

Abstract
Although the prostate gland is a rich source of α1-adreno- (α1-AR) and m1-cholino receptors (m1-AChR), the membrane processes associated with their activation in glandular epithelial cells is poorly understood. We used the whole-cell patch-clamp technique to show that the agonists of the respective receptors, phenylephrine (PHE) and carbachol (CCh), activate cationic membrane currents in lymph node carcinoma of the prostate (LNCaP) human prostate cancer epithelial cells, which are not dependent on the filling status of intracellular IP3-sensitive Ca2+ stores, but directly gated by diacylglycerol (DAG), as evidenced by the ability of its membrane permeable analogue, OAG, to mimic the effects of the agonists. The underlying cationic channels are characterized by the weak field-strength Eisenman IV permeability sequence for monovalent cations (PK(25) > PCs(4.6) > PLi(1.4) > PNa(1.0)), and the following permeability sequence for divalent cations: PCa(1.0) > PMg(0.74) > PBa(0.6) > PSr(0.36) > PMn(0.3). They are 4.3 times more permeable to Ca2+ than Na+ and more sensitive to the inhibitor 2-APB than SK&F 96365. RT-PCR analysis shows that DAG-gated members of the transient receptor potential (TRP) channel family, including TRPC1 and TRPC3, are present in LNCaP cells. We conclude that, in prostate cancer epithelial cells, α1-ARs and m1-AChRs are functionally coupled to Ca2+-permeable DAG-gated cationic channels, for which TRPC1 and TRPC3 are the most likely candidates.
Calcium is a universal second messenger, regulating many important cellular processes, including growth, differentiation, and programmed cell death (Berridge, 1995; Berridge et al. 1998). The effectiveness of Ca2+ as a universal regulator is greatly enhanced thanks to sophisticated mechanisms that control its transport into cells, translating external stimuli into intracellular events. The significance of Ca2+ in coupling extra- and intracellular signalling highlights the role of Ca2+ entry pathways in normal and pathological cell functions. In non-excitable cells, Ca2+ entry is provided by voltage-independent cationic channels probably from the so-called transient receptor potential (TRP) channel family, initially found in Drosophila photoreceptors and then identified in various mammalian tissues by homologue screening (Clapham et al. 2001). These channels are characterized by diverse terminal activation mechanisms, which are, nevertheless, generally triggered by a common initial receptor-mediated breakdown of inositol phospholipids. This breakdown is catalysed by the enzyme phospholipase C (PLC), coupled to surface receptors via G-proteins, resulting in a derivation of second messengers, inositol 1,4,5 trisphosphate (IP3) and diacylglycerol (DAG). IP3 releases Ca2+ from intracellular stores, and the concomitant store depletion activates store-operated Ca2+ channels (SOCs), which in many cases have been tentatively identified as TRP family members (Clapham et al. 2001; Zitt et al. 2002). DAG, the IP3 receptor, and second messengers such as cyclic ADP-ribose are also capable of activating some other TRPs directly, without depleting intracellular Ca2+ stores (e.g. Hoffmann et al. 1999; Perraud et al. 2001; Tang et al. 2001).
Although prostate growth is controlled primarily by endocrine means, the autonomic nervous system plays an important role in its functions as well, regulating the prostate gland via noradrenergic and cholinergic innervation (McVary et al. 1998; Pennefather et al. 2000). Noradrenergic nerves primarily innervate the prostatic fibromuscular stroma, and their stimulation causes contractions of the prostate smooth muscle via the activation of α1-adrenoreceptors (α1-ARs). Inhibition of these receptors underlies the therapeutic action of α1-AR antagonists in benign prostatic hyperplasia (BHP). The role of cholinergic nerves is less defined, but the fact that they are more closely related to the glandular epithelium suggests that they may be involved in secretory functions. The human prostate is characterized by the predominant expression of the m1 subtype of muscarinic acetylcholine receptors (m1-AChR) (Ruggieri et al. 1995; Luthin et al. 1997).
The action of the subtypes of G-protein-coupled α1-adrenoreceptors and muscarinic acetylcholine receptors (m-AChRs) is known to be mediated via a phosphoinositide hydrolysis signalling pathway (e.g. Exton, 1988; Wu et al. 1992; Caulfield, 1993). Moreover, recent data suggest that activation of members of the TRP-channel family may be an essential component of the action of these receptors (Inoue et al. 2001; Zhang & Saffen, 2001). Although the prostate gland is a rich source of α1-ARs and m-AChRs, neither the mechanism of their action nor the functional link between these receptors and transmembrane Ca2+ entry has been clearly established. The role of TRP and TRP-like channels in receptor-mediated Ca2+ entry in prostate cells is also far from clear, although it is of special interest, in view of the fact that all TRPs discovered so far in the prostate become strongly overexpressed during progression to prostate cancer (Peng et al. 2001; Tsavaler et al. 2001; Wissenbach et al. 2001).
In this study, we investigated the biophysical properties and pharmacological sensitivity of agonist-activated cationic channels in androgen-dependent LNCaP (lymph node carcinoma of the prostate (Horoszewicz et al. 1983; Pousette et al. 1997)) prostate cancer epithelial cell line for the first time. We show that the α1-AR agonist, phenylephrine, and the m-AChR agonist, carbachol, activate non-specific cationic channels in these cells, which can also be activated directly by a membrane-permeable analogue of diacylglycerol - OAG (1-oleoyl-2-acetyl-sn-glycerol). Our data suggest the existence of receptor-activated Ca2+ entry in prostate cells that functions independently of intracellular Ca2+ store depletion, and provide a basis for identifying the TRP proteins responsible for this phenomenon.
METHODS
Cell cultures
LNCaP cells from the American Type Culture Collection were cultured in RPMI 1640 medium (Biowhittaker, Fontenay sous Bois, France) supplemented with 5 mml-glutamine (Sigma, L'Isle d'Abeau, France) and 10 % fetal bovine serum (Seromed, Poly-Labo, Strasbourg, France). The culture medium also contained 50 000 IU l−1 penicillin and 50 mg l−1 streptomycin. Cells were routinely grown in 50 ml flasks (Nunc, Poly-Labo) and kept at 37 °C in a humidified incubator in an air/CO2 (95/5 %) atmosphere. For electrophysiology, the cells were subcultured in Petri dishes (Nunc) and used within 3 to 6 days.
Electrophysiology
Macroscopic membrane ionic currents in LNCaP cells (average whole-cell membrane capacitance, Cm = 25.5 ± 1.2 pF, n = 46) were recorded in the whole-cell configuration of the patch-clamp technique, using a computer-controlled EPC-9 amplifier (HEKA Electronic, Germany). To prevent contamination of agonist-stimulated current in LNCaP cells with endogenous voltage-activated K+ current (Skryma et al. 1997), we used Cs+ as the major cation in our intracellular pipette solution (mm): CsCl 50, Cs(OH) 90, MgCl2 2, CaCl2 3 (calculated [Ca2+]free = 280 nm), glucose 5, Hepes 10, EGTA 4, Na-ATP 2, pH 7.3 (adjusted with glutamic acid, osmolarity 310 mosmol l−1). The normal extracellular solution contained (mm): NaCl 130, KCl 5, glucose 10, CaCl2 2, MgCl2 2, Hepes 10, TEA(OH) 20, pH 7.3 (adjusted with HCl, osmolarity 340 mosmol l−1). However, in order to enhance the amplitude of agonist-induced current, most of the experiments were performed in saline solution, in which NaCl had been replaced with an equimolar amount of CsCl. Receptor agonists, and channel inhibitors were added directly to the extracellular solution from respective stocks at concentrations that did not change the osmolarity.
