

Abstract
Modulation of K+ channels by hypoxia is a common O2-sensing mechanism in specialised cells. More recently, acid-sensitive TASK-like background K+ channels, which play a key role in setting the resting membrane potential, have been implicated in O2-sensing in certain cell types. Here, we report a novel O2 sensitivity mediated by a weakly pH-sensitive background K+ conductance in nitric oxide synthase (NOS)-positive neurones of the glossopharyngeal nerve (GPN). This conductance was insensitive to 30 mm TEA, 5 mm 4-aminopyridine (4-AP) and 200 μm Cd2+, but was reversibly inhibited by hypoxia (O2 tension (PO2) = 15 mmHg), 2–5 mm halothane, 10 mm barium and 1 mm quinidine. Notably, the presence of halothane occluded the inhibitory effect of hypoxia. Under current clamp, these agents depolarised GPN neurones. In contrast, arachidonic acid (5–10 μm) caused membrane hyperpolarisation and potentiation of the background K+ current. This pharmacological profile suggests the O2-sensitive conductance in GPN neurones is mediated by a class of background K+ channels different from the TASK family; it appears more closely related to the THIK (tandem pore domain halothane-inhibited K+) subfamily, or may represent a new member of the background K+ family. Since GPN neurones are thought to provide NO-mediated efferent inhibition of the carotid body (CB), these channels may contribute to the regulation of breathing during hypoxia via negative feedback control of CB function, as well as to the inhibitory effect of volatile anaesthetics (e.g. halothane) on respiration.
Oxygen (O2) plays a pivotal role in cell homeostasis and survival, and not surprisingly many organisms have evolved strategies for ensuring adequate supply of O2 to the tissues. While all cells probably have some ability to sense O2, over the last ∼15 years there has been a major focus on the more specialised O2-sensing cells, including carotid body chemoreceptors (Gonzalez et al. 1994; Peers, 1997; Prabhakar, 2000; Lopez-Barneo et al. 2001), pulmonary neuroepithelial bodies (Youngson et al. 1993; Fu et al. 2000), adrenal chromaffin cells (Thompson & Nurse, 1998), vascular smooth muscle cells (Archer et al. 1993, 1998; Lopez-Barneo et al. 2001) and central neurones (Haddad & Jiang, 1993, 1997; Henrich et al. 2002; Plant et al. 2002). However, with the exception of pulmonary neuroepithelial bodies (Fu et al. 2000), the molecular identity of the O2 sensor is unclear though the transduction step in most cases involves modulation of various K+ channel subtypes (Haddad & Jiang, 1997; Peers, 1997; Lopez-Barneo et al. 2001; Plant et al. 2002).
In the last few years an emerging group of ‘leak‘ or background K+ channels, called two-pore (2P) domain K+ channels, has attracted special interest as targets for hypoxic modulation (Buckler 1997; Buckler et al. 2000; Hartness et al. 2001; Plant et al. 2002). These channels have subunits that contain four transmembrane regions surrounding two pore-forming loops in which lies the consensus sequence for the K+ selectivity filter (Lesage & Lazdunski, 2000; Goldstein et al. 2001; Patel & Honoré, 2001b). In addition, these channels lack voltage dependence and play a key role in setting the resting membrane potential and input resistance of the cell. A family of genes that encode background K+ channels has been recently identified, leading to the cloning and characterisation of a number of family members (Lesage & Lazdunski, 2000; Goldstein et al. 2001; Patel & Honoré, 2001a,b; Talley & Bayliss, 2002). The subfamilies include the acid-sensitive channels TASK-1 to TASK-5 (Duprat et al. 1997; Reyes et al. 1998; Kim et al. 2000; Ashmole et al. 2001; Decher et al. 2001; Vega-Saenz et al. 2001), the mechanosensitive channels TREK-1, TREK-2 and TRAAK, the weak inward rectifiers TWIK-1 and TWIK-2 (Lesage & Lazdunski, 2000; Patel & Honoré, 2001a,b), TALK-1 (Girard et al. 2001), and the halothane-inhibited channels THIK-1 and THIK-2 (Rajan et al. 2001). The background channels differ from other major K+ channel families (e.g. the voltage-gated Kv channels), which consist of subunits with only a single pore-forming domain.
