontaneous firing pattern of Purkinje neurons is Ca2+ dependent.

A, current clamp recordings showing the spontaneous firing pattern of a Purkinje neuron under control conditions (left trace), when Ca2+ channels were blocked by cadmium chloride (middle trace) and following removal of the cadmium by perfusion (right trace). B, inter-spike interval (ISI) distributions and log-normal fits are shown for each trace in panel A. Firing frequency components were determined from the peaks of the fits. For the ACSF trace, the peak was at 20 ms, corresponding to a firing frequency of 50 Hz. For the cadmium trace, three peaks could be discerned, corresponding to frequencies of 161, 88 and 56 Hz. After the wash period, the distribution again showed a single peak corresponding to a firing frequency of 45 Hz. C and D, recordings and ISI distributions from a different Purkinje neuron show how the firing pattern and frequency changed as 10 mm BAPTA diffused into the cell from the patch pipette. A few seconds after the whole-cell configuration was established, the cell fired at 84 Hz (left trace). After only 30 s, the firing pattern was irregular, displaying both Na+ and Ca2+ spikes, with an intra-burst firing frequency of 293 Hz (middle trace). An additional 2.5 min of BAPTA diffusion time had little effect on the intra-burst firing frequency, increasing it to 296 Hz (right trace). E, firing rates (means ±s.e.m.) are shown for each condition. Black bars represent the baseline firing rate for each group while hatched bars represent firing rates under treatment conditions. BAPTA increased the firing rate from 174 ± 40 to 534 ± 70 Hz as it diffused into the cell (n = 5), while cadmium changed the firing rate from 53 ± 11 to 187 ± 26 Hz (n = 4). The firing rate of the control group remained stable over time, changing from 41 ± 16 initially to 47 ± 19 Hz after 10 min in ACSF (n = 6). The BAPTA group differed significantly from the control group both in its baseline rate (P < 0.0001) and in the difference between the treatment condition and the baseline (P < 0.0001).
A second mechanism of inhibiting KCa channel activity, one that does not also block Ca2+ influx, is to include a high concentration of the fast mobile Ca2+ buffer BAPTA (10 mm) in the pipette solution and observe the change in spontaneous activity as BAPTA diffuses throughout the cytoplasm (Naraghi & Neher, 1997). An example of one such experiment is shown in Fig. 2C and D. For the first few seconds after the whole-cell configuration was established with a BAPTA-containing electrode, this neuron had a tonic activity pattern similar to control recordings. Within 30 s, however, the neuron showed a bursting pattern featuring both Na+ and Ca2+ spikes and an extremely fast intra-burst firing frequency of 293 Hz. Similar effects were always observed using BAPTA-containing electrodes (n = 5).
The effects of cadmium and BAPTA on the spontaneous firing rate are summarized in Fig. 2E. Both treatments substantially increased the intra-burst spontaneous firing frequency, cadmium by 253 % and BAPTA by 207 %. By comparison, the control group, which consisted of Purkinje cells monitored under baseline conditions for at least 10 min (mean = 20.5 min, n = 6) to reflect washout or rundown effects, showed little change in the spontaneous firing rate over time. These data constitute strong evidence that Ca2+-dependent conductances are active during normal spontaneous activity, and that blocking them either by preventing Ca2+ influx or by chelating intracellular Ca2+ severely disrupts the spontaneous firing pattern and increases the firing frequency.
To gain a better understanding of when Ca2+-dependent conductances are active and why they are so important for normal Purkinje cell firing patterns, the effects of cadmium and BAPTA were first examined in greater detail. Figure 3A shows how blocking Ca2+ influx with cadmium affects the shape of spontaneous Na+ spikes. In the presence of cadmium, Na+ spikes were essentially unchanged from control spikes with one exception: the AHP that normally follows each spike was significantly attenuated. The initial membrane potential, rising phase, spike width and repolarization were not altered, but the undershoot was abnormally brief, suggesting that a Ca2+-dependent conductance active immediately following each Na+ spike had been blocked.
Figure 3. Ca2+-dependent conductances contribute to the AHPs following Na+ and Ca2+ spikes.

A, the Na+ spike AHP requires Ca2+ influx. Left panel, reducing Ca2+ influx with 100 μm cadmium chloride (thick trace) attenuated the Na+ spike AHP without affecting spike repolarization. Right panel, when spike repolarization was first slowed with 100 μm 4-AP (thick continuous trace), Ca2+-dependent conductances became important for repolarization; if they were subsequently blocked with 100 μm cadmium added in the presence of 4-AP (dotted trace), spike repolarization was significantly delayed. The inset shows the same traces at an expanded scale (scale bar = 25 mV × 0.5 ms for inset). The vertical line in the inset is 0.4 ms after the beginning of the spike. B, the Ca2+ spike AHP involves Ca2+-activated conductances. Ca2+ spikes were evoked using the same protocol as in Fig. 1 A but with 10 mm BAPTA in the pipette solution. Representative Ca2+ spikes are shown from recordings taken immediately after establishing the whole-cell configuration (thin traces) and 12 min later, when BAPTA had diffused from the patch pipette into the cell (thick traces). The expanded traces in the right-hand panel show the effect of BAPTA on the AHP at different time points. At 1 ms after the peak of the spike (dashed line), there was no difference between the two conditions. However, the AHP of the later trace ended within about 3 ms of the spike peak (dotted line), while the control trace AHP showed a slower component that was still approaching its minimum value at 5 ms after the spike peak (continuous line). At the 5 ms time point, the baseline condition trace was 14 mV more negative than the trace recorded later.
This was confirmed by the experiment shown in the right-hand panel of Fig. 3A. In this experiment, a subset of delayed rectifier K+ channels was first blocked with 100 μm 4-AP (thick continuous trace), delaying repolarization of the action potential compared to the control (thin continuous trace). The subsequent application of cadmium in addition to the 4-AP (dotted trace) substantially compromised spike repolarization in this case. The deviation of the dotted trace from the thick trace in this figure (vertical line in the inset) indicates that the Ca2+-dependent current became a significant fraction of the total current about 0.4 ms after the beginning of the spike, a time at which a control action potential would just be completing the repolarization phase (thin trace in inset). This is consistent with our previous observation that Ca2+-dependent conductances are not required for repolarization, but rather modulate the magnitude and time course of the AHP.