We used 135 mm NaCl, LiCl, CsCl or KCl in the extracellular solution to measure monovalent cation permeability. Divalent cation permeability was measured in solutions containing 10 mm CaCl2, SrCl2, BaCl2, MgCl2 or MnCl2 and 125 mmN-methyl-d-glucamine (NMDG) chloride instead of NaCl and KCl. The permeability ratios were calculated from the shifts in reversal potential, using formulae presented in Watanabe et al. (2002). All voltages were corrected for a liquid junction potential of 13 mV between external and internal solutions. The resistance of the patch pipette varied between 3 and 5 MΩ, and series resistance compensation was used to improve voltage-clamp performance. External solutions were changed using a multibarrel puffing micropipette with common outflow positioned in close proximity to the cell under investigation. During the experiment, the cell was continuously superfused with the solution via a puffing pipette to reduce possible artifacts related to the switches from static to moving solution and vice versa. The external solution was changed completely in less than 1 s.
All reagents were from Sigma except SK&F 96365, which was supplied by Calbiochem. All stock solutions were prepared in water except OAG and 2-APB (2-aminoethoxydiphenyl borate), which were prepared in DMSO and methanol, respectively (final concentration of both solvents in the experimental solution did not exceed 0.05 %).
RT-PCR analysis
Total RNA from the LNCaP cell line was isolated by the guanidium thiocyanate-phenol-chloroform extraction procedure. After deoxyribonuclease I (0.1 U μl−1, 1 h at 25 ° C; Life Technonologies, Inc.) treatment, to eliminate genomic DNA, total RNA was reverse transcribed into cDNA as described by Roudbaraki et al. (1999). For the PCR reaction, specific sense and antisense primers (synthesized by Life Technologies) were selected based on GenBank TRPs sequences using Genejockey II (Biosoft, Cambridge, UK). Human TRPC1 splice variant TRPC1A-specific sense and antisense primers (GenBank accession no. P19334) were: 5′-TTCCTCTCCATCCTCTTCCTCG-3′ (nucleotides 795–816) and 5′-CATAGTTGTTACGATGAGCAGC-3′ (nucleotides 1231–1252). The predicted size of the PCR-amplified product was 457 base pairs (bp) for TRPC1 and 355 bp for the TRPC1A splice variant. Human TRPC3-specific sense and antisense primers (GenBank accession no. U47050) were: 5′-GGAAAAACATTACCTCCACCTTTCA-3′ (nucleotides 2260–2284) and 5′-CTCAGTTGCTTGGCTCTTGTCTTCC-3′ (nucleotides 2619–2643). The predicted size of the PCR-amplified TRPC3 product was 383 bp. Primers used to amplify the 625 bp part of the human TRPC6 coding sequence (GenBank accession no. AJ271066) were: 5′-GAACTTAGCAATGAACTGGCAGT-3′ (sense, nucleotides 895–917) and 5′-CATATCATGCCTATTACCCAGGA-3′ (antisense, nucleotides 1499–1521) and to amplify the 477 bp part of human TRPC7 cDNA (GenBank accession no. NM020389) were: 5′-GTCCGAATGCAAGGAAATCT-3′ (sense, nucleotides 1356–1375) and 5′-TGGGTTGTATTTGGCACCTC-3′ (antisense, nucleotides 1814–1833). Each sample was amplified by Ampli-Taq Gold DNA Polymerase (Perkin Elmer) in an automated thermal cycler (GenAmp 2400, Perkin Elmer). DNA amplification conditions included an initial 7 min denaturation step at 95 ° C (which, at the same time, activated the Taq Gold) and 40 cycles of 30 s at 95 ° C, 30 s at 58 ° C, 40 s at 72 ° C and a final 7 min elongation at 72 ° C. The RT-PCR samples were electrophoresed on a 1.5 % agarose gel, stained with ethidium bromide (0.5 μg ml−1), and photographed under ultraviolet illumination. In order to study band identity, the RT-PCR products were subjected to restriction enzyme analysis.
Data analysis
Each experiment was repeated several times, and results were expressed as means ±s.e.m. where appropriate. Data were analysed, and plots were produced using Origin 5.0 software (Microcal, Northampton, MA, USA). When analysing the time courses of the currents activated by various agents, the period required for current to increase 5 % above the baseline level was considered as delay and the period when current changed from 5 % to 95 % of its maximal amplitude was considered as development time.
RESULTS
α1-Adrenoreceptor stimulation induces cationic current in LNCaP cells
Under experimental conditions that ensured effective suppression of voltage-activated K+ current (intracellular Cs+, extracellular TEA) and minimization of store-dependent processes (relatively high [Ca2+]in = 280 nm), stimulating LNCaP cells bathed in normal extracellular solution with stepwise voltage pulses of variable amplitude or pulses that included voltage ramps between the levels of −100 mV and +50 mV revealed only a tiny baseline current, most probably reflecting the activity of some background channels (Fig. 1B and C). Exposing the cell to the α1-AR agonist, phenylephrine (PHE, 100 μm), however, elicited the development of additional current above the baseline, which reached a maximal steady level about 3 min after application (Fig. 1A), and had a reversal potential around −25 mV (Fig. 1D). Equimolar substitution of extracellular Cs+ for Na+ notably augmented the current, especially in the inward direction (around 5- to 6-fold at −100 mV), and shifted its reversal potential close to 0 mV (e.g. Fig. 6B), suggesting that: (1) the α1-AR agonist PHE obviously activates the current, which has cationic nature and (2) in view of the nearly symmetric Cs+ conditions following Na+ replacement (remember, we used Cs+-based intracellular solution) the underlying cationic channels apparently possess moderate inwardly rectifying properties. The PHE-activated current showed no voltage- or time-dependent gating. Inclusion of GTP (100 μm) in the pipette solution did not produce statistically different responses to PHE, suggesting that intracellular levels of GTP remain high enough to support adequate functioning of G protein-mediated signal transduction. Therefore, in most experiments GTP was not included.
Figure 1. Simulation of α 1-adrenoreceptors induces cationic current in LNCaP cells.
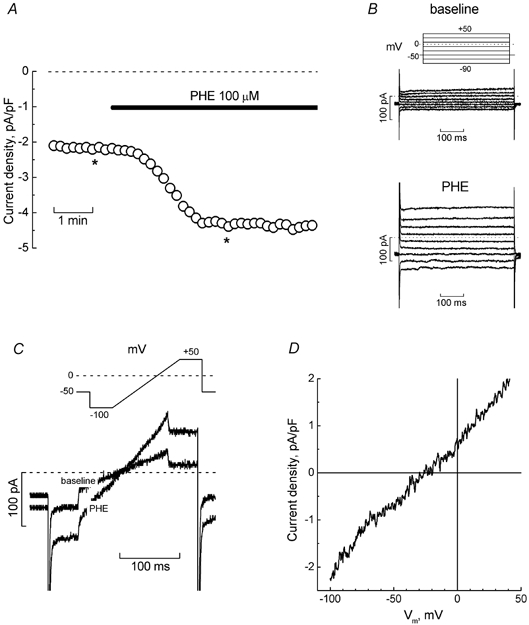
A, time course of the development of inward current (measured at −100 mV) in a representative LNCaP cell bathed in normal extracellular solution, in response to phenylephrine (PHE, 100 μm, marked by horizontal bar). B, original baseline (top) and PHE-induced (bottom) currents recorded at the times marked by asterisks on the time course of panel A; pulse protocol is depicted above the baseline currents. C, superimposed baseline and PHE-induced currents from the same cell in response to the pulse containing a linear voltage-ramp portion (shown above the records). D, I-V relationship of PHE-induced current derived from ramps in panel C.
