

Abstract
Adenosine modulates nociceptive processing in the superficial dorsal horn of the spinal cord. In other tissues, membrane transporters influence profoundly the extracellular levels of adenosine. To investigate the putative role of nucleoside transporters in the regulation of excitatory synaptic transmission in the dorsal horn, we employed immunohistochemistry and whole-cell patch-clamp recording of substantia gelatinosa neurons in slices of rat spinal cord in vitro. The rat equilibrative nucleoside transporter (rENT1) was revealed by antibody staining to be abundant in neonatal and mature dorsal horn, especially within laminae I-III. This was confirmed by immunoblots of dorsal horn homogenate. Nitrobenzylthioinosine (NBMPR), a potent non-transportable inhibitor of rENT1, attenuated synaptically evoked EPSCs onto lamina II neurons in a concentration-dependent manner. Application of an adenosine A1 antagonist 1,3-dipropyl-8-cyclopentylxanthine produced a parallel rightward shift in the NBMPR concentration-effect curve. The effects of NBMPR were partially reversed by adenosine deaminase, which facilitates the metabolic degradation of adenosine. The modulation by NBMPR of evoked EPSCs was mimicked by exogenous adenosine or the selective A1 receptor agonist, 2-chloro-N6-cyclopentyl adenosine. NBMPR reduced the frequency but not the amplitude of spontaneous miniature EPSCs and increased the paired-pulse ratio of evoked currents, an effect that is consistent with presynaptic modulation. These data provide the first direct evidence that nucleoside transporters are able to critically modulate glutamatergic synaptic transmission.
Nucleosides such as adenosine have a plethora of actions in mammalian tissues, many of which are mediated by extracellular binding to cell-surface receptors. The extracellular levels of nucleosides are controlled, in part, through production and degradation by extracellular enzymes. However, they are also regulated by uptake or loss from cells via nucleoside transport processes (Griffith & Jarvis, 1996). These can be classified as either concentrative (sodium dependent) or equilibrative (sodium independent). The latter can be further divided into those that are sensitive to inhibition by the nucleoside analogue nitrobenzylthioinosine (NBMPR; equilibrative sensitive, es) and those that are not (equilibrative insensitive, ei). Transporters possessing such activities have recently been cloned from both rat (r) and human (h) and are designated r/hENT1 (es) and r/hENT2 (ei), respectively (Griffiths et al. 1997a,b; Yao et al. 1997). Functional studies have revealed both es- and ei-type transport activity in the mammalian brain (Lee & Jarvis, 1988), and localisation studies at the mRNA and protein levels have shown that ENT1 and ENT2 are widely expressed in brain (Anderson et al. 1999a,b; Jennings et al. 2001). Although neither transporter has been directly localised to the spinal cord, the substantia gelatinosa (SG) of the rat superficial dorsal horn is rich in NBMPR binding sites, suggesting the presence of rENT1 (Geiger & Nagy, 1985). Nothing is known about the potential regulatory role of nucleoside transporters in CNS areas enriched with adenosine receptors.
The SG receives extrinsic nociceptive input from small diameter (Aδ and C fibre) primary afferents, and is the first relay station in the nociceptive pathway. The predominant excitation of SG neurons occurs through the release of glutamate from small-diameter primary afferents. While the nucleoside adenosine plays a critical neuromodulatory role throughout the CNS, in the dorsal horn of the spinal cord its actions are in particular associated with antinociception in vivo (Dunwiddie, 1985; Sawynok, 1998). For instance, intrathecal adenosine A1 receptor agonists produce antinociception (Sawynok et al. 1986), as do inhibitors of adenosine breakdown (Keil & DeLander, 1992; Poon & Sawynok, 1998). Adenosine acts via A1 receptors to reduce inflammation-induced Fos expression (Honore et al. 1998) and A1 receptor agonists reduce C-fibre-evoked discharges and wind-up in extracellular recording from dorsal horn neurons (Reeve & Dickenson, 1995).
Adenosine A1 receptors presynaptically inhibit glutamatergic synaptic transmission in the periaqueductal grey (Bagley et al. 1999), hippocampus (Lupica et al. 1992) and the lateral horn of the thoracic spinal cord (Deuchars et al. 2001). Recent evidence suggests that similar mechanisms occur in the superficial dorsal horn (Lao et al. 2001; Patel et al. 2001). In this case, it is possible that nucleoside transporters might function to modulate excitatory transmission in the SG by controlling extracellular adenosine levels. In the current study we have used immunohistochemistry and whole-cell patch clamping from neurons in an in vitro spinal cord slice preparation to test the hypothesis that es-type nucleoside transporters influence excitatory transmission in this fashion.
METHODS
Immunohistochemistry
Antibodies against the central cytoplasmic loop regions (residues 227–290) of rENT1 (anti-rENT1227–290) and hENT1 (anti-hENT1227–290), and against a synthetic peptide corresponding to residues 236–252 of hENT1 (anti-hENT1236–252), were produced in rabbits and affinity-purified using previously described methods (Sundaram et al. 2001; Musa et al. 2002). Rabbit antibodies against a synthetic peptide corresponding to residues 309–323 of the rat adenosine A1 receptor (anti-A1R309–323; Deuchars et al. 2001; Smith et al. 2001) were kindly provided by Dr Michael Yates (School of Biomedical Sciences, University of Leeds, UK). Adult Wistar rats (180–260 g; n = 3) or neonatal rats (aged 14 days) were given a lethal dose of anaesthetic (intraperitoneal dose of 210–300 mg kg−1 Sagatal for adults and 2 mg kg−1 urethane for neonates) prior to transcardiac perfusion with fixative (4 % paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4). All experiments were performed under a UK Home Office License and were in accordance with the regulations of the UK Animals (Scientific Procedures) Act 1986. Spinal cords were dissected and placed in 4 % paraformaldehyde in 0.1 m PB for 24 h before being placed into 30 % sucrose (diluted in 0.1 m PB) for 48 h. The lumbar cord was sectioned at 30 μm on a cryotome (Shandon Scientific, Cheshire, UK) and placed in 0.01 m phosphate buffered saline (PBS). Sections were permeabilised using 0.3 % Triton X-100 for 1 h and then immersed in primary antibody (1 μg ml−1 for anti-rENT1227–290, anti-hENT1227–290 and anti-hENT1236–252; 2.5 μg ml−1 for anti-A1R309–323) in PBS with 0.2 % sodium azide for 48 h at room temperature. To verify rENT and hENT antibody specificity, control sections were incubated in the absence of primary antibody or with primary antibody that had been pre-absorbed with 3 μg ml−1 antigen. Following incubation with primary antibody, sections were washed three times for 10–20 min each in PBS and then treated with biotinylated goat anti-rabbit IgG (5 μg ml−1; Vector Laboratories, Peterborough, UK) in PBS for 2 h at room temperature. Sections were washed and placed in Vectastain Elite ABC reagent (1:100; Vector Laboratories) in PBS for 1 h at room temperature. Sections were then washed in PBS for 30 min and incubated in diaminobenzidine solution (0.5 μg ml−1 in PBS containing 0.025 % H2O2) for 3–5 min. Once mounted and coverslipped, sections were viewed at the light-microscope level and images were captured using an integrating analog CCD camera (JVC KYF 55B) attached to an Acquis image capture system (Synoptics, Cambridge, UK).