In addition to their importance for the Na+ spike AHP, Ca2+-activated conductances are also essential for the pronounced AHP that follows Ca2+ spikes (Fig. 3B). Ca2+ spikes were evoked by depolarizing current steps (as in Fig. 1A), but the pipette solution contained 10 mm BAPTA. Similar to control recordings (Fig. 1A), each Ca2+ spike evoked at the beginning of the recording period was followed by a large AHP and a subsequent burst of Na+ spikes (thin traces in Fig. 3B). After BAPTA was allowed to diffuse into the cell for 12 min (thick traces), evoked Ca2+ spikes were virtually identical to those from the initial records in all but two respects: the peak amplitude was higher by 4 mV, and although spike repolarization was normal for the first 1 ms after the peak (dashed line in Fig. 3B), a slow component of the AHP was absent and the AHP was complete within 3 ms of the spike peak (dotted line in Fig. 3B). Na+ spikes were no longer observed following the Ca2+ spikes even though the neuron fired Na+ spikes spontaneously after the end of the depolarizing current step. One possible explanation is that in the absence of the slower component, the AHP may have been too brief to effectively relieve the inactivation of voltage-gated Na+ channels. In three Purkinje cells, BAPTA increased the amplitude of Ca2+ spikes by 45 %± 25 % and reduced the AHP measured at 5 ms after the peak by 44 ± 20 %. Both of these effects were significant (P < 0.05) compared to the group of five control cells, which showed a decrease of 3 ± 7 % in the spike amplitude and an increase of 4 ± 12 % in the AHP at 5 ms after the peak.
Effects of specific KCa channel blockers
Three different aspects of Purkinje neuron firing properties involving Ca2+-dependent conductances were identified: the frequency and pattern of spontaneous firing, the AHP following each Na+ spike, and the AHP following each Ca2+ spike. To determine which ion channels underlie these conductances, specific channel blockers were applied to the bath solution, and their effects on electrical properties were assessed. All of the properties measured remained stable over time in the control group of Purkinje neurons, demonstrating that the effects were attributable to the toxins and not to cytosolic washout or rundown processes.
Firing pattern
Figure 4A shows the effect of the specific SK channel blocker apamin (200 nm) on a Purkinje neuron that was firing tonically at 35 Hz under control conditions. Shortly after apamin-containing solution began replacing the control solution (time course shown in Fig. 4E, top), the neuron's firing pattern became irregular, alternating between bursts of spontaneous action potentials lasting 3.8 ± 1 s and quiescent periods lasting 4.7 ± 0.9 s. During the bursts, the average frequency increased more than threefold compared to the neuron's baseline firing rate (Fig. 4D, top). These were consistent effects in all cells tested: tonic firing patterns became irregular, with firing bursts lasting 2.1 ± 0.7 s and quiescent periods lasting 3.9 ± 1.9 s, while the mean firing frequency during bursts increased by more than 100 Hz in the presence of apamin.
Figure 4. SK and BK channels regulate Purkinje cell firing patterns.
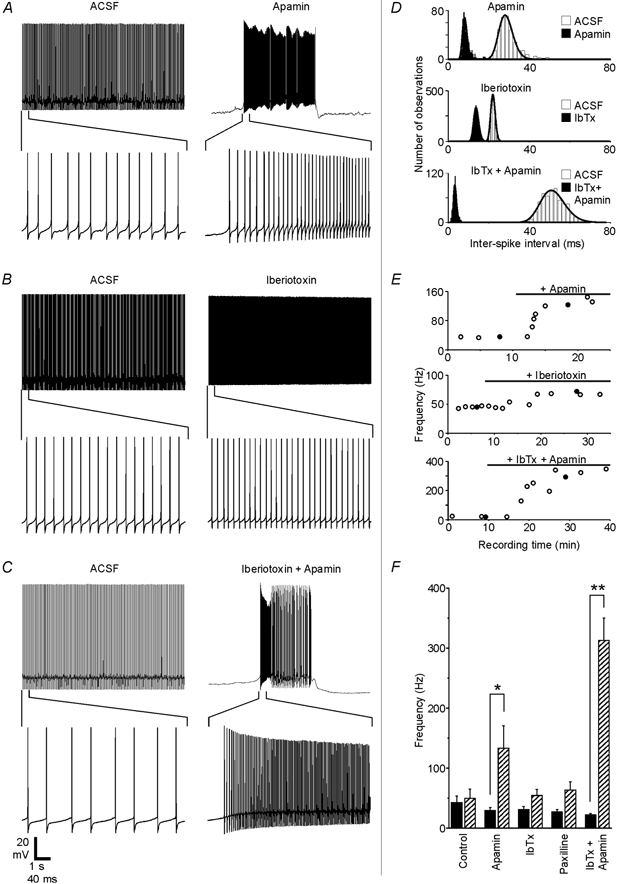
A-C, recordings from three different Purkinje neurons show how spontaneous firing was affected by 200 nm apamin, 100 nm iberiotoxin or the combination of the two blockers. The same scale bar applies to each set of traces. D, ISI distributions and fits are shown for the traces in A-C. E, spontaneous firing frequency plotted as a function of recording time for each experiment. •, the traces shown in A-C. Black horizontal bars show when toxins were present in the bath solution. Response delays partly reflect the time needed for residual control solution to empty out of the perfusion tubing and inline solution heater. F, the effects of specific KCa channel blockers on the firing frequency are summarized. Black bars show the baseline frequency for each group (means ±s.e.m.), while hatched bars show the frequency in the presence of the blocker. The firing frequency increased from 29 ± 5 to 133 ± 37 Hz with the application of apamin (n = 7 cells), from 29 ± 5 to 50 ± 12 Hz with iberiotoxin (n = 6), from 27 ± 4 to 63 ± 14 Hz with 1 μm paxilline (n = 5) and from 22 ± 3 to 313 ± 37 Hz when both iberiotoxin and apamin were applied together (n = 5). For the control group, the firing rate increased from 41 ± 16 initially to 47 ± 19 Hz after 10 min of control recording time (n = 6). Apamin had a significant effect compared to the control (P < 0.05), and the simultaneous application of iberiotoxin and apamin differed significantly from both the control group (P < 0.0001) and from the apamin group (P < 0.01). When applied alone, neither iberiotoxin nor paxilline had a statistically significant effect compared to the control group (P > 0.5).
While the SK channel blocker apamin always caused obvious changes in the firing frequency and pattern, iberiotoxin and paxilline, two specific BK channel blockers (Galvez et al. 1990; Knaus et al. 1994; Gribkoff et al. 1996), had much smaller effects. This is illustrated by Fig. 4B, which shows traces from a Purkinje cell that increased its average firing rate by only 28 Hz (inter-spike interval (ISI) distributions shown in Fig. 4D, middle) and did not adopt a bursting pattern even after more than 20 min in the presence of iberiotoxin (Fig. 4E, middle). Despite a consistent trend toward higher firing rates in the presence of BK channel blockers, neither iberiotoxin nor paxilline had a statistically significant effect on the firing rate compared to the control group (P > 0.5). BK channels do appear to play some role in spontaneous firing, however, as four out of five neurons that had tonic activity patterns under baseline conditions switched to oscillating patterns in the presence of iberiotoxin. For these four cells, firing periods lasted 10.6 ± 7.1 s and quiescent periods lasted 3.8 ± 2.0 s. By comparison, only one of six control cells that initially exhibited tonic firing patterns switched to an oscillating pattern (data not shown).