Interestingly, two members of the 2P K+ channel family, TASK-1 (Kcnk3) and TASK-3 (Kcnk9), have been found to be O2 sensitive in different tissues. A TASK-1-like conductance has been shown to be O2 sensitive in rat carotid body chemoreceptors (Buckler et al. 2000) and cerebellar granule neurones (Plant et al. 2002), whereas TASK-3 appears to be O2 sensitive in the lung carcinoma cell line H146, which is a model for pulmonary neuroepithelial bodies (Hartness et al. 2001).
In the present study we have characterised a novel O2-sensing mechanism in neurones from the rat glossopharyngeal nerve (GPN). These autonomic neurones, together with a population of sensory neurones located in the petrosal ganglion, give rise to an extensive plexus of nitric oxide (NO)-synthesising nerve fibres that innervate the carotid body and are thought to play a key role in chemoreceptor adaptation to hypoxia (Wang et al. 1993, 1994a,b, 1995a,b; Höhler et al. 1994; Grimes et al. 1994). Interestingly, our electrophysiological and pharmacological characterisation of the O2-sensitive K+ conductance in GPN neurones revealed properties inconsistent with those of TASK-1 or TASK-3. Rather, the pharmacological profile suggested a new class of O2-sensitive background K+ channels that appear more closely related to the tandem pore domain, halothane-inhibited K+ (THIK) channel family (Rajan et al. 2001), members of which are expressed in brain where their physiological function is unknown. Alternatively, the O2-sensitive background K+ channel expressed in GPN neurones may belong to a new subfamily not yet described, or to a new variant of one of the already described 2P domain members.
METHODS
Cell culture
A section of the glossopharyngeal nerve (GPN), extending from its intersection with the carotid sinus nerve (CSN) to a region ∼5 mm distal to the intersection, was dissected from Wistar rat pups (10–14 days old, Charles River Laboratory, St Constant, QC, Canada). Pups were first stunned by a blow to the head that rendered them unconscious, and then killed by decapitation before removing the carotid bifurcation, surrounding ganglia and attached nerves. All procedures for animal handling were carried out according to the guidelines of the Canadian Council on Animal Care (CCAC). The distal GPNs were pooled, digested with enzyme (0.1 % collagenase, Invitrogen Canada Inc., Burlington, ON, Canada; 0.1 % trypsin, Sigma; and 0.01 % DNase, Invitrogen) and mechanically dissociated to produce dispersed GPN neurones that were grown on a thin layer of Matrigel (Collaborative Research, Bedford, MA, USA). The solutions and culture procedures were identical to those described elsewhere for the carotid body (Zhong et al. 1997). Cultures were grown at 37 °C in a humidified atmosphere of 95 % air-5 % CO2, and used either for patch-clamp experiments or immunofluorescence studies ∼24–48 h following isolation.
NADPH-diaphorase (NADPHd) cytochemistry
GPN whole-mounts were rinsed in pre-warmed phosphate buffered saline (PBS) and fixed in 4 % para-formaldehyde overnight at 4 °C. After washing in PBS (3 × 5 min) the tissue was permeabilised in 0.5 % Triton X-100-Tris-HCl overnight and incubated in NADPH (Sigma)-nitrobluetetrazolium-0.5 % Triton X-100-Tris-HCl, for 30 min at 37 °C in the dark. Whole-mounts were examined under brightfield optics after rinsing (3 × 5 min) with Tris-HCl.
Immunofluorescence
Whole-mounts of the GPN or cultured GPN neurones were fixed in 4 % para-formaldehyde overnight at 4 °C or for 15 min at room temperature (RT), respectively. After washing in PBS (3 × 5 min) whole-mounts were incubated for 72 h at 4 °C with primary antibody; an overnight incubation was used for cultures. Primary antibodies were: polyclonal rabbit anti-rat neuronal nitric oxide synthase (nNOS; 1:200; ImmunoStar Inc., Hudson, WI, USA) or monoclonal mouse anti-rat nNOS (1:200; Sigma), anti-neurofilament, a monoclonal antibody against NF68 kDa (1:5; Boehringer Mannheim, Montreal, QC, Canada) and a polyclonal antibody against vesicular ACh transporter (VAChT) raised in goat (1:200; Chemicon International, Temecula, CA, USA). The primary antiserum was diluted in PBS containing 1 % BSA and 0.5 % Triton X-100. After incubation, the samples were washed in PBS (3 × 5 min) and incubated in the dark for 1 h at room temperature with the secondary antibody conjugated to Alexa 594 (1:500, Molecular Probes, OR, USA), Cy3 (1:50; Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA) or fluorescein isothiocyanate (FITC; 1:50; Cappel, Malvern, PA, USA). The secondary antiserum was diluted in PBS containing 1 % BSA and 0.5 % Triton X-100. Samples were covered with a photobleaching reagent (Vectashield; Vector Laboratories, Burlingame, CA, USA) and viewed with a confocal microscope (Bio-Rad Microradiance 2000) equipped with argon (two lines, 488 and 514 nm) and helium-neon (543 nm) lasers. Lasersharp software was used for image acquisition. In control experiments incubation with secondary antibody alone resulted in complete abolition of staining.