Immunoblotting
Adult Wistar rats (female 180–260 g; n = 3) were given a lethal intraperitoneal dose of Sagatal (210–300 mg kg−1) and perfused trancardially with 0.1 m PBS. Neonatal rats received a lethal dose of 2 mg kg−1 urethane before the 0.1 m PBS perfusion. The spinal cord was rapidly removed and placed into ice-cold normal artificial cerebrospinal fluid (ACSF). The latter contained (mm): 126 NaCl, 2.5 KCl, 1.4 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 25 NaHCO3, 11 glucose, and was equilibrated with 95 % O2-5 % CO2 to pH 7.4. Approximately 1 mm3 pieces of spinal dorsal horn were dissected, homogenised in gel sample buffer containing protease inhibitors (protease inhibitor cocktail for mammalian cell extracts, Sigma-Aldrich, Poole, UK) and 1 % SDS, and then centrifuged to remove insoluble material. For immunoblotting, samples were resolved on 10 % SDS-polyacrylamide gels (Laemmli, 1970). The electrophoresed proteins were transferred to nitrocellulose membranes and probed with affinity-purified anti-rENT1227–290 (2 μg ml−1). Blots were then incubated with horseradish-peroxidase-conjugated goat anti-rabbit antibody (Jackson Immunoresearch Laboratories, USA; 1:40 000) and developed using a chemiluminescence protocol (Supersignal West; Pierce, Rockford, IL, USA). As a control for antibody specificity, immunoblots included membrane samples (50 ng) prepared from Spodoptera frugiperda (Sf)9 cells infected with recombinant baculovirus encoding rENT1, and from uninfected Sf9 cells. A sample (5 ng) of a cellulose binding domain (CBD) fusion protein bearing the central cytoplasmic loop region (residues 227–290) of rENT1 (37 kDa) was also used as a positive control. As a further negative control, the primary antibody was pre-incubated with the CBD fusion protein at a weight ratio of 10:1 (CBD fusion protein: antibody) for 1 h before the immunoblot of the dorsal horn sample.
Patch-clamp recording
Neonatal rats aged 14–16 days were terminally anaesthetised with 2 g kg−1 urethane. Spinal cords were removed and placed into ice-cold ACSF in which sucrose (126 mm) was substituted for NaCl to minimise excitotoxicity during the slicing procedure. Once the spinal cords had been removed, the dura was dissected away and a section of the spinal cord from L4–L6 was embedded in 3 % agar. Slices (300 μm) were cut using a vibratome (Intracell, Royston, UK) and placed into normal ACSF that was maintained at 35 °C for 1 h. Slices were subsequently submerged in the recording chamber and constantly superfused with ACSF at a flow rate of 3 ml min−1. Neurons in lamina II of the dorsal horn were visualised using infrared differential-interference contrast (IR-DIC) imaging on an Olympus BX50WI microscope (Optivision, Osset, UK). Neurons were recorded using whole-cell voltage clamp using an Axopatch 1-D amplifier (Axon instruments, La Jolla, CA, USA). Electrodes of resistance 3–5 MΩ contained (mm): 140 potassium gluconate, 15 NaCl, 1 MgCl2, 11 EGTA, 10 Hepes, 2 ATP, 0.25 GTP; pH 7.4, 310 mosmol l−1. Biocytin (0.1 %) was included in the electrode for subsequent recovery and visualisation of the recorded neuron in order to confirm localisation within lamina II. Excitatory postsynaptic currents (EPSCs) were evoked with a concentric bipolar stimulating electrode placed at the dorsal root entry zone. Single stimuli of intensity 1.0–10 mA; 0.1–0.5 ms (frequency of 0.1 Hz), adequate for high-threshold nociceptive afferent excitation in the rat spinal slice, were used to evoke short, fixed-latency (< 2 ms) monosynaptic EPSCs (Yoshimura & Nishi, 1993). All EPSCs were recorded in the presence of 30 μm bicuculline methiodide and 10 μm strychnine to block inhibitory components of the evoked current. EPSCs were recorded using Strathclyde Electrophysiology Software (SES; Dr J. Dempster, Strathclyde University, UK). For analysis, six consecutive EPSCs were averaged and the peak amplitude of the EPSC was measured using SES. Drug effects were measured at apparent equilibrium and expressed as the percentage change from the averaged amplitude of six EPSCs evoked in the absence of drug. For all of the figures, the average of six consecutive traces is shown and stimulus artefacts have been removed. Concentration-effect relationships were constructed using non-linear regression. The EC50 was defined as the concentration that produced one-half of the maximal effect. For paired-pulse protocols, paired EPSCs were evoked with a latency of 50 ms. The paired-pulse ratio is expressed as the amplitude of the second EPSC/amplitude of the first EPSC (Kim & Alger, 2001). Spontaneous miniature EPSCs were recorded in the presence of 30 μm bicuculline methiodide, 10 μm strychnine and 1 μm tetrodotoxin (TTX) and low-pass filtered at 5 kHz. Data were sampled over 2 min epochs at a frequency of 10 kHz. Events were automatically detected and verified by visual analysis. A threshold of 1.5 × baseline noise was used for the detection of events. Events were ranked by amplitude and interevent interval (a measure of the frequency of events) and plotted using cumulative probability distributions for each individual neuron. The Kolmogorov-Smirnov (K-S) test was used to compare cumulative distributions in the absence or presence of NBMPR for each neuron. All data are expressed as the mean ±s.e.m.
NBMPR, a highly selective and potent non-transportable inhibitor of ENT1 that is inactive at any known adenosine receptor subtype (Geiger et al. 1985), nitrobenzylthioguanosine (NBTGR), a structurally distinct but less potent ENT1 inhibitor (Geiger et al. 1985), adenosine deaminase, the selective A1 receptor agonist 2-chloro-N6-cyclopentyl adenosine (CCPA), strychnine hydrochloride, bicuculline methiodide and TTX were all obtained from Sigma-Aldrich (Poole, UK). The highly selective A1 receptor antagonist 1,3-dipropyl-8-cyclopentylxanthine (DPCPX) was obtained from Tocris Cookson (Bristol, UK). All drugs were stored as 1 mm aliquots at −20 °C, apart from adenosine deaminase, which was stored as a 3 mg ml−1 solution at 4 °C. Drugs were superfused for 5–10 min to allow complete exchange of the recording chamber contents. In some experiments, drugs were applied for up to 20 min to ensure that a stable maximal drug effect was achieved. Recovery of EPSC amplitude after wash verified that drug-induced attenuation of EPSCs did not simply reflect time-dependent ‘run-down’. NBMPR, NBTGR, adenosine or CCPA were applied first, until apparent equilibrium was reached, and subsequently DPCPX or adenosine deaminase was co-applied.
