

Abstract
Respiratory activity phasically alters membrane potentials of preganglionic vagal and sympathetic motoneurones and continuously modulates their responsiveness to stimulatory inputs. The most obvious manifestation of this ‘respiratory gating’ is respiratory sinus arrhythmia, the rhythmic fluctuations of electrocardiographic R–R intervals observed in healthy resting humans. Phasic autonomic motoneurone firing, reflecting the throughput of the system, depends importantly on the intensity of stimulatory inputs, such that when levels of stimulation are low (as with high arterial pressure and sympathetic activity, or low arterial pressure and vagal activity), respiratory fluctuations of sympathetic or vagal firing are also low. The respiratory gate has a finite capacity, and high levels of stimulation override the ability of respiration to gate autonomic responsiveness. Autonomic throughput also depends importantly on other factors, including especially, the frequency of breathing, the rate at which the gate opens and closes. Respiratory sinus arrhythmia is small at rapid, and large at slow breathing rates. The strong correlation between systolic pressure and R–R intervals at respiratory frequencies reflects the influence of respiration on these two measures, rather than arterial baroreflex physiology. A wide range of evidence suggests that respiratory activity gates the timing of autonomic motoneurone firing, but does not influence its tonic level. I propose that the most enduring significance of respiratory gating is its use as a precisely controlled experimental tool to tease out and better understand otherwise inaccessible human autonomic neurophysiological mechanisms.
A large and burgeoning literature is based on the notion that an understanding of autonomic rhythms informs the neurophysiological mechanisms that generate them. Humans hold great attraction as subjects for research into autonomic rhythms because they can be studied without anaesthesia, and they can cooperate. Since respiratory-frequency rhythms often dominate other rhythms, human cooperation is particularly valuable: subjects can breathe as rapidly and as deeply as required, or they can hold their breaths and not breathe at all. This critical review treats neurally mediated human respiratory-frequency rhythms, and the insights into autonomic physiology they convey. (I do not treat pathophysiology in this review.) My review considers several elements of what Lopes & Palmer (1976) termed the ‘respiratory gate’: the gate itself; the qualities of the input signals that are gated; the influence of the frequency of gating on effector responses; and the interrelations among central and peripheral rhythms, as they all are affected by respiratory activity.
Methods of knowing
The tools available for study of human autonomic rhythms are quite powerful. Finger photoplethysmographic estimates of beat-by-beat arterial pressure correlate well with intraarterial pressure recordings (Imholz et al. 1998). Although vagal-cardiac nerve traffic has not been recorded in humans, changes of electrocardiographic R–R intervals may serve as adequate substitutes. In spontaneously breathing dogs, moderate increases and decreases of arterial pressure provoke parallel, and linearly correlated changes of vagal-cardiac nerve traffic and R–R intervals (Katona et al. 1970). Human sympathetic nerve traffic to the muscle and skin vascular beds can be measured directly (Vallbo et al. 1979). Although sympathetic nerve traffic to the heart has not been measured in humans, cardiac noradrenaline spillover, a surrogate for sympathetic activity, has been measured (Wallin et al. 1992). Noradrenaline spillover provides steady-state (over minutes) measurements that correlate well with muscle sympathetic nerve traffic during rest, mental stress, and exercise (Wallin et al. 1992). Therefore, in minimally invaded humans, respiration can be controlled, arterial pressure can be estimated accurately, vagal-cardiac nerve activity can be measured indirectly, and sympathetic nerve traffic can be measured directly.
The wide availability of personal computers and sophisticated analytical software places elegant analysis of human time series within reach of many laboratories. Traditional time-domain metrics, such as mean and standard deviation, yield information regarding tonic levels of autonomic nerve activity, but do not describe or quantitate rhythms. However, other methods, including fast Fourier transformation (Hyndman et al. 1971; de Boer et al. 1985b), autoregressive analysis (Pagani et al. 1986), and Wigner-Ville transformation (Novak et al. 1993) quantitate rhythms and their frequencies. Moreover, such analyses can be moved through data sets to identify fluctuations (or rhythms) of the rhythms. Correlations between two signals can be documented with cross-spectral analysis (de Boer et al. 1985a), and the influence of a third signal can be extracted from two other signals with partial coherence analysis (Kocsis et al. 1993). (For example, the respiratory rhythm can be removed from arterial pressure and R–R interval rhythms, so that correlations between arterial pressures and R–R intervals can be gauged independent of respiratory influences.)
Respiratory imprints on human autonomic signals
Figure 1 shows expired carbon dioxide concentrations and R–R intervals, and a horizontal section of a sliding fast Fourier transformation of the R–R interval time series, from one healthy supine subject who steadily slowed his breathing rate from 15 to 3 breaths min−1. At the most rapid breathing rate (extreme left) R–R interval excursions were small and spectral power was low. As the breathing rate slowed, R–R interval fluctuations and spectral power increased. These data document a profound influence of breathing on R–R interval fluctuations. Breathing exerts smaller, but qualitatively similar influences on arterial pressure and muscle sympathetic nerve activity (Badra et al. 2001; not shown in Fig. 1).
Figure 1. Expiratory carbon dioxide concentrations, R–R intervals, and a horizontal section through a sliding fast Fourier transformation of R–R intervals during ramped frequency breathing.
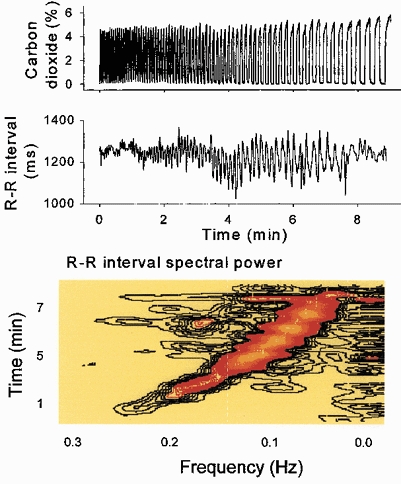
Respiratory sinus arrhythmia (middle and bottom panels, black to orange areas) was small at the extremes of breathing frequency. Low frequency spectral power (bottom panel, extreme right) was present throughout the period of ramped breathing. These data indicate that some R–R interval fluctuations closely track breathing.
Figure 2 shows power spectra (right) and horizontal sections of sliding fast Fourier transformations of R–R intervals, during spontaneous breathing (upper panels) and apnoea (lower panels). During apnoea, R–R interval spectral power was nearly absent at the former breathing frequency. These data suggest that it is respiration itself that drives respiratory-frequency autonomic rhythms - they are not secondary to some other oscillation which, in turn, drives respiration. Figure 2 also suggests that low frequency rhythms, which are prominent during breathing, are not affected by the absence of respiration. (Low frequency rhythms hold great fascination, but are beyond the scope of this review.)
Figure 2. R–R interval spectral power during frequency-controlled breathing and apnoea.
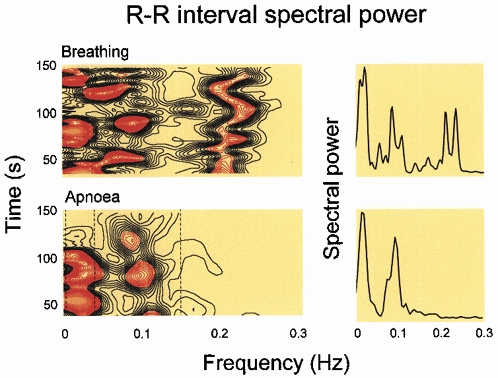
Respiratory-frequency R–R interval spectral power, shown as horizontal sections of a sliding fast Fourier transformation (lower left) or power spectrum (lower right) disappeared during apnoea. Breathing is necessary for the occurrence of respiratory-frequency R–R interval fluctuations; they are absent during apnoea.
In 1976, Lopes and Palmer advanced the provocative notion that respiration ‘gates’ autonomic responsiveness. We and others have explored respiratory gating of human autonomic outflows with a variety of research protocols.
