

Abstract
Unlike many other native and cloned K+ channels, human ether-à-go-go-related K+ (HERG) channels show significant Cs+ permeability with a PCs/PK (the permeability of Cs+ relative to that of K+) of 0.36 ± 0.03 (n = 10). Here, we find that raising the concentration of external Cs+ (Cs0+) dramatically slows HERG channel inactivation without affecting activation. Replacement of 5 mm K0+ by 135 mm Cs0+ increased both inactivation and recovery time constants and shifted the mid-point of the steady-state inactivation curve by 25 mV in the depolarized direction (n = 6, P < 0.01). Raising [Cs+]o also modulated the voltage sensitivity of inactivation gating. With 130 8mm Cs1+ and 135 mm NMDGo+, the inactivation time constant decreased e-fold per 47.5 ± 1.1 mV (n = 5), and when 20 mm Cs+ was added to the bath solution, the inactivation time constant decreased e-fold per 20.6 ± 1.3 mV (n = 5, P < 0.01). A quantitative analysis suggests that Cs0+ binds to a site in the pore that is influenced by the transmembrane electrical field, so that Cs0+-induced slowing of HERG inactivation is less prominent at strong depolarizations. K0+ has effects that are similar to Cs0+ and their effects were additive, suggesting Cs0+ and K0+ may share a common mechanism of action. The strong effects of Cs+ on inactivation but not on activation highlight the importance of ion and channel interactions during the onset of inactivation in the HERG channel.
The human ether-à-go-go-related (HERG) gene encodes the pore forming subunit of a K+ channel that exists in a number of cell types including neurons, cardiac myocytes and tumour cells (Trudeau et al. 1995; Faravelli et al. 1996; Bianchi et al. 1998). In the heart, HERG channels are partly responsible for a delayed rectifier K+ current (IKr) that is important for cardiac action potential repolarization (Sanguinetti & Jurkiewicz, 1990; Sanguinetti et al. 1995). Reduction of IKr by mutations of the HERG gene or by drug block causes some congenital and acquired forms of human long-QT syndrome (Sanguinetti et al. 1995; Zhang et al. 2001).
HERG inactivation is basically C-type in nature (Trudeau et al. 1995; Sanguinetti et al. 1995; Smith et al. 1996; Spector et al. 1996), although the gating behaviour is distinctive. First, channel inactivation is much faster than voltage-dependent activation, resulting in its characteristic rectification. Second, unlike C-type inactivation in Shaker-type channels (Hoshi et al. 1991; Ogielska et al. 1995; Fedida et al. 1999), HERG inactivation displays intrinsic voltage dependence (Smith et al. 1996; Spector et al. 1996). It has been proposed that HERG channels possess a voltage sensor for inactivation (Wang et al. 1996; Johnson et al. 1999), but this has not been identified and the molecular mechanisms of HERG inactivation are not yet completely understood. C-type inactivation is known to be regulated by permeant ions, and in Shaker and Kv1 channels, raising the concentration of external K+ (K0+) slows C-type inactivation (Lopez-Barneo et al. 1993), an effect that appears to be due to occupancy of the pore selectivity filter by K+ (Baukrowitz & Yellen, 1995). Other ions, such as Cs+, are much less effective than K+ in their ability to modulate inactivation (Lopez-Barneo et al. 1993). Raising [K+]o also slows HERG channel inactivation (Wang et al. 1996, 1997) but the mechanism remains poorly understood. A ‘foot-in-the-door’ mechanism was initially proposed to account for the slowing of deactivation of K+ channels in the presence of external K+ and Rb+ (Swenson & Armstrong, 1981; Matteson & Swenson, 1986). The same mechanism was also proposed to account for the slowing of K+ current inactivation in molluscan neurons (Ruben & Thompson, 1984) and for the slowing of C-type inactivation of Shaker K+ current by external K+ (Baukrowitz & Yellen, 1995, 1996). It is not known whether monovalent cations such as Cs+ can affect HERG inactivation in the same way as they do in Shaker and Kv1 channels.
Cs+ has been used in HERG channel studies for two major purposes. First, Cs+ is used to block HERG channels (Trudeau et al. 1995; Pancrazio et al. 1999). Block of inward HERG current by Cs+ is voltage dependent which is thought to reflect Cs+ entering the pore from the outside and interfering with K+ permeation (Trudeau et al. 1995). Second, in the presence of high concentrations of external Cs+ (Cs0+), large outward currents with an inactivation phase can be recorded during depolarizing pulses, in contrast to the small outward HERG currents recorded in the presence of K0+ (Schönherr & Heinemann, 1996; Pennefather et al. 1998; Barros et al. 1998). However, the changes of gating kinetics responsible for such large outward current with high [Cs+]o are not known and a detailed analysis of Cs+ effects on HERG channels has not been reported. Although the blocking properties of Cs+ on HERG channels suggest that Cs+ may enter the channel pore (Trudeau et al. 1995), the permeation of Cs+ through HERG channels has not been studied in detail. Here, we have studied Cs0+ modulation of HERG channels. We found that HERG channels readily conduct Cs+, and that high [Cs+]o dramatically slows HERG channel inactivation but does not affect HERG activation properties. The effect of Cs0+ on inactivation is twofold: first, it increases the inactivation time constant (τinact) and second, it modulates the voltage sensitivity of HERG channel inactivation. A quantitative analysis of the data indicates that Cs0+ binding to a site located in the transmembrane electrical field can explain both effects.
METHODS
Cells and solutions
HERG currents were recorded from channels stably expressed in a human embryonic kidney cell line, HEK 293 (American Type Culture Collection, Rockville, MD, USA). Cells were maintained in Dulbecco's modified Eagle's medium plus 10 % fetal bovine serum with 1 % penicillin-streptomycin and G418 (0.5 mg ml−1) to select for transfected cells. The pipette solution contained (mm): KCl or CsCl, 130; EGTA, 5; MgCl2, 1; Hepes, 10; and was adjusted to pH 7.2 with KOH or CsOH.
The bath solution contained (mm): Hepes, 10; MgCl2, 1; CaCl2, 2; the balance of ions was made up with 140 mmN-methyl-d-glucamine (NMDG+), and was adjusted to pH 7.4 with HCl. For recordings in the presence of different external Na+, K+, or Cs+ concentrations, the NMDG+-based external solution was used and the concentration of NMDG+ was reduced as the cation concentration was elevated to maintain constant osmolarity, and the pH was adjusted to 7.4 with the appropriate hydroxide solution. All chemicals were from Sigma. Throughout the text the subscripts ‘i’ and ‘o’ denote, respectively, intra- or extracellular ion concentrations.