When both BK and SK channels were blocked simultaneously, the effect on the firing rate was much greater than when either channel type was blocked individually. This is illustrated by Fig. 4C, in which a Purkinje neuron that was firing tonically at 19 Hz under control conditions switched to a bursting pattern with an intra-burst frequency of 292 Hz when the bath solution contained iberiotoxin and apamin (ISI distributions in Fig. 4D, bottom; time course in Fig. 4E, bottom). All neurons exposed to iberiotoxin and apamin together adopted a bursting pattern, and the average intraburst frequency increased by 291 Hz, a 14-fold change over the baseline.
Na+ spike AHP
One possible explanation for the increase in the firing frequency that was observed when Ca2+-dependent conductances were blocked is that under normal circumstances, the Ca2+-dependent AHP following each Na+ spike creates a refractory period during which the neuron is unable to reach threshold and fire the next spike. If this AHP were the primary determinant of the spontaneous firing frequency, the SK channel blocker apamin would be expected to have a greater inhibitory effect on the Na+ spike AHP than the BK channel blockers iberiotoxin and paxilline, in accordance with apamin's effect on the firing rate. However, as shown in Fig. 5, this was clearly not the case. Both of the BK channel blockers significantly attenuated the Na+ spike AHP (Fig. 5A and B), while blocking SK channels with apamin had no detectable effect (Fig. 5C). Still, the greatest effects were again observed when both BK and SK channels were blocked simultaneously (Fig. 5D), indicating that SK channels did contribute to the Na+ spike AHP when BK channels were blocked. The Na+ spikes observed in the combined presence of iberiotoxin and apamin occurred in short bursts that were very similar to those observed when Purkinje cells were filled with BAPTA (compare Fig. 5D with 5E).
Figure 5. BK channels are major contributors to the Na+ spike AHP.

A-D, spontaneous Na+ spikes were recorded in ACSF (thin traces), then in the presence of one or more channel blockers (thick traces). Each trace is an average of 10 action potentials except the thick trace in panel D, which shows a single spike burst characteristic of spontaneous activity in this condition. E, spontaneous Na+ spikes were recorded in ACSF while 10 mm BAPTA diffused into the cell from the pipette. The thin trace is an average of 10 action potentials recorded immediately after establishing the whole-cell configuration. The thick trace is a representative action potential burst recorded 3 min later. F, Na+ spike AHP amplitude was quantified as the difference between the initial voltage and the voltage 1 ms after the peak. G, the degree to which each treatment reduced the Na+ spike AHP amplitude is summarized for all groups (means ±s.e.m.). Apamin did not significantly affect the AHP, reducing it by only 2 ± 5 % (n = 5 cells). Iberiotoxin reduced the AHP by 53 ± 6 % (n = 7), paxilline by 57 ± 6 % (n = 5), cadmium by 71 ± 14 % (n = 5), BAPTA by 148 ± 29 % (n = 5) and the combination of iberiotoxin and apamin reduced the Na+ spike AHP by 106 ± 17 % (n = 5). All treatments except apamin differed significantly (P < 0.05) from the control, in which the AHP decreased in amplitude by 0 ± 9 % during 10 min in ACSF (n = 7).
To quantitatively compare the effects of different channel blockers on the Na+ spike AHP, the AHP amplitude was measured as the difference between the spike initial voltage (see Methods), and the membrane potential 1 ms after the peak (Fig. 5F). Ten action potentials were averaged for each AHP measurement. For treatments that induced high frequency bursting, the first spike from each of 10 different bursts was used to generate the average instead of 10 sequential spikes. This was done because with high frequency bursts, only the first spike of the burst began at an initial voltage that was comparable to that of the baseline condition action potentials; subsequent spikes began from more depolarized voltages, where the activation and inactivation states of voltage-dependent channels were likely to differ from the control. Using this method, we found that the BK channel blockers iberiotoxin and paxilline each reduced the AHP by more than 50 % (P < 0.01), while apamin had no significant effects (P≈ 0.92). The combination of iberiotoxin and apamin had the greatest effect, completely eliminating the AHP (P < 0.0001 vs. control, P < 0.05 vs. iberiotoxin and paxilline). None of the treatments caused a broadening of the action potential (ANOVA, P≈ 0.58)
Ca2+ spike amplitude and AHP
Using the same specific channel blockers described above, the contributions of BK channels and SK channels to Ca2+ spikes were assessed. Na+ spikes were blocked with TTX, and Ca2+ spikes were evoked by depolarization (Fig. 6A). As can be seen from the superimposed spikes in Fig. 6B, blocking SK channels with apamin (blue traces) did not substantially alter the Ca2+ spike AHP compared to the control (black traces). The combined block of BK and SK channels (green traces), however, increased the spike peak amplitude by 9 mV and blocked all but a fast component of the AHP.
Figure 6. Both BK and SK channels contribute to the Ca2+ spike AHP.

A, three traces from the same cell show Ca2+ spikes evoked under control conditions (left trace), in the presence of apamin (middle trace) and in the combined presence of iberiotoxin and apamin (right trace). Spikes were evoked by applying a 2 nA depolarizing current step for 3 s. B, (upper panel) five sequential Ca2+ spikes from each trace in A (spikes indicated by brackets) are shown aligned to peak time. Control traces are black, apamin traces are blue and iberiotoxin + apamin traces are green. The lower panel shows one trace from each condition on an expanded time scale. By 3 ms after the peak of the spike (dashed line), the iberiotoxin + apamin trace was 20 mV more depolarized than the apamin-only trace. C, iberiotoxin by itself (100 nm, red traces) increased the peak amplitude of Ca2+ spikes and reduced the AHP compared to control (black traces), but the subsequent addition of apamin to the iberiotoxin-containing bath (green traces) further attenuated the AHP (same scale as the upper traces in B.) D1, for all cells, Ca2+ spike AHP amplitudes were measured as the difference between the membrane potential 3 ms before the peak and the membrane potential 3 ms after the peak (when control spikes typically reached minimum voltages). Measurements are compared before and after the addition of a blocker or, in the case of the control group, before and after a 10 min time period. The effect of the different treatments is expressed as the change in AHP amplitude as a percentage of the initial AHP amplitude (100 × (treatment – control)/control). For the control group, the AHP amplitude changed by 0 ± 4 % (n = 5) over the 10 min recording period. Apamin increased the AHP amplitude by 10 ± 9 % (n = 4), but this did not differ significantly from the control group (P > 0.3). Iberiotoxin reduced the AHP amplitude by 31 ± 4 % (n = 5), while the combination of iberiotoxin and apamin reduced the AHP by 45 ± 14 % (n = 4), and both of these differed significantly from the control group (P < 0.05). D2, when BK channels were first blocked with iberiotoxin, the subsequent addition of apamin caused a further reduction of the AHP amplitude by 18 ± 5 % (n = 3) compared to iberiotoxin alone (green and black hatched bar), which was a significant effect (P < 0.05).
