

Summary
Cardiac fibroma is more commonly encountered in patients with Gorlin syndrome than the general population. Mutations of the tumor suppressor gene PTCH1 localized to 9q22.3 are the underlying cause of Gorlin syndrome. In this study, homozygous or heterozygous loss of the PTCH1 locus was identified in three nonsyndromic cardiac fibromas. These data support a somatic role of the PTCH1 tumor suppressor gene in sporadic cardiac fibroma.
Background
Cardiac fibroma is a rare benign tumor that is poorly characterized genetically. Cardiac fibroma is more commonly encountered in patients with Gorlin syndrome (3%) than the general population. Mutations of the tumor suppressor gene PTCH1 are the underlying cause of Gorlin syndrome.
Methods
Conventional cytogenetic analysis was performed on a peripheral blood and a cardiac fibroma sample from a 2-week-old male. In addition, FISH studies were performed to assess the copy number of the PTCH1 gene locus (9q22.3) on metaphase and interphase cells from these same specimens using YAC probe 891G1 and on representative paraffin-embedded tissue sections of two additional cardiac fibromas (one arising in a 2-month-old female and the other in a 13-week-old male). None of the patients had Gorlin syndrome.
Results
Karyotypically, the following abnormal chromosomal complement was detected in the 2-week-old male’s cardiac fibroma: 46,XY,del(9)(q22q34)[15]. FISH studies revealed homozygous loss of the PTCH1 locus in the cytogenetically analyzed cardiac fibroma and in the cardiac fibroma arising in the 13-week-old male. Heterozygous loss of this locus was identified in the remaining cardiac fibroma from the 2-month-old female. A mutational mechanism other than deletion may be responsible for PTCH1 inactivation on the other locus in this latter patient. Conventional cytogenetic and FISH studies of the peripheral blood sample from the 2-week-old male were normal.
Conclusion
These data support a tumor suppressor gene role for PTCH1 in nonsyndromic or sporadic cardiac fibromas.
Keywords: Cardiac fibroma, Gorlin syndrome, PTCH1 gene
Introduction
Primary cardiac tumors are rare. In the pediatric population, cardiac fibromas are the second most common primary cardiac tumor following rhabdomyoma. Cardiac fibromas are typically solitary, intramural tumors with a predilection for the left ventricular free wall or interventricular septum [1]. Depending on location and size, cardiac fibromas may be asymptomatic or may result in arrhythmias or cardiac failure due to valvular obstruction and chamber abolition [2]. Benign cardiac tumors in childhood have an excellent prognosis when completely excised and appear to have a good short-term prognosis even when excision is incomplete [3,4]. Cardiac fibroma most frequently arises sporadically, but it may also occur as a feature of the autosomal dominant disorder Gorlin syndrome.
The clinical manifestations of Gorlin syndrome or nevoid basal cell carcinoma (NBCC) syndrome were first well characterized in 1960 [5]. This phenotypically diverse disorder is characterized by distinctive congenital malformations and a variety of benign and malignant neoplasms, including cardiac fibroma. The gene for Gorlin syndrome, PTCH1, has been localized to 9q22.3 and is characterized as a tumor suppressor gene encoding for a transmembrane protein that functions as a receptor for sonic hedgehog [6,7]. Few nonsyndromic cardiac fibromas have been characterized genetically [8,9] and to the best of our knowledge there are no genetic studies of cardiac fibromas associated with Gorlin syndrome. The cytogenetic and/or molecular cytogenetic findings of three sporadic cardiac fibromas suggesting a pathogenetic role for the PTCH1 gene in these neoplasms are presented in the current study.
Methods
Tumor Samples
All tumors were diagnosed according to the World Health Organization histological criteria for cardiac fibroma [10]. The clinicopathologic and genetic findings of the three cases included in this study as well as two cases previously subjected to cytogenetic analysis [8,9] are summarized in Table 1. None of the tumors arose in patients with Gorlin syndrome. Grossly, each cardiac fibroma was firm, white and seemingly well-circumscribed. Two lesions arose in the right ventricle and one in the right atrium; all were approximately 3-4 cm in size. Histologically, the cardiac fibromas were composed of interweaving bundles of spindle cells extending into surrounding myocardium. Most of the tumor cells showed slender, bland-appearing nuclei with faintly eosinophilic cytoplasms, but occasional cells exhibited plumper nuclei with vesicular chromatin and occasional nucleoli. The neoplastic cells were immunoreactive for vimentin but negative for CD34, myogenin and ALK1. Necrosis was absent and mitoses were rare, averaging less than 2 per 10 hpf. The stroma was variably myxoid and collagenous and scattered lymphocytes and plasma cells were present in addition to rare foci of extramedullary hematopoiesis.
