

Abstract
In addition to exerting long-term neurotrophic influences on developmental process such as neuronal survival and neuritic outgrowth, brain-derived neurotrophic factor (BDNF) has been reported to modulate synaptic transmission in the short-term. Considerable evidence indicates that BDNF acutely modulates NMDA receptor-mediated synaptic activity. However, whether BDNF modulates inhibitory synaptic transmission remains to be firmly established. In the present study, we examined the effect of acute BDNF exposure on GABA-evoked whole-cell responses as well as GABAergic synaptic activity in cultured mouse cerebellar granule cells. GABA-evoked responses were reduced by 39.5 ± 4.7 % upon acute and focal application of BDNF (100 ng ml−1). The reduction of the GABA response recovered only partially even minutes after removal of BDNF. TrkB-IgG and K252a, but not K252b, prevented the BDNF-induced attenuation of the GABA response. BDNF exposure shifted the cumulative peak amplitude distribution leftward for both spontaneous IPSCs (sIPSCs) and miniature IPSCs (mIPSCs) without affecting the rise time and decay time constants. Acute exposure to BDNF also resulted in internalization of GABAA receptors in cultured cerebellar granule cells, as reflected by diminished immunostaining with an antibody against the GABAA receptor β2/3 subunit. Although the BDNF-induced GABAA receptor internalization was sensitive to K252a, it did not become manifest until 5 min after exposure to BDNF. Therefore, receptor internalization alone cannot account for the prompt BDNF-induced attenuation of GABA-mediated activity. We conclude that BDNF modulates GABAA receptor-mediated activity through TrkB receptor signalling that triggers a kinase-dependent short latency effect and a delayed longer latency effect hallmarked by receptor internalization.
Brain-derived neurotrophic factor (BDNF) plays a pivotal role in regulating synaptic plasticity in the central nervous system (see McAllister et al. 1999 for review). It is present in both the developing and mature central nervous system, and exerts its effects through the activation of the tyrosine receptor kinase B (TrkB) receptor, which, in turn, triggers a multitude of downstream signalling events (Huang & Reichardt, 2001). The expression and release of BDNF are regulated by synaptic activity. For example, activation of glutamate receptors or depolarization by high [K+] increases the expression of BDNF mRNA in cultured rat hippocampal neurons and cortical neurons (Lu et al. 1991; Zafra et al. 1992; Lindholm et al. 1994; Ghosh et al. 1994). Repetitive seizure activity augments BDNF mRNA levels in the hippocampus (Zafra et al. 1990). In hippocampal slices, patterned electrical stimulation or K+-induced depolarization results in increased secretion of BDNF (Blochl & Thoenen, 1995; Goodman et al. 1996).
Acute exposure to BDNF enhances the frequency and amplitude of glutamate-mediated excitatory postsynaptic currents (EPSCs) in the hippocampus (Lessmann et al. 1994; Kang & Schuman, 1995; Levine et al. 1996, 1998). In addition, disruption of BDNF or TrkB receptor expression results in deficits in long-term potentiation in hippocampal slices (Korte et al. 1995; Patterson et al. 1996; Kang et al. 1997). Thus, beyond its role as a permissive survival factor, the neurotrophin has been implicated in modulating synaptic transmission and plasticity. One proposed mechanism is that BDNF increases Ca2+ in presynaptic glutamatergic terminals to facilitate transmitter release (Kang & Schuman, 1995; Li et al. 1998). However, other studies propose mechanisms favouring a postsynaptic enhancement of NMDA receptor-mediated synaptic transmission (Levine et al. 1998). With regard to GABA-mediated inhibitory transmission, BDNF either attenuates or potentiates the amplitude of IPSCs, and both pre- and postsynaptic mechanisms have been independently proposed (Takana et al. 1997; Frerking et al. 1998; Boxall, 2000; Brunig et al. 2001). Overall, elucidating the nature of a BDNF-induced modulation of synaptic transmission will contribute toward identifying mechanisms underlying the cross-talk between BDNF, TrkB receptor signalling and neurotransmitter receptor function.
In this study, we investigated whether acute exposure to BDNF modulates responses of cerebellar granule cells to GABA. We report that BDNF suppresses GABA-mediated responses, hallmarked by a reduction in GABA-activated whole-cell current response as well as suppression of inhibitory synaptic events. Mechanistically, our results suggest that a sequel of at least two phases account for the BDNF-induced attenuation of GABAergic synaptic transmission. Specifically, by triggering the TrkB receptor-coupled intracellular cascade, BDNF suppresses postsynaptic GABA-mediated activity in an early phase and internalization of GABAA receptors in a later phase.
METHODS
Cerebellar granule cell culture
All procedures involving animals were carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals. C57BL/6J mouse pups (5–6 days postnatal) were decapitated and cerebella were isolated and immersed in ice-cold PBS. The cerebellar tissue was minced and enzymatically digested with trypsin (0.025 %; Worthington, Lakewood, NJ, USA) for 12 min at 37 °C. Cells were dissociated by triturating in 0.1 % DNAse (Sigma, St Louis, MO, USA), plated onto laminin-coated 12 mm round glass coverslips at 5 × 104 cells coverslip−1, and maintained at 37 °C in a humidified 95 % O2-5 % CO2 environment. The culture medium was Neurobasal medium (Gibco) to which was added 2 mm l-glutamine, B-27 supplement and penicillin-streptomycin-fungizone (Gibco). The concentrations of glucose and KCl in the medium were adjusted to 5 mg ml−1 and 25 mm, respectively. Forty-eight hours after plating, the cultures were treated with 6 μm fluorodeoxyuridine for 24–48 h to suppress the growth of glia (Rieff et al. 1999). Thereafter, the medium was changed every 2 days until used for experiments. After 7–10 days in vitro (DIV) cerebellar granule cells were used for the experiments reported in the present study.
Whole-cell patch-clamp recording, data acquisition and analysis
Patch electrodes were pulled from borosilicate capillary glass (1.5 mm o.d.; Sutter Instrument Co., Novato, CA, USA) and fire-polished to a bubble number of 4.2. The electrodes had resistances ranging between 8 and 10 MΩ when filled with internal solution. The internal (recording) solution was composed of (mm): 140 KCl, 4 NaCl, 2 MgCl2, 10 potassium EGTA, 10 Hepes, 2 magnesium ATP and 3 magnesium GTP (pH 7.3 with KOH). The external (bath) solution contained (mm): 140 NaCl, 2.8 KCl, 1 MgCl2, 3 CaCl2 10 Hepes, 10 glucose (pH 7.4 with NaOH). Recordings were made using an AxoPatch 200A amplifier (Axon Instruments, Foster City, CA, USA). Membrane currents were filtered at 5 kHz, digitized using Clampex v8.0 and analysed with Clampfit v8.0 (Axon Instruments) and Mini Analysis software (Version 5.1, Synaptosoft, Decatur, GA, USA) software. Analog signals were monitored on-line using a chart recorder (Gould, Valley View, OH, USA). Statistical analysis was performed using Sigmaplot 5.1 (SPSS Inc., Chicago, IL, USA). Mean peak current and frequency were analysed using Student's paired t test. The peak amplitude of GABA-mediated synaptic events was analysed using the Kolmogorov-Smirnov two-sample distribution test. Data are reported as mean ±s.e.m.