DiI retrograde labelling
Whole-mounts of the carotid bifurcation were washed with pre-warmed PBS and fixed overnight in 4 % paraformaldehyde at 4 °C. After washing (3 × 5 min each) in 0.1 m phosphate buffer, the tissue was attached by stainless steel minutien pins (Fine Science Tools Inc., Austria) to a Petri dish coated with a Sylgard bed. After immersion in PBS, an opening was made in the carotid body and a small crystal of DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate; Molecular Probes Inc.) was inserted and left in place for several weeks at 4 °C. Finally, the tissue was placed on a glass slide and covered with a photobleaching reagent, before viewing with the confocal microscope.
Electrophysiology
The methods for obtaining nystatin perforated-patch recordings of membrane potential (current-clamp) and ionic currents (voltage-clamp) from GPN neurones were identical to those previously described for petrosal neurones (Stea & Nurse, 1992; Zhong et al. 1997). Some experiments were carried out using conventional whole-cell recording that resulted in intracellular dialysis (Hamill et al. 1981). Patch pipettes were made from Corning no. 7052 glass (A-M Systems Inc., Carlsborg, WA, USA) or borosilicate glass (WPI, Sarasota, FL, USA) using a Brown-Flaming horizontal puller (Model P-97, Sutter Instruments Co., San Francisco, CA, USA) and were fire-polished. When filled with intracellular recording solution, micropipettes had a resistance of 5–10 MΩ and formed gigaseals between 2 and 12 GΩ. Approximately 75 % of the series resistance (range, 10–20 MΩ) was compensated. Data were corrected for changes in junction potentials in different extracellular K+ solutions using pCLAMP 8.0 software. Whole-cell currents were recorded at 22–25 °C using an Axopatch 1D Amplifier (Axon Instruments Inc., Union City, CA, USA) equipped with a 1 GΩ headstage feedback resistor. Signals were filtered at 1 kHz and stored on a Pentium II PC with the aid of a Digidata 1200A/B computer interface and pCLAMP 8.0 software (Axon Instruments). Hypoxia (PO2∼15 mmHg) was generated by bubbling 100 % N2 into the perfusion reservoir and PO2 measurements were obtained with the aid of a carbon-fibre electrode (10 μm diameter, Dagan Corporation, Minneapolis, MN, USA) and a VA-10 NPI Amplifier (NPI Electronic, Hauptstrasse, Tamm, Germany). Control experiments were performed by bubbling the extracellular recording solution with compressed air instead of N2 gas. The solution in the recording chamber (volume, 0.75–1 ml) was exchanged by perfusion under gravity and simultaneous removal by suction at a rate of 6 ml min−1 (Thompson & Nurse, 1998). The effects of hypoxia were examined by comparing peak currents from the average of three records at each step potential, over the range of −100 to +70 mV (10 mV increments) during 50 ms, or ramps from −100 to 25 mV during 350 ms, from a holding potential of −60 mV. In some traces high frequency noise was filtered with the help of Clampfit 8.0 software.
Solutions
For perforated-patch recording patch pipettes were filled with intracellular recording solution of the following composition (mm): potassium gluconate 110, KCl 25, Hepes 10, NaCl 5, CaCl2 2, pH adjusted to 7.2 with KOH, plus 500 μg ml−1 nystatin. For recordings during whole-cell configuration patch pipettes were filled with solution of the following composition (mm): potassium gluconate 110, KCl 25, Hepes 10, NaCl 5, CaCl2 1, EGTA 11, pH adjusted to 7.2 with KOH. The extracellular recording solution contained (mm): NaCl 135, KCl 5, Hepes 10, glucose 10, CaCl2 2, MgCl2 2, pH adjusted to 7.4 with NaOH at RT. Some experiments, designed to dissect the O2-sensitive K+ current (IK,O2), were carried out in Ca2+-free extracellular recording solution containing different concentrations of K+ (Na+ substituted) and (mm): Hepes 10, glucose 10, MgCl2 2, NiCl2 2, pH adjusted to 7.4 with KOH or NaOH at RT.