Recovery of filled neurons
At the termination of the experiments, slices were placed into 4 % paraformaldehyde in 0.1 m PB overnight and then transferred to 30 % sucrose in PBS. Slices were resectioned at 25 μm on a cryotome (Shandon Scientific), washed in PBS and placed into 0.1 % Triton X-100 in 0.1 m PBS for 1 h. Sections were transferred to Vectastain Elite ABC reagent (1:100; Vector Laboratories) for 2.5 h before being reacted with diaminobenzidine and cobalt acetate to visualise the neuron. Sections were viewed at the light-microscope level and images were captured using an integrating analog CCD camera (JVC KYF 55B) attached to an Acquis image capture system (Synoptics, Cambridge, UK).
RESULTS
Localisation of rENT1 and the A1 receptor in the dorsal horn
Immunohistochemical analysis of adult and neonatal spinal cord sections using anti-rENT1227–290 revealed dense staining for rENT1 in the dorsal horn (Fig. 1A–C). In the adult, rENT1 staining was especially intense within laminae I and outer II (IIo), declining progressively through deep dorsal horn laminae (Fig. 1A). Dense staining was also observed at the most medial aspect of the superficial dorsal horn. At higher magnification, the staining in laminae I and II appeared punctate in nature with some evidence for staining on cell bodies (Fig. 1B). The specificity of the staining for rENT1 was demonstrated by its complete abolition by pre-incubation of the antibodies with antigen or by removal of the primary antibody (Fig. 1A2). Moreover, essentially identical staining patterns were revealed using two antibodies raised against the homologous human protein hENT1, anti-hENT1227–290 and anti-hENT1236–252 (data not shown). These antibodies are crossreactive with rENT1 because the corresponding regions of the two proteins are 69 % and 76 % identical in amino acid sequence, respectively. In neonatal dorsal horn, rENT staining was more diffuse across laminae I-III (Fig. 1C). Immunohistochemical analysis of spinal cord sections using anti-A1R309–323 showed a dense band of staining within lamina II of the dorsal horn (Fig. 1D). This distribution of the adenosine A1 receptor is identical to that reported for thoracic spinal cord by others using the same antibody (Deuchars et al. 2001).
Figure 1. Distribution of the rat equilibrative nucleoside transporter, rENT1, and the adenosine A1 receptor in the adult and neonatal dorsal horn.
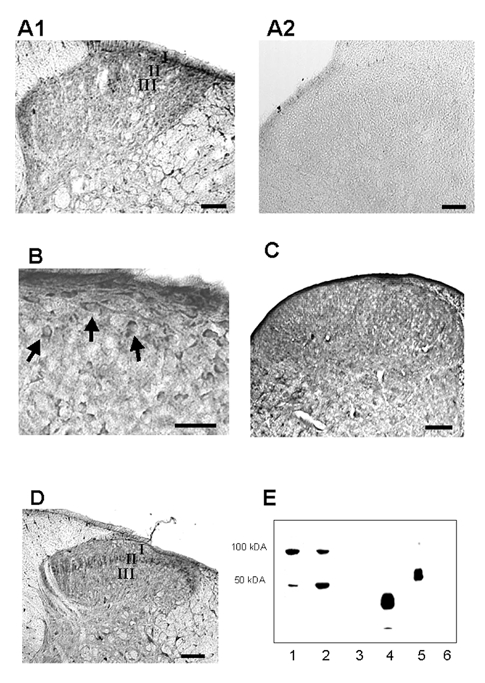
A1, low-magnification image of a dorsal horn section stained with anti-rENT1227–290. The most dense labelling is evident in lamina I and outer lamina II of the superficial dorsal horn. A2, no immunostaining is evident when the primary antibody is omitted. Scale bar = 100 μm. B, high-magnification image of the same section showing labelling of some cell bodies by the anti-rENT1 antibodies (arrows). Scale bar = 30 μm. C, diffuse anti-rENT1227–290 staining revealed in a dorsal horn section taken from a neonatal rat. Scale bar = 100 μm. D, low-magnification image of a dorsal horn section stained with anti-A1R309–323, showing the presence of the A1 receptor predominantly in lamina II. Scale bar = 100 μm. E, immunoblots of homogenate from neonatal (lane 1) or adult (lane 2) rat dorsal horn, homogenate of rat dorsal horn where the primary antibody had been preincubated with a CBD fusion protein bearing the rENT1 epitopes (lane 3), immunoblots of the CBD fusion protein (lane 4) and of membranes from Spodoptera frugiperda (Sf)9 cells infected with the baculovirus encoding rENT1 (lane 5) or from uninfected Sf9 cells (lane 6). The mobilities of protein standards with known molecular weights are indicated on the left.
Additional evidence for the specificity of the anti-rENT1227–290 antibody and for the presence of rENT1 was provided by immunoblotting of dorsal horn samples from neonatal and adult rats (Fig. 1E). These showed a strongly stained band of apparent size ∼50 kDa, consistent with the size of 49 984 Da predicted from the sequence of rENT1 (Yao et al. 1997). Additional bands at ∼100 kDa represent dimeric forms of the transporter; bands of size similar to the presumed monomeric and dimeric forms of the transporter in dorsal horn samples were seen in membrane samples from insect cells expressing recombinant rENT1. Both bands were completely absent from the dorsal horn samples when the primary antibody was preincubated with the CBD fusion protein (Fig. 1E). Dimers and higher-molecular-weight oligomers of the rENT1 homologue hENT1 have previously been detected in photoaffinity labelling studies, and may reflect SDS-induced aggregation of these very hydrophobic membrane proteins (Wu et al. 1983). No staining on immunoblots was seen for samples from uninfected Sf9 cells not expressing rENT1 (Fig. 1E), confirming the specificity of the antibodies for rENT1. Furthermore, a band of approximately 37 kDa was observed when the CBD fusion protein (37 kDa) was placed on the gel.
Characterisation of synaptic currents in lamina II neurons
Monosynaptic EPSCs were characterised by having a graded amplitude and constant, short latency in response to varying stimulus intensity. The EPSC amplitude was stable and did not run-down over the time course of the experiments. Application of the AMPA/kainate antagonist, 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulphonamide (NBQX; 10 μm) almost completely abolished the evoked EPSC at a holding potential of −60 mV (96.3 ± 3.6 % inhibition, P < 0.001, n = 5). This indicates that the EPSCs were due to release of glutamate acting predominantly at AMPA and/or kainate receptors. Subsequently, 21/38 biocytin-filled neurons were recovered histologically, and all had somata within lamina II.