Figure 6. Sensitivity of PHE-evoked current in LNCaP cells to ruthenium red and polyvalent cations.
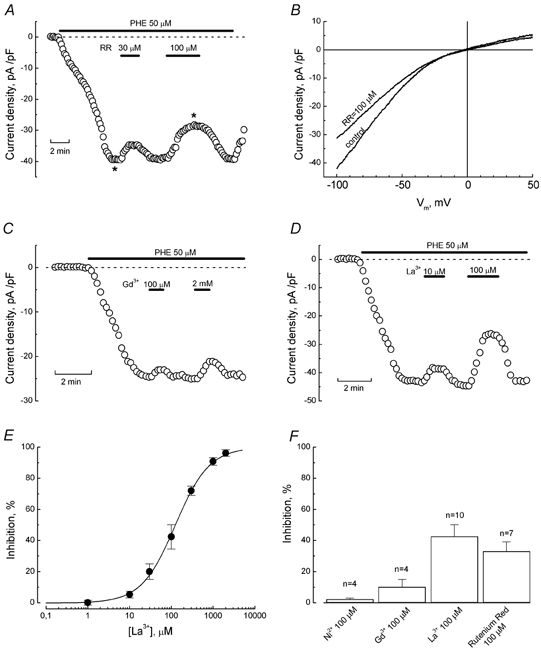
A, time course of PHE-evoked inward current (at −100 mV) in a representative cell and its response to 30 μm and 100 μm ruthenium red (RR); interventions are marked with horizontal bars. B, ramp-derived I-V relationships of control PHE-induced current and PHE-induced current in the presence of 100 μm RR, acquired at times marked by asterisks on the time course in panel A. C and D, inhibition of PHE-induced current in representative cells by Gd3+ (100 μm and 2 mm, C) and La3+ (10 μm and 100 μm, D); interventions are marked with horizontal bars. E, dose-response relationship for the inhibitory action of La3+ on PHE-evoked current; data points - mean ±s.e.m., n = 4–10, smooth curve fit of the data points by Langmuir's isotherm; IC50 value is 124 μm. F, comparison of the inhibitory action (mean ±s.e.m.) of Ni2+ (100 μm), Gd3+ (100 μm), La3+ (100 μm), and ruthenium red (100 μm) on phenylephrine-evoked current.
In order to quantify the relative permeability of monovalent cations through α1-AR-coupled membrane channels, we replaced all Na+ and K+ in the divalent cation-free (DVC-free) extracellular solution with equimolar amounts of Na+, Cs+, Li+, or K+ and measured the shifts in current reversal potential (Erev) relative to the one in the presence of Na+. Figure 2A shows representative I-V plots derived from a ramp recording of PHE-evoked current in Na+-, Cs+-, Li+-, or K+-containing solutions. Based on the shifts in Erev, the following order of relative permeability was deduced (n = 6): PK (25) > PCs (4.6) > PLi (1.4) > PNa (1.0), whereas the maximal Na+-, Cs+-, Li+- and K+-carried inward currents (at −90 mV) were related to each other as: IK (7.6) > ICs (5.5) > ILi (1.6) > INa (1.0). As Cs+ is much more permeable and able to carry much higher current than Na+ we, used Cs+ as a major charge carrier for further characterization of α1-AR-coupled cationic channel, unless otherwise specified. Cs+ was preferred to K+ to ensure suppression of any contribution from voltage-gated K+ channels.
Figure 2. Selective properties of α1-AR-coupled membrane channels in LNCaP cells.
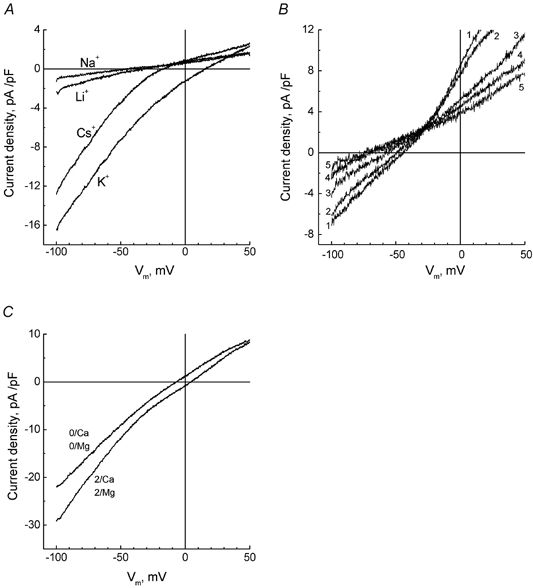
A, ramp-derived I-V relationships of PHE-evoked current in a representative cell sequentially exposed to divalent cation-free extracellular solution containing 135 mm Na+, Cs+, Li+ or K+. B, same as in A, but for another cell sequentially exposed to NMDG-based extracellular solution containing 10 mm Ca2+ (1), Mg2+ (2), Ba2+ (3), Sr2+ (4) or Mn2+ (5). C, I-V relationships of PHE-evoked current in representative cell in standard Cs+-based extracellular solution (2/Ca, 2/Mg) and following omission of Ca2+ and Mg2+ (0/Ca, 0/Mg).
α1-Adrenoreceptor-coupled channels are permeable to divalent cations
We also investigated whether PHE-activated cationic channels were permeable to divalent cations, particularly Ca2+. Figure 2C shows that removal of Ca2+ and Mg2+ from the Cs+-based extracellular solution resulted in a decrease of about 30 % in the inward current at −100 mV, suggesting that divalent cations either transfer a portion of the net current or regulate the underlying channels. To check possible permeation of various divalent cations we formulated an extracellular solution in which most of the Cs+ was replaced with impermeable NMDG and raised the concentration of the divalent cation tested - Ca2+, Mg2+, Ba2+, Sr2+ or Mn2+ - to 10 mm (see Table 1). In the presence of any of the divalent cations tested, application of PHE was still able to induce current above the baseline level, suggesting permeation through the channel. The I-V relationships for PHE-induced current in the presence of specified divalent charge carriers is presented in Fig. 2B. Tabulating the shifts in reversal potential relative to Ca2+ yielded the following order of relative permeability (n = 4): PCa (1.0) > PMg (0.74) > PBa (0.6) > PSr (0.36) > PMn (0.3).
Table 1.
Compositions of bath and pipette solutions used forelectrophysiological recordings
| Bath | Pipette | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Normal | MVC solution | DVC solution | |||||||||
| Basic | Na+-free | K+ | Li+ | Cs+ | Ca2+ | Ba2+ | Sr2+ | Mg2+ | Mn2+ | ||
| NaCl | 135 | — | — | — | — | — | — | — | — | — | — |
| KC1 | 5 | 5 | 135 | — | — | — | — | — | — | — | — |
| LiCl | — | — | — | 135 | — | — | — | — | — | — | — |
| NMDG | — | — | — | — | — | 125 | 125 | 125 | 125 | 125 | — |
| BaCl2 | — | — | — | — | — | — | 10 | — | — | — | — |
| SrCl2 | — | — | — | — | — | — | — | 10 | — | — | — |
| MnCl2 | — | — | — | — | — | — | — | — | — | 10 | — |
| CsCl | — | 130 | — | — | 135 | — | — | — | — | — | 50 |
| Cs(OH) | — | — | — | — | — | — | — | — | — | — | 90 |
| CaCl2 | 2 | 2 | — | — | — | 10 | — | — | — | — | 3 |
| MgCl2 | 2 | 2 | — | — | — | — | — | — | 10 | — | 2 |
| Glucose | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 5 |
| Hepes | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| EGTA | — | — | — | — | — | — | — | — | — | — | 4 |
| TEA(OH) | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | — |
| Na-ATP | — | — | — | — | — | — | — | — | — | — | 1 |
Concentrations are given in mM and the pH of all solutions was adjusted to 7.3. The osmolarities of the bath and pipette solutions were approximately 340 and 310 mosmol 1−1, respectively. MVC, monovalent cations; DVC, divalent cations.