Drugs
Various types of K+ currents (as indicated in the text) were blocked using the following drugs: tetraethylammonium (TEA, 5–30 mm), 4-aminopyridine (4-AP, 2–5 mm), Ba2+ (BaCl, 10 mm) and quinidine (0.01–1 mm) (all obtained from Sigma). In some experiments cadmium (CdCl2, 200 μm; Sigma) was used to block indirectly Ca2+-dependent K+ currents. In addition, some experiments were carried out in the presence of iberiotoxin (IbTx, 100 nm; Alomone) and apamin (100 nm, Alomone) to block large and small Ca2+-dependent K+ conductances, respectively. To block voltage-dependent Na+ currents, tetrodotoxin (TTX, 1 μm, Sigma) was added to the bathing solution. The volatile anaesthetic 2bromo-2chloro-1,1,1trifluoroethane (halothane; 2–5 mm, Fluka), arachidonic acid (AA, 5 μm, Sigma), ruthenium red (RR, 1–10 μm, Sigma) and anandamide (AN, 1–10 μM, Sigma) were added to the extracellular solution in some experiments.
Statistics
Current densities (pA pF−1) and membrane potentials (mV) were compared using paired Student's t tests and percentages were compared using a paired non-parametric test (Mann-Whitney), with the level of significance set at P < 0.05. In addition ANOVA was used for comparing difference currents, from independent groups (Fig. 2), in various experimental conditions.
Figure 2. Electrophysiological responses of cultured GPN neurones to hypoxia.
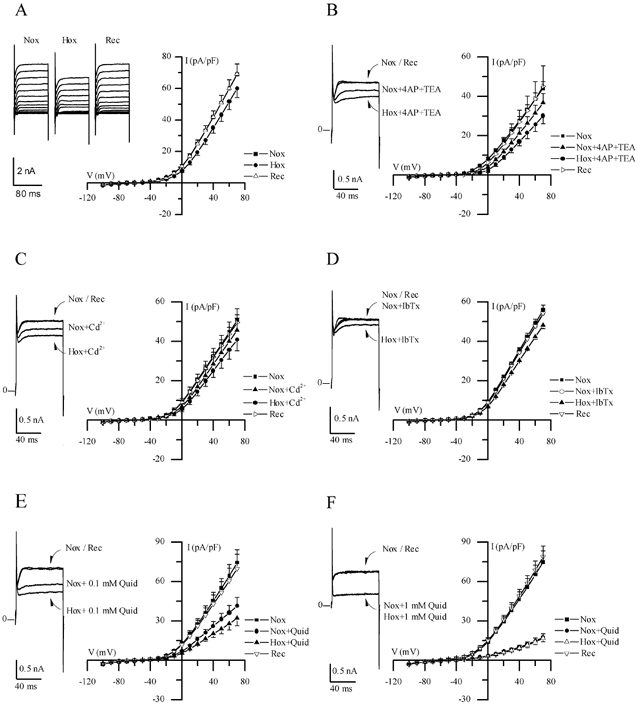
A, traces show a representative example of the reversible reduction of macroscopic outward current recorded in a GPN neurone after exposure to hypoxia in physiological extracellular K+ (5 mm) solution, during various step potentials from −100 to +70 mV. Current-voltage (I-V) plots of the current density (mean ±s.e.m.; n = 11) during normoxia (Nox), hypoxia (Hox) and recovery (Rec) are shown on the right; hypoxia caused a significant reduction in current density (P < 0.05) at potentials > 30 mV. B, traces show a representative example of the response to hypoxia in physiological solution containing 4-AP (2 mm) and TEA (5 mm), during a step to +40 mV; I-V plots show mean ±s.e.m. (n = 6) current density during normoxia and hypoxia in the presence of 4-AP and TEA. Note that hypoxic inhibition of the macroscopic outward currents persisted in physiological solution containing these classical voltage-dependent K+ channel blockers. Similar results were obtained in the presence of 200 μm Cd2+ (n = 6), 100 nm IbTx (n = 5) and 0.1 mm quinidine (Quid: n = 5) as shown in C, D and E, respectively. However, as shown in F, higher concentrations of quinidine (1 mm) caused further suppression of the outward current and, moreover, abolished the response to hypoxia (n = 7).