Inhibition of evoked EPSCs by inhibition of nucleoside transport
Application of the selective inhibitor of rENT1, NBMPR, inhibited evoked EPSCs compared to control EPSCs recorded in drug-free ACSF over the same period (Fig. 2 and Fig. 3). This synaptic inhibition was maintained at a constant level over a period of 20 min with no evidence of desensitisation or time-dependent diminution of the EPSC amplitude (Fig. 2A and C, n = 3). NBMPR-induced inhibition slowly reversed following wash (10–35 min), due to the high affinity of NBMPR for rENT1 (Fig. 2A). The action of NBMPR was dose dependent (1–1000 nm, n = 4–9), with a maximal effect (95 % confidence limits) of 58.1 % (44.5–71.7 %) inhibition of control (Fig. 3). The effective concentration at 50 % of the maximal effect (EC50; 95 % confidence limits) was 9.0 nm (1.7–47.4 nm; Fig. 3C). In the presence of the adenosine A1 receptor antagonist, DPCPX (10 μm), the effect of NBMPR was partially reversed and the concentration-effect curve was shifted sevenfold to the right in a parallel manner, with an EC50 of 63.5 nm (16.1–250.0 nm; n = 4–6, Fig. 3A–C), consistent with an inhibitory effect that is mediated through actions at the A1 receptor. DPCPX alone (10 μm) had no effect on the evoked EPSC (1.7 ± 2.5 % inhibition; n = 9; P > 0.05), indicating that tonic levels of adenosine were insufficient to modulate synaptic transmission through the A1 receptor. NBMPR alone had no effect on baseline current or on input resistance in any of the neurons tested.
Figure 2. The rENT1 inhibitors nitrobenzylthioinosine (NBMPR) and nitrobenzylthioguanosine (NBTGR) attenuate evoked EPSCs.
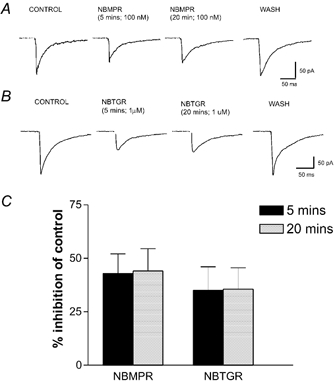
A, EPSCs evoked in control artificial cerebrospinal fluid (ACSF), in the presence of 100 nm NBMPR (5 min and 20 min) and after drug washout (10 min). B, EPSCs evoked in control ACSF, in the presence of 1μm NBTGR (5 min and 20 min) and after drug washout (15 min). C, quantified data for the percentage inhibition of evoked EPSC amplitude produced by NBMPR and NBTGR measured at two time points, 5 and 20 min. Note that there was no significant difference between the two.
Figure 3. The rENT1 inhibitor NBMPR elicits a dose-related inhibition of evoked EPSCs that is antagonised by DPCPX.
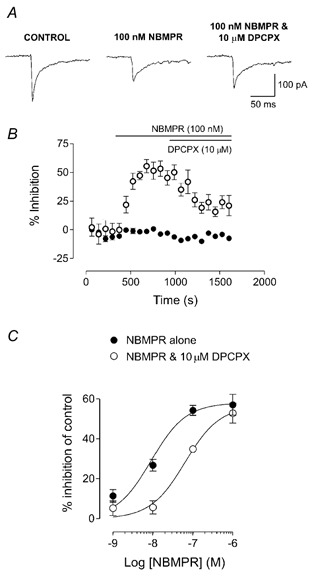
A, EPSCs evoked in the absence of any drug (control), in the presence of 100 nm NBMPR and in the presence of 100 nm NBMPR plus the A1 receptor antagonist 1,3-dipropyl-8-cyclopentylxanthine (DPCPX; 10 μm). B, time course of the inhibition of EPSCs produced by NBMPR (open circles) compared to control EPSCs recorded in drug-free ACSF (filled circles). Each point represents the mean ±s.e.m. of six consecutive EPSCs. Bars show the period of application of 100 nm NBMPR and 10 μm DPCPX. C, concentration-effect relationship for the inhibition of EPSCs by NBMPR (filled circles, n = 4–9). The curve is shifted to the right in the presence of 10 μm DPCPX (open circles, n = 4–6). Data in the presence of NBMPR and DPCPX are expressed as the percentage inhibition of the control EPSC.
To confirm that EPSC inhibition by NBMPR was via rENT1, an additional inhibitor of ENT1, NBTGR, was tested. NBTGR (1 μm; n = 8, 5 min application) attenuated the amplitude of the evoked EPSCs by 27.4 ± 4.3 % of control (Fig. 2B). As for NBMPR, the effect of NBTGR was stable over a prolonged application period (20 min, n = 3) and reversed after washing (Fig. 2B and C). This synaptic inhibition was reversed to a 14.5 ± 5.2 % inhibition of control by co-application of 10 μm DPCPX (P < 0.05; n = 8; Fig. 4), confirming a role for adenosine A1 receptors.
Figure 4. The inhibition of evoked EPSCs by the rENT1 inhibitor NBTGR is antagonised by DPCPX.
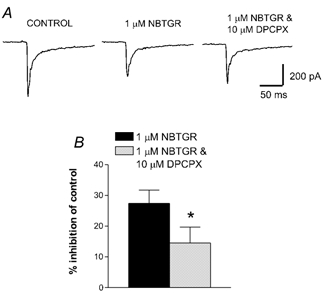
A, EPSCs are shown in the absence of any drug (control), in the presence of 10 μm NBTGR and in the presence of 10 μm NBTGR plus the A1 receptor antagonist DPCPX (10 μm). B, pooled data showing the inhibition of EPSCs by 1 μm NBTGR alone and 1 μm NBTGR in the presence of 10 μm DPCPX (n = 8; *P < 0.05).
The evoked EPSC was also partly inhibited following application of 1 μm adenosine (39.7 ± 2.7 % inhibition of control, 5 min application, n = 18, Fig. 5A). Prolonged application of adenosine (20 min, n = 3) produced an equivalent level of reversible inhibition consistent with an absence of desensitisation and with maximal drug effect. This adenosine-induced inhibition was partially reversed to 12.5 ± 12.7 % (P < 0.01) by co-application of DPCPX (10 μm, n = 9, Fig. 5B). Application of the selective A1 agonist CCPA (5 min, 1 μm) resulted in a more pronounced inhibition of the EPSC (Fig. 5A and B). This level of inhibition was constant over a 20 min application period (Fig. 5A, n = 3) and was partially reversed by co-application of DPCPX to a 34.4 ± 12.5 % inhibition of control (P < 0.005, n = 5; Fig. 5B). Neither adenosine nor CCPA had any effect on the baseline current or input resistance in any of the neurons tested.
Figure 5. Inhibition of the EPSC was mimicked by adenosine and an adenosine A1 receptor agonist, 2-chloro-N6-cyclopentyl adenosine (CCPA).

A, EPSCs evoked in the absence of drug, in the presence of 1 μm adenosine (5 and 20 min) and after wash (6 min). The histogram presents quantified data for the percentage inhibition of evoked EPSC amplitude produced by adenosine or CCPA measured at two time points, 5 and 20 min. Note that there was no significant difference between the two. B, EPSCs evoked in control ACSF, in the presence of 1 μm adenosine or in the presence of both 1 μm adenosine and 10 μm DPCPX. The histograms present pooled data for inhibition of EPSCs by adenosine (n = 18) and CCPA (1 μm, n = 5) in the absence and in the presence of 10 μm DPCPX (n = 9 for adenosine, n = 5 for CCPA). *P < 0.01, **P < 0.005, unpaired t test).