Electrophysiological procedures and analysis
Coverslips containing cells were removed from the incubator before experiments and placed in a superfusion chamber containing the control bath solution at 22–23 °C. The bath solution was constantly flowing through the chamber and the solution was exchanged by switching the perfusates at the inlet of the chamber, with complete bath solution changes taking 10 s. Whole-cell current recording and data analysis were performed using an Axopatch 200A amplifier and pCLAMP6 software (Axon Instruments, Foster City, CA, USA). Patch electrodes were fabricated using thin-walled borosilicate glass (World Precision Instruments, FL, USA). The electrodes had resistances of ∼2 MΩ when filled with the pipette solutions. Data were filtered at 5–10 kHz and sampled at 20–50 kHz for all protocols. Typically, 80 % series resistance (Rs) compensation was used. Leak subtraction was not used. Data are shown as means ±s.e.m. Statistical significance was determined using the Student's unpaired t test. All data analysis and curve fitting were done with Clampfit (Axon Instruments). Conductance and voltage data were fitted to a single Boltzmann function:
| (1) |
where y is the current normalized with respect to the maximal current, V1/2 is the half-activation or inactivation potential (mid-point of the activation or inactivation curve), V is the test voltage and k is the slope factor, in millivolts, reflecting the steepness of the voltage dependence of gating.
Model of Cs0+ effects on HERG inactivation rate
The HERG inactivation rate is intrinsically voltage dependent. We used the following equation to describe the rate of inactivation (α):
| (2) |
where α(V) is the inactivation rate at any voltage, α0 is the inactivation rate at 0 mV, Z′ is the valence of the charges moved by depolarization, δ′ is the electrical distance from the external side of the membrane, F, R and T have their usual meanings, and RT/F is 25 mV at the room temperature at which all experiments were carried out. There is also a site at which cation binding affects inactivation. The probability that this modulatory site does not have Cs0+ bound is:
| (3) |
where B is the number of channels with Cs+ bound, and O is the number of unbound channels. If the site is located within the transmembrane electrical field, the binding of cations is likely to be voltage dependent (Woodhull, 1973) such that:
| (4) |
where Kd(V) is the affinity at voltage V, Kd(0) is the equilibrium dissociation constant at 0 mV, Z for Cs+ or K+ is 1 and δ is the electrical distance of the cation binding site from the external side of the membrane.
We found that, as with K0+, raising [Cs+]o slowed the time course of the inactivation (see Results). The simplest explanation for this effect, which is also seen with external TEA+ (Smith et al. 1996), is a gating scheme in which Cs+ must dissociate from the modulatory site to allow inactivation (I) to proceed from the open (O) state:
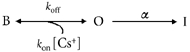 |
If binding and dissociation of Cs+ are rapid (at equilibrium) relative to the rate of inactivation (α), then the apparent rate of inactivation will be:
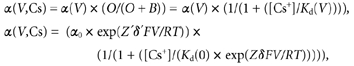 |
(5) |
where the equilibrium dissociation constant, Kd, is equal to koff/kon; α(V,Cs) is the inactivation rate in the presence of Cs+ at voltage V and α(V) is the inactivation rate in the absence of Cs+ at voltage V. α(V,Cs) is thus a function of both voltage (V) and Cs+ concentration [Cs+]o. We assumed a 1:1 stoichiometry for the interaction between the channel and Cs0+.
It should be noted that Z′δ′ is the apparent charge associated with the intrinsic inactivation rate in the absence of external Cs+ and K+, and Zδ is the apparent charge in the presence of external Cs+ or K+. Thus, α0 and Z′δ′ are the ‘external ion-independent’ components. The values for α0 are measured experimentally. Z′δ′ was considered a single parameter and obtained from fitting the τinact-voltage relationship in the absence of Cs0+ or K0+. These numbers were then constrained to be the same at all concentrations of Cs0+ or K0+. It should also be noted that eqn (5) does not predict perfectly linear relationships between log(τinact) and voltage (V), and the curvature can be seen in Fig. 7C and D especially for concentrations near the Kd(0). Since HERG steady-state inactivation is nearly complete at positive voltages at which the time constant for inactivation (τinact) was measured in the present study, the τinact is considered the reciprocal of α(V,Cs), and is used throughout the text and figures.
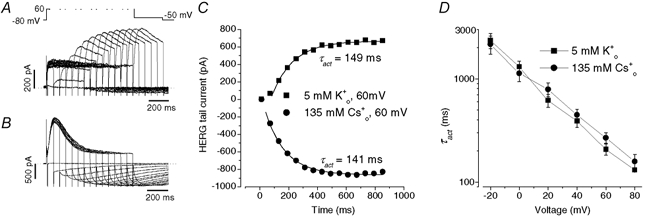
Figure 7. The effects of [Cs+]o and [K+]o on the voltage dependence of τinact of outward Cs+ current recorded with 130 mm Cs1+.
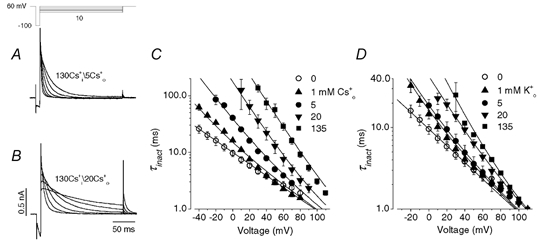
A and B, HERG Cs+ currents elicited by the voltage protocol shown above the current traces in external solution containing 5 mm Cs0+ (A) or 20 mm Cs0+ (B). HERG channels were initially inactivated at the holding potential of +60 mV. As with data in Fig. 4, a 10 ms repolarizing step to −100 mV allows HERG channels to recover from inactivation, and steps to a range of test potentials allows the inactivation time course to be observed. C, voltage dependence of time constant of the decay of outward Cs+ current at different [Cs+]o. Data were fitted (continuous lines) to eqn (5). Cs0+ increased τinact and modulated the voltage sensitivity of the τinact−V relationship. The free parameters for each fit are shown in Table 1. The mean Kd(0) and δ values are 1.7 ± 0.2 mm and 0.66 ± 0.05, respectively (n = 4). D, as in C, but with varying [K+]o. The mean Kd(0) and δ values are 6.8 ± 2.8 mm and 0.63 ± 0.04, respectively (n = 4). Thus, with Cs0+ and K0+ the location of the binding site in the electrical field is similar but the affinity of the site for K+ is slightly lower.
RESULTS
External Cs+ modifies HERG current
Figure 1A shows typical HERG currents in 130 mm K1+ and 5 mm K0+, elicited by 4 s depolarizing steps from the holding potential of −80 mV to between −70 and +70 mV. Tail current peak amplitudes were measured during the subsequent step to −50 mV where the inactivated channels recover quickly to the open state and then slowly deactivate (Trudeau et al. 1995; Sanguinetti et al. 1995; Smith et al. 1996; Spector et al. 1996). The current-voltage (I-V) relationship for HERG peak outward current measured during the depolarizing steps is shown as open circles in Fig. 1C and the normalized tail current peak amplitude at −50 mV versus depolarizing voltage is shown as open triangles in Fig. 1D. HERG current activated at voltages positive to −50 mV, maximum current was reached for steps to −10 mV and at more positive voltages inward rectification resulted from rapid voltage-dependent inactivation (Smith et al. 1996; Spector et al. 1996; Zhang et al. 1999). The tail currents reached a saturating amplitude following voltage steps positive to 20 mV.