Table 1.
Clinicopathologic, Cytogenetic, and FISH Findings
| Case # | Age/Sex | Location (Size) | Karyotype | FISH | Reference |
|---|---|---|---|---|---|
| 1 | 2 wk/M | R. Ventricle (1.0 × 0.5 × 0.3 cm) | 46,XY,del(9)(q22.2q33.1)[15]/46,XY[5] | Homozygous Loss | Current |
| 2 | 2 mth/F | R. atrium (3.2 × 3.0 × 2.3 cm) | NP | Heterozygous Loss | Current |
| 3 | 13 wk/M | R. Ventricle (N.A.) | NP | Homozygous Loss | Current |
| 4 | 11 mth/M | Subepicardial (5.0 × 3.0 × 2.5 cm) | 46,XY,t(1;9)(q32;q22),inv(9)(p11q12)c[15] | NP | Ferguson et al. (1996) |
| 5 | 13 mth/F | Ventricular Septum (N.A.) | 46,XX,t(1;9;5)(q24;q22;q22) | NP | Viswanathan et al. (2003) |
N.A = Not available
N.P. = not performed
Conventional Cytogenetics
A fresh, representative tissue sample of Case 1 was received within 24 hours from surgery for conventional cytogenetic analysis. A peripheral blood sample from this patient was also obtained. Culturing, harvesting, and slide preparation were performed as previously described [11]. Metaphase cells were banded with Giemsa trypsin and the karyotypes were expressed according to the International System for Human Cytogenetic Nomenclature (ISCN 2005) [12].
Molecular Cytogenetics
Bicolor FISH analysis with YAC probe 891G1 localized to the 9q22.3 PTCH1 gene locus [13] in green (Research Genetics, Huntsville, AL) and argininosuccinate synthetase (ASS) gene locus probe in aqua (9q34.1; Vysis, Des Plaines, IL) was performed on clonally abnormal metaphase cells and cytologic touch preparations of Case 1 as previously described [14]. [YAC probe 891G1 was directly labeled by nick translation with Spectrum Green-dUTP utilizing the manufacturer’s protocol (Vysis).] Similarly, bicolor FISH analysis with YAC probe 891G1 in green and a chromosome 9 centromeric probe in red (CEP 9; Vysis) was also performed on cytologic touch preparations of Case 1.
Tricolor FISH assays were conducted on formalin fixed, paraffin-embedded tissue sections of Cases 2 and 3 using the following DNA probe set: CEP9 (green), 891G1 (PTCH1 locus, red), and ASS (aqua). After pretreatment of the slides, the cells and probes were codenatured at 75°C for 1 min and incubated at 37°C overnight using the HYBrite denaturation/hybridization system (Vysis). Posthybridization washing was performed in 0.4X SSC/0.3% NP-40 at 72°C for 2 min, followed by 2X SSC/0.1% NP-40 at room temperature for 1 min. The slides were air-dried in the dark and counterstained with 4’, 6-diamidino-2-phenylindole (DAPI II; Vysis).
Hybridization signals were assessed in five metaphase cells or 100 interphase nuclei with strong and well delineated signals. An interphase cell specimen was interpreted as abnormal if the copy number for any of the chromosome 9 probes [CEP 9, 891G1 (PTCH1 gene locus) and ASS (9q34.1)] was less than two signals per probe in 20% of the cells evaluated (more than two standard deviations above the average false-positive rate). As a negative control, normal peripheral blood lymphocytes were simultaneously hybridized with these probes.
Results
Traditional karyotypic analysis of the cardiac fibroma from Case 1 revealed the following abnormal clone exhibiting an interstitial deletion of the long arm of chromosome 9 in all fifteen metaphase cells examined: 46,XY,del(9)(q22q34), Figure 1. Cytogenetic analysis of a peripheral blood sample from this patient was normal.
Figure 1.