To ascertain whether the inhibitory effects of NBMPR on the EPSC were through accumulation of extracellular adenosine, adenosine deaminase was applied to facilitate rapid adenosine breakdown. Application of 30 μg ml−1 adenosine deaminase alone had no effect on the evoked EPSC (2.4 ± 2.3 % potentiation; n = 4; P > 0.05). Application of 100 nm NBMPR resulted in a 38.8 ± 6.3 % inhibition of the EPSC. Subsequent application of adenosine deaminase (30 μg ml−1) partially reversed the effect, resulting in a 25.6 ± 6.2 % inhibition of control (P < 0.001; n = 6; Fig. 6A and C). Adenosine deaminase also attenuated the effects of adenosine on the EPSC. Application of adenosine inhibited the EPSC by 35.8 ± 2.8 %. This was reversed to a 10.0 ± 4.1 % inhibition of control in the presence of 30 μg ml−1 adenosine deaminase (P < 0.001; n = 8; Fig. 6B and C).
Figure 6. The inhibitory effects of NBMPR are partially reversed by the breakdown of extracellular adenosine.
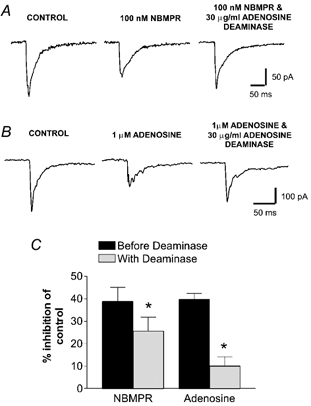
A, EPSCs evoked in the absence of drug (control), in the presence of 100 nm NBMPR or in the presence of both 100 nm NBMPR and 30 μg ml−1 adenosine deaminase. B, EPSCs evoked in the absence of drug (control), in the presence of 1 μm adenosine or in the presence of both 1 μm adenosine and 30 μg ml−1 adenosine deaminase. C, pooled data showing the percentage inhibition of the control EPSC produced by either 100 nm NBMPR (n = 6) or 1 μm adenosine (n = 18) alone, and following application of 30 μg ml−1 adenosine deaminase (n = 6 for NBMPR, n = 9 for adenosine; *P < 0.001, paired t test).
The effect of NBMPR on the EPSC involves action at a presynaptic locus
To confirm that the inhibition of the EPSC by NBMPR was through a presynaptic mechanism, a paired-pulse protocol and analysis of miniature EPSCs were performed. Spontaneous miniature EPSCs were recorded in the presence of 1μm TTX from five neurons (Fig. 7A). These miniature EPSCs had a mean inter-event interval of 1802 ± 36 ms (3.3. ± 0.1 Hz) and a mean amplitude of 40.0 ± 0.5 pA. Application of 1 μm NBMPR shifted the distribution curve for cumulative interevent intervals to the right such that the mean value increased to 2060 ± 37 ms (2.5 ± 0.1 Hz, P < 0.05, each neuron). The cumulative amplitudes remained unchanged (P > 0.05, each neuron), with a mean of 38.4 ± 0.6 pA. In response to paired stimuli (n = 8, Fig. 7B), the EPSCs exhibited paired-pulse depression with a mean ratio of 0.5 ± 0.1. Following application of NBMPR (1 μm), the amplitude of the first EPSC was inhibited by 41.9 ± 5.1 % of control. The paired-pulse ratio was significantly increased to 0.8 ± 0.1 (P < 0.05, 102 ± 45 % increase). This indicates a reduced probability of neurotransmitter release following NBMPR, a result consistent with a presynaptic mechanism of action.
Figure 7. The inhibitory effect of NBMPR on excitatory neurotransmission is through a presynaptic mechanism.
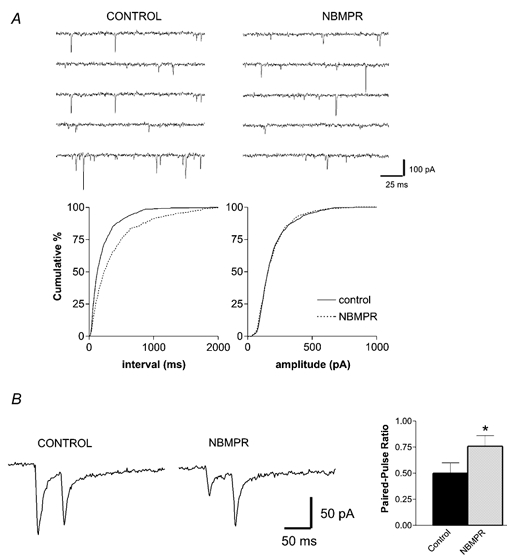
A, NBMPR reduced the frequency but not the amplitude of spontaneous miniature EPSCs. Consecutive traces from a typical recording, in the absence and presence of 1 μm NBMPR, are shown in the upper panels. The lower panels depict cumulative frequency plots of the interevent interval and the amplitude of spontaneous events recorded in the same neuron. A total of 595 events were recorded in the control and 311 in the presence of NBMPR. B, paired-pulse records (50 ms interval) of evoked EPSCs display paired-pulse depression in control ACSF and paired-pulse facilitation after 1 μm NBMPR. Pooled data reveals that the paired-pulse ratio is increased after NBMPR, an effect that is consistent with a presynaptic mechanism of action (n = 7; *P < 0.05, paired t test).
DISCUSSION
This study has provided the first direct evidence for the localisation of the equilibrative, NBMPR-sensitive nucleoside transporter, rENT1, in the immature and adult rat spinal cord. Immunohistochemistry of spinal cord sections, together with immunoblotting of tissue samples, revealed that this transporter is abundantly distributed within the dorsal horn. In the adult dorsal horn, rENT1 was localised especially to laminae I and IIo, with lower expression levels in deep dorsal horn laminae. In neonatal rats, more diffuse rENT1 staining was present across laminae I-III. These results are consistent with the findings of previous studies on the spinal distribution of adenosine transporter sites labelled by NBMPR binding, which were especially concentrated in the SG, with a large proportion of the sites located on the terminals of unmyelinated primary afferents (Geiger & Nagy, 1985). In rat whole brain, NBMPR binding sites and adenosine accumulation are evident from birth and both decline during development (Geiger, 1987). No firm conclusions can be drawn regarding the cellular origin of rENT1 from these data and ultrastructural studies will be required to further elucidate the precise subcellular localisation of ENT1. Immunohistochemistry of adult spinal cord sections confirmed previous reports that the adenosine A1 receptor localised to the superficial dorsal horn (Deuchars et al. 2001), with a dense band of staining for this receptor evident in the SG. Developmentally, adenosine receptors are expressed in rat spinal cord at postnatal day 1, and thereafter levels steadily increase until final adult expression is achieved (Geiger et al. 1984).