When the toxins were applied in the reverse order (Fig. 6C, different cell), the Ca2+ spike height was increased and the AHP reduced by iberiotoxin alone (red traces), demonstrating that BK channels make important contributions to the shape of Ca2+ spikes in Purkinje neurons. However, the AHP was still further reduced by the subsequent addition of the SK channel blocker apamin to the bath solution (Fig. 6C, green traces), indicating that both BK and SK channels must be blocked to fully eliminate the Ca2+-dependent components of the Ca2+ spike AHP.
Figure 6D summarizes the effects of apamin and iberiotoxin on the Ca2+ spike AHP. The AHP amplitude was measured as the difference between the spike initial voltage and the membrane potential 3 ms after the peak, when the fast, BAPTA-insensitive component of the AHP was largely complete (dashed line in Fig. 6B; also see Fig. 3B). Statistical comparisons confirmed that iberiotoxin alone, and iberiotoxin in combination with apamin, significantly reduced the Ca2+ spike AHP, while apamin alone did not. Because the iberiotoxin group did not differ statistically from the iberiotoxin plus apamin group in this comparison, we conducted a separate set of experiments to determine whether SK channels made any significant contribution. BK channels were blocked with iberiotoxin throughout the course of the experiment. Ca2+ spikes were first recorded in the presence of iberiotoxin alone, apamin was then added to the bath, and its effects on Ca2+ spikes were measured. Under these conditions, the block of SK channels by apamin caused a small but significant reduction in the amplitude of the Ca2+ spike AHP (Fig. 6D2).
In addition to their effects on the AHP, both iberiotoxin and apamin significantly increased the amplitude of the Ca2+ spikes. Iberiotoxin increased the average spike amplitude by 56 ± 9 % (P < 0.01), and apamin by 50 ± 27 % (P < 0.05), while the control group showed no change (−0.4 ± 0.9 %). The combined application of iberiotoxin and apamin did not have a greater effect than either toxin applied individually in this case, increasing the spike amplitude by 40 ± 18 %.
Effects of iberiotoxin on Purkinje cells from older animals
It was recently demonstrated by Cingolani and colleagues (2002) that SK channel mRNA and protein is developmentally downregulated in rat Purkinje neurons, from high levels at birth to low levels by postnatal day (P)24. SK channel expression was still high at day P12, indicating that a significant degree of SK channel downregulation occurs between P12 and P24. As we have found that BK channels become much more important regulators of the firing frequency when SK channels are blocked, we hypothesized that BK channel blockers would have greater effects on the firing rate of Purkinje neurons from older animals. To test this prediction, we assessed the effects of iberiotoxin on four Purkinje neurons from 24- to 31-day-old rats (data not shown). The group of Purkinje neurons from older animals was virtually identical to a group of Purkinje neurons from 13- to 15-day-old rats (n = 6) in both its baseline firing rate (38 ± 7 Hz for the older group, 33 ± 5 Hz for the younger group), and its firing rate in iberiotoxin (63 ± 16 Hz for the older group, 61 ± 9 Hz for the younger group, P ≈ 0.49). The effects of iberiotoxin on the Na+ spike AHP were also comparable for the two groups (53 ± 3 % reduction for the older group, 63 ± 4 % reduction for the younger group, P≈ 0.11). Therefore, there is no indication that the role of BK channels in Purkinje neurons changes over the time period during which SK channel expression is downregulated.
Effects of specific Ca2+ channel blockers
These data demonstrate that at least two types of KCa channels are active in Purkinje cells, and that each channel type makes unique contributions to the electrical properties of these neurons. It is likely that some of the differences in functional contributions arise from the distinct intrinsic channel properties. In particular, the voltage dependence of BK channels limits their activity to a much smaller range of conditions compared to SK channels. It is also possible, however, that different KCa channel family members are linked to distinct routes of Ca2+ entry, and that the properties of the Ca2+ sources contribute to differences in the KCa channel activity profiles. To investigate this possibility, we assessed the effects of specific blockers of voltage-gated Ca2+ channels on Purkinje neuron firing properties.
Three different voltage-gated Ca2+ channel blockers were tested: ω-Aga-IVA (100 nm) to block P-type channels, nifedipine (10 μm) to block L-type channels, and SNX-482 (200 nm) to block channels formed from the α1E subunit. The results, summarized in Fig. 7, show that blocking P-type Ca2+ channels with ω-Aga-IVA largely reproduced the effects of blocking all Ca2+ influx with cadmium. In the presence of ω-Aga-IVA a Purkinje neuron that was firing tonically at 40 Hz under control conditions switched to an irregular bursting pattern with a frequency of 259 Hz (Fig. 7A and B), and showed a substantial reduction in the Na+ spike AHP (Fig. 7C and D), similar to the effects of cadmium. In contrast to ω-Aga-IVA, the bath application of nifedipine, a blocker of L-type Ca2+ channels, or SNX-482, a blocker of R-type channels formed by the Ca2+ channel α1E subunit (Newcomb et al. 1998), had no detectable effect on either the firing rate (Fig. 7D) or the Na+ spike AHP (Fig. 7E). From this it can be concluded that P-type Ca2+ channels are the primary source of Ca2+ for both BK and SK channels in Purkinje neurons.
Figure 7. P-type voltage-gated Ca2+ channels contribute to the activity of both BK and SK channels.

A and B, application of the P-type Ca2+ channel blocker ω-Aga-IVA (100 nm) caused a Purkinje neuron to switch from a regular spontaneous firing pattern at a frequency of 40 Hz to a bursting pattern with an intra-burst frequency of 259 Hz. In contrast to cadmium experiments, Ca2+ spikes could still be observed in this condition (arrows in A). C, the AHP of spontaneous Na+ spikes was reduced in the presence of ω-Aga-IVA (thick trace, representative action potential from the beginning of a burst) compared to Na+ spikes recorded from the same neuron before the addition of the blocker (thin trace, average of 10 sequential action potentials in ACSF). D, the Na+ spike AHP was measured using the same method as in Fig. 5 F. ω-Aga-IVA significantly reduced the AHP by 54 ± 12 % (n = 4 cells, P < 0.05), while neither 10 μm nifedipine (8 ± 4 % increase in AHP, n = 3 cells) nor 200 nm SNX-482 (6 ± 20 % increase in AHP, n = 3 cells) had a significant effect. E, ω-Aga-IVA significantly increased the spontaneous firing frequency of Purkinje neurons from their baseline rate of 55 ± 11 to 201 ± 48 Hz in the presence of the blocker (n = 5, P < 0.05). The spontaneous firing rate was not affected by nifedipine (36 ± 9 Hz baseline, 33 ± 9 Hz in nifedipine, n = 3 cells) or SNX-482 (41 ± 26 Hz baseline, 41 ± 18 Hz in SNX-482, n = 3 cells).