As Na+ and Ca2+ permeation through the channel is the most relevant from a physiological perspective, we also estimated the relative permeability of Ca2+ and Na+, based on Erev shift following replacement of NMDG-based solution containing 10 mm Ca2+ for 135 mm Na+ DVC-free solution. These experiments provided the following permeability ratio: PCa:PNa = 4.3:1.0, whereas the maximal amplitudes of Na+- and Ca2+-carried PHE-induced currents at −100 mV were related to each other as ICa:INa = 1.2:1.0. Thus, under physiological conditions, stimulating α1-ARs apparently results in the activation of a transmembrane Ca2+ influx via α1-AR-coupled non-selective cationic channels.
Stimulating muscarinic cholinoreceptors activates similar cationic current
As previously mentioned, in addition to noradrenergic innervation, the prostate gland also has cholinergic innervation, which acts via m1-muscarinic cholinoreceptors. As the general mechanism of action of both receptors involves activating a PLC-catalysed inositol phospholipid hydrolysis signalling pathway, it was interesting to verify whether the m-AChR agonist, carbachol (CCh), was also able to activate cationic current in LNCaP cells and to examine the similarities, if any, between α1-AR and m-AChR-coupled channels.
Figure 3A and B shows that application of CCh (2 μm) in Cs+-based extracellular solution elicited a current in LNCaP cell superficially similar to the one activated by phenylephrine (see Fig. 1). This current showed no voltage- or time-dependent gating, exhibited inwardly rectifying properties and had a reversal potential close to 0 mV (Fig. 3C and D).
Figure 3. Muscarinic cholinoreceptor agonist, carbachol, induces cationic current in LNCaP cells similarly to PHE.
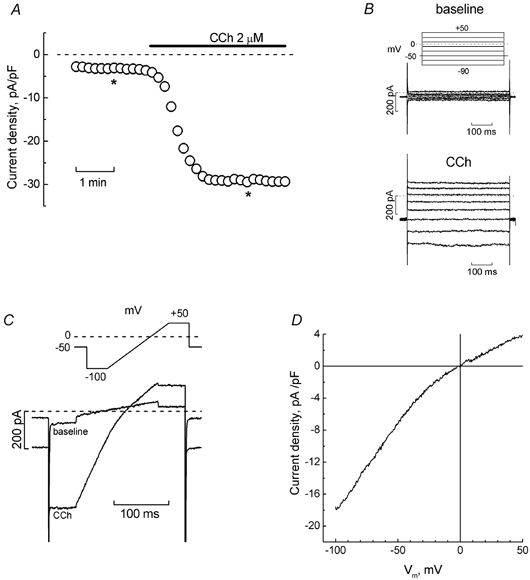
A, development time course of inward current (measured at −100 mV) in the representative LNCaP cell in Cs+-based extracellular solution in response to carbachol (CCh, 2 μm, marked by horizontal bar). B, original baseline (top) and CCh-induced (bottom) currents recorded at the times marked by asterisks on the time course in panel A; pulse protocol is depicted above the baseline currents. C, superimposed baseline and CCh-induced currents from the same cell in response to the pulse containing a linear voltage-ramp portion (shown above the records). D, I-V relationship of CCh-induced current derived from ramps in panel C.
Stimulation of muscarinic and α1-adrenoreceptors converges on diacylglycerol in activating cationic channels
Both α1-ARs and m-AChRs are coupled via a specific G protein to the PLC-catalysed inositol phospholipid breakdown signalling pathway (Brauner-Osborne & Brann, 1996; Marshall et al. 1999; Zhong & Minneman, 1999). Stimulation of this pathway may eventually result in the activation of IP3 store-dependent and store-independent membrane channels, both of which are thought to belong to the TRP-channel family (Clapham et al. 2001). One of the messengers implicated in store-independent gating of these channels is the second after IP3 product of PLC-catalysed phospholipid turnover, DAG, which has been shown to gate at least such members of TRP-channel family as TRP3 and TRP6 directly (Hofman et al. 1999; Tesfai et al. 2001).
As our experimental conditions did not favour SOC activation, we hypothesized that DAG may be an active element in the agonist-evoked cationic current in LNCaP cells. To verify this hypothesis, we used the membrane-permeable DAG analogue, 1-oleoyl-2-acetyl-sn-glycerol (OAG). Figure 4A-D shows that application of OAG (100 μm) to LNCaP cells elicited a membrane current, virtually undistinguishable from the currents evoked by PHE or CCh. Moreover, co-application of any of these agonists during maximal OAG-induced response failed to activate more current (Fig. 4A-D), suggesting not only that both agonists target the same channels as OAG, but also that, at least under the experimental conditions used, activation of the endogenous α1-AR- or m-AChR-coupled signalling pathways is unable to recruit either SOCs or other store-independent channels.
Figure 4. Stimulation of muscarinic and α1-adrenoreceptors converge on diacylglycerol-gated cationic channels.
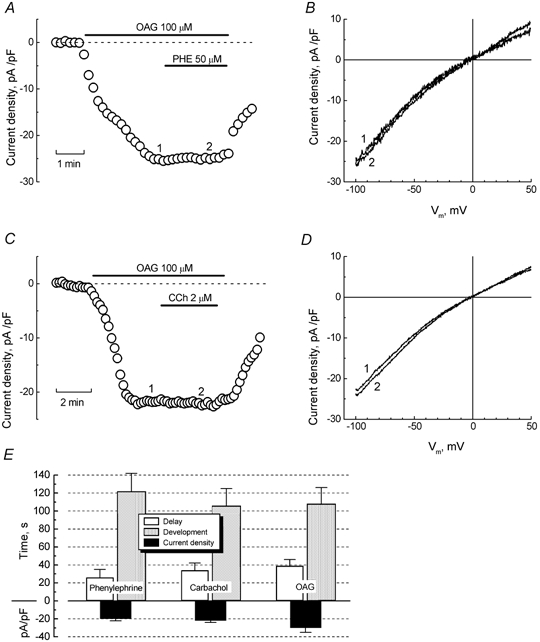
A, development time course of inward current (measured at −100 mV) in the representative LNCaP cell in Cs+-based extracellular solution in response to the membrane-permeable DAG analogue, 1-oleoyl-2-acetyl-sn-glycerol (OAG, 100 μm) alone, and following co-application with PHE (50 μm); interventions marked with horizontal bars. B, ramp-derived I-V relationships of OAG (1) and OAG + PHE (2) currents acquired at the respectively numbered times on the time course in panel A, showing virtually no difference in amplitude or reversal potential. C and D, same as in A and B, respectively, but with the interaction of OAG and CCh in another representative LNCaP cell. E, quantification of the delay, development periods, and maximal densities for PHE-, CCh- and OAG-induced currents (mean ±s.e.m., n = 10–15).
To further confirm the noninvolvement of IP3-dependent store depletion in the agonist-evoked activation of cationic current in LNCaP cells, we performed a series of experiments in which agonists were applied to cells pre-dialysed with heparin (1 mg ml−1), a known IP3 receptor inhibitor. Pre-dialysis did not affect the time course of current development or its maximal amplitude (at −100 mV) in response to PHE or CCh (data not shown). These findings are consistent with a solely store-independent current activation mechanism.