Focal applications of drugs
All drugs, including GABA (20 μm) and BDNF (20 or 100 ng ml−1), were dissolved in external solution, stored as frozen stock solutions and diluted to working concentrations with external solution immediately prior to each recording session. Drug solutions were loaded into separate barrels of an 8-barrel glass pipette assembly. The multibarrel assembly was constructed using fibre-filled borosilicate glass capillary tubes (Sutter Instrument Co.), pulled to a fine point, filled with the desired drug solutions and the tip was broken under microscopic control such that the outer diameter of each barrel was approximately 1.5 μm. The multibarrel assembly was mounted onto a hydraulically driven micromanipulator and navigated to within 10 μm of the cell under study for focal delivery of drugs. One of the barrels of the multibarrel assembly was routinely filled with external solution, which was applied between drug applications to clear drugs from the vicinity of the cell, and to control for mechanical artifacts due to bulk flow. In addition, since all drugs were dissolved in external solution, this also served as vehicle control to confirm drug effects. The application of drugs was always directed at the soma of cerebellar granule cells under study. Drug solutions were ejected by regulated pulses of pressure (< 3 p.s.i.; Picospritzer, General Valve Company, Fairfield, NJ, USA), with timing and duration controlled by a digital timing unit (Pulsemaster A300, World Precision Instruments, New Haven, CT, USA). Recombinant human BDNF was purchased from Peprotech Inc. (Rocky Hill, NJ, USA); TrkB-Fc protein was a gift from Regeneron (Tarrytown, NY, USA); K252a and K252b were purchased from Alomone Labs (Jerusalem, Israel); all other chemicals were purchased from Sigma.
Immunohistochemistry and analysis of immunofluorescence
Cerebellar granule cell cultures were fixed in 4 % paraformaldehyde in PBS (PFA-PBS) for 20 min and then incubated in 10 % normal goat serum for 30 min under either non-permeabilized or permeabilized conditions (0.2 % Triton X-100 in PBS). All subsequent reactions were carried out in the presence of 2 % normal goat serum. Double immunohistochemical labelling of TrkB receptor and GABAA receptor β2/3 subunit proteins was accomplished by overnight incubation with both mouse anti-β2/3 antibody (1:100) (Upstate Technology, Lake Placid, NY, USA) and rabbit anti-TrkB receptor antibody (1:1000) (Chemicon, Temecula, CA, USA) at 4 °C followed by exposure for 1 h at room temperature to anti-mouse IgG-Alexa 488 and anti-rabbit IgG-Alexa 594 secondary antibodies (Molecular Probes, both at 1:1000). Sister cultures processed in parallel without the addition of primary antibodies served as negative controls.
Immunofluorescence images were viewed and captured using the Olympus confocal microscope system FV300 (Olympus, Melville, NY, USA), under × 60 or × 100 oil objectives (aperture: 1.4), and edited using Photoshop 5.5 (Adobe System Incorporated, San Jose, CA, USA). The images were then digitized to a 256 grey-scale level, and the relative intensity of immunofluorescence was analysed based on a threshold segmentation algorithm using the MCID elite software (Image Research Inc., Ontario, Canada). Density thresholds were set such that immunoreactivity was distinguished from background, and subtracted from each measure. The median density levels were normalized to control. For each treatment, randomly selected neurites from 30–40 neurons were measured.
RESULTS
BNDF attenuates GABA-evoked whole-cell current in cultured cerebellar granule cells
We first assessed whether BDNF alters GABAA receptor-mediated current response. Whole-cell current responses of cerebellar granule cells (7–10 DIV) to 1 s pressure pulses of GABA (20 μm) applied at 20 s intervals for periods of up to 10 min were monitored continuously before, during and after exposure to BDNF (100 ng ml−1). Figure 1A illustrates a penwriter record taken from a cerebellar granule cell. Acute and focal application of BDNF resulted in a prompt attenuation in the amplitude of GABA-evoked current response that stabilized within 1 min of BDNF exposure. Overall, BDNF exposure attenuated GABA responses in 28 of 35 cells tested, with the magnitude of attenuation ranging from 18 to 56 % (39.5 ± 4.7 %; mean ±s.e.m.; P < 0.01, Student's t test) (Fig. 1C). A consistent observation was that recovery of responses to GABA from BDNF exposure was only partial (approximately 70 %), even after 5 min of removal of BDNF. The degree of recovery appeared to be dependent on the duration of exposure to BDNF insofar as shorter exposure times resulted in more complete recovery of GABA response to control levels. Two lines of evidence indicate that the observed attenuation of GABA response and partial recovery are not due to run-down or desensitization under our experimental protocol and recording conditions. Firstly, as illustrated in Fig. 1A, whole-cell current responses to GABA were reproducible in amplitude prior to exposure to BDNF. Secondly, in control experiments in which GABA responses were monitored continuously without exposure to BDNF over equivalent lengths of time, the GABA responses remained stable throughout the recording, displaying only 1–5 % fluctuation in amplitude (Fig. 2A).
Figure 1. Acute exposure to brain-derived neurotrophic factor (BDNF) attenuates GABA-evoked whole-cell current in cerebellar granule cells.
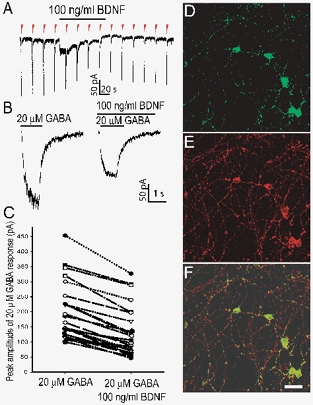
A, representative penwriter record displays current responses evoked by a 1 s application of GABA (20 μm) delivered every 20 s as denoted by the red arrowheads. Exposure to BDNF (100 ng ml−1) (continuous line above the arrowheads) reduces the peak amplitude of GABA responses. Note the depolarizing current during acute BDNF exposure in this example. B, digitized traces recorded in the absence (left) and presence (right) of BDNF are shown in expanded time scale. C, scatter plot summarizing the change in peak amplitude of GABA responses upon exposure to BDNF monitored in 20 granule cells. Each pair of connected points represents data derived from one cell. Confocal images of cultured cerebellar granule cells stained with antibodies against TrkB receptor (D) and synaptophysin (E), using green and red fluorescence respectively. F, overlay of D and E. Scale bar = 20 μm applies to D, E and F.