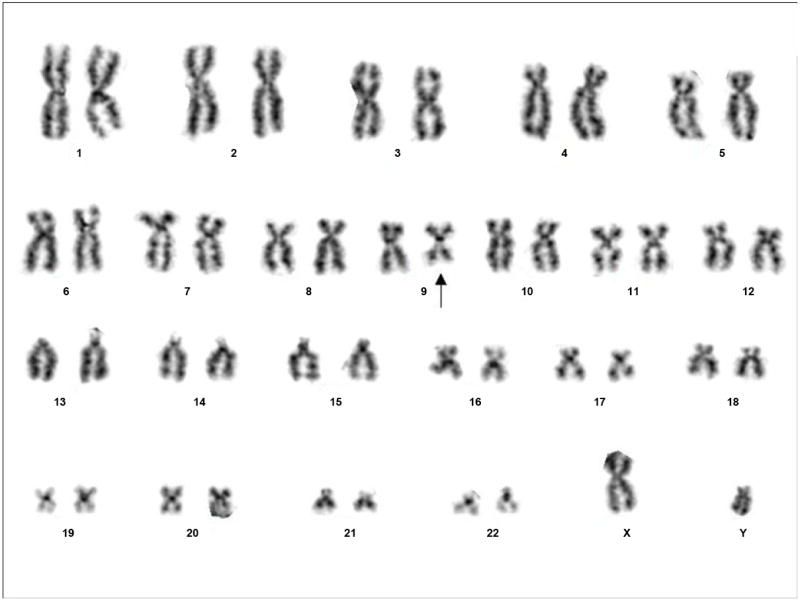
Representative karyotype of Case 1 showing the following abnormal complement: 46,XY,del(9)(q22q34).
FISH studies revealed homozygous loss of the PTCH1 locus in the cardiac fibromas of Cases 1 and 3 and heterozygous loss in the remaining cardiac fibroma, Case 2 (Figure 2). Specifically, loss of both PTCH1 locus specific probe signals were observed in 37% and 40% of interphase cells examined from Cases 1 and 3 respectively and loss of one PTCH1 locus specific probe in 33% of cells examined from Case 2. (Note, the predominant abnormal FISH pattern in Case 2 was represented by loss of one set of chromosome 9 probes [CEP 9, 891G1 (PTCH1 gene locus) and ASS (9q34.1)] consistent with monosomy 9 or possibly a structural alteration leading to loss of the long arm of chromosome 9 including the centromeric region.) A mutational mechanism other than deletion such as a submicroscopic mutation resulting in a truncated or inactive protein may be responsible for PTCH1 inactivation on the other locus in this latter patient. FISH studies of the peripheral blood sample from Case 1 were normal.
Figure 2.

Inverted DAPI metaphase cell image of Case 1 showing the cytogenetically deleted chromosome 9 (upper left). Bicolor FISH analysis with a PTCH1 gene locus (9q22.3) probe in red and ASS gene locus (9q34.1) in aqua shows loss of both PTCH1 gene loci including signal loss on the cytogenetially “normal-appearing” chromosome 9 homologue (indicated by “9” and arrow), (upper right). Bicolor interphase FISH analyses of cytologic touch preparations of Case 1 with the PTCH1 gene locus specific probe in green coupled with a chromosome 9 specific centromeric probe in orange and the PTCH1 gene locus specific probe in green coupled with the ASS gene locus specific probe in aqua confirms loss of both PTCH1 loci in abnormal tumor cells (middle left and right respectively). Case 2, tricolor FISH analysis performed on paraffin-embedded tissue sections using the following DNA probe set: CEP9 (green), PTCH1 locus (red), and ASS (aqua) shows loss of one set of chromosome 9 signals in a significant portion of cells (monosomy 9), (lower left). Using the same tricolor FISH approach described for Case 2, analysis of Case 3 revealed homozygous loss of the PTCH1 gene locus (red signal) in a significant portion of cells (lower right).
Discussion
Gorlin syndrome, also known as nevoid basal-cell carcinoma syndrome, is an autosomal dominant disorder characterized by an increased predisposition to basal cell carcinoma (BCC) among other neoplasms such as medulloblastoma, meningioma, fetal rhabdomyoma, and cardiac and ovarian fibromas [5]. The median age of onset of BCC in Gorlin syndrome is 25 years of age (arising in children as young as 2 years of age) in contrast to age 61 in the general population [15]. Congenital malformations in affected individuals may include broad facies, odontogenic keratocysts, calcified falx cerebri, palmar or plantar pits, and skeletal abnormalities.