We also sought to further validate the identity of agonist- and OAG-activated channels by quantifying the latter's relative permeabilities. This was done based on Erev shifts in response to permeating ion substitution, exactly as reported above for PHE-induced currents. As a result, we obtained the sequences of relative permeability of OAG-gated channels for mono- and divalent cations (n = 4, for each ion): PK (22.0) > PCs (5.2) > PNa (1.0) and PCa (1.0) > PMg (0.3) > PBa (0.3), nearly identical to the ones determined for PHE-activated channels. This provided us with an additional strong argument that both PHE and OAG target the same cationic channels in LNCaP cells.
Moreover, we also quantified the maximal densities and temporal parameters of current activation in response to PHE, CCh, and OAG. Figure 4E shows that, consistent with the notion of a common origin, all three agents evoked currents with statistically similar delay and development periods as well as densities.
Pharmacology of agonist-activated cationic current in LNCaP cells
The pharmacology of agonist- and OAG-induced cationic current in LNCaP cells was examined using a number of organic and inorganic compounds, which have been shown in numerous previous studies to act either on endogenous cationic currents in different cell types or on cationic currents induced by the heterologous expression of various TRPs. The organic compounds included SK&F 96365, a known blocker of receptor-operated Ca2+ entry in non-excitable cells (Merritt et al. 1990), 2-APB, an inhibitor of IP3 receptors and store-operated Ca2+ channels (e.g. Braun et al. 2001; Diver et al. 2001), and ruthenium red (RR), a blocker of the vanilloid subfamily of TRP channels (Nilius et al. 2001; Watanabe et al. 2002). Inorganic blockers included three polyvalent cations: Ni2+, La3+, and Gd3+.
Figure 5A-C shows that both SK&F 96365 and 2-APB at their typically used concentrations of 25 μm and 100 μm, respectively, quite rapidly and reversibly inhibited PHE-, CCh-, and OAG-induced currents. Inspection of individual time courses and comparison of the average percentage blockade (at −100 mV, Fig. 5E) suggests that, in general, 2-APB is more potent, blocking the currents by 60 % to 90 % compared to only about 20 % to 30 % for SK&F 96365. It should be noted that the differences in the blockade of PHE-, CCh- and OAG-induced currents by the same agent were statistically insignificant, providing further proof of the underlying channels' common origin. Furthermore, application of SK&F 96365 on top of 2-APB did not produce additional inhibition, suggesting that both agents target the same channels. Fitting the dose-response relationship for the blockade of phenylephrine-induced current by 2-APB to the Hill equation yielded an IC50 value for 2-APB action of 28.2 ± 1.3 μm (Fig. 5D).
Figure 5. 2-APB and SK&F 96365 sensitivity of PHE-, CCh-, and OAG-evoked currents in LNCaP cells.
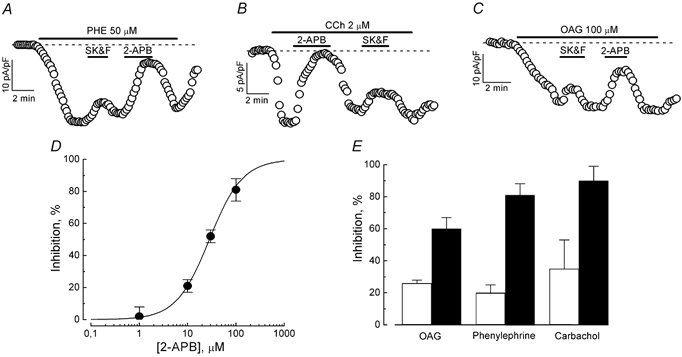
A-C, time courses of PHE-, CCh-, and OAG-evoked inward currents (at −100 mV) in representative cells and responses to the application of 100 μm 2-APB and 25 μm SK&F 96365; interventions marked with horizontal bars. D, dose-response relationship for the inhibitory action of 2-APB on PHE-evoked current; data points - mean ±s.e.m., n = 4–7, smooth curve fit of the data points by Langmuir's isotherm; IC50 value is 28.2 μm. E, comparison of the inhibitory action of 100 μm 2-APB (▪) and 25 μm SK&F 96365 (□) on phenylephrine-, carbachol- and OAG-evoked currents (mean ±s.e.m., n = 4–7).
PHE-induced current was also sensitive to RR, which, upon increasing its concentration from 30 μm to 100 μm, enhanced the current blockade from about 15 % to nearly 30 % (Fig. 6A and B). Among the polyvalent cations tested (Fig. 6C-F), the most potent was La3+, which at 100 μm inhibited about 50 % of PHE-induced current compared to only about 10 % and 5 % for Gd3+ and Ni2+ (both at 100 μm), respectively (Fig. 6F). Quantification of the dose-response relationship for the blocking action of La3+ gave an IC50 value of 124 μm (Fig. 6E).
Expression of receptor-operated TRP channels in human prostate cancer epithelial cells
Four members of the short subfamily of TRP channels, TRPC1, TRPC3, TRPC6, and TRPC7, are known to be directly gated by DAG (Clapham et al. 2001). Therefore, in order to determine potential candidates for the α1-AR- and m-AChR-coupled cationic channel(s) in LNCaP cells, we used RT-PCR to analyse the expression of the specific transcripts for the human isoforms of these DAG-gated TRP members in these cells. Figure 7 shows that LNCaP cells expressed only the TRPC1A splice variant and TRPC3 transcripts (Fig. 7A and B), whereas TRPC6 and TRPC7 were undetectable (Fig. 7C and D). Thus, on the basis of the expression pattern, TRPC1 and TRPC3 are the most likely candidates for the formation of agonist-activated cationic channels in LNCaP cells.
Figure 7. Expression of DAG-gated TRP members in LNCaP cells.
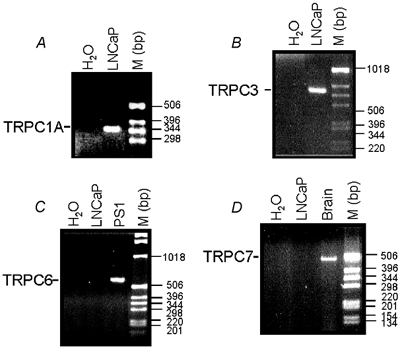
RT-PCR analysis of the expression of human TRPC1A (A), TRPC3 (B), TRPC6 (C), and TRPC7 (D) transcripts in LNCaP cells. The expression products were obtained using the primers described in the Methods. Smooth muscle PS1 cells and brain tissue were used as positive controls for the detection of TRPC6 and TRPC7, respectively. M: DNA ladder.
DISCUSSION
In this study, we show, for the first time, that stimulation of α1-adrenoreceptors and m1-cholinoreceptors in the human prostate cancer epithelial cells activates transmembrane current that includes an essential Ca2+ component, and that activation of this current involves a store-independent, DAG-mediated mechanism.
α1-Adrenoraceptors and muscarinic cholinoreceptors in the prostate
Four subtypes of α1-adrenoceptors (α1A, α1B, α1C and α1L) have been identified in the human prostate (Price et al. 1993; Chapple et al. 1994; Fukasawa et al. 1998; Takeda et al. 1999; Walden et al. 1999). These receptors are mostly localized in the fibromuscular stroma, where they mediate the contraction of prostate smooth muscle (Takeda et al. 1999). While the role of α1-ARs in maintaining the contractile properties of the prostate has been intensively investigated, and even received widespread recognition in the clinical treatment of BHP, the representation and function of these receptors in the glandular epithelium had not previously been determined. At the same time, there is growing evidence that α1-ARs may support the direct mitogenic effect of catecholamines on prostate growth (McVary et al. 1998).