Figure 2. TrkB-IgG and K252a, but not K252b, prevent BDNF-induced reduction of GABA response.
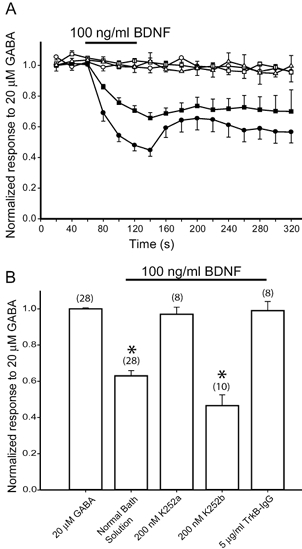
A, peak amplitude of responses to GABA (20 μm) plotted as a function of time before, during and after exposure to 100 ng ml−1 BDNF. The continuous line denotes time and duration of BDNF application. GABA responses were monitored in normal bath solution without exposure to BDNF during recording (□), in normal bath solution with a 1 min exposure to BDNF (▪), and with added K252a (200 nm; ○), K252b (200 nm; •) and TrkB-IgG (5 μg ml−1; ▵). Each point represents data averaged from 8–10 cells. B, bar graph summarizing mean peak amplitude of GABA responses as a function of the conditions in A.
Acute BDNF exposure also induced a depolarization current in 20 of 35 granule cells tested. Although not examined in detail in the present study, this is likely to be due to the opening of Na+ channels, as been reported previously (Kafitz et al. 1999; Urbano & Buno, 2000). Since an attenuation of GABA response has been observed in cells that do not display the depolarizing current response (data not shown), these two BDNF-induced effects appear to be independent of each other.
The effect of BDNF is mediated by TrkB receptor
In a second series of experiments, we asked whether the observed effect of BDNF was mediated by the TrkB receptor. Both in situ hybridization and immunohistochemistry revealed the presence of TrkB receptor in cerebellar granule cells (Klein et al. 1990; Segal et al. 1995). We confirmed the expression of TrkB receptor in cultured cerebellar granule cells. Figure 1D, E and F shows that TrkB immunoreactivity overlaps with that of synaptophysin, a presynaptic protein used as synaptic marker, indicating that TrkB receptor is present in both somatic and synaptic membranes. Our imaging system precluded identification of presynaptic versus postsynaptic locus of the TrkB receptors.
We then tested whether the addition of 200 nm K252a, an inhibitor of autophosphorylation of tyrosine receptor kinases, or 200 nm K252b, its non-functional analogue (Knusel & Hefti, 1992), alters the effect of BDNF on GABA-evoked current responses. Granule cell cultures were pre-incubated with either K252a or K252b for 15 min. Then experiments were conducted in the presence of either K252a or K252b. In the presence of K252a, the peak response to 20 μm GABA fluctuated about 4.5 ± 0.6 % around the mean value but was not reduced upon addition of 100 ng ml−1 BDNF. In contrast, the reduction of the GABA response induced by BDNF persisted in the presence of 200 nm K252b (Fig. 2A and B). In Fig. 2B, the reduction in the mean peak amplitude of GABA response appeared to be greater upon exposure to K252b and BDNF than to BDNF alone. However, this difference was not statistically significant. Since K252a also inhibits tyrosine receptor kinases other than TrkB, we confirmed further the involvement of the TrkB receptor by using the TrkB receptor antibody (TrkB-IgG), which competes with native TrkB receptors to sequester BDNF and prevent TrkB receptor activation (Davis et al. 1994). TrkB-IgG (5 μg ml−1) has been shown to be effective in blocking BDNF-dependent long-term potentiation in hippocampal slices (Kang et al. 1997). At this concentration of TrkB-Fc, GABA-evoked responses remained unaltered after exposure to 100 ng ml−1 BDNF. Thus, consistent with the experiments employing K252a, TrkB-Fc blocked BDNF-induced modulation. These results indicate that the acute effect of BDNF depends on activation of the TrkB receptor.
BDNF decreases the amplitude and frequency of spontaneous IPSCs
To determine whether the BDNF-induced attenuation of GABA-mediated responses could be resolved at the synaptic level, we recorded spontaneous GABAergic inhibitory postsynaptic currents (sIPSCs) in the absence and presence of BDNF in the same cerebellar granule cells. The antagonists 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μm) and dl-2-amino-5-phosphonovaleric acid (APV, 20 μm) were added to the external solution to block AMPA and NMDA receptor-mediated synaptic currents, respectively. Under each condition, sIPSC events were collected at 34 °C over a period of at least 5 min in order to obtain sufficient number of events (> 100 events) for analysis. Figure 3A shows inwardly directed events recorded in the presence of CNQX and APV from a cultured cerebellar granule cell held at −60 mV. These sIPSCs were completely and reversibly blocked by bicuculline (10 μm), indicating mediation by synaptic GABAA receptors (data not shown). In the absence of BDNF, the frequency of spontaneous IPSCs ranged between 0.3 and 1.8 Hz, with a mean amplitude of 36.5 ± 3.7 pA (mean ±s.e.m.). Upon a 5 min focal exposure to BDNF (20 ng ml−1), the amplitude of sIPSCs were reduced to 27.3 ± 2.3 pA and remained at this level for at least 10 min (Fig. 3B). Cumulative distribution of amplitude (Fig. 3C) revealed a leftward shift during exposure to BDNF (n = 9, P < 0.05 by two-sample Student's t test and Kolmogorov-Smirnov two-sample test). The frequency of sIPSCs decreased from 0.94 ± 0.2 to 0.34 ± 0.1 Hz. Cumulative distribution of inter-event interval (Fig. 3D) revealed a rightward shift during exposure to BDNF (n = 9, P < 0.05 by two-sample Student's t test and Kolmogorov-Smirnov two-sample test).
Figure 3. BDNF decreases the amplitude and frequency of spontaneous IPSCs (sIPSCs) in cerebellar granule cells.