RESULTS
Location and projection of GPN neurones
The neurones investigated in this study were distributed in two groups along the rat glossopharyngeal nerve (GPN) near its branch point with the carotid sinus nerve (CSN) and at a more distal location (see Fig. 1A; rectangle). Together with a population of sensory neurones located in the rostral part of the petrosal ganglion (Wang et al. 1993) they give rise to a plexus of neuronal nitric oxide synthase (nNOS)-positive fibres that are thought to provide efferent inhibitory innervation to the carotid body (CB; Wang et al. 1993, 1994a,b, 1995a, b; Prabhakar, 1999; Fung et al. 2001). These neurones were revealed by retrograde labelling after placement of the lipophilic dye DiI in the carotid body (Fig. 1B), and by NADPHd activity (Fig. 1C) or positive nNOS-immunoreactivity (Fig. 1D) in whole-mounts of GPN. Interestingly, and as previously described by Wang et al. (1993) for neurones involved in efferent inhibition of CB chemoreceptors, GPN neurones expressed the cholinergic marker, vesicular ACh transporter (VAChT, Fig. 1E), which was co-localised with nNOS (Fig. 1F). Enzymatic dissociation of GPN yielded dispersed neurones that survived for several days in culture and were easily identified under phase contrast microscopy (Fig. 1G). These cultured neurones also stained positively for NADPHd activity (Fig. 1H), nNOS-immunoreactivity (Fig. 1I and J) and neurofilament immunoreactivity (NF 68 kDa; Fig. 1K and L). In addition, cultured GPN neurones expressed vesicular ACh transporter (VAChT, Fig. 1M). All VAChT immunoreactive cells in culture were found to co-express NF (e.g. Fig. 1L-N).
Figure 1. Localisation and properties of neurones of the rat glossopharyngeal nerve (GPN) in situ and in culture.
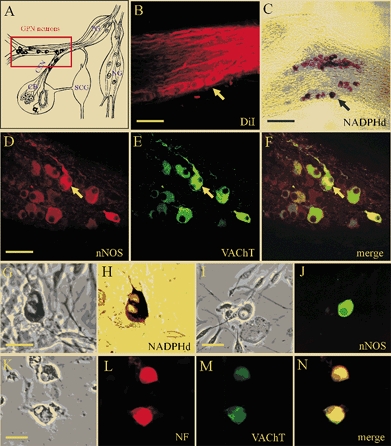
A, schematic drawing of the carotid bifurcation showing localisation of GPN neurones (red rectangle) embedded in the glossopharyngeal and carotid sinus (CSN) nerves (modified from Wang et al. 1993). Petrosal ganglion (PG), nodose ganglion (NG), superior cervical ganglion (SCG) and carotid body (CB) are indicated in the diagram. B, the GPN neurones (yellow arrow) are retrogradely labelled following injection of the lipophilic dye DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate) into the CB; whole-mount of glossopharyngeal nerve visualised with confocal microscopy. C, GPN neurones (black arrow) are visualised in situ in a whole-mount of glossopharyngeal nerve after staining for NADPH-diaphorase (NADPHd) activity. D-F, same microscopic field of GPN neurones in situ showing positive immunostaining for nNOS (D; Cy3) and the cholinergic marker VAChT (E; fluorescein isothiocyanate (FITC)); note co-localisation of the two markers (e.g. yellow arrow) in the same neurones (F). G and H, corresponding phase and brightfield images of positive NADPHd activity in two adjacent neurones after 2 days in culture. I and J, corresponding phase and nNOS immunofluorescence (FITC) in a cultured GPN neurone, 2 days after isolation. K-N, corresponding phase and fluorescence micrographs showing a pair of cultured GPN neurones that were immunopositive for both neurofilament (NF, L; Alexa 594) and VAChT (M; FITC); overlap of the two markers is shown in N. Scale bars represent 50 μm in B and D; 150 μm in C and 30 μm in G, I and K.