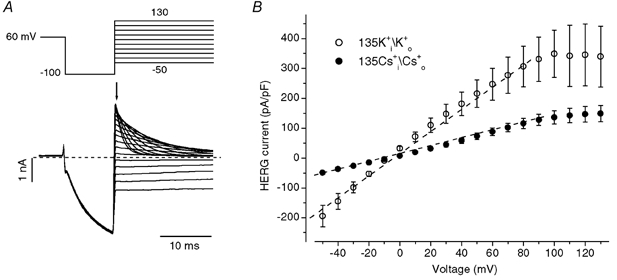
Figure 1. External Cs+ modifies human ether-à-go-go-related K+ (HERG) current inactivation.
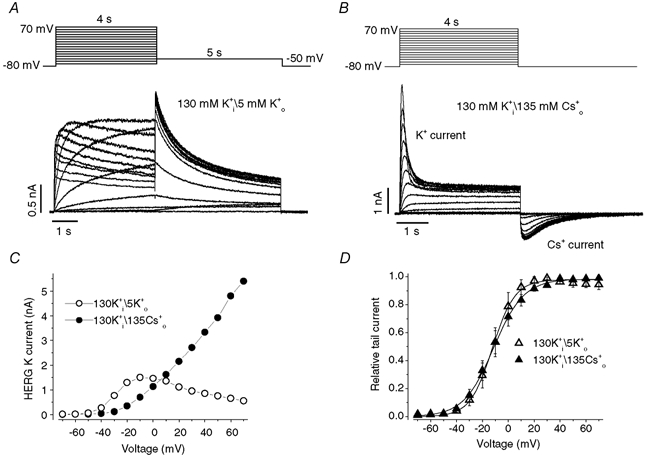
A and B, families of HERG currents elicited by the voltage protocol (10 mV steps) shown above the current traces in external solution containing 5 mm K0+ (A) or 135 mm Cs0+ (B) with an internal solution containing 130 mm K1+. C, current-voltage (I-V) relationships for peak outward HERG current during depolarizing pulses in A (○) and B (•). D, voltage dependence of HERG activation. Amplitudes of tail currents were normalized to the largest current and plotted versus prepulse potentials. The normalized data for each cell were fitted to a Boltzmann function. The averaged half-activation voltage (V1/2) and the slope factor (k) in 5 mm K0+ are, respectively, −11.1 ± 4.0 mV and 7.9 ± 0.7 mV (n = 4). The corresponding values in 135 mm Cs0+ solution are −9.8 ± 3.5 mV and 9.3 ± 0.4 mV (n = 6).
Replacement of 5 mm K0+ by 135 mm Cs0+ resulted in an increase of outward K+ currents that inactivated quickly as previously reported (Schönherr & Heinemann, 1996). In Fig. 1B, HERG currents were elicited by depolarizing steps to between −70 and +70 mV for 4 s. HERG channels displayed a significant Cs+ conductance, with visible inward Cs+ tail currents upon repolarization to −80 mV. The I-V relationship for maximal HERG current measured during depolarizing steps is shown as filled circles in Fig. 1C, and the normalized tail current peak amplitude at −80 mV versus the depolarizing voltage used to activate the current is shown as filled triangles in Fig. 1D. In 135 mm Cs0+, the relationship between the peak outward current and voltage was no longer bell-shaped but showed a monotonic increase with depolarizing potential (Fig. 1C). Despite this dramatic change of the I-V relationship, the voltage dependence of HERG channel activation in solutions with 5 mm K0+ and 135 mm Cs0+ was found to be similar (Fig. 1D). Therefore, although replacement of 5 mm K0+ by 135 mm Cs0+ affected the peak I-V relationship, this was not the result of an action on the voltage dependence of HERG channel activation.
Cs+ readily permeates HERG channels
From the inward tail currents in Fig. 1B, it is clear that Cs+ permeates HERG channels. We studied the permeability of Cs+ relative to that of K+ (PCs/PK) by determining reversal potentials under bi-ionic conditions in which 135 mm K+ was present on one side and 135 mm Cs+ was present on the other side of the membrane. Fully activated HERG open channel instantaneous I-V relationships derived with the protocol shown in Fig. 2A were plotted, and reversal potentials were obtained. The PCs/PK was then calculated (Hille, 2001). We found that PCs,out/PK,in was 0.34 ± 0.03 (n = 6) and PCs,in/PK,out was 0.38 ± 0.04 (P > 0.05, n = 4). There is apparently no preferential direction for Cs+ to permeate the channel, and the pooled data give a PCs/PK of 0.36 ± 0.03 (n = 10). To compare the conduction properties of Cs+ and K+ current through HERG channels, I-V relationships were obtained in either symmetrical Cs+ or K+ conditions (Fig. 2). The I-V relationships are nearly linear to +80 mV under both conditions. The slope conductance of the linear fit to the relationships in symmetrical Cs+ or K+ was 1.23 ± 0.17 and 3.66 ± 0.77 nS pF−1, respectively.
Figure 2. The HERG channels conduct K+ better than Cs+.
A, HERG currents elicited by the voltage protocol above the current traces in symmetrical 135 mm K+. B, fully activated HERG open channel (instantaneous, signified by the arrow in A) current-voltage relationships in either symmetrical K+ (○) or Cs+ (•). The conductance was determined from the slope of a linear fit to current amplitudes at potentials between −50 and +80 mV.