Given the expression of rENT1 and the A1 receptor within the superficial laminae, we hypothesised that the transporter, via regulation of extracellular adenosine levels, might play a novel role in modulating excitatory synaptic transmission in the rat superficial spinal dorsal horn. Experimentation provided direct evidence for this, in that selective inhibition of rENT1 led to a potent attenuation of EPSCs evoked within the SG. Several pieces of evidence indicated that the inhibitory effects of NBMPR on synaptic transmission stemmed from the activation of presynaptic A1 receptors, secondary to an increase in the extracellular adenosine concentration. The attenuation of EPSC amplitude by NBMPR was competitively inhibited by DPCPX, an A1 antagonist, suggesting that the inhibition was due to actions at the A1 receptor. It is unlikely that the effects of NBMPR are through direct activation of the A1 receptor, as NBMPR is inactive at adenosine receptor sites (Geiger et al. 1985). Also, a second, structurally distinct inhibitor of ENT1, NBTGR, mimicked the inhibitory effect of NBMPR on the EPSC. These compounds are firmly established to be selective inhibitors of ENT1 with very high affinity (Hyde et al. 2001). In addition, the effect of NBMPR was attenuated by the adenosine-degrading enzyme adenosine deaminase, suggesting that the action of NBMPR is principally via the accumulation of extracellular adenosine. NBMPR reduced the frequency but not the amplitude of spontaneous miniature EPSCs, and increased the paired-pulse ratio. Both of these results indicate that NBMPR, presumably through its inhibition of rENT1-mediated transport, reduced the probability of neurotransmitter release at a presynaptic locus. All of these effects are consistent with the previously reported and well-established presynaptic actions of adenosine (Deuchars et al. 2001; Lao et al. 2001; Patel et al. 2001). The possibility of an additional postsynaptic A1 receptor effect cannot be completely ruled out, although we found no evidence of direct postsynaptic actions for any of the drugs tested. Two possibilities may account for the lack of postsynaptic effects seen in our preparation. Firstly, washout of vital second messengers may occur by diffusion of cytoplasmic content through the patch pipette in whole-cell mode. This is unlikely, as similar methods were used in previously published studies with adenosine (Patel et al. 2001). Secondly, inhibition of rENT1 may lead to local build up of adenosine within the vicinity of the transporter. If rENT1 expression is compartmentalised to regions close to presynaptic terminals, then one may speculate that presynaptic A1 receptors may be preferentially activated following inhibition of adenosine uptake.
NBMPR has been shown to potentiate the effects of adenosine in the olfactory cortex (Sanderson & Scholfield, 1986) and accelerate anoxia-induced synaptic inhibition in the hippocampus (Cassar et al. 1998). However, the present data are the first to establish that selective inhibition of the nucleoside transporter rENT1 with NBMPR can profoundly modulate excitatory synaptic transmission. Exogenous application of adenosine or the A1 agonist CCPA was also sufficient to activate presynaptic A1 receptors, thereby resulting in an inhibition of EPSCs. Adenosine inhibits excitatory transmission by activating presynaptic A1 receptors in the dorsal horn (Lao et al. 2001; Patel et al. 2001). However, the ion channel involved in the inhibition of the EPSC is unclear. Inhibition of voltage-dependent calcium channels has been seen in dorsal root ganglion neurons (Haas & Selbach, 2000). Indeed, A1 receptors appear to inhibit N-type calcium channels directly via G-proteins independently from cAMP (see Cunha, 2001 for review). It is possible that a similar mechanism may lead to the inhibition of calcium entry into synaptic terminals within the dorsal horn and subsequent reduction in neurotransmitter release.
Application of neither DPCPX nor adenosine deaminase alone had any effect on the EPSC amplitude, suggesting that there was no tonic activation of A1 receptors in the experimental preparation. Adenosine is rapidly removed from the synapse by uptake and breakdown, and normal levels of adenosine may not be sufficient to activate the presynaptic A1 receptors in this region (Dunwiddie, 1985). Levels of adenosine are normally elevated during hypoxia or ischaemia, where it plays an important neuroprotective role (Fowler, 1989; Rudolphi et al. 1992). It is possible that accumulation of extracellular adenosine recruits novel presynaptic receptor populations. In the hippocampus (Scanziani et al. 1997) and spinal dorsal horn (Bird et al. 2001), diminished glutamate uptake by transporter inhibition recruits presynaptic glutamate metabotropic receptors that are normally inactive.
SG neurons are a projection target of glutamatergic nociceptive C and Aδ primary afferents (Yoshimura & Nishi, 1993), and this was confirmed in the present study by abolition of the evoked EPSCs by the AMPA/kainate receptor antagonist, NBQX. Although neurokinins such as substance P are released from nociceptive primary afferents terminating in lamina II, they do not have an excitatory effect on SG neurons (Bleazard et al. 1994). In this regard, it seems likely that inhibition of glutamate release would result in attenuation of the nociceptive drive onto SG neurons and a subsequent antinociceptive effect. Indeed, intrathecal administration of adenosine receptor agonists reduces nociception and hyperalgesia in rats (Sawynok et al. 1986). In addition, adenosine acts at A1 receptors to reduce the Fos expression induced by carageenan in experimental inflammation (Honore et al. 1998). Finally, transgenic mice that lack the A1 receptor show enhanced responses to thermal nociceptive stimuli (Johansson et al. 2001). As central adenosine receptors are clearly involved in the modulation of nociceptive processing, increasing endogenous levels of adenosine might be expected to have profound effects on nociception. Indeed, recent advances have been made with the use of inhibitors of adenosine metabolism as potential therapeutic agents in the treatment of pain. For example, adenosine kinase inhibitors produce antinociception (Keil & DeLander, 1992; Poon & Sawynok, 1998; Jarvis et al. 2000) as well as being antihyperalgesic in various inflammatory models (Poon & Sawynok, 1998; Kowaluk et al. 1999, 2000; Boyle et al. 2001). In theory, the targeting of transporter proteins that increase levels of endogenous adenosine extracellularly may have significant clinical utility. Pertinent to this is the fact that nucleoside transport inhibitors significantly enhance opioid-mediated antinociception (Keil & Delander, 1995).
The results presented here demonstrate that inhibition of adenosine uptake effectively reduces primary-afferent-mediated neurotransmission in SG neurons. We suggest that inhibition of rENT1 by NBMPR results in an accumulation of extracellular adenosine that subsequently acts at presynaptic A1 receptors to attenuate glutamate release. These data provide firm evidence that the presence of functional nucleoside transporters is vital for normal neurotransmission at these synapses and that synaptic transmission can be critically modulated by inhibition of nucleoside transporters.