Vagal-cardiac motoneurones
In the first of our studies (Eckberg & Orshan, 1977), we stretched carotid baroreceptive arteries (Kober & Arndt, 1970) with brief neck suction pulses during controlled frequency breathing. This experiment confirmed results from animal studies published earlier by Iriuchijima & Kumada (1964) and Haymet & McCloskey (1975), and showed that vagal responses are greater when baroreceptor stimuli are applied in expiration than inspiration. In a second study (Eckberg et al. 1980), we applied brief neck suction at six times during the normal breathing cycle. Figure 3 shows average R–R interval responses, plotted as functions of the times neck suction pulses were begun.
Figure 3. Changes of P–P intervals provoked by brief neck suction, applied at different times in the breathing cycle.
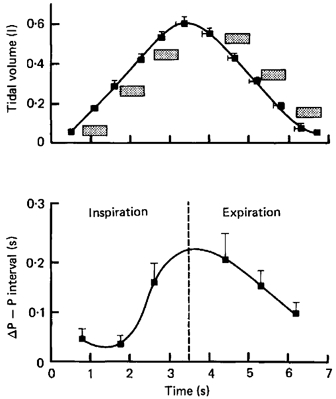
The stippled boxes show the times and durations of applications of baroreceptor stimulation. Changes of P–P intervals were calculated by subtracting measurements made during breathing from those made at the same times in the breathing cycle after neck suction. Respiratory gating of vagal-cardiac motoneurone responsiveness is continuously variable throughout the breathing cycle. Adapted from Eckberg et al. (1980).
These static measurements suggest that respiratory gating of human vagal-cardiac motoneurone responsiveness varies sinusoidally: at the breathing rate of the subjects studied, vagal-cardiac motoneurones are most responsive when baroreceptor stimuli begin in late inspiration and early expiration. These data belie at least three notions. First, that the respiratory gate is binary - either open or closed; respiratory gating of baroreceptor stimulation is continuously variable. Second, that the respiratory gate is closed in inspiration and open in expiration; the respiratory gate is (variably) open during both phases of breathing. Third, that the gate is closed completely; moderate baroreceptor stimuli elicit responses, albeit small, even in early inspiration.
In an elegant experiment performed in conscious cats, Gilbey and colleagues (1984) worked out the electrophysiological basis for respiratory gating of vagal-cardiac motoneurones. They showed that neurones in the ambiguus nucleus receive baroreceptor inputs which modulate their membrane potentials, and that such baroreceptor influences vary according to other, ongoing respiratory membrane potential fluctuations. Gilbey et al. reported that the ability of vagal-cardiac motoneurones to fire in response to excitant amino acids fluctuates in parallel with these respiratory fluctuations of vagal-cardiac motoneurone membrane potentials.
Sympathetic-muscle motoneurones
We (Eckberg et al. 1985) examined respiratory gating of sympathetic motoneurones by applying brief neck pressure pulses at different times in the breathing cycle. (Neck pressure compresses the carotid sinuses (Kober & Arndt, 1970), reduces carotid baroreceptor activity, and increases muscle sympathetic nerve activity (Wallin & Eckberg, 1982).) We found that sympathetic-muscle motoneurone responsiveness to reductions of baroreceptor input is broadly similar to vagal-cardiac motoneurone responsiveness to increases of baroreceptor input; neck pressure is most likely to provoke muscle sympathetic nerve firing when it is delivered in early expiration. This and the experiments described above, indicate that respiration gates both vagal and sympathetic motoneurone responsiveness to changes of baroreceptor input. (This review focuses on baroreceptor mechanisms. Respiration also gates responses to other neurosensory inputs, including those from chemoreceptors (Haymet & McCloskey, 1975).)
Very recently, we (Rothlisberger et al. 2003) identified a new physiological expression of respiratory gating of human autonomic outflow. In 1985, Bertinieri and his coworkers, and in 1986, our group called attention to spontaneously occurring parallel increases and decreases of systolic pressures and R–R intervals in cats (Bertinieri et al. 1985) and humans (Fritsch et al. 1986), and suggested that these fluctuations reflect baroreflex physiology - pressure changes trigger parallel R–R interval changes. Rothlisberger's study (Rothlisberger et al. 2003) showed that respiration orders the occurrence of such spontaneous ‘baroreflex sequences’.
The upper panel of Fig. 4 shows muscle sympathetic nerve activity signal-averaged on the beginning of expiration, and the lower panel shows baroreflex sequences, also triggered on early expiration. The upper panel indicates clearly that (as shown earlier: Eckberg et al. 1985; Seals et al. 1990; Macefield & Wallin, 1995) respiration gates muscle sympathetic nerve activity. The lower panel of Fig. 4 indicates that respiratory gating also determines the timing of spontaneous upgoing and downgoing baroreflex sequences. Thus, breathing initiates a cascade: (1) the respiratory gate opens and sympathetic bursts appear; (2) the sympathetic bursts increase arterial pressure, and pressure elevations trigger baroreflex R–R interval prolongations; (3) the increase of arterial pressure also silences sympathetic motoneurones, arterial pressure falls, and pressure reductions lead to baroreflex R–R interval shortening.
Figure 4. Muscle sympathetic nerve activity and up and down baroreflex slopes, signal-averaged on the beginning of expiration.
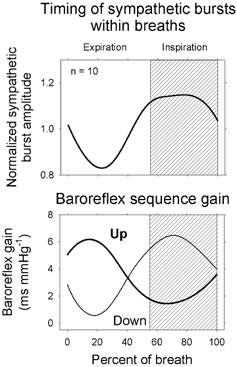
Respiratory gating of spontaneous upgoing and downgoing baroreflex sequences may be a consequence of respiratory gating of muscle sympathetic neurone firing. Adapted from Rothlisberger et al. (2003).
Notably, in Rothlisberger's study (Rothlisberger et al. 2003) cited above, respiration did not appear to affect the gain of baroreflex sequences; gain was comparable during breathing and apnoea. Moreover, although respiration strongly affects the timing of muscle sympathetic nerve bursts (Eckberg et al. 1985; Seals et al. 1990; Macefield & Wallin, 1995), it does not affect the quantity of sympathetic traffic; tonic sympathetic neural outflow is the same during slow and rapid breathing (Seals et al. 1990). Very recently Lehrer et al. (2003) studied the effects of biofeedback on cross-spectral vagal baroreflex gain (Robbe et al. 1987). In their study, subjects were shown a real-time display of their respiratory sinus arrhythmia, and told to increase it. Subjects breathed more slowly to increase their sinus arrhythmia, as discussed below. However, there was no significant correlation between the increased baroreflex gain that occurred during biofeedback and breathing frequency.
Factors that determine respiratory gate throughput
The stimulatory inputs that are gated
Implicit in the notion that respiration gates vagal-cardiac and sympathetic-muscle motoneurone responsiveness, is the corollary that for gating to occur, stimulatory inputs must be present. The relation between the level of baroreceptor stimulation and respiratory gating (of vagal R–R interval responses) was worked out superbly in dogs in an article published by Anrep et al. (1936a).
Figure 5 (adapted from Eckberg et al. 1988) documents the importance of stimulus intensity on respiratory gating. These data were obtained from one healthy supine subject, who was given intravenous phenylephrine to raise his arterial pressure (top row), saline (second row), and graded nitroprusside to lower his arterial pressure (third and fourth rows). All responses were signal-averaged on early expiration.
Figure 5. Signal-averaged mean ±s.e.m. measurements obtained from one supine subject at different arterial pressure levels.
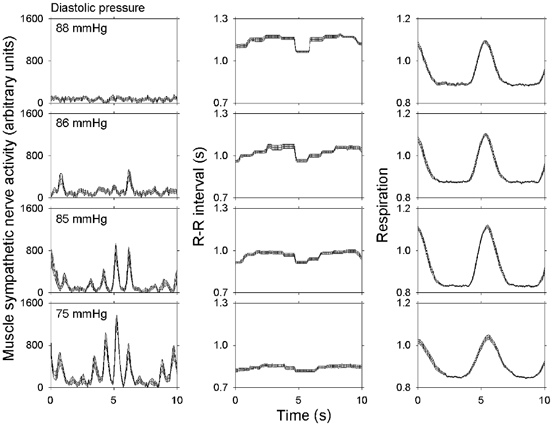
The magnitude of respiratory gating depends importantly on level of stimulation of sympathetic-muscle and vagal-cardiac motoneurones. Adapted from Eckberg et al. (1988).