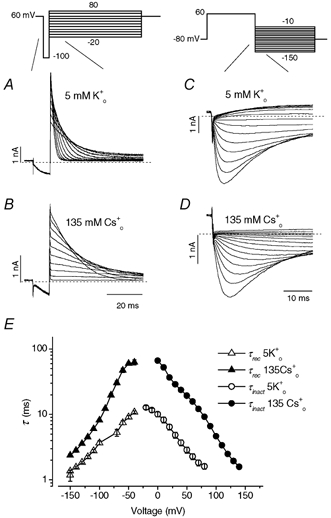
High Cs0+ does not affect HERG activation kinetics
The enhanced peak outward current in 135 mm Cs0+ (Fig. 1B and C) was not due to an increased driving force because the reversal potential in 130 mm K1+ and 135 mm Cs0+ was more positive (−28.3 ± 2.4 mV, n = 6) than that with 130 mm K1+ and 5 mm K0+ (−81.2 ± 2.9 mV, n = 4). The instantaneous I-V relationship also gave no indication of impaired outward K+ current (Fig. 2). Thus, enhanced outward K+ current recorded with 135 mm Cs0+ may result either from an acceleration of activation and/or a slowing of inactivation. We therefore studied the activation time course in solutions containing either 5 mm K0+ or 135 mm Cs0+ (Fig. 3). Activation time constants (τact) were determined by fitting the envelope of the extrapolated (see below) peak amplitudes of tail current recorded at repolarizing steps to −50 mV after varying duration depolarizations to between −20 and +80 mV from a holding potential of −80 mV. During the subsequent step to −50 mV, HERG channels reopen (recover from inactivation) much faster than they close (deactivate). Consequently, the tail current rises to a peak and then decays. Thus, deactivation overlaps recovery from inactivation. To estimate the peak current amplitude immediately (time 0) following repolarization, the deactivating phase of tail current for each trace was fitted to a single exponential function and extrapolated to time 0 of repolarization. The current amplitude at time 0, which is assumed to be proportional to the total number of activated channels, is plotted against the depolarizing duration and fitted to a single exponential function to obtain the time constant of activation. It should be noted that because HERG activation is sigmoidal in nature, the fit to a single exponential function represents a simplified approximation. Data in Fig. 3A and B show the original tail current traces obtained after 60 mV depolarizing steps in external solution containing 5 mm K+ and 135 mm Cs+, respectively. The extrapolated peak tail currents from Fig. 3A (▪) and B (•), and single exponential fits to them (continuous lines) are shown in Fig. 3C. The values for τact were similar in 5 mm K0+ or 135 mm Cs0+. The voltage dependences of τact in 5 mm K0+ (▪) or 135 mm Cs0+ (•) are plotted in Fig. 3D. It can be seen that there is little difference in the macroscopic activation rate of HERG current in external solutions containing either 5 mm K+ or 135 mm Cs+.
Figure 3. HERG current activation is similar in 5 mm K0+ and 135 mm Cs0+.
The values for activation time constants (τact) were determined by fitting the envelopes of the extrapolated peak amplitude of tail currents recorded at −50 mV after varying duration depolarizations to between −20 and +80 mV from a holding potential of −80 mV. The protocol for a depolarization to +60 mV is shown above panel A. The internal solution contained 130 mm K+. A and B show envelopes of tail currents elicited by a depolarization to 60 mV with variable duration in external solution containing 5 mm K0+ (A) or 135 mm Cs0+ (B). C, τact was measured from single exponential fits to the extrapolated peak tail current envelope from A (▪) and B (•). The values for τact were 149 and 141 ms, respectively. D, the voltage dependence of τact in external solution containing 5 mm K0+ (▪) or 135 mm Cs0+ (•) (n = 5–8).
High [Cs+]o slows HERG inactivation
We next studied the effects of replacement of 5 mm K0+ by 135 mm Cs0+ on HERG channel inactivation and discovered that 135 mm Cs0+ slowed HERG current inactivation. To measure the inactivation time constant (τinact), we used the protocol shown above the current traces in panel A of Fig. 4. HERG current was fully activated and inactivated at the holding potential of +60 mV. The cell was then repolarized to −100 mV for 10 ms to allow recovery from inactivation and to minimize deactivation of HERG channels (Smith et al. 1996; Spector et al. 1996). A test step was then applied to different voltages to observe the inactivation time course. The τinact was obtained by fitting the currents to a single exponential function. Data in Fig. 4A and B show HERG current traces in external solutions containing 5 mm K0+ (A) or 135 mm Cs0+ (B). The averaged τinact in 5 mm K0+ (○) and 135 mm Cs0+ (•) in nine cells are plotted as a function of test potential in Fig. 4E. A concentration of 135 mm Cs0+ slowed HERG inactivation at all voltages tested. For example, τinact at 0 mV increased from 10.1 ± 0.1 ms in 5 mm K0+ to 66.4 ± 4.8 ms in 135 mm Cs0+ (n = 9, P < 0.01).
Figure 4. 135 mm Cs0+ slows both onset of and recovery from HERG inactivation.
A and B, HERG currents elicited by the voltage protocol shown above the current traces in external solution containing 5 mm K0+ (A) or 135 mm Cs0+ (B) with an internal solution containing 130 mm K+. Starting from a holding potential of +60 mV, a 10 ms repolarizing step to −100 mV allowed HERG channels to recover from inactivation, and steps to a range of test potentials allowed the inactivation time course to be observed. Current traces represent responses in the time interval demarcated by the slanted lines in the voltage protocol. C and D, the time and voltage dependence of recovery from inactivation was measured by the voltage protocol shown above the current traces in external solution containing 5 mm K0+ (C) or 135 mm Cs0+ (D) and with 130 mm K1+. HERG was activated by a 400 ms pulse to 60 mV, followed by a test pulse to different potentials to record HERG tail currents. The time constant of recovery (τrec) from inactivation was measured as the single exponential fit to the rising phase (> −80 mV) or as the fast time constant of a double exponential fit (≤−80 mV) to HERG tail current (Sanguinetti et al. 1995; Spector et al. 1996). E, voltage dependence of the τrec and the inactivation time constant (τinact) in external solution containing 5 mm K0+ (▵, O) or 135 mm Cs0+ (▴, •). Each point is the mean ±s.e.m. for eight to nine cells.
The effects of replacement of 5 mm K0+ by 135 mm Cs0+ on HERG channel recovery from inactivation were also examined. As shown above Fig. 4C, the voltage protocol consisted of a depolarization to 60 mV for 400 ms to inactivate HERG channels, followed by repolarization to potentials between −150 and −10 mV to elicit a tail current. The rising phase of the tail current represents the rapid recovery of HERG channels from inactivated to open state and is followed by channel deactivation. The current traces were fitted to a single (increasing phase, > −80 mV) or double exponential function (increasing and decreasing phases, ≤−80 mV), and the time constant for recovery (τrec) was obtained (Sanguinetti et al. 1995; Spector et al. 1996). Replacement of 5 mm K0+ (Fig. 4C) by 135 mm Cs0+ (Fig. 4D) significantly slowed the recovery from inactivation (P < 0.01, n = 8). Thus, 135 mm Cs0+ slows the time course of both inactivation and recovery from inactivation.
To study the effects of replacement of 5 mm K0+ by 135 mm Cs0+ on the voltage dependence of steady-state inactivation, a triple-pulse protocol was used (Fig. 5). After a 500 ms pulse to + 60 mV to activate and then inactivate HERG channels, a pulse to −100 mV was applied for 20 ms to allow channels to recover from inactivation. The third pulse (P3) consisted of steps to potentials between from −100 mV to + 90 mV to record the voltage dependence of current inactivation. Following the approach of Wang et al. (1997), the steady-state inactivation relationship was calculated as the ratio of the currents measured at 100 ms to the instantaneous current of P3. The ratio was normalized to the maximal value, plotted against the P3 voltage and fitted to a Boltzmann function (Fig. 5). Replacement of 5 mm K0+ with 135 mm Cs0+ shifted the mid-point of the steady-state inactivation curve to more positive potentials by 25 mV (n = 6, P < 0.01) and decreased the slope factor from 15.8 ± 1.2 mV to 12.8 ± 0.7 mV (n = 6, P < 0.05).