Discussion
Our results demonstrate that in cerebellar Purkinje neurons, Ca2+ entering through P-type Ca2+ channels activates both BK and SK channels, and each KCa channel type plays a unique functional role in the generation of Purkinje cell electrical behaviour. In general, SK channels control the firing frequency, while BK channels contribute to the Na+ spike and Ca2+ spike AHPs and are likely to contribute to climbing fibre responses in vivo. Despite this functional separation, however, the greatest effects on the firing frequency and spike AHPs were observed following the simultaneous block of both BK and SK channels.
Electrical properties of Purkinje neurons in this study
The basic electrical properties of the Purkinje neurons in this study are comparable to those observed in vivo. Most neurons were spontaneously active, firing Na+ spikes at a median rate of 30 Hz. Some displayed spontaneous oscillatory bursting activity, although the majority did not. In most neurons, Ca2+ spikes could be evoked by current injections. Na+ spikes and Ca2+ spikes are each followed by a characteristic AHP, and both of these AHPs as well as the frequency and pattern of spontaneous activity are critically dependent upon KCa channels.
Contribution of BK channels
In Purkinje neurons, BK channels are activated by action potentials and give rise to a fast AHP after each spike. They do not appear to contribute substantially to repolarization, since blockers of BK channels have no detectable effect on spike width when applied alone. In this regard, Purkinje neurons differ from other neurons in which the role of BK channels has been investigated (Marsh & Brown, 1991; Zhang & McBain, 1995; Shao et al. 1999; Pedarzani et al. 2000; Faber & Sah, 2002). When repolarization is slowed with 4-AP, the addition of cadmium does result in broader spikes; however, the broadening is due to an abrupt slowing of repolarization that starts about 0.4 ms after the beginning of the spike, a time at which a control action potential would just be crossing below the initial threshold voltage and beginning the AHP. This is somewhat surprising, given that BK channels are voltage dependent and would be expected to be most active during the peak of the spike and much less active near resting potential. However, the time course of BK channel activity will be determined by multiple factors including the kinetics and proximity of the Ca2+ channels that supply Ca2+ for BK channel activity, the rate at which Ca2+ ions are bound by intracellular buffers or transported across the membrane, and the activation and deactivation kinetics of the BK channels themselves. Previous work from Raman & Bean (1999) has demonstrated that in acutely dissociated Purkinje cell bodies, somatic P-type Ca2+ current peaks near the end of the falling phase of simulated action potential voltage commands, raising the possibility that P-type Ca2+ channel activation represents a rate-limiting step for BK channel activation. However, the possibility that BK channels activate earlier during the spike but don't account for a significant fraction of the total current until near the end of the spike, when many purely voltage-gated channels have deactivated, cannot be ruled out.
As for the duration of BK channel activity after the action potential, we suspect that the channels remain active only long enough to bring the AHP to its minimum value, and that the time course of recovery from the AHP is determined by other conductances. Consistent with this hypothesis, Raman & Bean (1999) found that the total somatic K+ current blocked by 1 mm TEA, which should include most of the BK current (Reinhart et al. 1989), deactivated with very fast kinetics (τ = 280 μs) and was absent between spikes. This may explain the finding that BK channels do not significantly affect the firing rate.
Contribution of apamin-sensitive SK channels
Apamin-sensitive SK channels control the firing frequency of Purkinje neurons. In the presence of apamin, Purkinje cells are unable to maintain a stable, tonic firing pattern and instead oscillate between high frequency bursts and periods of quiescence. The increase in the firing rate indicates that SK channels are active during normal tonic firing even though they do not contribute substantially to action potential waveforms. It may be that the total SK channel current is small, such that it is of little significance during action potentials, but remains active between spikes while almost all of the voltage-gated current shuts off (Raman & Bean, 1999). In this case, apamin would block a major portion of the outward current active between spikes and might be expected to cause high frequency firing. The reason for the quiescent period following each burst is unknown, but has been proposed to be the activation of a slow, Ca2+-dependent hyperpolarizing current (Llinas & Sugimori, 1980a; Hounsgaard & Midtgaard, 1989).
As they do in Purkinje neurons, SK channels also regulate the firing rate of hippocampal pyramidal neurons (Stocker et al. 1999; Pedarzani et al. 2001) and dorsal vagal neurons (Sah, 1996; Pedarzani et al. 2000); however, the effect of SK channel block on the activity of Purkinje neurons is much more pronounced, suggesting that Purkinje neurons rely on SK channels to a greater extent than these other neuron types.
Cingolani and colleagues (2002) recently demonstrated that SK channel expression is developmentally regulated in rat Purkinje neurons, with high expression levels at birth giving way to lower expression levels by postnatal day 24. They furthermore found that apamin increased the spontaneous firing frequency of Purkinje neurons from young (p12) rats, but not adult (P50–60) rats. The downregulation of SK channels approximately corresponds with the temporal upregulation of BK channel expression in the same neurons (Muller et al. 1998), raising the possibility that SK channel functions may be taken over by BK channels later in development. If such a switch does occur, however, it happens later than would be expected from the temporal pattern of SK channel expression, because BK channel block does not have any greater effects on the firing rate of Purkinje cells in 24- to 31-day-old rats than in 13- to 15-day-old rats. In addition, preliminary results suggest that the effects of apamin do not differ significantly between these two age ranges (data not shown), but this is a matter of ongoing investigation.
We did not observe any effect of apamin by itself on the Ca2+ spike AHP. Apamin does increase the amplitude of Ca2+ spikes, though, and when applied in the presence of iberiotoxin, apamin reduces a slow component of the Ca2+ spike AHP. These results indicate that SK channels are normally active during Ca2+ spikes, but that their total current amplitude is so much smaller than that of BK channels and voltage-gated channels that it does not have a large effect on the membrane potential unless the membrane resistance is increased, as is the case when BK channels are blocked. It may be that the SK channel current density is much lower than that of BK channels, or SK channels may be isolated from the sites of dendritic Ca2+ influx so that most are not activated by dendritic Ca2+ spikes.
P-type Ca2+ channels are critical for both BK and SK channel activity
Both BK and SK channels appear to receive Ca2+ predominantly from the same Ca2+ channel type in Purkinje neurons, unlike the situation reported for some other neuron types (Davies et al. 1996; Marrion & Tavalin, 1998). Blocking P-type Ca2+ channels with ω-Aga-IVA in Purkinje neurons largely mimics the effects of cadmium on both the Na+ spike AHP and the overall firing pattern, while the L-type channel blocker nifedipine and the α1E channel blocker SNX-482 are without effect.