These results indicate that the magnitude of respiratory gating depends critically on the level of stimulation of sympathetic and vagal motoneurones. At high arterial pressures, sympathetic nerve activity and respiratory gating are absent (top left panel), and at low arterial pressures, sympathetic nerve activity is high and respiratory modulation of sympathetic activity is large (bottom left panel). The opposite pattern is found with vagally mediated R–R interval responses: at high pressures, respiratory gating is large (top two middle panels), and at low pressures, respiratory gating is almost absent (bottom middle panel). These data document proportionality between the level of stimulation and respiratory fluctuations of neural outflow.
The data shown in Fig. 5 do not indicate how very high levels of stimulation might influence respiratory gating. However, other studies indicate clearly that the ability of respiration to gate both sympathetic-muscle and vagal-cardiac motoneurone responsiveness is finite. Although moderate carotid baroreceptor stimulation with neck suction provokes more cardiac slowing when stimuli are delivered in expiration than inspiration, intense baroreceptor stimulation provokes equal vagal motoneurone responses in expiration and inspiration (Eckberg & Orshan, 1977). When arterial pressure is increased to high levels pharmacologically, respiratory sinus arrhythmia diminishes (Goldberger et al. 1994) or disappears altogether (Anrep et al. 1936a). Figure 6 shows that when muscle sympathetic nerve activity is increased physiologically by graded passive upright tilt, the significant inspiratory - expiratory differences of sympathetic outflow present in the supine position and lower tilt angles also disappear (Cooke et al. 1999).
Figure 6. Inspiratory (□) and expiratory ( ) muscle sympathetic nerve activity during supine rest and passive upright tilt.
) muscle sympathetic nerve activity during supine rest and passive upright tilt.
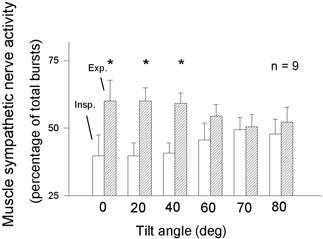
* Significant (P ≤ 0.05) differences between supine and tilt measurements. Insp. = inspiration; Exp. = expiration. Respiratory gating of sympathetic firing declines steadily as the level of sympathetic stimulation increases. Adapted from Cooke et al. (1999).
Figure 7 depicts the relation between the levels of baroreceptor stimulation of motoneurones and the ability of respiration to gate motoneurone responsiveness. (In this scheme, the height of the water before it passes through the gate indicates the level of stimulation; low baroreceptor activity stimulates sympathetic-muscle motoneurones and high baroreceptor activity stimulates vagal-cardiac motoneurones.) In each example, opening and closing of the gate with breathing continues apace, and occurs without regard to the level of upstream stimulation. Gating is most apparent at usual levels of stimulation (middle), and is weak or absent at the extremes of stimulation.
Figure 7. Schematic representation of the respiratory gate.
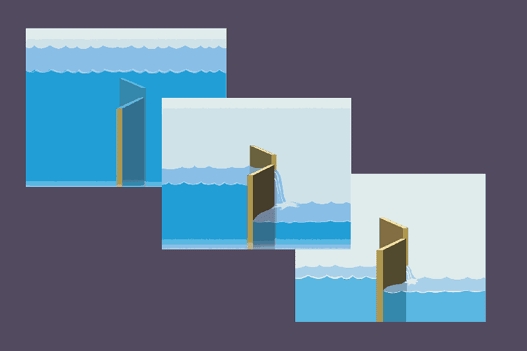
In this scheme, the level of stimulation of sympathetic or vagal motoneurones is represented as the height of the water to the left of the gates. Fluctuations of neural outflow are greatest at usual levels of stimulation, and are less at low or high levels of stimulation.
Properties of the gate itself
Although the scheme depicted in Fig. 7 may inform respiratory gating of autonomic outflow in a population, it provides no information on the heights of individual gates in relation to ongoing levels of stimulation, and how completely individual gates close. Such information is necessary, if fluctuations of autonomic neural outflow, such as respiratory sinus arrhythmia, are used as surrogates for vagus nerve traffic (Eckberg, 1983). This practice began with Katona & Jih (1975), who studied anaesthetized spontaneously breathing dogs and documented a highly linear relation between directly measured vagal-cardiac nerve activity and respiratory, peak minus valley R–R interval differences at different arterial pressures. Kollai & Mizsei (1990) continued this line of enquiry in healthy volunteers studied after β-adrenergic blockade, by measuring their respiratory peak minus valley R–R interval differences before and after large dose atropine. (The difference between R–R intervals after β-adrenergic blockade and after addition of atropine was taken as an index of vagal-cardiac nerve traffic (or ‘parasympathetic control’).)
Kollai & Mizsei (1990) concluded that peak minus valley R–R interval differences yield significant, but imperfect indices of vagal-cardiac nerve activity (average linear regression coefficient: 0.61). These studies indicate that the physiology underlying use of simple, non-invasive measures of respiratory sinus arrhythmia as surrogates for human vagal-cardiac nerve activity is complex. Measurements of respiratory sinus arrhythmia usually are not made before and after autonomic blockade, and no effort is made to determine the position of individuals' gates in relation to stimulatory inputs (Gonschorek et al. 2001). There is a third major complexity, breathing rate, which is discussed next.
Frequency of respiratory oscillation
The above discussion focuses on three elements of the respiratory gate: the timing of gate opening and closing within the breathing cycle, the stimuli that are gated, and the location of the gate in relation to resting levels of stimulation. There is a fourth important element in this physiology - the frequency of respiratory gate opening and closing. Breathing frequency is not fixed in healthy humans: it varies widely at rest and during normal activities (Lenfant, 1967). Therefore, respiratory-frequency fluctuations of sympathetic and vagal motoneurone activity release noradrenaline and acetylcholine peripherally, at varying intervals. It follows that ongoing effector responses must in some way reflect not only changes provoked by the most recent boluses of noradrenaline and acetylcholine released by sympathetic and vagus nerve volleys, but also the past history of such neurotransmitter release. That is, effector responses to respiration-related, episodic neurotransmitter release betray influences of the quantity of the neurotransmitters, the timing of their release and clearance, and the kinetics of effector responses.
Figure 8 shows average expiratory and inspiratory peak and valley P-P interval responses of six healthy supine subjects who breathed at different respiratory intervals at nearly constant tidal volumes (Eckberg, 1983). (Electrocardiographic P–P intervals equal R–R intervals when atrioventricular conduction is constant.) The top panel shows that the longest P–P intervals are longer, and the shortest P–P intervals are shorter at slow (right) than rapid (left) breathing rates. These respiration-related fluctuations of cholinergic inhibition are asymmetrical, such that sinoatrial node inhibition provoked by successive boluses of acetylcholine begins more abruptly than it decays. The difference between the two, respiratory sinus arrhythmia, increases asymptotically as breathing rate slows (lower panel).
Figure 8. Peak and valley P–P intervals at different breathing frequencies.
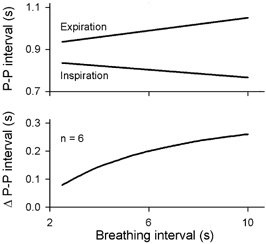
Maximum heart periods become longer, and minimum (Inspiration) heart periods become shorter as breathing rate slows. Adapted from Eckberg (1983).