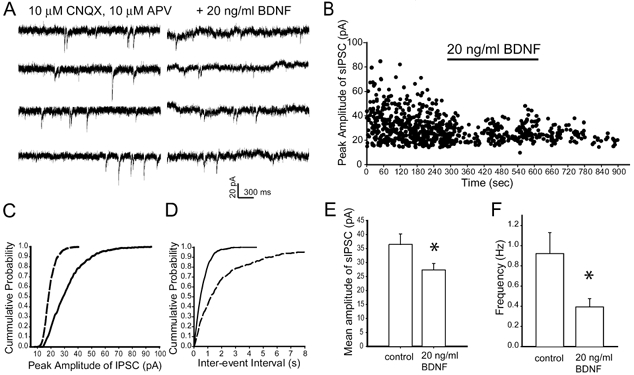
A, representative traces recorded in the presence of 10 μm CNQX and 10 μm APV, without (left panel) or with (right panel) BDNF (20 ng ml−1); holding potential −60 mV. B, peak amplitude of sIPSCs plotted as a function of time. Upon a 5 min exposure to BDNF, both amplitude and frequency of sIPSCs declined. C, cumulative distribution of peak amplitude of sIPSCs in the presence of BDNF (dashed line) shifted leftward compared to that obtained under control conditions (continuous line). D, exposure to BDNF (dashed line) shifted rightward the cumulative distribution of inter-event interval of sIPSCs compared to that obtained under control conditions (continuous line). E and F, histograms summarizing mean amplitude (E) and frequency (F) of sIPSCs derived from 8 cells; the mean amplitude decreased from 37 + 3.7 to 26 + 2.3 pA; frequency decreased from 0.94 ± 0.2 to 0.34 ± 0.1 Hz. * Statistically significant (Student's paired t test, P < 0.05).
While the attenuation of sIPSC amplitude is consistent with data on whole-cell GABA response, the decrease in frequency of sIPSCs, which is typically attributed to reduction of presynaptic transmitter release, could also be due to a suppression of postsynaptic GABAA receptor. To address this issue, we included K252a (200 nm) in the internal solution and recorded sIPSCs before, during and after exposure to BDNF. With K252a present intracellularly, the BDNF-induced attenuation of GABA response amplitude was no longer observed (data not shown). These results suggest that postsynaptic mechanisms contribute to the observed BDNF-induced effect.
BDNF decreases amplitude and frequency of miniature IPSCs
To assess further the BDNF-induced postsynaptic modification of GABA-mediated synaptic activity, we examined the properties of miniature IPSCs (mIPSCs) that are independent of action potential-evoked presynaptic release of neurotransmitter (Otis & Mody, 1992). In the absence of BDNF, the frequency of mIPSCs ranged from 0.2 to 1.5 Hz, with a mean amplitude of 25.8 ± 2.1 pA. Multiexponential fits to averaged events revealed a fast rise time (trise = 2.9 ± 0.2 ms) and a biexponential decay time course (τdec1 = 7.0 ± 0.3 ms and τdec2 = 38.2 ± 1.9 ms) (n = 8). In the presence of BDNF (20 ng ml−1), the amplitude of mIPSCs recorded differed significantly from those obtained under the control condition (20.7 ± 1.8 pA) (paired t test, P < 0.05). The frequency of mIPSCs decreased to 0.3 ± 0.1 Hz (Fig. 4). However, the kinetic profiles of these events were indistinguishable from control (trise = 3.2 ± 0.3 ms, τdec1 = 6.5 ± 0.3 ms and τdec2 = 33.8 ± 1.4 ms; n = 8; P < 0.5 by two-sample Student's t test) (Table 1). These results indicate that BDNF attenuates the peak amplitude and decreases the frequency of mIPSCs through a postsynaptic alteration of GABAA receptor properties. Nonetheless, it should be noted that our results do not exclude the possibility that presynaptic mechanisms regulating probability of transmitter release may also contribute to the synaptic effects of BDNF.
Figure 4. Exposure to BDNF decreases the amplitude and frequency of miniature IPSCs (mIPSCs).
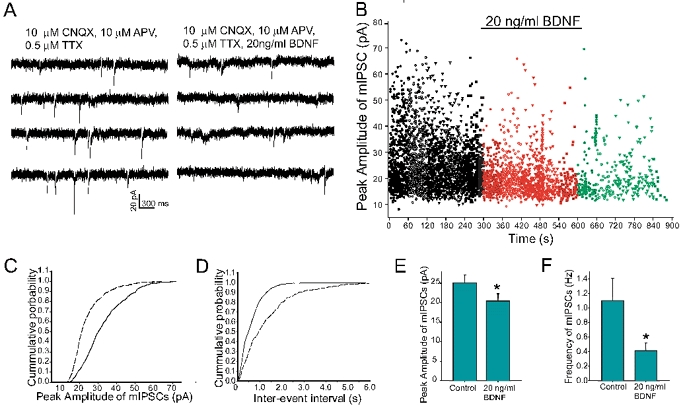
A, representative traces recorded in the presence of 10 μm CNQX, 10 μm APV and 0.5 μm TTX, without (lefthand traces) or with (righthand traces) BDNF (20 ng ml−1); holding potential −60 mV. B, peak amplitude of mIPSCs plotted as a function of time before, during and after exposure to BDNF. There are fewer large-size amplitude events during BDNF exposure (red dots) than during the control period (black dots). C, cumulative distribution of peak amplitude of mIPSCs in the presence of BDNF (dashed line) shifted leftward compared to that obtained under control conditions (continuous line). D, exposure to BDNF (dashed line) shifted rightward the cumulative distribution of inter-event interval of mIPSCs compared to that obtained under control conditions (continuous line). E and F, histograms summarizing data derived from 8 cells; exposure to BDNF decreased the mean amplitude (E) and frequency (F) of mIPSCs from 25.8 ± 2.1 to 20.4 ± 0.8 pA and 1.1 ± 0.3 to 0.41 ± 0.3 Hz, respectively. * Statistically significant (Student's paired t test, P < 0.05).
Table 1.
Properties of IPSCs in the absence and presence of 20 ng ml−1 BDNF
| Mean | sIPSC | sIPSC + BDNF | mlPSC | mlPSC + BDNF |
|---|---|---|---|---|
| Amplitude (pA) | 36.5 ± 3.7 | 27.3 ± 2.3* | 25.8 ± 2.1 | 20.4 ± 1.8* |
| Frequency (Hz) | 0.94 ± 0.2 | 0.34 ± 0.1* | 1.1 ± 0.3 | 0.41 ± 0.3* |
| trise (ms) | 3.2 ± 0.4 | 3.3 ± 0.4 | 3.1 ± 0.2 | 2.9 ± 0.3 |
| Tdec1, (ms) | 3.7 ± 0.6 | 3.3 ± 0.7 | 2.9 ± 0.5 | 2.7 ± 0.4 |
| Tdec2(ms) | 18.2 ± 2.8 | 16.9 ± 3.1 | 14.7 ± 2.3 | 12.9 ± 1.8 |
| No. of cells | 9 | 9 | 8 | 8 |
BDNF, brain-derived neurotrophic factor; sIPSC, spontanteous IPSC; mlPSC miniature IPSC; trise, the 10–90% rise time; Tdecl and Tdec2 are time constants of the fast and slow phases of the biexponential decay, respectively. Data are presented as means ±s.e.m.