Purkinje neurons are also known to express low-threshold, T-type Ca2+ channels of the α1G subtype (Regan, 1991; Mouginot et al. 1997; Pouille et al. 2000) and to have multiple mechanisms for release of Ca2+ from intracellular stores (Llano et al. 1991, 1994; Khodakhah & Armstrong, 1997; Finch & Augustine, 1998), and it is possible that these Ca2+ entry pathways can contribute to KCa channel activity in certain circumstances. However, the effects of ω-Aga-IVA on the Na+ spike AHP are very similar to the effects of iberiotoxin, paxilline and cadmium, suggesting that BK channel activity is largely dependent upon P-type Ca2+ channels. Furthermore, the effects of ω-Aga-IVA on the spontaneous firing frequency and pattern are comparable to the effects of apamin, indicating that SK channel activity is also dependent upon P-type Ca2+ channels. Therefore, the combined activity of L-type, T-type and α1E voltage-gated channels as well as release of Ca2+ from intracellular stores is insufficient to compensate for the loss of Ca2+ through P-type channels. It should be noted, however, that all of the present recordings were made using electrodes placed at the soma, so these experiments are unlikely to reflect the activity of KCa channels expressed at more distal dendritic sites. The possibility that additional populations of KCa channels exist in the distal dendrites that are linked to Ca2+ sources other than P-type channels can therefore not be excluded.
While the effects of ω-Aga-IVA on the spontaneous firing rate and the Na+ spike AHP were quantitatively similar to those of cadmium, the combined application of iberiotoxin and apamin or the presence of 10 mm BAPTA in the pipette resulted in significantly greater effects on both the firing frequency and the Na+ spike AHP than did either of the Ca2+ channel blockers. This may be an indication that the cytoplasmic Ca2+ concentration in the presence of ω-Aga-IVA or cadmium is still high enough to allow for significant KCa channel activation. Given that SK channels are substantially active at sub-micromolar concentrations of Ca2+ (Kohler et al. 1996), and BK channels can be activated at low [Ca2+] by membrane depolarizations (Latorre, 1994), it is very likely that at least some KCa channel activity persists even when the majority of the Ca2+ channels are blocked. It is also possible that when KCa channels are selectively blocked, inward Ca2+ currents significantly depolarize the membrane potential, reducing the Na+ spike AHP and increasing the firing frequency compared to conditions where the Ca2+ currents are also blocked.
Implications for Purkinje cell activity in vivo
The similarity of Purkinje cell firing rates in vivo to the baseline firing rates observed in this study suggest that SK channels are active and acting to regulate firing rates in the intact cerebellum. Currents driving the spontaneous firing of spikes include persistent and resurgent Na+ currents (Llinas & Sugimori, 1980a; Raman & Bean, 1997), low-threshold Ca2+ currents (Crepel & Penit-Soria, 1986; Raman & Bean, 1999) and Ih, the hyperpolarization-activated cation current (Crepel & Penit-Soria, 1986; Williams et al. 2002). Our results and those of Cingolani and colleagues (2002) demonstrate that SK channels play a major role in counterbalancing these excitatory currents, probably in concert with a voltage dependent, inactivating K+ conductance that is active near rest (Hounsgaard & Midtgaard, 1988). Dynamic modulation of SK channels could then be used to scale the overall excitability level of Purkinje cells when necessary.
In contrast to SK channels, BK channels are expected to be activated by each Na+ and Ca2+ spike but to remain active for no more than a few milliseconds because of their voltage dependence. This is consistent with the finding that BK channel block does not dramatically affect the firing rate when SK channels are functional. Instead, BK channel activity modulates the waveform of Na+ and Ca2+ spikes, particularly the AHPs. The importance of BK channels to the amplitude of the Ca2+ spike and Ca2+ spike AHP suggest that they will contribute to the climbing fibre response in vivo. The climbing fibre input normally fires at about 1 Hz and evokes a burst of three to five Na+ spikes and a dendritic Ca2+ spike in the target Purkinje neuron (Eccles et al. 1966; Llinas & Sugimori, 1980a; Lang et al. 1999). The Ca2+ spike prevents parallel fibre EPSPs from reaching the soma and results in a suppression of spiking lasting for tens of milliseconds, probably because of the large AHP current that follows the spike (McBevitt et al. 1982; Rawson & Tilokskulchai, 1982). Given the effect of BK channel block on the Ca2+ spike AHP, our results indicate that BK channels will contribute substantially to this AHP current. It is also possible that through their effects on the waveform of the Ca2+ spike, BK channels may regulate the amount of Ca2+ influx that accompanies each climbing fibre-evoked spike. In this case, BK channel activity could affect the induction of synaptic depression at parallel fibre-Purkinje cell synapses (Ito, 1989).
Overall, our experiments have shown that Ca2+ entering through P-type voltage-gated Ca2+ channels activates two types of Ca2+-activated K+ channels in Purkinje neurons from young rats: small-conductance (SK) and large-conductance (BK) KCa channels. Each of these channel types plays a unique role in shaping the electrical properties of the Purkinje cell – SK channels in setting the baseline firing frequency, and BK channels in regulating action potential shape and modulating the climbing fibre response. Together, these two channel types mediate most of the Ca2+-dependent electrical responses of cerebellar Purkinje cells.
Acknowledgments
We are grateful to Dr E. A. Finch for sharing her technical expertise and to Dr J. F. Wesseling and Mr J. M. Welch for their helpful comments on the manuscript. This work was supported by NIH grants to P.H.R. and J.R.E.