In contrast to α1-ARs, a predominantly epithelial location of muscarinic cholinoreceptors in the prostate is well recognized (e.g. Ventura et al. 2002). It has been suggested that these receptors are involved in the secretory function, although the mechanisms have not yet been elucidated. The situation is further confused by the fact that the most abundant m1 subtype of m-AChRs in the prostate (Ruggieri et al. 1995; Luthin et al. 1997) is coupled to the same PLC-catalysed inositol phospholipid breakdown signalling pathway as α1-ARs, raising the possibility that the functions of both receptors in epithelial cells may largely overlap.
Indeed, we show in the present study that the α1-AR agonist, phenylephrine, and the m-AChR agonist, carbachol, activate cationic membrane currents in LNCaP prostate cancer epithelial cells with nearly identical major biophysical properties, suggesting that they may be transferred through the same population of ion channels. The strongest argument that both receptors target the same channel is that basically indistinguishable current was activated by one of the products of the PLC-catalysed inositol phospholipid breakdown signalling pathway common to both receptors, DAG. Moreover, the fact that neither of these agonists were able to elicit additional current when applied in addition to the membrane-permeable DAG analogue, OAG, proves not only the common origin of the channels, but also that, at least under our experimental conditions, these agonists did not activate any channels other than those directly gated by DAG.
Agonist-activated cationic channels in prostate cancer epithelial cells and their probable molecular counterparts
The hallmark properties of agonist- and DAG-activated cationic channels in LNCaP cells can be summarized as follows: (1) lack of voltage-dependent gating; (2) moderate inward rectification; (3) a monovalent cation permeability - PK > PCs > PLi > PNa - very similar to the Eisenman IV sequence for a weak-field-strength (Hille, 2001); (4) a divalent cation permeability sequence: PCa > PMg > PBa > PSr > PMn; (5) noticeably higher permeability for Ca2+ than Na+; and (6) higher sensitivity to 2-APB than SK&F 96365.
As far as finding the molecular counterparts for these endogenous channels is concerned, the potential candidates are probably limited to the members of the short subfamily of TRP channels (Clapham et al. 2001) such as TRPC1 (Lintschinger et al. 2000), TRPC3 (Zhu et al. 1998; Hofmann et al. 1999; Lintschinger et al. 2000; Wu et al. 2000), TRPC6 (Hofmann et al. 1999; Inoue et al. 2001; Tesfai et al. 2001), and TRPC7 (Okada et al. 1999), all of which exhibit distinct DAG dependency of activation. The issue of correlation is complicated, however, by the fact that heteromeric assembly of several TRPs may generate a channel with functionally distinct properties from any of the monomeric ones, and that some of the TRPs may display dual, store-dependent, and DAG-dependent gating. There is also a possibility that expression system-specific endogenous TRPs may modulate the properties of heterologously expressed ones.
Among all the potential candidates mentioned above, our RT-PCR analysis showed the presence of the specific mRNA for only TRPC1 and TRPC3 in LNCaP cells. Heterologous expression of TRPC1 or TRPC3 alone, or their combination in HEK 293, has been shown to induce carbachol- and OAG-activated conductances with quite distinct Ca2+ permeability, Ca2+-dependent regulation and agonist-sensitivity characteristics (Lintschinger et al. 2000). Depending on the expression system, TRPC3 is capable of producing a current that is largely constitutively activated under basal conditions (Zitt et al. 1997, Kamouchi et al. 1999, Lintschinger et al. 2000), but can be further stimulated by OAG (Hofmann et al. 1999, Lintschinger et al. 2000), or even a current that exhibits a distinct IP3 store-dependent activation mode (Kiselyov et al. 1998, Vazquez et al. 2001). The involvement of TRPC1 in the formation of a store-dependent Ca2+ entry pathway has also been demonstrated in a number of studies (e.g. Wu et al. 2000; Brough et al. 2001).
Correlation of the permeation and pharmacological profile of the endogenous agonist- and DAG-gated cationic channels in LNCaP cells with those described for heterologously expressed TRPC1 and TRPC3 shows a number of notable differences. We did not observe, for instance, constitutively activated current under non-stimulated conditions in LNCaP cells. However, this type of current is clearly evident in TRPC3-expressing HEK 293 cells (Lintschinger et al. 2000) and bovine pulmonary artery endothelial (CPAE) cells (Kamouchi et al. 1999) and it further increases after co-expression of TRPC1 (Lintschinger et al. 2000). Moreover, the weak-field Eisenman IV monovalent cation-permeability profile found in our experiments seems to differ from the stronger-field profile (i.e. PNa > PCs∼PK) displayed by TRPC3 (Kamouchi et al. 1999), but is rather similar to the one reported for the volume-sensitive TRP, TRPV4 (Watanabe et al. 2002). In addition, despite the fact that reversal potential shifts suggest about 1.6 times higher Ca2+ than Na+ TRPC3 permeability, an increase in [Ca2+]out actually reduces the TRPC3-induced current (Kamouchi et al. 1999; Lintschinger et al. 2000), whereas in LNCaP cells not only was the permeability to Ca2+ higher than to Na+, but agonist- and OAG-stimulated currents were also directly dependent on [Ca2+]out. Anomalous [Ca2+]out dependence was reported for TRPC1- and TRPC1/ TRPC3-induced currents as well (Lintschinger et al. 2000).
Although the specificity of two popular cationic channel blockers, SK&F 96365 and 2-APB, still remains questionable, and their pharmacological profile may largely overlap, there are indications that 2-APB may be a preferential blocker of SOCs (Braun et al. 2001; Voets et al. 2001) whereas SK&F 96365 is probably less specific, inhibiting channels activated by store-independent mechanisms as well. Our results show that 2-APB is capable of blocking nearly 100 % PHE-induced current in LNCaP cells with a relatively low IC50 of 28.2 μm, compared to only about 20 % of maximal blockade produced by SK&F 96365, suggesting that, with respect to high 2-APB sensitivity, the underlying cationic channels are closely related to classic calcium release activated channels (CRAC). On the other hand, the PHE-induced current also appeared to be quite sensitive to RR, a potent inhibitor of such members of the vanilloid subfamily of TRP channels as volume-regulated TRPV4 (Watanabe et al. 2002) and Ca2+-regulated TRPV5 (ECaC1 or CaT2; Nilius et al. 2001). La3+, a non-specific cationic channel blocker, fully inhibited PHE-induced current in LNCaP with an IC50 of 124 μm, about five times larger than that reported for human TRPC3 expressed in CPAE cells (Kamouchi et al. 1999).
These findings indicate that the DAG-gated cationic channel(s) in LNCaP cells are probably heterotetramultimers, that necessarily include TRPC1 and/or TRPC3, which confer the property of DAG gating, and some other TRP members that, in combination, determine the resultant permeation and pharmacological characteristics of the whole channel.
Practical implications
Although α1-ARs and m1-AChRs are abundantly expressed in the prostate and the expression of some of the TRPs such as CaT1 (TRPV6) and Trp-p8 (TRPM8) increase considerably during the progression to prostate cancer (Peng et al. 2001; Wissenbach et al. 2001; Tsavaler et al. 2001), neither the functional link between these receptors and TRPs nor their physiological role in prostate cancer epithelial cells has yet been firmly established. Although direct DAG gating for prostate cancer-specific TRPs has not been demonstrated, it is plausible that by association with such DAG-sensitive members as TRPC1 and/or TRPC3 that they may participate in the formation of the resultant α1-AR- and m1-AChR-coupled channel. This implies that stimulation of these receptors provides a variable Ca2+ influx, depending on the stage of the cancer, suggesting differential autonomic regulation not only of the secretory response of epithelial cells, but also of such important Ca2+-dependent processes as proliferation and apoptosis. As targeting cell proliferation and apoptosis in an attempt to control prostate growth is emerging as a potentially powerful therapeutic approach for the effective treatment of prostate cancer (Plonowski et al. 2002; Crescioli et al. 2002), further studies of the intimate mechanisms of agonist-stimulated Ca2+ entry may aid in a search for novel targets for therapeutic interventions.