Significant difference between the absence and presence of BDNF (20 ng ml−1) by paired Student's t test at P < 0.05.
BDNF reduces immunoreactivity of cell surface GABAA receptor β2/3 subunits
A recent study (Brunig et al. 2001) reported that a 30 min exposure to BDNF reduced the surface immunoreactivity of GABAA receptor α2 subunits in cultured hippocampal neurons. We asked whether receptor internalization could account mechanistically for the observed postsynaptic attenuation of GABA responses by BDNF. Cerebellar granule cells were exposed to BDNF for 1, 5, 15, 30 and 60 min, fixed with 4 % PFA-PBS and subsequently processed for double-label immunohistochemistry using the GABAA receptor β2/3 subunit and TrkB antibodies. As illustrated in Fig. 5A1, GABAA receptor β2/3 subunit immunoreactivity appeared punctate, outlining the cell body and neurites of granule cells. TrkB immunostaining revealed somatic and neuritic distribution, and overlay of the two staining patterns indicated co-localization of GABAA receptors and TrkB receptors. No immunostaining was observed in negative control cultures with the primary antibodies omitted.
Figure 5. Exposure to BDNF reduces GABAA receptor β2/3 subunit immunoreactivity in a time-dependent manner.
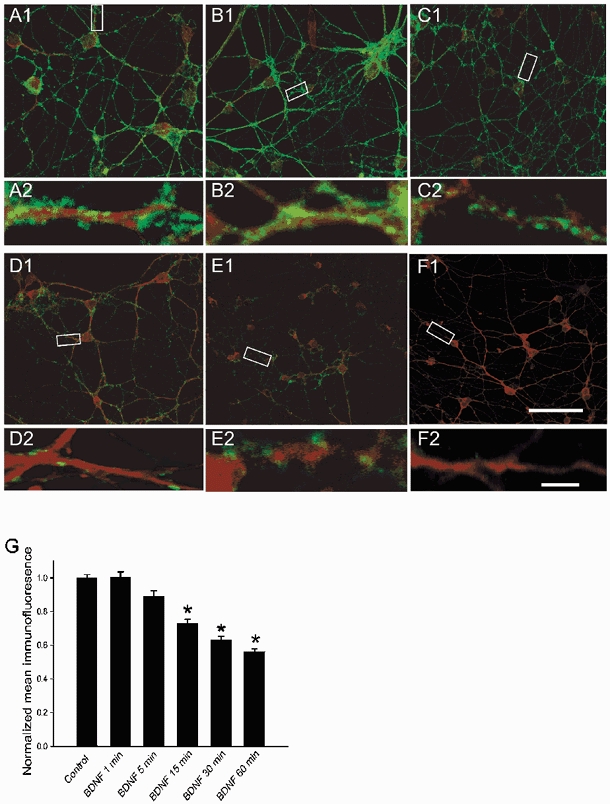
Confocal images of cultured cerebellar granule cells before (A1) and following a 1 (B1), 5 (C1), 15 (D1), 30 (E1) and 60 (F1) min exposure to BDNF (100 ng ml−1). The cultures were fixed and double-labelled with antibodies against GABAA receptor β2/3 subunits (green fluorescence) and TrkB receptors (red fluorescence). A2, B2, C2, D2, E2 and F2, GABAA receptor β2/3 subunit immunoreactive profiles corresponding to the boxed areas in A1-F1 are resolved at higher resolution (× 60 oil) to illustrate decreased immunoreactivity along neurites following a > 5 min exposure to BDNF. Scale bars, 50 μm in A1-F1 and 5 μm in A2-F2. The images illustrated above were digitized; relative median density level was determined for each treatment and normalized to control. G, data are presented as means ±s.e.m. * Significant difference between control and the corresponding times of exposure to BDNF (100 ng ml−1) (Student's t test at P < 0.01).
After a 1 min incubation with 100 ng ml−1 BDNF, there was no apparent change in GABAA receptor β2/3 subunit immunoreactivity (Fig. 5B1). However, after a 5 min exposure to BDNF, the reduction in GABAA receptor β2/3 subunit immunoreactivity became evident in so far as the number and size of the puncta decreased. This can be discerned to better advantage at a higher magnification (Fig. 5C2). With longer periods of treatment with BDNF, the reduction of GABAA receptor β2/3 subunit immunoreactivity became increasingly striking. In contrast, the TrkB receptor staining remained unchanged at all time points tested. Performing the experiments under either non-permeabilized or permeabilized conditions yielded similar results. Figure 5G summarizes the change in mean immunofluorescence as a function of duration of exposure to BDNF (1–60 min). A statistically significant decrease in immunofluorescence was first detected after 15 min of exposure to BDNF.
Pretreatment of cerebellar granule cell cultures with K252a (200 nm), but not with K252b (200 nm), prevented the reduction of GABAA receptor β2/3 subunit immunoreactivity (Fig. 6). Thus, as is the case with attenuation of GABA responses, BDNF reduces GABAA receptor β2/3 subunit immunoreactivity through a tyrosine receptor kinase-dependent pathway.
Figure 6. K252a blocks BDNF-induced reduction of cell surface GABAA receptor β2/3 subunit immunoreactivity.
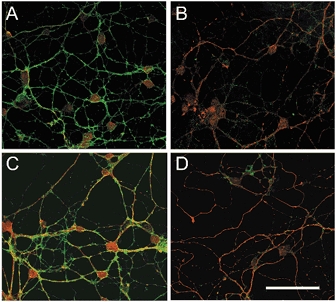
Confocal images of cultured cerebellar granule cells before (A) and following a 60 min exposure to BDNF (100 ng ml−1) (B, C and D). The cultures were fixed and co-stained with antibodies against GABAA receptor β2/3 subunits (green fluorescence) and TrkB receptors (red fluorescence). GABAA receptor β2/3 subunit immunoreactivity, illustrated in A, is markedly reduced following exposure to BDNF (B). C and D, addition of K252a (200 nm) prevented the BDNF-induced reduction of GABAA receptor β2/3 subunit immunoreactivity (C), which persisted in the presence of K252b (200 nm; D). Scale bar (applies to all panels), 50 μm.