The kinetics of sinoatrial node responses to abrupt changes of vagal-cardiac neuronal activity are understood well. The latency between the onset of baroreceptor or electrical carotid sinus nerve stimulation and the earliest sinoatrial node response is short (between about 0.25 and 0.50 s: Pickering & Davies, 1973; Eckberg, 1976; Borst & Karemaker, 1983; Seidel et al. 1997), and maximum responses occur after about 1.5–2.0 s (Eckberg, 1980). The rate of decay of vagal baroreflex responses is particularly relevant to respiratory sinus arrhythmia. We (Eckberg & Eckberg, 1982) studied this physiology by delivering trains of 0.6 s repetitive, ramped, R-wave-coupled neck suction pulses to healthy supine subjects. Figure 9 shows average P–P interval responses to trains of one (10 mmHg), two (10 and 20 mmHg), three (10, 20 and 30 mmHg), and four (10, 20, 30 and 40 mmHg) neck suction pulses delivered during held expiration. The left panel documents latencies (zero intercepts) of slightly less than 0.5 s for all trains, and similar R–R interval rise times after the beginnings of stimulus trains. The right panel shows average decay times, calculated from linear regression analyses of the descending P–P interval slopes shown in the left panel. This analysis indicates that the decay of vagally mediated R–R interval prolongation lasts about 2.0 s, and is nearly independent of the amount of acetylcholine released.
Figure 9. Average P–P intervals after trains of one, two, three, or four brief neck suction pulses.
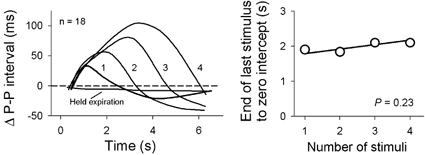
The right panel shows average times from the peak of P–P interval prolongation to the baseline, calculated with least squares linear regression. The decay of vagal baroreflex inhibition was nearly constant, and its rate was nearly independent of the amount of inhibition. Adapted from Eckberg & Eckberg (1982).
Fluctuations of efferent vagal-cardiac neuronal activity secondary to physiological respiratory gating are more complex than fluctuations provoked by experimental square wave (maximum pressure change ∼3000 mmHg s−1; Eckberg, 1976) neck suction. Respiratory gating is a continuous function (Fig. 3) that variably modulates bursts of vagal neuronal activity, according to the levels of successive arterial pressures. An additional complexity is that changing R–R intervals during breathing move the timing of the arrival of vagal volleys within the cardiac cycle closer to or farther from the times when maximum sinoatrial node responses might be elicited (Eckberg, 1976; Seidel et al. 1997). Nonetheless, the net effect of all of these influences can be gauged by simple measures of peak minus valley R–R interval changes (Fig. 8 of Anrep et al. 1936a) or spectral power at the breathing frequency (Angelone & Coulter, 1964; Brown et al. 1993).
Figure 10 (Taylor et al. 2001) shows average respiratory-frequency R–R interval spectral powers measured during three recording sessions from ten healthy supine men and women who breathed at 13 different frequencies, during saline infusion, and after cholinergic blockade with atropine or β-adrenergic blockade with atenolol. These results yield several insights into respiratory gating of human autonomic outflow. First, although respiratory sinus arrhythmia is determined importantly by breathing rate, this is not a simple linear function. Sinus arrhythmia is small at usual breathing frequencies (righthand side of left panel of Fig. 10; 0.25 Hz, or one breath every 4 s), maximal at frequencies of about 0.1 Hz (Hirsch & Bishop, 1981; Saul et al. 1991; Brown et al. 1993), and intermediate at very low breathing frequencies (lefthand side of left panel of Fig. 10). It is likely that this complex pattern of responses importantly reflects the kinetics of sinoatrial node responses to fluctuating levels of acetylcholine.
Figure 10. Average R–R interval spectral power and R–R intervals during breathing over a range of breathing frequencies, before (Saline) and after β-adrenergic (Atenolol) or cholinergic (Atropine) blockade.
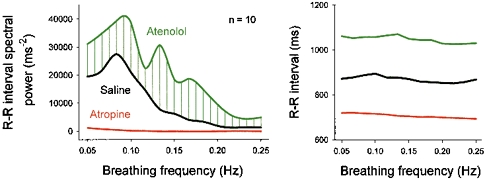
This analysis documents the importance of vagal fluctuations in generating R–R interval fluctuations, and indicates that sympathetic stimulation opposes vagal inhibition at all breathing frequencies. Adapted from Taylor et al. (2001).
Second, Fig. 10 indicates that sympathetic stimulation reduces vagal inhibition at all breathing frequencies, including the most rapid studied (righthand side of left panel of Fig. 10). However, sympathetic opposition to vagal effects is not uniform - inhibition is small at rapid breathing frequencies, and does not reach maximum levels until breathing rates fall below about 0.15 Hz (a respiratory frequency of nine breaths min−1). Third, respiratory (and all other) R–R interval fluctuations are mediated importantly by vagal-cardiac nerve fluctuations; R–R intervals are short, and nearly monotonic after cholinergic blockade with atropine. Fourth, if mean R–R interval (Fig. 10, right) is taken as an index of vagal-cardiac nerve traffic (Katona et al. 1970; Raczkowska et al. 1983), breathing frequency does not appreciably influence net vagal-cardiac neuronal activity (Brown et al. 1993; Cooke et al. 1998), or sympathetic opposition to vagal restraint. Over a wide range of breathing rates, mean R–R intervals are nearly constant.
The location of the gate
There is an abundant literature from animals and humans that dichotomizes mechanisms responsible for respiratory gating, and poses the questions, Is respiratory gating central, secondary to efferent respiratory motoneurone activity? or Is it peripheral, secondary to afferent neural activity from pulmonary and thoracic stretch receptors?Frédéricq (1882) was among the first to ask these questions. He showed that respiratory sinus arrhythmia continues in animals when their chests are open and no lung motions are discernible. Respiratory sinus arrhythmia disappears, however, when inspiratory motoneurone activity is silenced by hyperventilation. Heymans (1929) showed that respiratory sinus arrhythmia continues after lung denervation.
In the second of their two articles based on highly invasive experiments with anaesthetized dogs, Anrep et al. (1936b) concluded that both central and peripheral mechanisms are operative. However, Joels & Samueloff (1956) studied anaesthetized dogs and cats with ‘diffusion respiration’ (which maintains normal oxygenation, but leads to hypercapnia), and measured respiratory arterial pressure oscillations under two experimental conditions: deep thiopentone anaesthesia, which abolishes central respiratory motoneurone activity, and succinylcholine paralysis which abolishes respiratory movements. Since deep thiopentone anaesthesia abolished respiratory arterial pressure fluctuations, and paralysis did not, Joels & Samueloff (1956) concluded that arterial pressure fluctuations are caused by efferent respiratory motoneurone activity, and not by afferent inputs from pulmonary and thoracic stretch receptors. Shykoff et al. (1991) studied anaesthetized dogs and used constant flow ventilation (which ventilates without rhythmic lung inflations and deflations) and showed that fluctuations of heart rate parallel fluctuations of phrenic nerve activity, and persist in the absence of lung movements.
We (Koh et al. 1998) performed a similar experiment in healthy young volunteers, lightly anaesthetized prior to elective orthopaedic surgery, and showed that respiratory sinus arrhythmia is much greater when subjects are studied during spontaneous breathing than during high frequency jet ventilation (which provides adequate ventilation, without the usual lung inflations and deflations). One limitation of our study is that respiratory sinus arrhythmia and vagal outflow may have been diminished by the vagolytic effects of anaesthesia (Hicks et al. 1981). A second limitation is that although we assumed that high frequency jet ventilation abolished respiratory drive, we did not record any index of phrenic nerve activity. However, it is well established that in conscious volunteers, mechanical ventilation, which presumably suppresses respiratory motoneurone activity but preserves phasic inputs from pulmonary and thoracic stretch receptors, nearly abolishes respiratory sinus arrhythmia (Freyschuss & Melcher, 1975; Macefield & Wallin, 1995).
In another study (Badra et al. 2001), we measured autonomic neural outflows during apnoea, after hyperventilation with 100 % oxygen. All subjects studied could hold their breaths for at least 180 s. However, unbeknownst to either the subjects or the investigators during the experiments, pneumograph recordings (pressure transducers connected to abdominal bellows) documented erratic, very small changes during apnoea. (The transducer gain was so high that minute aortic pulsations were registered.) Figure 11 shows six of these movements (the small notches in the pneumograph recording) recorded in one subject, as well as systolic pressure, R–R intervals, and sympathetic nerve activity, signal-averaged on these pneuomgraph movements. The bottom panels indicate that systolic pressure, R–R interval, and sympathetic nerve activity responses to these small pneumograph changes were large. Since pneumotachograph recordings did not document any movement of air, we assumed that the changes shown in Fig. 11 were secondary to inspiratory motoneurone activity, and reflected aborted, minute gasps. If our interpretation is correct, these anecdotal data suggest that central respiratory motoneurone activity plays a large role in generation of respiration-related fluctuations of autonomic outflow.