Electrophysiological properties of the hypoxia-sensitive K+ current (IK,O2) in GPN neurones
Perforated-patch recordings were made from GPN neurones usually within 1–2 days of isolation. In this study voltage- and current-clamp recordings were obtained from ∼100 neurones and of these 85 (i.e. ∼85 %) responded to hypoxia (PO2∼15 mmHg). As shown in Fig. 2A, hypoxia reversibly inhibited outward currents in GPN neurones at positive test potentials (n = 11); for a step to +40 mV, the mean outward current density was 42.0 ± 3.8 pA pF−1 before, 35.1 ± 3.5 pA pF−1 during (P < 0.05), and 41.9 ± 3.9 pA pF−1 after hypoxia. This effect was absent after intracellular dialysis during recording in the whole-cell configuration (not shown, n = 5) and in control experiments where the extracellular solution was bubbled with compressed air instead of N2 gas (not shown, n = 3). Notably, the inhibitory effect of hypoxia on the macroscopic outward currents persisted in physiological solution containing 5 mm TEA plus 2 mm 4-AP (n = 6; P < 0.05, Fig. 2B), or 200 μm cadmium (n = 6; Fig. 2C). In addition, 100 nm iberiotoxin (n = 5; Fig. 2C), a selective blocker of large conductance Ca2+-activated K+ channels, and apamin (n = 4; not shown), a selective blocker for small conductance Ca2+-activated K+ channels, had little or no effect on the magnitude of the macroscopic outward current, and similarly a full hypoxic response remained in the presence of these blockers. In contrast, 1 mm quinidine caused a dose-dependent inhibition of the outward current (Fig. 2E and F; n = 5 and 7, respectively) and also occluded the hypoxic response. Statistical analysis revealed that in the presence of the above K+ channel blockers (with the exception of 1 mm quinidine) hypoxic inhibition of outward current was significant (P < 0.05) at potentials > +30 mV. Moreover, the magnitude of the hypoxia-sensitive ‘difference’ current density (obtained by subtracting the current density during hypoxia from that during normoxia) remained unaffected in the presence of these blockers. These combined data suggest that background K+ channels were likely to be the mediators of hypoxic sensitivity in these neurones rather than voltage-gated, delayed rectifier-type or Ca2+-dependent K+ channels.
To enhance the magnitude of ‘leak’ currents and facilitate quantification of the hypoxia-sensitive component, voltage-clamp recordings were carried out in high K+ external solutions containing 1 μm TTX, 30 mm TEA, 5 mm 4-AP and 2 mm Ni2+ to block voltage-dependent currents. Representative current traces, typical of instantaneous ‘leak’ currents, and corresponding current-voltage (I-V) plots are shown in Fig. 3A for a cell exposed to this solution in symmetrical K+ (135 mm) conditions. The hypoxia-sensitive difference current (IK,O2) was obtained by subtracting the current recorded in hypoxia from that in normoxia (Fig. 3B). In symmetrical (135 mm) K+ conditions IK,O2 reversed direction near 0 mV (n = 7, Fig. 3C), the calculated Nernst K+ equilibrium potential (EK), and the I-V relation displayed little voltage dependence as expected of a K+-selective background conductance (Fig. 3B and C). To confirm K+ selectivity, current density versus voltage plots for IK,O2 were obtained using various extracellular K+ concentrations. When extracellular [K+] was 75 mm (n = 4), the reversal potential of IK,O2 shifted to ∼−14 mV as predicted for EK (Fig. 3C). In normal extracellular K+ (5 mm), the current density (pA pF−1) versus voltage plot for IK,O2 showed moderate outward rectification and reversed near −83 mV, the calculated K+ equilibrium potential (n = 4; Fig. 3C).
Figure 3. Electrophysiological properties of the background hypoxia-sensitive K+ current (IK,O2) in GPN neurones.
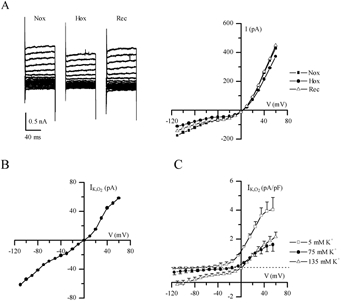
A, traces show a representative example of background or ‘leak’ currents before (Nox), during (Hox) and after (Rec) hypoxia, recorded in symmetrical high K+ solution (135 mm) containing TTX (1 μm), 4-AP (5 mm), TEA (30 mm) and Ni2+ (2 mm; substituted for Ca2+), during various voltage steps from −100 to +70 mV. Corresponding I-V plots are shown on the right. B, I-V plot shows a representative example of the hypoxia-sensitive ‘difference’ current (IK,O2), obtained by subtracting the current density during hypoxia from that during normoxia (see A). C, IK,O2 current density was obtained from groups of cells at three different extracellular K+ concentrations as follows: 5 mm (n = 4), 75 mm (n = 4) and 135 mm (n = 7); note that for each case the reversal potential of IK,O2 was similar to that predicted by the Nernst equation for K+ (i.e. −80, −14 and 0 mV, respectively).