Acknowledgments
This work was funded by the MRC, UK and by the Canadian Institutes of Health Research and the Alberta Cancer Board. JDY is an AHFMR Heritage Scientist and CEC is a Canada Research Chair in Oncology. Technical assistance provided by Mrs J. Daniel and Mrs J. C. Ingram. We thank Z. A. Berry and F. Abidi for anti-rENT1 antibodies.
references
- Anderson CM, Baldwin SA, Young JD, Cass CE, Parkinson FE. Distribution of mRNA encoding a nitrobenzylthioinosine-insensitive nucleoside transporter (ENT2) in rat brain. Brain Res Mol Brain Res. 1999a;70:293–297. doi: 10.1016/s0169-328x(99)00164-3. [DOI] [PubMed] [Google Scholar]
- Anderson CM, Xiong W, Geiger JD, Young JD, Cass CE, Baldwin SA, Parkinson FE. Distribution of equilibrative, nitrobenzylthioinosine-sensitive nucleoside transporters (ENT1) in brain. J Neurochem. 1999b;73:867–873. doi: 10.1046/j.1471-4159.1999.0730867.x. [DOI] [PubMed] [Google Scholar]
- Bagley EE, Vaughan CW, Christie MJ. Inhibition by adenosine receptor agonists of synaptic transmission in rat periaqueductal grey neurons. J Physiol. 1999;516:219–225. doi: 10.1111/j.1469-7793.1999.219aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird GC, Asghar AUR, Ackley MA, King AE. Modulation of primary afferent-mediated neurotransmission and Fos expression by glutamate uptake inhibition in rat spinal neurones in vitro. Neuropharmacology. 2001;41:582–591. doi: 10.1016/s0028-3908(01)00111-3. [DOI] [PubMed] [Google Scholar]
- Bleazard L, Hill RG, Morris R. The correlation between the distribution of the NK1 receptor and the actions of tachykinin agonists in the dorsal horn of the rat indicates that substance P does not have a functional role on substantia gelatinosa (lamina II) neurons. J Neurosci. 1994;14:7655–7664. doi: 10.1523/JNEUROSCI.14-12-07655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle DL, Kowaluk EA, Jarvis MF, Lee CH, Bhagwat SS, Williams M, Firestein GS. Anti-inflammatory effects of ABT-702, a novel non-nucleoside adenosine kinase inhibitor, in rat adjuvant arthritis. J Pharmacol Exp Ther. 2001;296:495–500. [PubMed] [Google Scholar]
- Cassar M, Jones MG, Szatkowski M. Reduced adenosine uptake accelerates ischaemic block of population spikes in hippocampal slices from streptozotocin-treated diabetic rats. Eur J Neurosci. 1998;10:239–245. doi: 10.1046/j.1460-9568.1998.00035.x. [DOI] [PubMed] [Google Scholar]
- Cuhna RA. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: different roles, different sources and different receptors. Neurochem Int. 2001;38:107–125. doi: 10.1016/s0197-0186(00)00034-6. [DOI] [PubMed] [Google Scholar]
- Deuchars SA, Brooke RE, Deuchars J. Adenosine A1 receptors reduce release from excitatory but not inhibitory synaptic inputs onto lateral horn neurons. J Neurosci. 2001;21:6308–6320. doi: 10.1523/JNEUROSCI.21-16-06308.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV. The physiological role of adenosine in the central nervous system. Int Rev Neurobiol. 1985;27:63–139. doi: 10.1016/s0074-7742(08)60556-5. [DOI] [PubMed] [Google Scholar]
- Fowler JC. Adenosine antagonists delay hypoxia-induced depression of neuronal activity in hippocampal brain slice. Brain Res. 1989;490:378–384. doi: 10.1016/0006-8993(89)90258-8. [DOI] [PubMed] [Google Scholar]
- Geiger JD. Adenosine uptake and [3H]nitrobenzylthioinosine binding in developing rat brain. Brain Res. 1987;436:265–272. doi: 10.1016/0006-8993(87)91670-2. [DOI] [PubMed] [Google Scholar]
- Geiger JD, Labella FS, Nagy JI. Ontogenesis of adenosine receptors in the central nervous system of the rat. Dev Brain Res. 1984;13:97–104. doi: 10.1016/0165-3806(84)90080-4. [DOI] [PubMed] [Google Scholar]
- Geiger JD, Labella FS, Nagy JI. Characterization of nitrobenzylthioinosine binding to nucleoside transport sites selective for adenosine in rat brain. J Neurosci. 1985;5:735–740. doi: 10.1523/JNEUROSCI.05-03-00735.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JD, Nagy JI. Localization of [3H]nitrobenzylthioinosine binding sites in rat spinal cord and primary afferent neurons. Brain Res. 1985;347:321–327. doi: 10.1016/0006-8993(85)90193-3. [DOI] [PubMed] [Google Scholar]
- Griffith DA, Jarvis SM. Nucleoside and nucleobase transport systems of mammalian cells. Biochem Biophys Acta. 1996;1286:153–181. doi: 10.1016/s0304-4157(96)00008-1. [DOI] [PubMed] [Google Scholar]
- Griffiths M, Beaumont N, Yao SY, Sundaram M, Boumah CE, Davies A, Kwong FY, Coe I, Cass CE, Young JD, Baldwin SA. Cloning of a human nucleoside transporter implicated in the cellular uptake of adenosine and chemotherapeutic drugs. Nat Med. 1997a;3:89–93. doi: 10.1038/nm0197-89. [DOI] [PubMed] [Google Scholar]
- Griffiths M, Yao SY, Abidi F, Phillips SE, Cass CE, Young JD, Baldwin SA. Molecular cloning and characterization of a nitrobenzylthioinosine-insensitive (ei) equilibrative nucleoside transporter from human placenta. Biochem J. 1997b;328:739–743. doi: 10.1042/bj3280739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas HL, Selbach O. Functions of neuronal adenosine receptors. Naunyn-Schmiedebergs Arch Pharmacol. 2000;362:375–381. doi: 10.1007/s002100000314. [DOI] [PubMed] [Google Scholar]
- Honore P, Buritova J, Chapman V, Besson JM. UP 202–56, an adenosine analogue, selectively acts via A1 receptors to significantly decrease noxiously-evoked spinal c-Fos protein expression. Pain. 1998;75:281–293. doi: 10.1016/s0304-3959(98)00006-2. [DOI] [PubMed] [Google Scholar]
- Hyde RJ, Cass CE, Young JD, Baldwin SA. The ENT family of eukaryote nucleoside and nucleobase transporters: recent advances in the investigation of structure/function relationships and the identification of novel isoforms. Mol Membr Biol. 2001;18:53–63. [PubMed] [Google Scholar]
- Jarvis MF, Yu H, Kohlhaas K, Alexander K, Lee CH, Jiang M, Bhagwat SS, Williams M, Kowaluk EA. ABT-702 (4-amino-5-(3-bromophenyl)-7-(6-morpholinopyridin-3-yl)pyrido[2,3-d]pyrimidine), a novel orally effective adenosine kinase inhibitor with analgesic and anti-inflammatory properties: I. In vitro characterization and acute antinociceptive effects in the mouse. J Pharmacol Exp Ther. 2000;295:1156–1164. [PubMed] [Google Scholar]