References
- Armstrong DM, Rawson JA. Activity patterns of cerebellar cortical neurones and climbing fibre afferents in the awake cat. J Physiol. 1979;289:425–448. doi: 10.1113/jphysiol.1979.sp012745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatz AL, Magleby KL. Single apamin-blocked Ca-activated K+ channels of small conductance in cultured rat skeletal muscle. Nature. 1986;323:718–720. doi: 10.1038/323718a0. [DOI] [PubMed] [Google Scholar]
- Chung YH, Shin C, Park KH, Cha CI. Immunohistochemical study on the distribution of neuronal voltage-gated calcium channels in the rat cerebellum. Brain Res. 2000;865:278–282. doi: 10.1016/s0006-8993(00)02288-5. [DOI] [PubMed] [Google Scholar]
- Cingolani LA, Gymnopoulos M, Boccaccio A, Stocker M, Pedarzani P. Developmental regulation of small-conductance Ca2+-activated K+ channel expression and function in rat Purkinje neurons. J Neurosci. 2002;22:4456–4467. doi: 10.1523/JNEUROSCI.22-11-04456.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crepel F, Penit-Soria J. Inward rectification and low threshold calcium conductance in rat cerebellar Purkinje cells. An in vitro study. J Physiol. 1986;372:1–23. doi: 10.1113/jphysiol.1986.sp015993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies PJ, Ireland DR, McLachlan EM. Sources of Ca2+ for different Ca2+-activated K+ conductances in neurones of the rat superior cervical ganglion. J Physiol. 1996;495:353–366. doi: 10.1113/jphysiol.1996.sp021599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow RS, Moruzzi G. The Physiology and Pathology of the Cerebellum. Minneapolis: Blackwell Science Inc; 1958. [Google Scholar]
- Ebner TJ. A role for the cerebellum in the control of limb movement velocity. Curr Opin Neurobiol. 1998;8:762–769. doi: 10.1016/s0959-4388(98)80119-0. [DOI] [PubMed] [Google Scholar]
- Eccles JC, Llinas R, Sasaki K. The excitatory synaptic action of climbing fibres on the Purkinje cells of the cerebellum. J Physiol. 1966;182:268–296. doi: 10.1113/jphysiol.1966.sp007824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES, Sah P. Physiological role of calcium-activated potassium currents in the rat lateral amygdala. J Neurosci. 2002;22:1618–1628. doi: 10.1523/JNEUROSCI.22-05-01618.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch EA, Augustine GJ. Local calcium signalling by inositol-1,4,5-trisphosphate in Purkinje cell dendrites. Nature. 1998;396:753–756. doi: 10.1038/25541. [DOI] [PubMed] [Google Scholar]
- Gahwiler BH, Llano I. Sodium and potassium conductances in somatic membranes of rat Purkinje cells from organotypic cerebellar cultures. J Physiol. 1989;417:105–122. doi: 10.1113/jphysiol.1989.sp017793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez A, Gimenez-Gallego G, Reuben JP, Roy-Contancin L, Feigenbaum P, Kaczorowski GJ, Garcia ML. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus. J Biol Chem. 1990;265:11083–11090. [PubMed] [Google Scholar]
- Gribkoff VK, Lum-Ragan JT, Boissard CG, Post-Munson DJ, Meanwell NA, Starrett JE, Jr, Kozlowski ES, Romine JL, Trojnacki JT, McKay MC, Zhong J, Dworetzky SI. Effects of channel modulators on cloned large-conductance calcium-activated potassium channels. Mol Pharmacol. 1996;50:206–217. [PubMed] [Google Scholar]
- Gruol DL, Jacquin T, Yool AJ. Single-channel K+ currents recorded from the somatic and dendritic regions of cerebellar Purkinje neurons in culture. J Neurosci. 1991;11:1002–1015. doi: 10.1523/JNEUROSCI.11-04-01002.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausser M, Clark BA. Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron. 1997;19:665–678. doi: 10.1016/s0896-6273(00)80379-7. [DOI] [PubMed] [Google Scholar]
- Hounsgaard J, Midtgaard J. Intrinsic determinants of firing pattern in Purkinje cells of the turtle cerebellum in vitro. J Physiol. 1988;402:731–749. doi: 10.1113/jphysiol.1988.sp017231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hounsgaard J, Midtgaard J. Synaptic control of excitability in turtle cerebellar Purkinje cells. J Physiol. 1989;409:157–170. doi: 10.1113/jphysiol.1989.sp017490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M. The Cerebellum and Neural Control. New York: Blackwell Science Inc; 1984. [Google Scholar]
- Ito M. Long-term depression. Annu Rev Neurosci. 1989;12:85–102. doi: 10.1146/annurev.ne.12.030189.000505. [DOI] [PubMed] [Google Scholar]
- Ivry R. Cerebellar timing systems. Int Rev Neurobiol. 1997;41:555–573. [PubMed] [Google Scholar]
- Jacquin TD, Gruol DL. Ca2+ regulation of a large conductance K+ channel in cultured rat cerebellar Purkinje neurons. Eur J Neurosci. 1999;11:735–739. doi: 10.1046/j.1460-9568.1999.00478.x. [DOI] [PubMed] [Google Scholar]
- Khodakhah K, Armstrong CM. Inositol trisphosphate and ryanodine receptors share a common functional Ca2+ pool in cerebellar Purkinje neurons. Biophys J. 1997;73:3349–3357. doi: 10.1016/S0006-3495(97)78359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus HG, McManus OB, Lee SH, Schmalhofer WA, Garcia-Calvo M, Helms LM, Sanchez M, Giangiacomo K, Reuben JP, Smith AB., III Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry. 1994;33:5819–5828. doi: 10.1021/bi00185a021. [DOI] [PubMed] [Google Scholar]
- Knaus HG, Schwarzer C, Koch RO, Eberhart A, Kaczorowski GJ, Glossmann H, Wunder F, Pongs O, Garcia ML, Sperk G. Distribution of high-conductance Ca(2+)-activated K+ channels in rat brain: targeting to axons and nerve terminals. J Neurosci. 1996;16:955–963. doi: 10.1523/JNEUROSCI.16-03-00955.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeppen AH. The hereditary ataxias. J Neuropathol Exp Neurol. 1998;57:531–543. doi: 10.1097/00005072-199806000-00001. [DOI] [PubMed] [Google Scholar]
- Kohler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 1996;273:1709–1714. doi: 10.1126/science.273.5282.1709. [DOI] [PubMed] [Google Scholar]
- Lancaster B, Nicoll RA. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J Physiol. 1987;389:187–203. doi: 10.1113/jphysiol.1987.sp016653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang EJ, Sugihara I, Welsh JP, Llinas R. Patterns of spontaneous purkinje cell complex spike activity in the awake rat. J Neurosci. 1999;19:2728–2739. doi: 10.1523/JNEUROSCI.19-07-02728.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorre R. Molecular workings of large conductance (maxi) ca2+-activated k+ channels. In: Peracchia C, editor. Handbook of Membrane Channels: Molecular and Cellular Physiology. San Diego, CA, USA: Blackwell Science Inc; 1994. pp. 79–102. [Google Scholar]
- Latorre R, Oberhauser A, Labarca P, Alvarez O. Varieties of calcium-activated potassium channels. Ann Rev Physiol. 1989;51:385–399. doi: 10.1146/annurev.ph.51.030189.002125. [DOI] [PubMed] [Google Scholar]
- Llano I, DiPolo R, Marty A. Calcium-induced calcium release in cerebellar Purkinje cells. Neuron. 1994;12:663–673. doi: 10.1016/0896-6273(94)90221-6. [DOI] [PubMed] [Google Scholar]