Pharmacological properties of IK,O2
In order to test for functional similarities to other members of the background 2P domain K+ channel family, we investigated the sensitivity of IK,O2 to various agents including barium, quinidine, the polyunsaturated fatty acid arachidonic acid (AA) and the volatile anaesthetic halothane. The effects of some of these agents on ‘leak’ currents and their interaction with hypoxia are illustrated in Fig. 4A-C and summarised in Fig. 4D. A representative example of the reversible inhibition of the background current by hypoxia under symmetrical K+ conditions is shown in Fig. 4A. Similarly, halothane (2–5 mm) inhibited the background K+ current at negative potentials and interestingly, the inhibitory effect of hypoxia was occluded in the presence of halothane (Fig. 4B). Quantitative comparison of the inhibitory effect of hypoxia and halothane, applied separately and in combination, on background K+ current is shown in Fig. 4D. These data suggest that IK,O2 is halothane-inhibitable in GPN neurones, in contrast to the background IK,O2 expressed in rat carotid body chemoreceptors (Buckler et al. 2000). In addition to halothane, both barium (10 mm) and quinidine (1 mm) occluded the effect of hypoxia in GPN neurones, indicating that IK,O2 was also barium- and quinidine-sensitive (see also Fig. 2E and F). In contrast, AA caused potentiation of the background current as exemplified in Fig. 4C and summarised in Fig. 4D. In symmetrical K+ solutions and at a membrane potential of −60 mV, whereas quinidine (1 mm), barium (10 mm) and halothane (2–5 mm) inhibited the background K+ current by 40–100 %, AA (5 μm) caused a marked potentiation (∼50 %) of this current (Fig. 4D). Hypoxia had little or no effect (n = 4) when applied to preparations in which the background current was previously potentiated by AA (Fig. 4C). While these results may indicate that AA activated IK,O2, the data are inconclusive since in voltage-clamp studies the potentiating effects of AA were poorly reversible and therefore the interaction between AA and hypoxia could not be satisfactorily studied. Nevertheless, these effects of halothane and AA are opposite to those expected of the O2-sensitive TASK-1-like background K+ channels implicated in hypoxia-sensing in rat carotid body (Buckler et al. 2000) and of O2-sensitive TASK-3 channels in the H-146 cell line, a model for pulmonary airway chemoreceptors (Hartness et al. 2001). In contrast, they more closely resemble those expected of the THIK-1 (Rajan et al. 2001) or TWIK-2 (Chavez et al. 1999) 2P domain channels.
Figure 4. Pharmacological characterisation of the background K+ current in cultured GPN neurones.
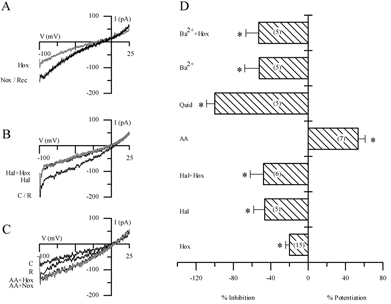
A-C, traces showing representative examples of the effect of hypoxia (A), halothane (B) and arachidonic acid (AA) (C) on the background current elicited during ramp depolarisation from −100 to +25 mV. Holding potential was −60 mV. Note the reversible inhibition of the current by hypoxia in A, and by halothane (2–5 mm) in B. In the presence of halothane, hypoxia produces no further inhibition of the current (B). In contrast, 5 μm AA potentiated the background current in C, and appeared to occlude the hypoxic response. D, effect of several agents on the background current in normoxia and hypoxia under symmetrical K+ conditions; for each agent bars represent the difference current at −60 mV expressed as a percentage of the control current (in normoxia); the difference current was obtained by subtracting the current during the particular treatment from the control current. The effects of hypoxia (Hox), halothane (Hal, 2–5 mm), quinidine (Quid, 1 mm) and barium (Ba2+, 10 mm) were all inhibitory. Note, however, that AA (5 μm) caused potentiation of the background current. In addition, when halothane and Ba2+ were applied in combination with hypoxia (Hal + Hox; Ba2++ Hox) there was occlusion of the response to hypoxia (P < 0.05). IK,O2 was also occluded in presence of 1 mm quinidine (Quid), which completely inhibited the background current. The sample size n is indicated in parentheses within the bars; *P < 0.05.