Our electrophysiological experiments demonstrating partial recovery from a BDNF-induced attenuation of the GABA response (Fig. 2) suggest that recovery is likely to be a slow process. A possible mechanism to account for such slow recovery is the time course of reversal of BDNF-induced internalization of GABAA receptors. To test this idea, cerebellar granule cell cultures were first incubated in medium containing BDNF (100 ng ml−1) for 5 and 60 min, simulating an acute and longer term exposure, respectively. The medium containing BDNF was subsequently replaced with normal medium and individual cultures were fixed for immunohistochemical processing at selected time points between 15 min and 18 h after removal of BDNF. Figure 7 illustrates that, with both 5 min (Fig. 7B1 and B2) and 60 min (Fig. 7C1 and C2) exposure, the diminution of GABAA receptor β2/3 subunit immunoreactivity was gradually reversible over 30 min after removal of BDNF. We have not previously recorded from cultured cerebellar granule cells for this length of time due to technical difficulties in maintaining a stable recording and reproducible responses to repeated applications of GABA. However, the relatively slow time course of recovery from receptor internalization could at least in part account for the observed incomplete recovery of GABA responses within the first 5 min following exposure to BDNF.
Figure 7. Recovery from BDNF-induced reduction of GABAA receptor β2/3 subunit immunoreactivity.
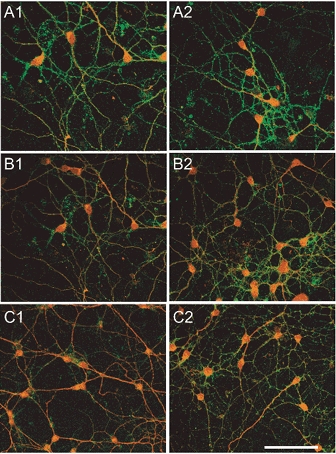
Before (A1 and A2) and following bath application of BDNF (100 ng ml−1) for 5 min (B1 and B2) and 60 min (C1 and C2), a set of cerebellar granule cell cultures were fixed and processed for GABAA receptor β2/3 subunit (green fluorescence) and TrkB receptor (red fluorescence) immunohistochemistry (A1, B1 and C1). Another set of sister cultures was returned to normal medium after the 5 and 60 min exposures to BDNF, and incubated for an additional 30 min before being processed immunohistochemically (A2, B2 and C2). In cultures exposed to BDNF for either 5 or 60 min, recovery of GABAA receptor β2/3 subunit immunoreactivity occurred by 30 min after washout of the neurotrophin. Scale bar (applies to all panels), 50 μm.
DISCUSSION
This study examined the interaction between acute exposure to BDNF and GABA-activated current responses in cultured cerebellar granule cells. In our culture system, as in vivo (Klein et al. 1990; Segal et al. 1995), cerebellar granule cells express an abundance of TrkB receptors. The major findings of our study are threefold. Firstly, focally applied BDNF acutely attenuates cerebellar granule cell responses to GABA delivered exogenously or released endogenously at synaptic sites. Secondly, the BDNF-induced attenuating effect is mediated by the TrkB receptor. Thirdly, exposure to BDNF results in internalization of GABAA receptors, as reflected by diminished cell surface immunostaining of the GABAA receptor β2/3 subunit. Both the BDNF-induced attenuation and internalization of GABAA receptors are dependent on kinase activity downstream of receptor activation. Taken together, our results demonstrate the presence of postsynaptic mechanisms by which BDNF attenuates GABA-mediated responses, including an early phase characterized by a tyrosine receptor kinase-dependent suppression of GABAA receptor-mediated activity and a later phase hallmarked by internalization of GABAA receptors.
BDNF attenuation of GABAergic synaptic activity
In the present study, BDNF reduced the frequency and amplitude of both the spontaneous and miniature IPSCs. Typically, a decrease in the amplitude of IPSCs implies a change in the properties and/or number of postsynaptic GABAA receptors. On the other hand, a decrease in the frequency of IPSCs implicates reduced presynaptic neurotransmitter release. We defined sIPSCs as inhibitory postsynaptic currents inherent in our culture system that were recorded without intentional manipulation or stimulation to induce transmitter release, and mIPSCs as those that were monitored in the presence of TTX and were thus independent of action potential-induced transmitter release. In fact, the amplitude and frequency of GABA-mediated synaptic currents were similar in the absence or presence of TTX, suggesting that action potential-dependent transmitter release is but a minor component of the overall spontaneous synaptic activity.
At first glance, our data on sIPSCs suggest involvement of both pre- and postsynaptic mechanisms. However, further analysis revealed that the decrease in frequency persisted even when mIPSCs were monitored. Since mIPSCs are not influenced by action potential-dependent presynaptic release processes, the apparent reduction in frequency is likely to be a consequence of reduced IPSC amplitude. In addition, direct intracellular delivery of K252a prevented the BDNF-induced attenuation of spontaneous IPSCs. These data lead us to conclude that the neurotrophin modifies properties of postsynaptic GABAA receptors, thereby reducing GABAA receptor-mediated synaptic events. Indeed, at the ultrastructural level, TrkB receptor immunoreactivity has been localized to the postsynaptic membrane (Aoki et al. 2001). In addition, BDNF has been reported to enhance glutamatergic transmission through postsynaptic modulation of NMDA receptor activity (Levine et al. 1996, 1998). These studies provide evidence that BDNF can modulate synaptic transmission through postsynaptic mechanisms. However, it should be noted that K252a itself reduced the frequency of postsynaptic currents, and that this confounded our ability to definitively differentiate between the contribution of presynaptic and postsynaptic components in the BDNF-induced process. Thus, in a strict sense, the presence of a presynaptic effect of BDNF in modulating GABA-mediated synaptic transmission cannot be excluded based on available data.
BDNF-GABAA receptor cross-talk: possible mechanisms
How does acute exposure to BDNF attenuate GABA-mediated activity? We considered two possibilities, namely, modulation of GABAA receptor properties by TrkB receptor-mediated signalling and BDNF-induced internalization of GABAA receptors.