Figure 11. Aborted respiratory movements during apnoea, and measurements obtained by signal-averaging on the small pneumograph changes shown as red notches in the upper panel.
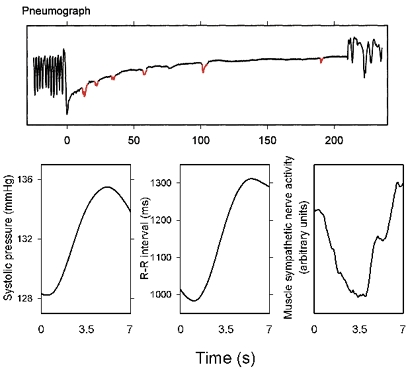
The subject was not aware of the minute abortive respiratory movements, and the pneumotachograph recording did not register airflow. These responses provide indirect evidence for the importance of respiratory motoneurone activity in modulating autonomic neural firing. Adapted from Badra et al. (2001).
This speculation iterates arguments advanced earlier by Snyder (1915). He also noted persistence of respiratory sinus arrhythmia in the absence of any visible respiratory activity, and considered that sinus arrhythmia could result from respiratory motoneurone discharges that were so insignificant that they did not provoke chest expansion, or even respiratory muscle contraction. Peňāz & Buriánek (1963) documented major arterial pressure and R–R interval changes when apnoeic subjects took isolated breaths. These haemodynamic changes appeared to be triggered by inspiration, since they occurred whether or not inspiration was followed by expiration. It is likely that pulmonary and thoracic stretch receptors make some contribution to respiratory-frequency autonomic fluctuations; however, other authors (Taha et al. 1995; St Croix et al. 1999) assign greater importance to this mechanism than I do.
Are autonomic and cardiovascular rhythms, therefore, driven by respiration?
The foregoing discussion establishes beyond argument the fact that breathing can exert important effects on human autonomic and cardiovascular rhythms. The remaining discussion focuses on two questions: Must breathing impress its mark on autonomic outflow? and, Might peripheral rhythms with respiratory frequencies, in fact, be caused by breathing?
Figure 12 shows systolic pressures and R–R intervals, recorded from one healthy young woman breathing spontaneously, in the supine position. In this, and virtually all other such recordings, systolic pressures and R–R intervals fluctuate deterministically, in parallel with breathing. (This figure also shows that the timing of spontaneous baroreflex slopes (shown as red lines in the arterial pressure signal) is not dictated simply by breathing; qualified (Parati et al. 1988) baroreflex slopes occur much less frequently than respiratory arterial pressure and R–R interval changes.) The association shown in Fig. 12 does not, however, prove that both systolic pressure and R–R interval fluctuations are due to respiration; indeed, a strong case has been made that breathing triggers a cascade, such that (1) intrathoracic pressure changes lead to changes of left ventricular stroke volume and arterial pressure (Karam et al. 1984; Guz et al. 1987), and (2) arterial pressure changes provoke parallel R–R interval changes by means of baroreflex physiology (de Boer et al. 1985a; Pagani et al. 1988; Baselli et al. 1994).
Figure 12. Systolic pressures, R–R intervals and spontaneous baroreflex slopes from one subject during quiet breathing.
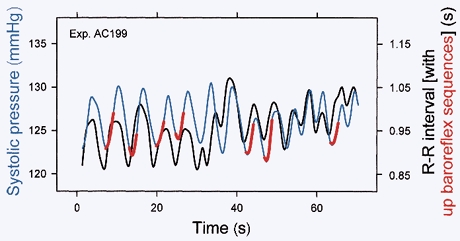
Systolic pressures and R–R intervals fluctuated together with every breath, and spontaneous baroreflex slopes (in red) occurred less frequently. Therefore, spontaneous baroreflex sequences do not result simply from breathing. Adapted from Rothlisberger et al. (2003).
We considered an alternative interpretation, that respiration, rather than baroreflex physiology, is responsible for parallel, respiratory-frequency systolic pressure and R–R interval changes. The task of proving causality in human research is daunting, and although it is a trivial matter to prove that two signals change at the same times (with signal-averaging or cross-spectral analysis and calculation of coherence), it is another matter altogether to prove that changes of one parameter cause changes of another. Badra and her colleagues (2001) attempted to deal with this issue by using partial coherence analysis (Bendat & Piersol, 1986). Partialization is a mathematical technique used to remove the influence of one signal from two other signals (in Badra's study, the influence of respiration was removed from systolic pressures and R–R intervals), and then determine how much residual coherence remains between those two signals. Figure 13 shows sliding cross-spectra from one spontaneously breathing supine subject before (upper panels) and after (lower panels) partialization. (This recording was made over 5 min; cross-spectral phase and coherence were measured over 90 s windows, and moved by 3 s steps through the data.)
Figure 13. Sliding systolic pressure and R–R interval coherence and phase from one subject during spontaneous breathing, before and after partialization.
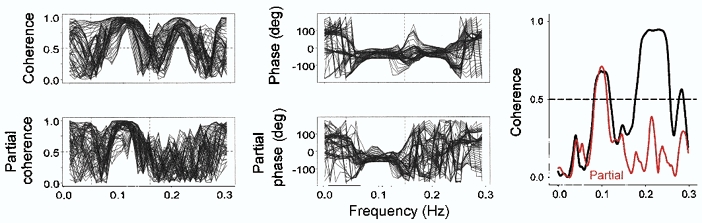
Removal of the influence of respiration from systolic pressures and R–R intervals by partialization (dashed line, see text) abolished the systematic coherence and phase differences that were present before partialization. This suggests that parallel respiratory-frequency systolic pressure and R–R interval fluctuations are secondary to breathing, and do not reflect baroreflex physiology. Adapted with permission (Badra et al. 2001).
The analysis in Fig. 13 illustrates several important features of human autonomic rhythms. First, as shown in the upper panels, systolic pressures and R–R intervals may be highly, and systematically coherent at low (∼0.1 Hz) and respiratory (in this case, ∼0.2 Hz) frequencies. (Perfect coherence is 1.0, and significant coherence is usually taken as above 0.5 (de Boer et al. 1985b; Taylor et al. 1998).) Second, fluctuations between systolic pressures and R–R intervals at coherent frequencies may have nearly constant phase angles (upper middle panel). In Badra's subjects, average phase angles over coherent low and respiratory frequencies were −50 and −32 deg. If arterial pressures lead R–R intervals as seems likely, these phase angles translate into actual latencies (to P waves) of 1.73 and 0.15 s.
Third, partialization convincingly abolishes the significant coherence and nearly fixed phase angles between systolic pressures and R–R intervals at breathing frequencies, but does not materially influence the phase and coherence between these signals at low frequencies (Fig. 8 left, lower panels; right panel). These data support the inference that parallel fluctuations of systolic pressures and R–R intervals at breathing frequencies are secondary to respiratory influences on both systolic pressures and R–R intervals, and do not result from baroreflex physiology.
Other observations also challenge the baroreflex hypothesis for respiratory-frequency arterial pressure and R–R interval rhythms.
The calculated latency between respiratory-frequency signals shown in Fig. 13, 0.15 s, is shorter than the shortest human vagal baroreflex latency measured ∼0.25 s (Eckberg, 1976).
A latency of 0.15 s is much shorter than the latency between an abrupt baroreflex stimulus and maximum sinoatrial node responses, which is about 1.5 – 2.0 s (Fig. 9, (Eckberg 1980; Eckberg & Eckberg, 1982)).
In the group of volunteers studied by Badra et al. (2001), five of nine subjects had positive phase angles at breathing frequencies; that is, R–R interval changes lead the systolic pressure changes that are thought to provoke them.