- Llano I, Dreessen J, Kano M, Konnerth A. Intradendritic release of calcium induced by glutamate in cerebellar Purkinje cells. Neuron. 1991;7:577–583. doi: 10.1016/0896-6273(91)90370-f. [DOI] [PubMed] [Google Scholar]
- Llinas R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell somata in mammalian cerebellar slices. J Physiol. 1980a;305:171–195. doi: 10.1113/jphysiol.1980.sp013357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. J Physiol. 1980b;305:197–213. doi: 10.1113/jphysiol.1980.sp013358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Sugimori M, Lin JW, Cherksey B. Blocking and isolation of a calcium channel from neurons in mammals and cephalopods utilizing a toxin fraction (FTX) from funnel-web spider poison. Proc Natl Acad Sci U S A. 1989;86:1689–1693. doi: 10.1073/pnas.86.5.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBevitt CJ, Ebner TJ, Bloedel JR. The changes in Purkinje cell simple spike activity following spontaneous climbing fiber inputs. Brain Res. 1982;237:484–491. doi: 10.1016/0006-8993(82)90460-7. [DOI] [PubMed] [Google Scholar]
- Magistretti J, Mantegazza M, Guatteo E, Wanke E. Action potentials recorded with patch-clamp amplifiers: are they genuine? Trends Neurosci. 1996;19:530–534. doi: 10.1016/s0166-2236(96)40004-2. [DOI] [PubMed] [Google Scholar]
- Marsh SJ, Brown DA. Potassium currents contributing to action potential repolarization in dissociated cultured rat superior cervical sympathetic neurones. Neurosci Lett. 1991;133:298–302. doi: 10.1016/0304-3940(91)90593-i. [DOI] [PubMed] [Google Scholar]
- Mouginot D, Bossu JL, Gahwiler BH. Low-threshold Ca2+ currents in dendritic recordings from Purkinje cells in rat cerebellar slice cultures. J Neurosci. 1997;17:160–170. doi: 10.1523/JNEUROSCI.17-01-00160.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller YL, Reitstetter R, Yool AJ. Regulation of Ca2+-dependent K+ channel expression in rat cerebellum during postnatal development. J Neurosci. 1998;18:16–25. doi: 10.1523/JNEUROSCI.18-01-00016.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naraghi M, Neher E. Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci. 1997;17:6961–6973. doi: 10.1523/JNEUROSCI.17-18-06961.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb R, Szoke B, Palma A, Wang G, Chen X, Hopkins W, Cong R, Miller J, Urge L, Tarczy-Hornoch K, Loo JA, Dooley DJ, Nadasdi L, Tsien RW, Lemos J, Miljanich G. Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas. Biochemistry. 1998;37:15353–15362. doi: 10.1021/bi981255g. [DOI] [PubMed] [Google Scholar]
- Ojakangas CL, Ebner TJ. Purkinje cell complex and simple spike changes during a voluntary arm movement learning task in the monkey. J Neurophysiol. 1992;68:2222–2236. doi: 10.1152/jn.1992.68.6.2222. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Kulik A, Muller M, Ballanyi K, Stocker M. Molecular determinants of Ca2+-dependent K+ channel function in rat dorsal vagal neurones. J Physiol. 2000;527:283–290. doi: 10.1111/j.1469-7793.2000.t01-1-00283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, Mosbacher J, Rivard A, Cingolani LA, Oliver D, Stocker M, Adelman JP, Fakler B. Control of electrical activity in central neurons by modulating the gating of small conductance Ca2+-activated K+ channels. J Biol Chem. 2001;276:9762–9769. doi: 10.1074/jbc.M010001200. [DOI] [PubMed] [Google Scholar]
- Pouille F, Cavelier P, Desplantez T, Beekenkamp H, Craig PJ, Beattie RE, Volsen SG, Bossu JL. Dendro-somatic distribution of calcium-mediated electrogenesis in purkinje cells from rat cerebellar slice cultures. J Physiol. 2000;527:265–282. doi: 10.1111/j.1469-7793.2000.00265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci. 1999;19:1663–1674. doi: 10.1523/JNEUROSCI.19-05-01663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson JA, Tilokskulchai K. Climbing fibre modification of cerebellar Purkinje cell responses to parallel fibre inputs. Brain Res. 1982;237:492–497. doi: 10.1016/0006-8993(82)90461-9. [DOI] [PubMed] [Google Scholar]
- Regan LJ. Voltage-dependent calcium currents in Purkinje cells from rat cerebellar vermis. J Neurosci. 1991;11:2259–2269. doi: 10.1523/JNEUROSCI.11-07-02259.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart PH, Chung S, Levitan IB. A family of calcium-dependent potassium channels from rat brain. Neuron. 1989;2:1031–1041. doi: 10.1016/0896-6273(89)90227-4. [DOI] [PubMed] [Google Scholar]
- Robitaille R, Charlton MP. Presynaptic calcium signals and transmitter release are modulated by calcium-activated potassium channels. J Neurosci. 1992;12:297–305. doi: 10.1523/JNEUROSCI.12-01-00297.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P. Ca(2+)-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci. 1996;19:150–154. doi: 10.1016/s0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- Schmahmann JD. Rediscovery of an early concept. Int Rev Neurobiol. 1997;41:3–27. doi: 10.1016/s0074-7742(08)60345-1. [DOI] [PubMed] [Google Scholar]
- Shao LR, Halvorsrud R, Borg-Graham L, Storm JF. The role of BK-type Ca2+-dependent K+ channels in spike broadening during repetitive firing in rat hippocampal pyramidal cells. J Physiol. 1999;521:135–146. doi: 10.1111/j.1469-7793.1999.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker M, Krause M, Pedarzani P. An apamin-sensitive Ca2+-activated K+ current in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 1999;96:4662–4667. doi: 10.1073/pnas.96.8.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker M, Pedarzani P. Differential distribution of three Ca2+-activated K+ channel subunits, SK1, SK2, and SK3, in the adult rat central nervous system. Mol Cell Neurosci. 2000;15:476–493. doi: 10.1006/mcne.2000.0842. [DOI] [PubMed] [Google Scholar]
- Storm JF. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol. 1987;385:733–759. doi: 10.1113/jphysiol.1987.sp016517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol. 1998;8:321–329. doi: 10.1016/s0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- Wallesch CW, Bartels C. Inherited cerebellar diseases. Int Rev Neurobiol. 1997;41:441–453. doi: 10.1016/s0074-7742(08)60364-5. [DOI] [PubMed] [Google Scholar]
- Williams SR, Christensen SR, Stuart GJ, Hausser M. Membrane potential bistability is controlled by the hyperpolarization-activated current I(H) in rat cerebellar Purkinje neurons in vitro. J Physiol. 2002;539:469–483. doi: 10.1113/jphysiol.2001.013136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama CT, Westenbroek RE, Hell JW, Soong TW, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of the neuronal class E calcium channel alpha 1 subunit. J Neurosci. 1995;15:6419–6432. doi: 10.1523/JNEUROSCI.15-10-06419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, McBain CJ. Potassium conductances underlying repolarization and after-hyperpolarization in rat CA1 hippocampal interneurones. J Physiol. 1995;488:661–672. doi: 10.1113/jphysiol.1995.sp020998. [DOI] [PMC free article] [PubMed] [Google Scholar]