Mediation by TrkB receptor signalling
Pretreatment of cultured granule cells with either K252a or TrkB-Fc prevented the BDNF-induced diminution of the GABA response. Since granule cells do not express immunohistochemically detectable levels of the p75 receptor (Anderson et al. 1995), the BDNF-induced effect would appear to be mediated primarily through the TrkB receptor. In light of this, activation of the TrkB receptor and its downstream signalling pathways may trigger phosphorylation cascades, leading to altered affinity or channel conductance of GABAA receptors. The outcome of such a BDNF-GABAA receptor interaction would be expected to depend on several factors, including cell type-dependent expression of GABAA receptors harbouring different subunit combinations or distinct BDNF signalling pathways, or both. Indeed, BDNF exerts an attenuating effect on GABA-mediated IPSCs in rat hippocampal neurons (Tanaka et al. 1997) but enhances them in cerebellar Purkinje cells (Boxall, 2000). Other studies report that protein tyrosine kinase receptors modulate response to GABA in opposite ways. In cultured superior cervical ganglion neurons, intracellular application of vanadate, a tyrosine phosphatase inhibitor, increases the amplitude of GABA-activated current (Moss et al. 1995). In contrast, platelet-derived growth factor binds to its receptor tyrosine kinase and activates phospholipase C-γ in rat hippocampal neurons, resulting in Ca2+-dependent decreases in both synaptic and somatic responses to GABA (Valenzuela et al. 1995). In recombinant GABAA receptors expressed in HEK 293 cells, the presence of the β1, β2 or β3 subunit dictates the direction of modulation by cyclic AMP (McDonald et al. 1998), underscoring the dependence of phosphorylation-mediated modulation of GABAA receptors on subunit expression.
In our experiments, recovery from a BDNF-induced attenuation of the GABA-mediated response was only partial during our electrophysiological recordings. Since we have experimentally ruled out GABAA receptor run-down and desensitization as possible confounding factors, phosphorylation of TrkB receptors is likely to account for the observed partial recovery. Indeed, once phosphorylated, the TrkB receptor and its associated downstream signalling proteins can remain in this state for hours beyond removal of BDNF (Choi et al. 2001).
Finally, the neurotrophin 4/5 (NT4/5), via activation of the TrkB receptors, exerts effects that mimic those of BDNF (Rabacchi et al. 1999; Fan et al. 2000; Brunig et al. 2001). It would be reasonable to postulate that NT4/5 also attenuates GABA responses in cerebellar granule cells through TrkB receptor-mediated signalling. These and other related issues await experimentation in future studies.
Internalization of GABAA receptors
Another possible mechanism to account for our findings is receptor internalization. Our electrophysiological data indicate that exposure to BDNF decreases IPSC amplitude but leaves kinetic properties of GABAA receptors unchanged, suggesting a reduction in GABAA receptor numbers. Indeed, our immunohistochemical results indicate quantitatively that exposure to BDNF diminishes cell surface immunofluorescence of GABAA receptor β2/3 subunits, consistent with receptor internalization. Trafficking of receptors as a means of regulating the efficacy of synaptic transmission is quite prevalent. Activation of the insulin receptor, a member of the family of tyrosine kinase receptors, recruits GABAA receptors to the cell surface (Wan et al. 1997). Activation of protein kinase C reduces GABAA receptor α1 subunit immunoreactivity in the short term (Chapell et al. 1998). In HEK 293 cells, recombinant GABAA receptors undergo clathrin-independent endocytosis (Cinar & Barnes, 2001). Activation of mGluR1 induces a loss of AMPA and NMDA receptors, resulting in decreased frequency and amplitude of mEPSCs (Snyder et al. 2001). In addition, BDNF exposure decreases the number of cell surface GABAA receptors in cultured hippocampal neurons (Brunig et al. 2001). However, in all these cases, the onset of receptor internalization is 5–60 min following BDNF exposure, which is too slow to account for the rapid suppression of the GABA response of cerebellar granule cells observed in our study. We nonetheless consider that a BDNF-induced internalization of GABAA receptors with a relatively long time course (> 30 min) of recovery could explain the partial recovery of GABA responses consistently observed in our electrophysiological studies following removal of BDNF.
Finally, our data suggest that activation of the TrkB receptor and its downstream signalling cascade may be mechanistically a point of convergence for the early and later phases of the acute action of BDNF demonstrated in this study, characterized by attenuation of GABA responses and internalization of GABAA receptors, respectively. Overall, we propose that the effects of acute exposure to BDNF of prompt attenuation of GABAA receptor-mediated activity and a longer-latency internalization of GABAA receptors are not mutually exclusive and, in fact, are complementary and occur in tandem to account for the sequence of events.
Acknowledgments
This work was supported by PHS grants NS24830 and NS41489. The authors wish to thank Drs Ted Begenisich and Roman Giger for critical reading of the manuscript, and Drs C.-H. Chan, M. Ericson and Ms P. Yeh for technical assistance.
REFERENCES
- Anderson KD, Alderson RF, Altar CA, DiStefano PS, Corcoran TL, Lindsay RM, Wiegand SJ. Differential distribution of exogenous BDNF, NGF, and NT-3 in the brain corresponds to the relative abundance and distribution of high-affinity and low-affinity neurotrophin receptors. J Comp Neurol. 1995;357:296–317. doi: 10.1002/cne.903570209. [DOI] [PubMed] [Google Scholar]
- Aoki C, Wu K, Elste A, Len G, Lin S, McAuliffe G, Black IB. Localization of brain-derived neurotrophic factor and TrkB receptors to postsynaptic densities of adult rat cerebral cortex. J Neurosci Res. 2000;59:454–463. doi: 10.1002/(SICI)1097-4547(20000201)59:3<454::AID-JNR21>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Blochl A, Thoenen H. Characterization of nerve growth factor (NGF) release from hippocampal neurons: evidence for a constitutive and an unconventional sodium-dependent regulated pathway. Eur J Neurosci. 1995;7:1220–1228. doi: 10.1111/j.1460-9568.1995.tb01112.x. [DOI] [PubMed] [Google Scholar]
- Boxall AR. GABAergic mIPSCs in rat cerebellar Purkinje cells are modulated by TrkB and mGluR1-mediated stimulation of Src. J Physiol. 2000;524:677–684. doi: 10.1111/j.1469-7793.2000.00677.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunig I, Penschuck S, Berninger B, Benson J, Fritschy JM. BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABA(A) receptor surface expression. Eur J Neurosci. 2001;13:1320–1328. doi: 10.1046/j.0953-816x.2001.01506.x. [DOI] [PubMed] [Google Scholar]