Taylor & Eckberg (1996) studied supine subjects during normal sinus rhythm, and during fixed rate left atrial pacing, and showed that respiratory fluctuations of systolic pressure are significantly greater when normal respiratory flucuations of R–R intervals are present, than when they are absent. This implies that heart period changes modulate systolic pressure, rather than the reverse (baroreflex physiology).
Di Rienzo and coworkers (1996) showed that in conscious cats, sino-aortic baroreceptor denervation substantially reduces coherence between systolic pressure and R–R intervals at low frequencies, but does not alter coherence at respiratory frequencies.
Across studies, phase angles at low frequencies, about 0.10 s, are nearly constant during different experimental circumstances, but latencies at breathing frequencies are fungible - they vary appreciably, and under some circumstances, systematically. One example is lower body suction, which changes the phase angle between arterial pressure and R–R intervals systematically (Blaber et al. 1995).
Figure 14 shows two other examples: phase angles at low (left panels) and respiratory frequencies (right panels) from two different studies. The upper panels show systolic pressure vs. R–R interval phase angles during stepwise, passive upright tilt (adapted from Cooke et al. 1999). Phase angles were large, negative, and nearly constant at low frequencies, and were small and variable at respiratory frequencies - the average phase changed systematically from positive at 0 deg tilt, to negative at higher angles of tilt. The lower panels of Fig. 14 (adapted from Koh et al. 1998) indicate that low frequency phase angles are similar, and respiratory-frequency phase angles are highly variable during spontaneous breathing and mechanical ventilation (average calculated latencies at low frequencies (lower left panel) were −2.7 and −3.0 s, and average calculated latencies at breathing frequencies (lower right panel) were −0.29 and 0.135 s).
Figure 14. Low- and respiratory-frequency phase angles derived from cross spectral analysis of systolic pressures and R–R intervals during passive upright tilt (upper panels) and spontaneous breathing or mechanical ventilation (lower panels).
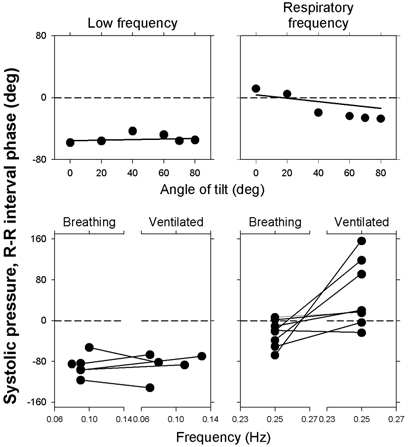
Nearly constant low frequency phase angles are consistent with baroreflex physiology. Conversely, highly variable respiratory-frequency phase angles during tilt and mechanical breathing point toward respiratory influences, and away from baroreflex mechanisms. Adapted from Koh et al. (1998) and Cooke et al. (1999).
These considerations suggest that it is respiration that is responsible for the correlation between arterial pressure and R–R intervals at breathing frequencies, rather than baroreflex physiology. Additional evidence of respiratory ordering of autonomic outflow is found in recordings of sympathetic activity. Macefield & Wallin (1995) showed that respiration-related fluctuations of muscle sympathetic nerve activity are the same during positive- and negative-pressure mechanical ventilation, notwithstanding the fact that the two modes of ventilation provoke opposite trending of arterial pressure.
Conclusions
Cardiovascular rhythms have been recognized since the Statical Essays published by Stephen Hales (1733), who reported fluctuations of the level of blood in a glass tube lodged in a crural artery of a ‘mare tied down alive on her back’. This review treats neurally mediated rhythms occurring at respiratory frequencies. It discusses the rhythmic gating of responsiveness of both vagal-cardiac and sympathetic-muscle motoneurones by breathing. It emphasizes the importance of the level of stimulation of those motoneurones to the gating process, and deals with other aspects of gating, not least, the frequency of opening and closing of the gate. A case is made that respiratory gating may have great usefulness as a tool to explore and understand otherwise inaccessible human autonomic neurophysiology.
Acknowledgments
The research from my laboratory cited in this review has been supported generously for many years by grants and contracts from the National Institutes of Health, the Department of Veterans Affairs, and the National Aeronautics and Space Administration.
REFERENCES
- Angelone A, Coulter NA., Jr Respiratory sinus arrhythmia: a frequency dependent phenomenon. J Appl Physiol. 1964;19:479–482. doi: 10.1152/jappl.1964.19.3.479. [DOI] [PubMed] [Google Scholar]
- Anrep GV, Pascual W, Rössler R. Respiratory variations of the heart rate. I-The reflex mechanism of the respiratory arrhythmia. Proc R Soc Lond B Biol Sci. 1936a;119 B:191–217. [Google Scholar]
- Anrep GV, Pascual W, Rössler R. Respiratory variations of the heart rate. II-The central mechanism of the respiratory arrhythmia and the inter-relations between the central and the reflex mechanisms. Proc R Soc Lond B Biol Sci. 1936b;119 B:218–230. [Google Scholar]
- Badra LJ, Cooke WH, Hoag JB, Crossman AA, Kuusela TA, Tahvanainen KUO, Eckberg DL. Respiratory modulation of human autonomic rhythms. Am J Physiol Heart Circ Physiol. 2001;280:H2674–2688. doi: 10.1152/ajpheart.2001.280.6.H2674. [DOI] [PubMed] [Google Scholar]
- Baselli G, Cerutti S, Badilini F, Biancardi L, Porta A, Pagani M, Lombardi F, Rimoldi O, Furlan R, Malliani A. Model for the assessment of heart period and arterial pressure variability interactions and respiration influences. Med Biol Eng Comput. 1994;32:143–152. doi: 10.1007/BF02518911. [DOI] [PubMed] [Google Scholar]
- Bendat JS, Piersol AG. Random Data. Analysis and Measurement Procedures. 2. New York, USA: Blackwell Science Inc; 1986. [Google Scholar]
- Bertinieri G, Di Rienzo M, Cavallazzi A, Ferrari AU, Pedotti A, Mancia G. A new approach to analysis of the arterial baroreflex. J Hypertens. 1985;3(suppl. 3):S79–S81. [PubMed] [Google Scholar]
- Blaber AP, Yamamoto Y, Hughson RL. Change in phase relationship between SBP and R–R interval during lower body negative pressure. Am J Physiol. 1995;268:H1688–1693. doi: 10.1152/ajpheart.1995.268.4.H1688. [DOI] [PubMed] [Google Scholar]
- Borst C, Karemaker JM. Time delays in the human baroreceptor reflex. J Auton Nerv Syst. 1983;9:399–409. doi: 10.1016/0165-1838(83)90004-8. [DOI] [PubMed] [Google Scholar]
- Brown TE, Beightol LA, Koh J, Eckberg DL. Important influence of respiration on human R–R interval power spectra is largely ignored. J Appl Physiol. 1993;75:2310–2317. doi: 10.1152/jappl.1993.75.5.2310. [DOI] [PubMed] [Google Scholar]
- Cooke WH, Cox JF, Diedrich AM, Taylor JA, Beightol LA, Ames JE, Hoag JB, Seidel H, Eckberg DL. Controlled breathing protocols probe human autonomic cardiovascular rhythms. Am J Physiol. 1998;274:H709–718. doi: 10.1152/ajpheart.1998.274.2.h709. [DOI] [PubMed] [Google Scholar]