Acknowledgments
This work was supported by grants from INSERM, Laboratoire d'Urologie du CH of Roubaix and INTAS-99-01248. Y. M. Shuba was supported by INSERM and the French Ministry of Science.
REFERENCES
- Berridge MJ. Calcium signalling and cell proliferation. Bioessays. 1995;17:491–500. doi: 10.1002/bies.950170605. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Lipp P. Calcium - a life and death signal. Nature. 1998;935:645–648. doi: 10.1038/27094. [DOI] [PubMed] [Google Scholar]
- Brough GH, Wu S, Cioffi D, Moore TM, Li M, Dean N, Stevens T. Contribution of endogenously expressed Trp1 to a Ca2+-selective, store-operated Ca2+ entry pathway. FASEB J. 2001;15:1727–1738. [PubMed] [Google Scholar]
- Braun FJ, Broad LM, Armstrong DL, Putney JW., Jr Stable activation of single Ca2+ release-activated Ca2+ channels in divalent cation-free solutions. J Biol Chem. 2001;276:1063–1070. doi: 10.1074/jbc.M008348200. [DOI] [PubMed] [Google Scholar]
- Brauner-Osborne H, Brann MR. Pharmacology of muscarinic acetylcholine receptor subtypes (m1-m5): high throughput assays in mammalian cells. Eur J Pharmacol. 1996;295:93–102. doi: 10.1016/0014-2999(95)00639-7. [DOI] [PubMed] [Google Scholar]
- Caulfield MP. Muscarinic receptors-characterization, coupling and function. Pharmacol Ther. 1993;58:319–379. doi: 10.1016/0163-7258(93)90027-b. [DOI] [PubMed] [Google Scholar]
- Chapple CR, Burt RP, Andersson PO, Greengrass P, Wyllie M, Marshall I. α 1-adrenoreceptor subtypes in the human prostate. Br J Urol. 1994;74:585–589. doi: 10.1111/j.1464-410x.1994.tb09188.x. [DOI] [PubMed] [Google Scholar]
- Clapham D, Runnels L, Strubing C. The TRP ion channel famely. Nat Rev Neurosci. 2001;2:387–396. doi: 10.1038/35077544. [DOI] [PubMed] [Google Scholar]
- Crescioli C, Maggi M, Luconi M, Vannelli GB, Salerno R, Sinisi AA, Bonaccorsi L, Ferruzzi P, Barni T, Forti G, Serio M. Vitamin D3 analogue inhibits keratinocyte growth factor signaling and induces apoptosis in human prostate cancer cells. Prostate. 2002;50:15–26. doi: 10.1002/pros.10028. [DOI] [PubMed] [Google Scholar]
- Diver JM, Sage SO, Rosado JA. The inositol trisphosphate receptor antagonist 2-aminoethoxydiphenylborate (2-APB) blocks Ca2+ entry channels in human platelets: cautions for its use in studying Ca2+ influx. Cell Calcium. 2001;30:323–329. doi: 10.1054/ceca.2001.0239. [DOI] [PubMed] [Google Scholar]
- Exton JH. The roles of calcium and phosphoinositides in the mechanism of α 1-adrenergic and other agonists. Rev Physiol Biochem Pharmacol. 1988;111:117–224. doi: 10.1007/BFb0033873. [DOI] [PubMed] [Google Scholar]
- Fukasawa R, Taniguchi N, Moriyama N, Ukai Y, Yamazaki S, Ueki T, Kameyama S, Kimura K, Kawabe K. The α1L-adrenoreceptor subtype in the lower urinary tract: a comparison of human urethra and prostate. Br J Urol. 1998;82:733–737. doi: 10.1046/j.1464-410x.1998.00845.x. [DOI] [PubMed] [Google Scholar]
- Hille B. Ion Channels of Exitable Membranes. Sunderland, MA, USA: 2001. pp. 343–344. [Google Scholar]
- Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, Mirand EA, Murphy GP. LNCaP model of human prostatic carcinoma. Cancer Res. 1983;43:1809–1818. [PubMed] [Google Scholar]
- Inoue R, Okada T, Onoue H, Hara Y, Shimizu S, Naitoh S, Ito Y, Mori Y. The transient receptor potential protein homologue TRP6 is the essential component of vascular α 1-adrenoceptor-activated Ca2+-permeable cation channel. Circ Res. 2001;88:325–332. doi: 10.1161/01.res.88.3.325. [DOI] [PubMed] [Google Scholar]
- Kamouchi M, Philipp S, Flockerzi V, Wissenbach U, Mamin A, Raeymaekers L, Eggermont J, Droogmans G, Nilius B. Properties of heterologously expressed hTRP3 channels in bovine pulmonary artery endothelial cells. J Physiol. 1999;518:345–358. doi: 10.1111/j.1469-7793.1999.0345p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselyov K, Xu X, Mozhayeva G, Kuo T, Pessah I, Mignery G, Zhu X, Birnbaumer L, Muallem S. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature. 1998;396:478–482. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- Lintschinger B, Balzer-Geldsetzer M, Baskaran T, Graier WF, Romanin C, Zhu MX, Groschner K. Coassembly of Trp1 and Trp3 proteins generates diacylglycerol- and Ca2+-sensitive cation channels. J Biol Chem. 2000;275:27799–27805. doi: 10.1074/jbc.M002705200. [DOI] [PubMed] [Google Scholar]
- Luthin GR, Wang P, Zhou H, Dhanasekaran D, Ruggieri MR. Role of m1 receptor-G protein coupling in cell proliferation in the prostate. Life Sci. 1997;60:963–968. doi: 10.1016/s0024-3205(97)00035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall I, Burt RP, Chapple CR. Signal transduction pathways associated with alpha1-adrenoceptor subtypes in cells and tissues including human prostate. Eur Urol. 1999;36:42–47. doi: 10.1159/000052317. [DOI] [PubMed] [Google Scholar]
- McVary KT, McKenna KE, Lee C. Prostate innervation. Prostate Suppl. 1998;8:2–13. [PubMed] [Google Scholar]
- Merritt JE, Armstrong WP, Benham CD, Hallam TJ, Jacob R, Jaxa-Chamiec A, Leigh BK, McCarthy SA, Moores KE, Rink TJ. SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem J. 1990;271:515–522. doi: 10.1042/bj2710515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Prenen J, Vennekens R, Hoenderop JG, Bindels RJ, Droogmans G. Pharmacological modulation of monovalent cation currents through the epithelial Ca2+ channel ECaC1. Br J Pharmacol. 2001;134:453–462. doi: 10.1038/sj.bjp.0704272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Inoue R, Yamazaki K, Maeda A, Kurosaki T, Yamakuni T, Tanaka I, Shimizu S, Ikenaka K, Imoto K, Mori Y. Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. Ca2+ -permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. J Biol Chem. 1999;274:27359–27370. doi: 10.1074/jbc.274.39.27359. [DOI] [PubMed] [Google Scholar]
- Peng JB, Zhuang L, Berger UV, Adam RM, Williams BJ, Brown EM, Hediger MA, Freeman MR. CaT1 expression correlates with tumor grade in prostate cancer. Biochem Biophys Res Commun. 2001;282:729–734. doi: 10.1006/bbrc.2001.4638. [DOI] [PubMed] [Google Scholar]
- Pennefather JN, Lau WA, Mitchelson F, Ventura S. The autonomic and sensory innervation of smooth muscle of the prostate gland: a review of pharmacological and histological studies. J Auton Pharmacol. 2000;20:193–206. doi: 10.1046/j.1365-2680.2000.00195.x. [DOI] [PubMed] [Google Scholar]