Figure 5. Replacement of 5 mm K0+ by 135 mm Cs0+ shifted the steady-state inactivation curve to more positive potentials.
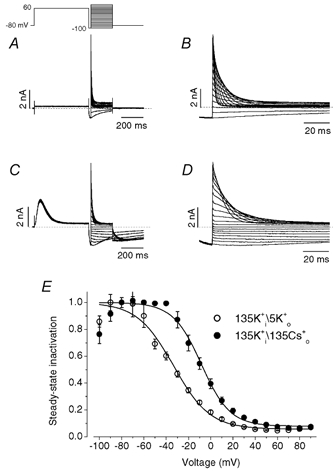
Steady-state inactivation was measured using a triple-pulse protocol, shown above the current traces, in bath solutions containing 5 mm K+ (A and B) or 135 mm Cs+ (C and D) and with 130 mm K1+. A 500 ms pulse (P1) to 60 mV to activate and then inactivate HERG was followed by a 20 ms pulse (P2) to −100 ms to remove the inactivation. In the third pulse (P3) the potential varied between −100 and +90 mV. Currents during the P3 pulses of A and C are expanded in B and D, respectively, to show the time course of inactivation. E, the steady-state inactivation was calculated as the ratio of the current measured 100 ms after the onset of P3 to the instantaneous current at the same potential in 5 mm K+ (○) or 135 mm Cs+ (•). The ratio was normalized to the maximal value, plotted as a function of voltage and fitted to a Boltzmann function. Such measurement represents an estimation of steady-state inactivation because of the overlapping deactivation at negative voltages. Data were obtained from six cells. The half-inactivation voltage and slope factor were −33.5 ± 2.5 mV and −15.8 ± 1.2 mV for 5 mm K0+, and −8.3 ± 2.6 mV and −12.8 ± 0.7 mV for 135 mm Cs0+, respectively. Both the change of half-inactivation voltage and slope factor are significant (n = 6, P < 0.01 and P < 0.05, respectively).
Concentration dependence of Cs0+ and K0+ effects on HERG inactivation
The effect of Cs0+ on the voltage dependence of HERG τinact was concentration dependent (Fig. 6A). The voltage dependence of HERG τinact at different [Cs+]o is plotted in panel A. Cs0+ not only increased τinact but also modulated the voltage sensitivity. When log10τinact -V was fitted to a linear function, it was found that the τinact decreased e-fold per 51.5 ± 1.3 mV (n = 9), 49.1 ± 1.0 mV (P > 0.05, n = 5), 39.3 ± 0.4 mV (P < 0.01, n = 6), 36.3 ± 0.6 mV (P < 0.01, n = 9), 36.2 ± 1.4 mV (P < 0.01, n = 6) and 35.7 ± 0.8 mV (P < 0.01, n = 9) at 0, 1, 5, 20, 67.5 and 135 mm Cs0+, respectively (one-way ANOVA, compared to that in zero Cs0+). To get insight into the effects of Cs0+, we used a gating scheme in which Cs0+-bound channel cannot inactivate, and used eqn (3) to describe the concentration dependence of Cs0+ binding to the channel (Methods). We reasoned that if the binding site is located in the membrane electrical field, the Kd would also be voltage dependent (Woodhull, 1973). Since HERG channel inactivation is intrinsically voltage dependent, HERG inactivation rate is a function of both voltage and Cs0+ concentrations (see Methods for details). Continuous lines superimposed on the data represent the least squares fit of eqn (5) (Methods) which combined the extracellular cation concentration- and voltage-dependent binding. The α0 was obtained experimentally and Z′δ′ was considered a single parameter and was obtained by fitting data in the absence of Cs0+. The values for α0 and Z′δ′ were 0.177 ms−1 and 0.5, respectively, and were fixed in fittings of the data at the different Cs0+ concentrations with eqn (5). Each fitting gave similar values for Kd(0) and δ (Table 1). The means for Kd(0) and δ were 7.8 ± 1.7 mm and 0.16 ± 0.01, respectively (based on the fits at five different [Cs+]o, 5–9 cells at each Cs0+ concentration).
Figure 6. The effects of [Cs+]o and [K+]o on the voltage dependence of τinact.
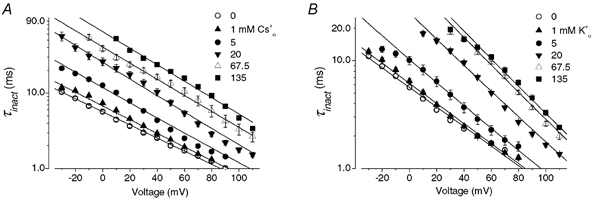
A, the voltage dependence of τinact with the indicated [Cs+]o and 130 mm K1+ is plotted. Continuous lines represent fits of the data to a model (see Methods, eqn (5)) in which binding is voltage dependent. The free parameters for each fit are shown in Table 1. The mean Kd(0) and δ values are 7.8 ± 1.7 mm and 0.16 ± 0.01, respectively (n = 5). B, as for A, but with changes of [K+]o. The mean Kd(0) and δ values of 8.7 ± 1.5 mm and 0.23 ± 0.01, respectively (n = 5), with K0+ are similar to those obtained for Cs0+.
Table 1.
Free parameters in the fitting of the effects of [Cs+]o and [K+]o on the voltage dependence of outward K+ and Cs+ current inactivation time constants
| Internal solution (mm) | Cso+ | Ko+ | Concentration (mm) | ||
|---|---|---|---|---|---|
| Kd(0) (mm) | (δ) | Kd(0) (mm) | δ | ||
| 130 K+ | 5.2 | 0.15 | 10.9 | 0.20 | 1 |
| 4.6 | 0.16 | 6.1 | 0.23 | 5 | |
| 5.6 | 0.15 | 5.1 | 0.24 | 20 | |
| 10.7 | 0.14 | 7.7 | 0.25 | 67.5 | |
| 12.8 | 0.14 | 13.5 | 0.25 | 135 | |
| 130 Cs+ | 1.9 | 0.58 | 1.4 | 0.68 | 1 |
| 1.2 | 0.75 | 5.2 | 0.60 | 5 | |
| 2.1 | 0.75 | 5.7 | 0.53 | 20 | |
| 1.7 | 0.58 | 14.9 | 0.71 | 135 | |
Kd(0) is the equilibrium dissociation constant at 0 mV; δ is the electrical distance of the cation binding site from the external side of the membrane.