Cardiac fibroma (CF) is the second most common primary cardiac tumor, following rhabdomyoma, in the general pediatric population. Cardiac fibromas arise predominantly in the interventricular septum or left ventricular free wall and appear to be well circumscribed. This gross demarcation is deceptive however, as microscopically tumor cells interdigitate among normal myocardium. Histopathologically, cardiac fibromas are composed of spindled cells with variable amounts of collagenized stroma and occasional foci of calcification. Vascularity is frequent and mitoses are few. Cardiac fibroma occurs in only 3-5% of patients with Gorlin syndrome but that is compared to an incidence of less than .02 % in a study of 27,640 children evaluated for cardiac disease by echocardiography [16]. Cardiac fibromas rarely cause symptoms in Gorlin syndrome patients, although they can reach significant size [17]. Occasionally, tumor growth may impede blood flow or displace or directly involve mitral and aortic valves and result in hemodynamically significant valvular stenosis or regurgitation or may be associated with arrhythmias [18].
The human homologue of the Drosphila patched gene, PTCH1, is a putative tumor suppressor gene that is mutated in both hereditary (Gorlin syndrome) and sporadic basal cell carcinomas [19]. Notably, this gene has been mapped to 9q22.3 [6]. Interestingly, in the current study, conventional cytogenetic analysis of a sporadic cardiac fibroma revealed a clonal interstitial deletion of the long arm of one chromosome 9 homologue to include the PTCH1 gene locus. Subsequent FISH studies of this case and two other nonsyndromic cardiac fibromas with a PTCH1 locus-specific probe revealed homozygous or heterozygous loss in all three neoplasms. Karyotypic studies of cardiac fibromas are few (only two cases reported) [8,9] and there are no molecular cytogenetic descriptions. Notably however, structural rearrangements of 9q22 were identified in both previous reports. Specifically, Ferguson, et al [8] detected a t(1;9)(q32;q22) and Viswanathan et al [9] a t(1;9;5)(q24;q22;q22). One might speculate that these karyotypic rearrangements of the 9q22 locus have resulted in loss or structural alteration of the PTCH1 gene.
In summary, the role of germline PTCH1 tumor suppressor gene mutations in Gorlin syndrome, to include the presence of cardiac fibromas, is well recognized. The data of the current study, ie. homozygous loss of the PTCH1 gene locus in two nonsyndromic cardiac fibromas and heterozygous loss in one, support a somatic role of the PTCH1 tumor suppressor gene in sporadic cardiac fibromas as well. Additional studies of this rare tumor type (sporadic and syndromic cardiac fibromas) to include other technical approaches may expose an even higher frequency of PTCH1 loss or mutation.
Acknowledgments
The authors would like to thank Patti Cattano for her expert technical assistance. This work was supported in part by NIH/NCI P30 CA 3672 and State of Nebraska LB595.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Burke Allen P, Rosado-de-Christenson Melissa, Templeton Philip A, Virmani Renu. Cardiac fibroma: clinicopathologic correlates and surgical treatment. J Thorac Cardiovasc Surg. 1994;108:862–870. [PubMed] [Google Scholar]
- 2.Becker Anton E. Primary heart tumors in the pediatric age group: a review of salient pathologic features relevant for clinicians. Pediatr Cardiol. 2000;21:317–323. doi: 10.1007/s002460010071. [DOI] [PubMed] [Google Scholar]
- 3.Reece Ian J, Cooley Denton A, Howard Frazier O, Hallman Grady L, Powers Patrick L, Montero Carlos G. Cardiac tumors: Clinical spectrum and prognosis of lesions other than classical benign myxoma in 20 patients. J Thorac Cardiovasc Surg. 1984;88(3):439–46. [PubMed] [Google Scholar]