- Jennings LL, Hao C, Cabrita MA, Vickers MF, Baldwin SA, Young JD, Cass CE. Distinct regional distribution of human equilibrative nucleoside transporter proteins 1 and 2 (hENT1 and hENT2) in the central nervous system. Neuropharmacology. 2001;40:722–731. doi: 10.1016/s0028-3908(00)00207-0. [DOI] [PubMed] [Google Scholar]
- Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Gimenez-Llort L, Escorihuela RM, Fernandez-Teruel A, Wiesenfeld-Hallin Z, Xu XJ, Hardemark A, Betsholtz C, Herlenius E, Fredholm BB. Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc Natl Acad Sci U S A. 2001;98:9407–9412. doi: 10.1073/pnas.161292398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil GJ, Delander GE. Spinally-mediated antinociception is induced in mice by an adenosine kinase-, but not by an adenosine deaminase-, inhibitor. Life Sci. 1992;51:171–176. doi: 10.1016/0024-3205(92)90566-8. [DOI] [PubMed] [Google Scholar]
- Keil GJ, Delander GE. Time-dependent antinociceptive interactions between opioids and nucleoside transport inhibitors. J Pharmacol Exp Ther. 1995;274:1387–1392. [PubMed] [Google Scholar]
- Kim J, Alger BE. Random response fluctuations lead to spurious paired-pulse facilitation. J Neurosci. 2001;21:9608–9618. doi: 10.1523/JNEUROSCI.21-24-09608.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaluk EA, Kohlhaas KL, Bannon A, Gunther K, Lynch JJ, Jarvis MF. Characterization of the effects of adenosine kinase inhibitors on acute thermal nociception in mice. Pharmacol Biochem Behav. 1999;63:83–91. doi: 10.1016/s0091-3057(98)00236-6. [DOI] [PubMed] [Google Scholar]
- Kowaluk EA, Mikusa J, Wismer CT, Zhu CZ, Schweitzer E, Lynch JJ, Lee CH, Jiang M, Bhagwat SS, Gomtsyan A, McKie J, Cox BF, Polakowski J, Reinhart G, Williams M, Jarvis MF. ABT-702 (4-amino-5-(3-bromophenyl)-7-(6-morpholino-pyridin-3-yl)pyrido[2, 3-d]pyrimidine), a novel orally effective adenosine kinase inhibitor with analgesic and anti-inflammatory properties. II. In vivo characterization in the rat. J Pharmacol Exp Ther. 2000;295:1165–1174. [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lao LJ, Kumamoto E, Luo C, Furue H, Yoshimura M. Adenosine inhibits excitatory transmission to substantia gelatinosa neurons of the adult rat spinal cord through the activation of presynaptic A(1) adenosine receptor. Pain. 2001;94:315–324. doi: 10.1016/S0304-3959(01)00367-0. [DOI] [PubMed] [Google Scholar]
- Lee CW, Jarvis SM. Nucleoside transport in rat cerebral-cortical synaptosomes. Evidence for two types of nucleoside transporters. Biochem J. 1988;249:557–564. doi: 10.1042/bj2490557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupica CR, Proctor WR, Dunwiddie TV. Presynaptic inhibition of excitatory synaptic transmission by adenosine in rat hippocampus: analysis of unitary EPSP variance measured by whole-cell recording. J Neurosci. 1992;12:3753–3764. doi: 10.1523/JNEUROSCI.12-10-03753.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musa H, Dobrzynski H, Berry Z, Abidi F, Cass JD, Young SA, Baldwin SA, Boyett MR. Immunocytochemical demonstration of the equilibrative nucleoside transporter rENT1 in rat sinoatrial node. J Histochem Cytochem. 2002;50:305–309. doi: 10.1177/002215540205000302. [DOI] [PubMed] [Google Scholar]
- Patel MK, Pinnock RD, Lee K. Adenosine exerts multiple effects in dorsal horn neurones of the adult rat spinal cord. Brain Res. 2001;920:19–26. doi: 10.1016/s0006-8993(01)02844-x. [DOI] [PubMed] [Google Scholar]
- Poon A, Sawynok J. Antinociception by adenosine analogs and inhibitors of adenosine metabolism in an inflammatory thermal hyperalgesia model in the rat. Pain. 1998;74:235–245. doi: 10.1016/s0304-3959(97)00186-3. [DOI] [PubMed] [Google Scholar]
- Reeve AJ, Dickenson AH. The roles of spinal adenosine receptors in the control of acute and more persistent nociceptive responses of dorsal horn neurones in the anaesthetized rat. Br J Pharmacol. 1995;116:2221–2228. doi: 10.1111/j.1476-5381.1995.tb15057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolphi KA, Schubert P, Parkinson FE, Fredholm BB. Neuroprotective role of adenosine in cerebral ischaemia. Trends Pharmacol Sci. 1992;13:439–445. doi: 10.1016/0165-6147(92)90141-r. [DOI] [PubMed] [Google Scholar]
- Sanderson G, Scholfield CN. Effects of adenosine uptake blockers and adenosine on evoked potentials of guinea-pig olfactory cortex. Pflügers Arch. 1986;406:25–30. doi: 10.1007/BF00582948. [DOI] [PubMed] [Google Scholar]
- Sawynok J. Adenosine receptor activation and nociception. Eur J Pharmacol. 1998;347:1–11. doi: 10.1016/s0014-2999(97)01605-1. [DOI] [PubMed] [Google Scholar]
- Sawynok J, Sweeney MI, White TD. Classification of adenosine receptors mediating antinociception in the rat spinal cord. Br J Pharmacol. 1986;88:923–930. doi: 10.1111/j.1476-5381.1986.tb16267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- Smith JA, Sivaprasadarao A, Munsey TS, Bowmer CJ, Yates MS. Immunolocalisation of adenosine A(1) receptors in the rat kidney. Biochem Pharmacol. 2001;61:237–244. doi: 10.1016/s0006-2952(00)00532-3. [DOI] [PubMed] [Google Scholar]
- Sundaram M, Yao SY, Ingram JC, Berry ZA, Abidi F, Cass CE, Baldwin SA, Young JD. Topology of a human equilibrative, nitrobenzylthioinosine (NBMPR)-sensitive nucleoside transporter (hENT1) implicated in the cellular uptake of adenosine and anti-cancer drugs. J Biol Chem. 2001;276:45270–45275. doi: 10.1074/jbc.M107169200. [DOI] [PubMed] [Google Scholar]
- Wu J-SR, Kwong FYP, Jarvis SM, Young JD. Identification of the erythrocyte nucleoside transporter as a band 4. 5 polypeptide. J Biol Chem. 1983;258:13745–13751. [PubMed] [Google Scholar]
- Yao SY, Ng AM, Muzyka WR, Griffiths M, Cass CE, Baldwin SA, Young JD. Molecular cloning and functional characterization of nitrobenzylthioinosine (NBMPR)-sensitive (es) and NBMPR-insensitive (ei) equilibrative nucleoside transporter proteins (rENT1 and rENT2) from rat tissues. J Biol Chem. 1997;272:28423–28430. doi: 10.1074/jbc.272.45.28423. [DOI] [PubMed] [Google Scholar]
- Yoshimura M, Nishi S. Blind patch-clamp recordings from substantia gelatinosa neurons in adult rat spinal cord slices: pharmacological properties of synaptic currents. Neurosci. 1993;53:519–526. doi: 10.1016/0306-4522(93)90216-3. [DOI] [PubMed] [Google Scholar]