Since increasing [K+]o has also been reported (Wang et al. 1996, 1997) to slow HERG channel inactivation, the concentration dependence of K0+ effects was also studied (Fig. 6B). K0+ had very similar effects to Cs0+: it increased τinact and decreased voltages required per e-fold change of τinact (data not shown). Fitting data at the different [K+]o to eqn (5) with the fixed α0 and Z′δ′ values gave similar Kd(0) and δ values at each K0+ concentration (Table 1). The mean Kd(0) and δ values were 8.7 ± 1.5 mm and 0.23 ± 0.01, respectively (based on fits at five different [K+]o, 5–11 cells at each K0+ concentration). The similar δ values with both Cs0+ and K0+ suggest that they may bind to the same depth within the membrane electric field.
Voltage- and ion-dependent modulation of HERG Cs0+ conductance
Unlike many other K+ channels, HERG channels conduct Cs+ readily even in the presence of K+ (Fig. 1B and Fig. 2). We also studied the voltage- and concentration-dependent effects of the extracellular cations on the HERG currents when outward current was carried by Cs+ ions. To do this we used a pipette solution containing 130 mm Cs+. By comparing current relaxations in panels A and B of Fig. 7 it can be seen that raising Cs0+ slows inactivation of outward Cs+ currents significantly. The concentration dependent slowing of the inactivation rate and the change of slope of the τinact−V relationships are similar to those observed for K+ currents (Fig. 4 and Fig. 6). The τinact−V curve is shown at different [Cs+]o in Fig. 7C. Cs0+ increased τinact and modulated the voltage sensitivity. When log10τinact−V was fitted to a linear function, it was found that the τinact decreased e-fold per 47.5 ± 1.2 mV (n = 5), 31.2 ± 1.2 mV (P < 0.01, n = 7), 24.7 ± 0.6 mV (P < 0.01, n = 6), 20.6 ± 1.3 mV (P < 0.01, n = 5) and 20.6 ± 0.2 mV (P < 0.01, n = 7) at 0, 1, 5, 20 and 135 mm Cs0+, respectively (one-way ANOVA, compared to that in zero Cs0+). Although we fitted the log10τinact−V relationship to a linear function to address the change of the voltage dependence of HERG channel inactivation, a careful inspection of the τinact-V relationship in Fig. 7C and D reveals that it is not perfectly linear especially for [Cs+]o and [K+]o at 5 mm. This slight non-linearity is consistent with our model described in eqn (5) which contains two exponential components, and so does not predict perfectly linear relationships between log10τinact and voltage, especially for concentrations near the Kd(0). In Fig. 7C, the data were fitted to eqn (5). The ‘external ion-independent’ components α0 and Z′δ′ of 0.104 ms−1 and 0.6, respectively, were obtained as for K+ current, and these values were fixed in fitting data at the different [Cs+]o (Fig. 7C), as well as [K+]o (Fig. 7D). The continuous lines represent the least squares fits and the number of free parameters is summarized in Table 1. The mean Kd(0) and δ values were 1.7 ± 0.2 mm and 0.66 ± 0.05, respectively (based on fits at four different [Cs+]o, 5–7 cells at each concentration). For comparison, the concentration dependence of K0+ effects was also studied (Fig. 7D, Table 1). The means for Kd(0) and δ were 6.8 ± 2.8 mm and 0.63 ± 0.04, respectively (based on fits at four different [K+]o, 5–7 cells at each concentration). Interestingly, although the model provides good fits to experimental data under both K+ (Fig. 6) and Cs+ permeating conditions (Fig. 7), the δ value is apparently greater (deeper, relative to the external surface, within the electric field) when the channels are conducting Cs+. Thus, our results showed that there are clear differences in gating kinetics when Cs+ or K+ is carrying the current. The model incorporated the differences in gating kinetics between Cs+- and K+-mediated current in terms of different δ values.
Effects of Cs0+ and K0+ on HERG inactivation are additive
Under the two different experimental conditions used to record outward K+ or Cs+ current we have shown that raising Cs0+ or K0+ has similar effects on HERG channel inactivation. Model analyses suggest similar Kd(0) and δ values for the two ions when the major internal cation is K+, and similar δ values when the major internal cation is Cs+. These observations support the idea that both ions, when added extracellularly, act through a common binding site. This idea was tested using the experiment illustrated in Fig. 8, which essentially tested whether the voltage dependent actions of Cs0+ and K0+ were additive. Concentrations of Cs0+ or K0+ near Kd(0) were used. In Fig. 8, the τinact−V relationships at 0 (○), 10 mm K0+ (•), 10 mm K0+ plus 10 mm Cs0+ (▪) were plotted. Data obtained with 20 mm K0+ (▵) and 20 mm Cs0+ (▿) are also shown. 10 mm K0+ increased τinact. An additional increase was caused by 10 mm K0+ plus 10 mm Cs0+, which was significant (P < 0.01). The increase caused by 10 mm K0+ plus 10 mm Cs0+ is not different from that induced by either 20 mm K0+ or 20 mm Cs0+ (P > 0.05), suggesting the effects of Cs0+ and K0+ were additive.
Figure 8. Effects of Cs0+ and K0+ on HERG inactivation were additive.
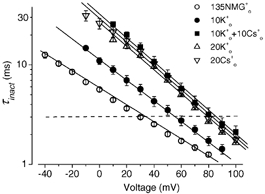
Voltage dependence of K+ current τinact with 130 mm K1+ and 0 (○), 10 mm K0+ (•), and 10 mm K0+ plus 10 mm Cs0+ (▪). Data with 20 mm K0+ (▵) and 20 mm Cs0+ (▿) (taken from Fig. 6) are also shown for comparison. When viewed as a voltage shift, at τinact of 3 ms, 10 mm K0+ shifted the τinact−V relationship to the right by 24.2 ± 1.7 mV (n = 6). 10 mm K0+ plus 10 mm Cs0+ shifted the relationship to the right by 51.1 ± 4.4 mV (n = 6), which is significantly greater than the 10 mm K0+-induced shift (P < 0.01), but not different from either 20 mm K0+- or 20 mm Cs0+-induced shifts (46.0 ± 2.1, n = 5 and 49.2 ± 2.7 mV, n = 9, respectively. P > 0.05). This indicates that Cs0+ and K0+ may share a common mechanism of action.
DISCUSSION
Selective effect of high [Cs+]o and [K+]o on inactivation
Raising Cs0+ (or K0+) significantly slows HERG channel inactivation, and the concentration- and voltage-dependent effects can be well expressed in terms of binding to a site located within the membrane electric field. Changing from 5 mm K0+ to 135 mm Cs0+ slowed both onset of and recovery from inactivation, shifted the mid-point of the steady-state inactivation by 25 mV, but did not affect either the steady-state voltage dependence or the kinetics of HERG activation (Fig. 1 and Fig. 2). Our finding highlights the importance of interactions between extracellular permeant cations and the channel during the onset of inactivation in the HERG channel.