- 4.Davies Ben, Oppido Guido, Brizard Christian P. Surgical management of symptomatic cardiac fibromas in children. J Thorac Cardiovasc Surg. 2007;133:254–55. doi: 10.1016/j.jtcvs.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Gorlin Robert J, Goltz Robert W. Multiple nevoid basal cell epithelioma, jaw cysts and bifid rib syndrome. N Engl J Med. 1960;262:908–912. doi: 10.1056/NEJM196005052621803. [DOI] [PubMed] [Google Scholar]
- 6.Farndon Peter A, Del Mastro Richard G, Gareth D, Evans R, Kilpatrick Michael W. Location of gene for Gorlin syndrome. Lancet. 1992;33:581–582. doi: 10.1016/0140-6736(92)90868-4. [DOI] [PubMed] [Google Scholar]
- 7.Chen Yu, Struhl Gary. Dual roles for Patched in sequestering and transducing Hedgehog. Cell. 1996;87:553–563. doi: 10.1016/s0092-8674(00)81374-4. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson Heather L, Hawkins Edith P, Cooley Linda D. Infant cardiac fibroma with clonal t(1;9)(q32;q22) and review of benign fibrous tissue cytogenetics. Cancer Genet Cytogenet. 1996;87(1):34–37. doi: 10.1016/0165-4608(95)00264-2. [DOI] [PubMed] [Google Scholar]
- 9.Viswanathan Sargeetha, Gibbs John L, Roberts Paul. Clonal translocation in a cardiac fibroma presenting with incessant ventricular tachycardia in childhood. Cardiol Young. 2003;13(1):101–102. doi: 10.1017/s1047951103000167. [DOI] [PubMed] [Google Scholar]
- 10.Travis William D, Brambilla Elisabeth, Konrad Muller-Hermelink H, Harris Curtis C., editors. World Health Organization Classification of Tumors Pathology and Genetics of Tumors of the Lung, Pleura, Thymus and Heart. Lyon: IARC Press; 2004. pp. 268–270. [Google Scholar]
- 11.Althof Pamela A, Ohmori Kazou, Zhou Ming, et al. Cytogenetic and molecular cytogenetic findings in 43 aneurysmal bone cysts: aberrations of 17p mapped to 17p13.2 by fluorescence in situ hybridization. Mod Pathol. 2004;17:518–25. doi: 10.1038/modpathol.3800090. [DOI] [PubMed] [Google Scholar]
- 12.Shaffer Lisa G, Tommerup Niels., editors. ISCN (2005) An International System for Human Cytogenetic Nomenclature. Karger; 2005. [Google Scholar]
- 13.Blair Ian P, Hulme Dennis J, Dawkins Jennifer L, Nicholson Garth A. A YAC-based transcript map of human chromosome 9q22.1-q22.3 encompassing the loci for hereditary sensory neuropathy type I and multiple self-healing squamous epithelioma. Genomics. 1998;51:277–281. doi: 10.1006/geno.1998.5373. [DOI] [PubMed] [Google Scholar]
- 14.Bridge Julia A, Weibolt Vines, Liu Jian, et al. Novel Genomic Imbalances in Embryonal Rhabdomyosarcoma Revealed by Comparative Genomic Hybridization and Fluorescence in situ Hybridization: An Intergroup Rhabdomyosarcoma Study. Genes Chromosom Cancer. 2000;27:337–344. doi: 10.1002/(sici)1098-2264(200004)27:4<337::aid-gcc1>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 15.Kimonis Virginia E, Goldstein Alisa M, Pastakia Behram, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299–308. [PubMed] [Google Scholar]
- 16.Beghetti Maurice, Gow Robert M, Haney Isabel, Mawson John, Williams William G, Freedom Robert M. Pediatric primary benign cardiac tumors: A 15-year review. Am Heart J. 1997;134:1107–1114. doi: 10.1016/s0002-8703(97)70032-2. [DOI] [PubMed] [Google Scholar]
- 17.Gorlin Robert J. Nevoid basal cell carcinoma (Gorlin) syndrome. Genetics in Medicine. 2004;6(6):530–539. doi: 10.1097/01.gim.0000144188.15902.c4. [DOI] [PubMed] [Google Scholar]
- 18.Firtenberg Michael S, Thomas James D. [April 26, 2007];Benign Cardiac Tumors, E-medicine from WebMD. 2007 [Online] Available at: http://www.emedicine.com/med/topic2999.htm#section~miscellaneous and/or http://www.emedicine.com/med/topic299.htm.
- 19.Gailani Mae R, Ståhle-Bäckdahl Mona, Lefell David J, et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet. 1996;14(1):78–81. doi: 10.1038/ng0996-78. [DOI] [PubMed] [Google Scholar]