- Perraud AL, Fleig A, Dunn C, Bagley L, Launay P, Schmitz C, Stokes A, Zhu Q, Bessman M, Penner R, Kinet J-P, Scharenberg A. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–599. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- Plonowski A, Schally AV, Nagy A, Groot K, Krupa M, Navone NM, Logothetis C. Inhibition of in vivo proliferation of MDA-PCa-2b human prostate cancer by a targeted cytotoxic analog of luteinizing hormone-releasing hormone AN-207. Cancer Lett. 2002;176:57–63. doi: 10.1016/s0304-3835(01)00734-0. [DOI] [PubMed] [Google Scholar]
- Pousette A, Carlstrom K, Henriksson P, Grande M, Stege R. Use of a hormone-sensitive (LNCaP) and a hormone-resistant (LNCaP-r) cell line in prostate cancer research. Prostate. 1997;31:198–203. doi: 10.1002/(sici)1097-0045(19970515)31:3<198::aid-pros9>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Price DT, Schwinn DA, Lomasney JW, Allen LF, Caron MG, Lefkowitz RJ. Identification, quantification, and localization of mRNA for three distinct α 1 adrenergic receptor subtypes in human prostate. J Urol. 1993;150:546–551. doi: 10.1016/s0022-5347(17)35544-1. [DOI] [PubMed] [Google Scholar]
- Roudbaraki M, Lorsignol A, Langouche L, Callewaert G, Vankelecom H, Denef C. Target cells of gamma3-melanocyte-stimulating hormone detected through intracellular Ca2+ responses in immature rat pituitary constitute a fraction of all main pituitary cell types, but mostly express multiple hormone phenotypes at the messenger ribonucleic acid level. Refractoriness to melanocortin-3 receptor blockade in the lacto- somatotroph lineage. Endocrinology. 1999;140:4874–4885. doi: 10.1210/endo.140.10.7080. [DOI] [PubMed] [Google Scholar]
- Skryma RN, Prevarskaya NB, Dufy-Barbe L, Odessa MF, Audin J, Dufy B. Potassium conductance in the androgen-sensitive prostate cancer cell line, LNCaP: involvement in cell proliferation. Prostate. 1997;33:112–122. doi: 10.1002/(sici)1097-0045(19971001)33:2<112::aid-pros5>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Takeda M, Hatano A, Arai K, Obara K, Tsutsui T, Takahashi K. α 1- and α 2-adrenoreceptors in BPH. Eur Urol. 1999;36:31–34. doi: 10.1159/000052315. [DOI] [PubMed] [Google Scholar]
- Tang J, Lin Y, Zhang Z, Tikunova S, Birnbaumer L, Zhu MX. Identification of common binding sites for calmodulin and inositol 1,4,5-trisphosphate receptors on the carboxyl termini of TRP channels. J Biol Chem. 2001;276:21303–21310. doi: 10.1074/jbc.M102316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesfai Y, Brereton HM, Barritt GJ. A diacylglycerol-activated Ca2+ channel in PC12 cells (an adrenal chromaffin cell line) correlates with expression of the TRP-6 (transient receptor potential) protein. Biochem J. 2001;358:717–726. doi: 10.1042/0264-6021:3580717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsavaler L, Shapero MH, Morkowski S, Laus R. Trp-p8, a novel prostate-specific gene, is up-regulated in prostate cancer and other malignancies and shares high homology with transient receptor potential calcium channel proteins. Cancer Res. 2001;61:3760–3769. [PubMed] [Google Scholar]
- Vazquez G, Lievremont JP, St J, Bird G, Putney JW., Jr Human Trp3 forms both inositol trisphosphate receptor-dependent and receptor-independent store-operated cation channels in DT40 avian B lymphocytes. Proc Natl Acad Sci U S A. 2001;98:11777–11782. doi: 10.1073/pnas.201238198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura S, Pennefather J, Mitchelson F. Cholinergic innervation and function in the prostate gland. Pharmacol Ther. 2002;94:93–112. doi: 10.1016/s0163-7258(02)00174-2. [DOI] [PubMed] [Google Scholar]
- Voets T, Prenen J, Fleig A, Vennekens R, Watanabe H, Hoenderop JG, Bindels RJ, Droogmans G, Penner R, Nilius B. CaT1 and the calcium release-activated calcium channel manifest distinct pore properties. J Biol Chem. 2001;276:47767–47770. doi: 10.1074/jbc.C100607200. [DOI] [PubMed] [Google Scholar]
- Walden PD, Gerardi C, Lepor H. Localization and expression of the α 1A-1, α 1B and α 1D-adrenoreceptors in hyperplastic and non-hyperplastic human prostate. J Urol. 1999;61:635–640. [PubMed] [Google Scholar]
- Watanabe H, Davis JB, Smart D, Jerman JC, Smith GD, Hayes P, Vriens J, Cairns W, Wissenbach U, Prenen J, Flockerzi V, Droogmans G, Benham CD, Nilius B. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem. 2002;277:13569–13577. doi: 10.1074/jbc.M200062200. [DOI] [PubMed] [Google Scholar]
- Wissenbach U, Niemeyer BA, Fixemer T, Schneidewind A, Trost C, Cavalie A, Reus K, Meese E, Bonkhoff H, Flockerzi V. Expression of CaT-like, a novel calcium-selective channel, correlates with the malignancy of prostate cancer. J Biol Chem. 2001;276:19461–19468. doi: 10.1074/jbc.M009895200. [DOI] [PubMed] [Google Scholar]
- Wu D, Katz A, Lee CH, Simon MI. activation of phospholipase C by α 1-adrenergic receptors is mediated by the α subunit of Gq family. J Biol Chem. 1992;67:25798–25802. [PubMed] [Google Scholar]
- Wu X, Babnigg G, Villereal ML. Functional significance of human trp1 and trp3 in store-operated Ca2+ entry in HEK-293 cells. Am J Physiol Cell Physiol. 2000;278:C526–536. doi: 10.1152/ajpcell.2000.278.3.C526. [DOI] [PubMed] [Google Scholar]
- Zhang L, Saffen D. Muscarinic acetylcholine receptor regulation of TRP6 Ca2+ channel isoforms. Molecular structures and functional characterization. J Biol Chem. 2001;276:13331–13339. doi: 10.1074/jbc.M008914200. [DOI] [PubMed] [Google Scholar]
- Zhong H, Minneman KP. α 1-adrenoceptor subtypes. Eur J Pharmacol. 1999;375:261–276. doi: 10.1016/s0014-2999(99)00222-8. [DOI] [PubMed] [Google Scholar]
- Zhu X, Jiang M, Birnbaumer L. Receptor-activated Ca2+ influx via human Trp3 stably expressed in human embryonic kidney HEK293 cells. Evidence for a non-capacitative Ca2+ entry. J Biol Chem. 1998;273:133–142. doi: 10.1074/jbc.273.1.133. [DOI] [PubMed] [Google Scholar]
- Zitt C, Halaszovich CR, Luckhoff A. The TRP family of cation channels: probing and advancing the concepts on receptor-activated calcium entry. Progr Neurobiol. 2002;66:243–264. doi: 10.1016/s0301-0082(02)00002-3. [DOI] [PubMed] [Google Scholar]
- Zitt C, Obukhov AG, Strubing C, Zobel A, Kalkbrenner F, Luckhoff A, Schultz G. Expression of TRPC3 in Chinese hamster ovary cells results in calcium-activated cation currents not related to store depletion. J Cell Biol. 1997;138:1333–1341. doi: 10.1083/jcb.138.6.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]