Modulation of inactivation by extracellular cations
Slowing of HERG inactivation by K0+ has been noted previously, but the mechanism remains poorly understood (Wang et al. 1996, 1997). The marked effect of Cs0+ on HERG inactivation is surprising because Cs0+ has a much smaller effect on C-type inactivation in N-type inactivation-removed Shaker channels (Lopez-Barneo et al. 1993). We found that Cs0+, K0+ and Rbo+ (data not shown) all slow HERG inactivation to a similar extent and our analyses showed similar Kd(0) values for Cs0+ and K0+ when the major internal cation is K+ (Fig. 6). Compared with Cs0+ and K0+, we found that Nao+ had little effect on HERG inactivation (data not shown, see also Numaguchi et al. 2000). These results suggest that the external cation binding site selects strongly against the smaller Na+ ion, but only modestly among the three larger ions.
The ‘foot-in-the-door’ mechanism was originally proposed to account for the slowing by K+ and Rb+ of K+ channel deactivation in the squid giant axon and was extended to account for the effects on inactivation in K+ channels (Swenson & Armstrong, 1981; Ruben & Thompson, 1984; Matteson & Swenson, 1986; Baukrowitz & Yellen, 1995, 1996). Slowing of HERG inactivation by external TEA+ has also been explained by the same mechanism (Smith et al. 1996). In the present study, we applied the ‘foot-in-the-door’ concept to a model to account for the slowing of HERG channel inactivation by Cs0+ and K0+. We also reasoned that if the binding site is located in the membrane electrical field, the Kd would also be voltage dependent (Woodhull, 1973) and therefore we incorporated voltage dependent cation binding in the model. Although similar Kd(0) and δ values (≈0.2) for Cs0+ and K0+ were obtained when the major intracellular cation was K+ (Fig. 6), a different response to Cs0+ and K0+ was seen when the major intracellular cation was Cs+ such that extracellular regulation site was ‘deeper’ (Δ≈ 0.6) with Cs1+. When log10τinact−V relationships were fitted to a linear function, Cs0+ has a bigger effect on the slope factor with internal Cs+ than with internal K+. Thus, our model incorporates the different effects by Cs0+ on the slope factor with internal Cs+vs. K+ by way of different δ values. The mechanistic basis for the different δ is uncertain. Because the K+ channel is a multi-ion pore, the δ value from a Woodhull (1973) model could be complicated by interactions between ions. The change in the apparent δ may simply result from differences in cation binding sites within the channel between K+ and Cs+ (in the KcsA channel, four binding sites for K+vs. three binding sites for Cs+; Zhou & MacKinnon, 2002), and the coupling of charge movement (Spassova & Lu, 1999). Another possibility is that when physically bigger Cs+ permeates the channel, binding of ions of different sizes at sites deeper in the selectivity filter may induce different degrees of structural ‘deformation’ at external ion binding sites. Similar observations that internal ions can influence putative external binding sites have been made in Kv2.1 and ROMK1 channels (Immke et al. 1999; Spassova & Lu, 1999). As for the residues contributing to the external site, it seems that the position equivalent to threonine (T) 449 in the outer pore mouth of Shaker is a key site for regulation of C-type inactivation (Lopez-Barneo et al. 1993). Mutations at this position that alter inactivation of other K+ channels also change modulation by K0+ (Pardo et al. 1992; Kirsch et al. 1992; Lopez-Barneo et al. 1993). For example, replacement of T449 of a mutant Shaker B channel (ShBΔ6–46) with lysine or glutamate increased the rate of C-type inactivation 100-fold and made the channel availability sensitive to K0+ (Lopez-Barneo et al. 1993). Similarly, in Kv1.4, mutation of a single amino acid (K533Y) in a position homologous to T449 abolished K0+ modulation of these channels (Pardo et al. 1992). In HERG, strong effects of mutations at S631 (equivalent to T449 in Shaker) on inactivation have been reported (Smith et al. 1996; Schönherr & Heinemann, 1996). Mutation of serine 631 to alanine (S631A) also caused a dramatic shift in the voltage dependence of channel inactivation, but, reflecting the results reported here, did not affect the voltage dependence of activation (Zou et al. 1998). However, increasing extracellular K+ still slowed inactivation in HERG S631A mutant channels (Zou et al. 1998). Therefore, the binding site(s) at which K0+ and Cs0+ regulate inactivation is not known.
Implications for the molecular basis of HERG inactivation
Unlike C-type inactivation in Shaker channels, HERG inactivation is strongly voltage dependent and so the channel has been suggested to possess an independent voltage sensor (Wang et al. 1996; Johnson et al. 1999). However, Baukrowitz & Yellen (1996) proposed that in Shaker channels K+ ions controlled the C-type inactivation rate at a K+ binding site involved in permeation, and that it was the off-rate of the last ion from a binding site in the permeation pathway that controlled the inactivation rate (Baukrowitz & Yellen, 1996). From studies of chimeric Kv2.1/1.3 channels Kiss & Korn (1998) proposed a model in which the selectivity filter binding site is the primary site at which K+ modulates inactivation, such that C-type inactivation involves a constriction at the selectivity filter, and the constriction cannot proceed when the selectivity filter is occupied by K+. Our results on HERG channels suggest that Cs0+ not only slows inactivation, but also modulates the voltage sensitivity of the τinact−V relationship by binding to a site in the pore. This suggests that ion and channel interactions also contribute to the voltage dependence of HERG inactivation as is the case for Kv channels. However, the contribution of external cations to the voltage dependence (which, in terms of our model, depends on Kd(0) and δ) is less important than the ‘intrinsic’ voltage dependence (Z′δ′) because the external ion effect is rather small under physiological conditions. Our model does not explicitly consider effects of occupancy by internal cations. However, our results showed that there are clear differences in gating kinetics between Cs+- and K+-mediated current.
Conclusions
Raising the external Cs+ concentration dramatically slows HERG channel inactivation, but does not affect HERG activation properties. Cs0+ increases τinact and modulates the voltage sensitivity of τinact. Both effects are concentration dependent and well described by a simple model in which binding of Cs0+ or K0+ slows HERG inactivation in a voltage-dependent manner. Although the site interacts with K+ and Cs+ similarly, the apparent electrical location appears to depend on the nature of the internal cation. The strong effects of permeant ions on inactivation but not on activation suggest that permeant ion and channel interactions contribute significantly to HERG inactivation properties.
Acknowledgments
We thank Dr Zhuren Wang for helpful discussions and Sandy Wang and Linda Sui for preparing cells. The project was supported by grants from the Heart and Stroke Foundations of British Columbia and Yukon, and the CIHR to D.F. and a Heart and Stroke Foundation of Canada Research Fellowship Award to S.Z.