- Chapell R, Bueno OF, Alvarez-Hernandez X, Robinson LC, Leidenheimer NJ. Activation of protein kinase C induces gamma-aminobutyric acid type A receptor internalization in Xenopus oocytes. J Biol Chem. 1998;273:32595–32601. doi: 10.1074/jbc.273.49.32595. [DOI] [PubMed] [Google Scholar]
- Choi DY, Toledo-Aral JJ, Segal R, Halegoua S. Sustained signaling by phospholipase C-gamma mediates nerve growth factor-triggered gene expression. Mol Cell Biol. 2001;21:2695–2705. doi: 10.1128/MCB.21.8.2695-2705.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinar H, Barnes EM., Jr Clathrin-independent endocytosis of GABA(A) receptors in HEK 293 cells. Biochemistry. 2001;40:14030–14036. doi: 10.1021/bi011025t. [DOI] [PubMed] [Google Scholar]
- Davis S, Gale NW, Aldrich TH, Maisonpierre PC, Lhotak V, Pawson T, Goldfarb M, Yancopoulos GD. Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science. 1994;266:816–819. doi: 10.1126/science.7973638. [DOI] [PubMed] [Google Scholar]
- Fan G, Egles C, Sun Y, Minichiello L, Renger JJ, Klein R, Liu G, Jaenisch R. Knocking the NT4 gene into the BDNF locus rescues BDNF deficient mice and reveals distinct NT4 and BDNF activities. Nat Neurosci. 2000;3:350–357. doi: 10.1038/73921. [DOI] [PubMed] [Google Scholar]
- Frerking M, Malenka RC, Nicoll RA. Brain-derived neurotrophic factor (BDNF) modulates inhibitory, but not excitatory, transmission in the CA1 region of the hippocampus. J Neurophysiol. 1998;80:3383–3386. doi: 10.1152/jn.1998.80.6.3383. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Hefti F. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci. 1996;7:222–238. doi: 10.1006/mcne.1996.0017. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafitz KW, Rose CR, Thoenen H, Konnerth A. Neurotrophin-evoked rapid excitation through TrkB receptors. Nature. 1999;401:918–921. doi: 10.1038/44847. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- Klein R, Martin-Zanca D, Barbacid M, Parada LF. Expression of the tyrosine kinase receptor gene trkB is confined to the murine embryonic and adult nervous system. Development. 1990;109:845–850. doi: 10.1242/dev.109.4.845. [DOI] [PubMed] [Google Scholar]
- Knusel B, Hefti F. K-252 compounds: modulators of neurotrophin signal transduction. J Neurochem. 1992;59:1987–1996. doi: 10.1111/j.1471-4159.1992.tb10085.x. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. NeuroReport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-d-aspartic acid receptor activity. Proc Natl Acad Sci U S A. 1998;95:10235–10239. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR. Selective role for trkB neurotrophin receptors in rapid modulation of hippocampal synaptic transmission. Brain Res Mol Brain Res. 1996;38:300–303. doi: 10.1016/0169-328x(96)00025-3. [DOI] [PubMed] [Google Scholar]
- Li YX, Xu Y, Ju D, Lester HA, Davidson N, Schuman EM. Expression of a dominant negative TrkB receptor, T1, reveals a requirement for presynaptic signaling in BDNF-induced synaptic potentiation in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 1998;95:10884–10889. doi: 10.1073/pnas.95.18.10884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm D, Castren E, Berzaghi M, Blochl A, Thoenen H. Activity-dependent and hormonal regulation of neurotrophin mRNA levels in the brain - implications for neuronal plasticity. J Neurobiol. 1994;25:1362–1372. doi: 10.1002/neu.480251105. [DOI] [PubMed] [Google Scholar]
- Lu B, Yokoyama M, Dreyfus CF, Black IB. Depolarizing stimuli regulate nerve growth factor gene expression in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 1991;88:6289–6292. doi: 10.1073/pnas.88.14.6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- McDonald BJ, Amato A, Connolly CN, Benke D, Moss SJ, Smart TG. Adjacent phosphorylation sites on GABAA receptor beta subunits determine regulation by cAMP-dependent protein kinase. Nat Neurosci. 1998;1:23–28. doi: 10.1038/223. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Gorrie GH, Amato A, Smart TG. Modulation of GABAA receptors by tyrosine phosphorylation. Nature. 1995;377:344–348. doi: 10.1038/377344a0. [DOI] [PubMed] [Google Scholar]
- Otis TS, Mody I. Modulation of decay kinetics and frequency of GABAA receptor-mediated spontaneous inhibitory postsynaptic currents in hippocampal neurons. Neuroscience. 1992;49:13–32. doi: 10.1016/0306-4522(92)90073-b. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Rabacchi SA, Kruk B, Hamilton J, Carney C, Hoffman JR, Meyer SL, Springer JE, Baird DH. BDNF and NT4/5 promote survival and neurite outgrowth of pontocerebellar mossy fiber neurons. J Neurobiol. 1999;40:254–269. [PubMed] [Google Scholar]
- Rieff HI, Raetzman LT, Sapp DW, Yeh HH, Siegel RE, Corfas G. Neuregulin induces GABA(A) receptor subunit expression and neurite outgrowth in cerebellar granule cells. J Neurosci. 1999;19:10757–10766. doi: 10.1523/JNEUROSCI.19-24-10757.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal RA, Pomeroy SL, Stiles CD. Axonal growth and fasciculation linked to differential expression of BDNF and NT3 receptors in developing cerebellar granule cells. J Neurosci. 1995;15:4970–4981. doi: 10.1523/JNEUROSCI.15-07-04970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4:1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbano FJ, Buno W. Neurotrophin regulation of sodium and calcium channels in human neuroblastoma cells. Neuroscience. 2000;96:439–443. doi: 10.1016/s0306-4522(99)00552-7. [DOI] [PubMed] [Google Scholar]
- Valenzuela CF, Kazlauskas A, Brozowski SJ, Weiner JL, Demali KA, Mcdonald BJ, Moss SJ, Dunwiddie TV, Harris RA. Platelet-derived growth factor receptor is a novel modulator of type A gamma-aminobutyric acid-gated ion channels. Mol Pharmacol. 1995;48:1099–1107. [PubMed] [Google Scholar]
- Wan Q, Xiong ZG, Man HY, Ackerley CA, Braunton J, Lu WY, Becker LE, MacDonald JF, Wang YT. Recruitment of functional GABAA receptors to postsynaptic domains by insulin. Nature. 1997;388:686–690. doi: 10.1038/41792. [DOI] [PubMed] [Google Scholar]
- Zafra F, Hengerer B, Leibrock J, Thoenen H, Lindholm D. Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J. 1990;9:3545–3550. doi: 10.1002/j.1460-2075.1990.tb07564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra F, Lindholm D, Castren E, Hartikka J, Thoenen H. Regulation of brain-derived neurotrophic factor and nerve growth factor mRNA in primary cultures of hippocampal neurons and astrocytes. J Neurosci. 1992;12:4793–4799. doi: 10.1523/JNEUROSCI.12-12-04793.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]