- Cooke WH, Hoag JB, Crossman AA, Kuusela TA, Tahvanainen KUO, Eckberg DL. Human responses to upright tilt: a window on central autonomic integration. J Physiol. 1999;517:617–628. doi: 10.1111/j.1469-7793.1999.0617t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boer RW, Karemaker JM, Strackee J. Relationships between short-term blood-pressure fluctuations and heart-rate variability in resting subjects 1: a spectral analysis approach. Med Biol Eng Comput. 1985a;23:352–358. doi: 10.1007/BF02441589. [DOI] [PubMed] [Google Scholar]
- De Boer RW, Karemaker JM, Strackee J. Relationships between short-term blood-pressure fluctuations and heart-rate variability in resting subjects II: a simple model. Med Biol Eng Comput. 1985b;23:359–364. doi: 10.1007/BF02441590. [DOI] [PubMed] [Google Scholar]
- Di Rienzo M, Castiglioni P, Parati G, Mancia G, Pedotti A. Effects of sino-aortic denervation on spectral characteristics of blood pressure and pulse interval variability: a wide-band approach. Med Biol Eng Comput. 1996;34:133–141. doi: 10.1007/BF02520018. [DOI] [PubMed] [Google Scholar]
- Eckberg DL. Temporal response patterns of the human sinus node to brief carotid baroreceptor stimuli. J Physiol. 1976;258:769–782. doi: 10.1113/jphysiol.1976.sp011445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckberg DL. Nonlinearities of the human carotid baroreceptor-cardiac reflex. Circ Res. 1980;47:208–216. doi: 10.1161/01.res.47.2.208. [DOI] [PubMed] [Google Scholar]
- Eckberg DL. Human sinus arrhythmia as an index of vagal cardiac outflow. J Appl Physiol. 1983;54:961–966. doi: 10.1152/jappl.1983.54.4.961. [DOI] [PubMed] [Google Scholar]
- Eckberg DL, Eckberg MJ. Human sinus node responses to repetitive, ramped carotid baroreceptor stimuli. Am J Physiol. 1982;242:H638–644. doi: 10.1152/ajpheart.1982.242.4.H638. [DOI] [PubMed] [Google Scholar]
- Eckberg DL, Kifle YT, Roberts VL. Phase relationship between normal human respiration and baroreflex responsiveness. J Physiol. 1980;304:489–502. doi: 10.1113/jphysiol.1980.sp013338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckberg DL, Nerhed C, Wallin BG. Respiratory modulation of muscle sympathetic and vagal cardiac outflow in man. J Physiol. 1985;365:181–196. doi: 10.1113/jphysiol.1985.sp015766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckberg DL, Orshan CR. Respiratory and baroreceptor reflex interactions in man. J Clin Invest. 1977;59:780–785. doi: 10.1172/JCI108699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckberg DL, Rea RF, Andersson OK, Hedner T, Pernow J, Lundberg JM, Wallin BG. Baroreflex modulation of sympathetic activity and sympathetic neurotransmitters in humans. Acta Physiol Scand. 1988;133:221–231. doi: 10.1111/j.1748-1716.1988.tb08401.x. [DOI] [PubMed] [Google Scholar]
- Frédéricq L. De l'influence de la respiration sur la circulation. Les oscillations respiratoires de la pression artérielle chez le chien. Arch Biol Paris. 1882;3:55–100. [Google Scholar]
- Freyschuss U, Melcher A. Sinus arrhythmia in man: influence of tidal volume and oesophageal pressure. Scand J Clin Lab Invest. 1975;35:487–496. [PubMed] [Google Scholar]
- Fritsch JM, Eckberg DL, Graves LD, Wallin BG. Arterial pressure ramps provoke linear increases of heart period in humans. Am J Physiol. 1986;251:R1086–1090. doi: 10.1152/ajpregu.1986.251.6.R1086. [DOI] [PubMed] [Google Scholar]
- Gilbey MP, Jordan D, Richter DW, Spyer KM. Synaptic mechanisms involved in the inspiratory modulation of vagal cardio-inhibitory neurones in the cat. J Physiol. 1984;356:65–78. doi: 10.1113/jphysiol.1984.sp015453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberger JJ, Ahmed MW, Parker MA, Kadish AH. Dissociation of heart rate variability from parasympathetic tone. Am J Physiol. 1994;266:H2152–2157. doi: 10.1152/ajpheart.1994.266.5.H2152. [DOI] [PubMed] [Google Scholar]
- Gonschorek AS, Lu I-L, Halliwill JR, Beightol LA, Taylor JA, Painer JA, Warzel H, Eckberg DL. Influence of respiratory motor neurone activity on human autonomic and haemodynamic rhythms. Clin Physiol. 2001;21:323–334. doi: 10.1046/j.1365-2281.2001.00324.x. [DOI] [PubMed] [Google Scholar]
- Guz A, Innes JA, Murphy K. Respiratory modulation of left ventricular stroke volume in man measured using pulsed Doppler ultrasound. J Physiol. 1987;393:499–512. doi: 10.1113/jphysiol.1987.sp016836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales S. Statical Essays: Containing Haemastaticks; or, An Account of some Hydraulick and Hydrostatical Experiments made on the Blood and Blood-Vessels of Animals. London: Blackwell Science Inc; 1733. [Google Scholar]
- Haymet BT, McCloskey DI. Baroreceptor and chemoreceptor influences on heart rate during the respiratory cycle in the dog. J Physiol. 1975;245:699–712. doi: 10.1113/jphysiol.1975.sp010869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymans C. Über die Physiologie und Pharmakologie des Herz-Vagus-Zentrums. Ergeb Physiol. 1929;28:244–311. [Google Scholar]
- Hicks HC, Mowbray AG, Yhap EO. Cardiovascular effects of and catecholamine responses to high dose fentanyl-O2 for induction of anesthesia in patients with ischemic coronary artery disease. Anesth Analg. 1981;60:563–568. [PubMed] [Google Scholar]
- Hirsch JA, Bishop B. Respiratory sinus arrhythmia in humans: how breathing pattern modulates heart rate. Am J Physiol. 1981;241:H620–629. doi: 10.1152/ajpheart.1981.241.4.H620. [DOI] [PubMed] [Google Scholar]
- Hyndman BW, Kitney RI, Sayers BMcA. Spontaneous rhythms in physiological control systems. Nature. 1971;233:339–341. doi: 10.1038/233339a0. [DOI] [PubMed] [Google Scholar]
- Imholz BPM, Wieling W, Van Montfrans GA, Wesseling KH. Fifteen years experience with finger arterial pressure monitoring: assessment of the technology. Cardiovasc Res. 1998;38:605–616. doi: 10.1016/s0008-6363(98)00067-4. [DOI] [PubMed] [Google Scholar]
- Iriuchijima J, Kumada M. Activity of single vagal fibers efferent to the heart. Jap J Physiol. 1961;14:479–487. doi: 10.2170/jjphysiol.14.479. [DOI] [PubMed] [Google Scholar]
- Joels N, Samueloff M. The activity of the medullary centres in diffusion respiration. J Physiol. 1956;133:360–372. doi: 10.1113/jphysiol.1956.sp005592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karam M, Wise RA, Natarajan TK, Permutt S, Wagner HN. Mechanism of decreased left ventricular stroke volume during inspiration in man. Circulation. 1984;69:866–873. doi: 10.1161/01.cir.69.5.866. [DOI] [PubMed] [Google Scholar]
- Katona PG, Jih F. Respiratory sinus arrhythmia: noninvasive measure of parasympathetic cardiac control. J Appl Physiol. 1975;39:801–805. doi: 10.1152/jappl.1975.39.5.801. [DOI] [PubMed] [Google Scholar]
- Katona PG, Poitras JW, Barnett GO, Terry BS. Cardiac vagal efferent activity and heart period in the carotid sinus reflex. Am J Physiol. 1970;218:1030–1037. doi: 10.1152/ajplegacy.1970.218.4.1030. [DOI] [PubMed] [Google Scholar]
- Kober G, Arndt JO. Die Druck-Durchmesser-Beziehung der A. carotis communis des wachen Menschen. Pflugers Arch. 1970;314:27–39. doi: 10.1007/BF00587044. [DOI] [PubMed] [Google Scholar]