REFERENCES
- Barros F, Gomez-Varela D, Viloria CG, Palomero T, Giraldez T, De la Peña P. Modulation of human erg K+ channel gating by activation of a G protein coupled receptor and protein kinase C. J Physiol. 1998;511:333–346. doi: 10.1111/j.1469-7793.1998.333bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baukrowitz T, Yellen G. Modulation of K+ current by frequency and external [K+]: A tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Yellen G. Use-dependent blockers and exit rate of the last ion from the multi-ion pore of a K+ channel. Science. 1996;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- Bianchi L, Wible B, Arcangeli A, Taglialatela M, Morra F, Castaldo P, Crociani O, Rosati B, Faravelli L, Olivotto M, Wanke E. HERG encodes a K+ current highly conserved in tumors of different histogenesis - a selective advantage for cancer cells. Cancer Res. 1998;58:815–822. [PubMed] [Google Scholar]
- Faravelli L, Arcangeli A, Olivotto M, Wanke E. A HERG-like K+ channel in rat F-11 DRG cell line: Pharmacological identification and biophysical characterization. J Physiol. 1996;496:13–23. doi: 10.1113/jphysiol.1996.sp021661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedida D, Maruoka ND, Lin S. Modulation of slow inactivation in human cardiac Kv1. 5 channels by extra- and intra-cellular permeant cations. J Physiol. 1999;515:315–329. doi: 10.1111/j.1469-7793.1999.315ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. 3. Sunderland, MA, USA: Sinauer Associates; 2001. Ion selectivity; p. 21. [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: Effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Immke D, Wood M, Kiss L, Korn SJ. Potassium-dependent changes in the conformation of the Kv2. 1 potassium channel pore. J Gen Physiol. 1999;113:819–836. doi: 10.1085/jgp.113.6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JP, Jr, Mullins FM, Bennett PB. Human ether-à-go-go-related gene K+ channel gating probed with extracellular Ca2+. Evidence for two distinct voltage sensors. J Gen Physiol. 1999;113:565–580. doi: 10.1085/jgp.113.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch GE, Drewe JA, Taglialatela M, Joho RH, Debiasi M, Hartmann HA, Brown AM. A single nonpolar residue in the deep pore of related K+ channels acts as a K+:Rb+ conductance switch. Biophys J. 1992;62:136–144. doi: 10.1016/S0006-3495(92)81800-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss L, Korn SJ. Modulation of C-type inactivation by K+ at the potassium channel selectivity filter. Biophys J. 1998;74:1840–1849. doi: 10.1016/S0006-3495(98)77894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors & Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Matteson DR, Swenson RP. External monovalent cations that impede the closing of K+ channels. J Gen Physiol. 1986;87:795–816. doi: 10.1085/jgp.87.5.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numaguchi H, Mullins FM, Johnson JP, Jr, Johns DC, Po SS, Yang IC, Tomaselli GF, Balser JR. Probing the interaction between inactivation gating and d-sotalol block of HERG. Circ Res. 2000;87:1012–1018. doi: 10.1161/01.res.87.11.1012. [DOI] [PubMed] [Google Scholar]
- Ogielska EM, Zagotta WN, Hoshi T, Heinemann SH, Haab J, Aldrich RW. Cooperative subunit interactions in C-type inactivation of K channels. Biophys J. 1995;69:2449–2457. doi: 10.1016/S0006-3495(95)80114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancrazio JJ, Ma W, Grant GM, Shaffer KM, Kao WY, Liu QY, Manos P, Barker JL, Stenger DA. A role for inwardly rectifying K+ channels in differentiation of NG108–15 neuroblastoma x glioma cells. J Neurobiol. 1999;38:466–474. [PubMed] [Google Scholar]
- Pardo LA, Heinemann SH, Terlau H, Ludewig U, Lorra C, Pongs O, Stühmer W. Extracellular K+ specifically modulates a rat brain K+ channel. Proc Natl Acad Sci U S A. 1992;89:2466–2470. doi: 10.1073/pnas.89.6.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennefather PS, Zhou W, DeCoursey TE. Idiosyncratic gating of HERG-like K+ channels in microglia. J Gen Physiol. 1998;111:795–805. doi: 10.1085/jgp.111.6.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruben P, Thompson S. Rapid recovery from K current inactivation on membrane hyperpolarization in molluscan neurons. J Gen Physiol. 1984;84:861–875. doi: 10.1085/jgp.84.6.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jurkiewicz NK. Two components of delayed rectifier K+ current. J Gen Physiol. 1990;96:195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönherr R, Heinemann SH. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J Physiol. 1996;493:635–642. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- Spassova M, Lu Z. Tuning the voltage dependence of tetraethylammonium block with permeant ions in an inward-rectifier K+ channel. J Gen Physiol. 1999;114:415–426. doi: 10.1085/jgp.114.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector PS, Curran ME, Zou AR, Sanguinetti MC. Fast inactivation causes rectification of the IKr channel. J Gen Physiol. 1996;107:611–619. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson RP, Armstrong CM. K+ channels close more slowly in the presence of external K+ and Rb+ Nature. 1981;291:427–429. doi: 10.1038/291427a0. [DOI] [PubMed] [Google Scholar]
- Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- Wang S, Liu S, Morales MJ, Strauss HC, Rasmusson RL. A quantitative analysis of the activation and inactivation kinetics of HERG expressed in Xenopus oocytes. J Physiol. 1997;502:45–60. doi: 10.1111/j.1469-7793.1997.045bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Morales MJ, Liu S, Strauss HC, Rasmusson RL. Time, voltage and ionic concentration dependence of rectification of h-erg expressed in Xenopus oocytes. FEBS Lett. 1996;389:167–173. doi: 10.1016/0014-5793(96)00570-4. [DOI] [PubMed] [Google Scholar]
- Woodhull AM. Ionic blockage of sodium channels in nerve. J Gen Physiol. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Rajamani S, Chen Y, Gong Q, Rong Y, Zhou Z, Ruoho A, January CT. Cocaine blocks HERG, but not KvLQT1+minK, potassium channels. Mol Pharmacol. 2001;59:1069–1076. doi: 10.1124/mol.59.5.1069. [DOI] [PubMed] [Google Scholar]
- Zhang S, Zhou Z, Gong Q, Makielski JC, January CT. Mechanism of block and identification of the verapamil binding domain to HERG potassium channels. Circ Res. 1999;84:989–998. doi: 10.1161/01.res.84.9.989. [DOI] [PubMed] [Google Scholar]
- Zhou Y, MacKinnon R. Rate theory from Brownian dynamics: analysis of a simulated ion channel (abstract) Biophys J. 2002;82:350. [Google Scholar]
- Zou A, Xu Q, Sanguinetti MC. A mutation in the pore region of HERG K+ channels expressed in Xenopus oocytes reduces rectification by shifting the voltage dependence of inactivation. J Physiol. 1998;509:129–137. doi: 10.1111/j.1469-7793.1998.129bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]