- Kocsis B, Fedina L, Gylmesi-Pelczer K, Ladocsi T, Pasztor E. Differential sympathetic reactions during cerebral ischaemia in cats: the role of desynchronized nerve discharge. J Physiol. 1993;469:37–50. doi: 10.1113/jphysiol.1993.sp019803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh J, Brown TE, Beightol LA, Eckberg DL. Contributions of tidal lung inflation to human R–R interval and arterial pressure fluctuations. J Autonom Nerv Syst. 1998;68:89–95. doi: 10.1016/s0165-1838(97)00114-8. [DOI] [PubMed] [Google Scholar]
- Kollai M, Mizsei G. Respiratory sinus arrhythmia is a limited measure of cardiac parasympathetic control in man. J Physiol. 1990;424:329–342. doi: 10.1113/jphysiol.1990.sp018070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrer P, Vaschillo E, Vaschillo B, Lu S-E, Eckberg DL, Edelberg R, Shih WJ, Lin Y, Kuusela TA, Tahvanainen KUO. Psychosom Med. 2003. Biofeedback-induced neuroplasticity of vagal baroreflex and pulmonary control mechanisms. in the Press. [DOI] [PubMed] [Google Scholar]
- Lenfant C. Time-dependent variations of pulmonary gas exchange in normal man at rest. J Appl Physiol. 1967;22:675–684. doi: 10.1152/jappl.1967.22.4.675. [DOI] [PubMed] [Google Scholar]
- Lopes OU, Palmer JF. Proposed respiratory ‘gating’ mechanism for cardiac slowing. Nature. 1976;264:454–456. doi: 10.1038/264454a0. [DOI] [PubMed] [Google Scholar]
- Macefield VG, Wallin BG. Modulation of muscle sympathetic activity during spontaneous and artificial ventilation and apnoea in humans. J Autonom Nerv Syst. 1995;53:137–147. doi: 10.1016/0165-1838(94)00173-h. [DOI] [PubMed] [Google Scholar]
- Novak V, Novak P, De Champlain J, Le Blanc AR, Martin R, Nadeau R. Influence of respiration on heart rate and blood pressure fluctuations. J Appl Physiol. 1993;74:617–626. doi: 10.1152/jappl.1993.74.2.617. [DOI] [PubMed] [Google Scholar]
- Pagani M, Lombardi F, Guzzetti S, Rimoldi O, Furlan R, Pizzenelli P, Sandrone G, Malfatto G, Dell'Orto S, Piccaluga E, Turiel M, Baselli G, Cerutti S, Malliani A. Power spectral analysis of heart rate and arterial pressure variabilities as a marker of sympatho-vagal interaction in man and conscious dog. Circ Res. 1986;59:178–193. doi: 10.1161/01.res.59.2.178. [DOI] [PubMed] [Google Scholar]
- Pagani M, Somers V, Furlan R, Dell'Orto S, Conway J, Baselli G, Cerutti S, Sleight P, Malliani A. Changes in autonomic regulation induced by physical training in mild hypertension. Hypertension. 1988;12:600–610. doi: 10.1161/01.hyp.12.6.600. [DOI] [PubMed] [Google Scholar]
- Parati G, Di Rienzo M, Bertinieri G, Pomidossi G, Casadei R, Groppelli A, Pedotti A, Zanchetti A, Mancia G. Evaluation of the baroreceptor-heart rate reflex by 24-hour intra-arterial blood pressure monitoring in humans. Hypertension. 1988;12:214–222. doi: 10.1161/01.hyp.12.2.214. [DOI] [PubMed] [Google Scholar]
- Peňāz J, Buriánek P. Zeitverlauf und Dynamik der durch Atmung ausgelösten Kreislaufänderungen beim Menschen. Pflugers Arch. 1963;276:618–635. [Google Scholar]
- Pickering TG, Davies J. Estimation of the conduction time of the baroreceptor-cardiac reflex in man. Cardiovasc Res. 1973;7:213–219. doi: 10.1093/cvr/7.2.213. [DOI] [PubMed] [Google Scholar]
- Raczkowska M, Eckberg DL, Ebert TJ. Muscarinic cholinergic receptors modulate vagal cardiac responses in man. J Autonom Nerv Syst. 1983;7:271–278. doi: 10.1016/0165-1838(83)90080-2. [DOI] [PubMed] [Google Scholar]
- Robbe HWJ, Mulder LJM, Rüddel H, Langewitz WA, Veldman JBP, Mulder G. Assessment of baroreceptor reflex sensitivity by means of spectral analysis. Hypertension. 1987;10:538–543. doi: 10.1161/01.hyp.10.5.538. [DOI] [PubMed] [Google Scholar]
- Rothlisberger BW, Badra LJ, Hoag JB, Cooke WH, Kuusela TA, Tahvanainen KUO, Eckberg DL. Spontaneous ‘baroreflex sequences’ are deterministic functions of breathing phase. Clin Physiol. 2003 doi: 10.1046/j.1475-0961.2003.00489.x. in the Press. [DOI] [PubMed] [Google Scholar]
- St Croix CM, Satoh M, Morgan BJ, Skatrud JB, Dempsey JA. Role of respiratory motor output in within-breath modulation of muscle sympathetic nerve activity in humans. Circ Res. 1999;85:457–469. doi: 10.1161/01.res.85.5.457. [DOI] [PubMed] [Google Scholar]
- Saul JP, Berger RD, Albrecht P, Stein SP, Chen MH, Cohen RJ. Transfer function analysis of the circulation: unique insights into cardiovascular regulation. Am J Physiol. 1991;261:H1231–1245. doi: 10.1152/ajpheart.1991.261.4.H1231. [DOI] [PubMed] [Google Scholar]
- Seals DR, Suwarno NO, Dempsey JA. Influence of lung volume on sympathetic nerve discharge in normal humans. Circ Res. 1990;67:130–141. doi: 10.1161/01.res.67.1.130. [DOI] [PubMed] [Google Scholar]
- Seidel H, Herzel H, Eckberg DL. Phase dependencies of the human baroreceptor reflex. Am J Physiol. 1997;272:H2040–2053. doi: 10.1152/ajpheart.1997.272.4.H2040. [DOI] [PubMed] [Google Scholar]
- Shykoff BE, Naqvi SSJ, Menon AS, Slutsky AS. Respiratory sinus arrhythmia in dogs. Effects of phasic afferents and chemostimulation. J Clin Invest. 1991;87:1621–1627. doi: 10.1172/JCI115176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder CD. A study of the causes of respiratory change of heart rate. Am J Physiol. 1915;37:104–117. [Google Scholar]
- Taha BH, Simon PM, Dempsey JA, Skatrud JB, Iber C. Respiratory sinus arrhythmia in humans: an obligatory role for vagal feedback from the lungs. J Appl Physiol. 1995;78:638–645. doi: 10.1152/jappl.1995.78.2.638. [DOI] [PubMed] [Google Scholar]
- Taylor JA, Carr DL, Myers CW, Eckberg DL. Mechanisms underlying very-low-frequency RR-interval oscillations in humans. Circulation. 1998;98:547–555. doi: 10.1161/01.cir.98.6.547. [DOI] [PubMed] [Google Scholar]
- Taylor JA, Eckberg DL. Fundamental relations between short-term RR interval and arterial pressure oscillations in humans. Circulation. 1996;93:1527–1532. doi: 10.1161/01.cir.93.8.1527. [DOI] [PubMed] [Google Scholar]
- Taylor JA, Myers CW, Halliwill JR, Seidel H, Eckberg DL. Sympathetic restraint of respiratory sinus arrhythmia: implications for vagal-cardiac tone assessment in humans. Am J Physiol Heart Circ Physiol. 2001;280:H2804–2814. doi: 10.1152/ajpheart.2001.280.6.H2804. [DOI] [PubMed] [Google Scholar]
- Vallbo ÅB, Hagbarth K-E, Torebjörk HE, Wallin BG. Somatosensory, proprioceptive, and sympathetic activity in human peripheral nerves. Physiol Rev. 1979;59:919–957. doi: 10.1152/physrev.1979.59.4.919. [DOI] [PubMed] [Google Scholar]
- Wallin BG, Eckberg DL. Sympathetic transients caused by abrupt alterations of carotid baroreceptor activity in humans. Am J Physiol. 1982;242:H185–190. doi: 10.1152/ajpheart.1982.242.2.H185. [DOI] [PubMed] [Google Scholar]
- Wallin BG, Esler M, Dorward P, Eisenhofer G, Ferrier C, Westerman R, Jennings G. Simultaneous measurements of cardiac noradrenaline spillover and sympathetic outflow to skeletal muscle in humans. J Physiol. 1992;453:45–58. doi: 10.1113/jphysiol.1992.sp019217. [DOI] [PMC free article] [PubMed] [Google Scholar]