Evidence that IK,O2 is not carried by TASK-related channels
In order to confirm that acid-sensitive TASK channels did not mediate hypoxic sensitivity in GPN neurones we determined the pH sensitivity of IK,O2. These experiments revealed that extracellular acidification (pH 6.5 or 5.5) produced only a weak inhibition of IK,O2. A representative example is shown in Fig. 5A, where acidic pH (6.5 or 5.5) caused only ∼50 % inhibition of IK,O2 over a range of membrane potentials (−40 to −110 mV) under symmetrical K+ (135 mm) conditions. This pH sensitivity is relatively weak compared to the pronounced acid sensitivity of TASK-1 channels, but is not unlike that of THIK-1 (Kcnk13), the only functional member of the THIK family described to date (Rajan et al. 2001). For comparison, normalised values for IK,O2 in GPN neurones and for heterologously expressed TASK-1 in COS-7 cells (data from Kim et al. 1999) are plotted against pH in Fig. 5B, under symmetrical K+ solutions and at a membrane potential of −60 mV. Furthermore in GPN neurones, alkaline pH (8.5) produced only a weak potentiation of the normalised IK,O2 relative to neutral pH (Fig. 5B), a feature previously reported for THIK-1 channels (Rajan et al. 2001).
Figure 5. Regulation of the O2-sensitive current (IK,O2) expressed in GPN neurones by extracellular pH.
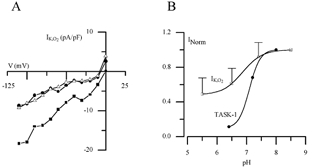
A, a representative example of the inhibition of IK,O2 by acidic pH in symmetrical 135 mm K+ solutions. Acidosis (pH 6.4 and 5.5) caused ∼50 % inhibition of IK,O2 at negative potentials. B, normalised pH sensitivity of IK,O2 in GPN neurones and TASK-1 current expressed in COS-7 cells (data from Kim et al. 1999) at −60 mV in symmetrical K+ solutions. Note the weak pH sensitivity of IK,O2 expressed in GPN neurones compared to TASK-1 current.
To further rule out the possible involvement of TASK-1 and TASK-3 channels in the O2-sensitive conductance expressed in GPN neurones, we used anandamide (AN, 1–10 μm) as a blocker for TASK-1 and ruthenium red (RR, 1–10 μm) as a blocker for TASK-3, while recording membrane potential in physiological K+ solutions. Figure 6 shows representative examples of the effect of these blockers. AN (Fig. 6A, n = 6) had no effect on the neuronal resting potential. Furthermore, when GPN neurones were exposed to hypoxia in the presence of AN the response to hypoxia was unaffected. In contrast, 2 μm RR caused hyperpolarisation of the neuronal resting potential (Fig. 6B, n = 5), from −46.3 ± 4.6 mV in control solution to −53.2 ± 5.8 mV (P < 0.05) and −48.4 ± 5.9 mV after washout. The cause of this hyperpolarisation is unknown but it may arise secondarily from an increase in intracellular Ca2+. Effects of RR on Ca2+ signalling include inhibition of the Ca2+-ATPase and mitochondrial Ca2+ uptake (Rigoni & Deana, 1986; Zhou & Bers, 2002), leading to an increase in cytosolic Ca2+ and activation of Ca2+-dependent K+ channels. Nonetheless, GPN neurones still responded to hypoxia in the presence of RR. These data strongly suggest that the O2-sensitive current expressed in GPN neurones is insensitive to both anandamide and RR, and therefore supports the non-involvement of TASK-1 and TASK-3 background K+ channels in hypoxic sensitivity.
Figure 6. Effect of anandamide and ruthenium red on membrane potential and hypoxic response.
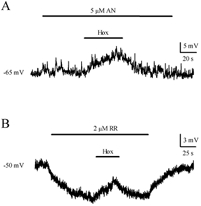
A, the TASK-1 channel blocker anandamide (AN, 5 μm) was without effect on the neuronal resting potential. In addition, the response to hypoxia was unaffected in the presence of AN (n = 6). B, application of the TASK-3 channel blocker ruthenium red (RR, 2 μm) caused hyperpolarisation of the resting potential probably due to an increase in intracellular Ca2+ and consequent activation of Ca2+-dependent K+ channels. However, in the presence of this agent GPN neurones still depolarised during hypoxia (n = 5).
Effects of hypoxia, halothane and AA on membrane potential
Since hypoxia inhibited a background K+ current in GPN neurones we predicted this would lead to membrane depolarisation and an increase in electrical excitability. Indeed, during current-clamp recordings hypoxia depolarised GPN neurones by 3–7 mV and increased membrane excitability, as illustrated in Fig. 7A and B. In physiological K+ solutions, the membrane potential depolarised from a mean ±s.e.m. resting level of −49.8 ± 1.6 mV (n = 18) in normoxia to −46.8 ± 1.6 mV during hypoxia (P < 0.001), followed by recovery to −49.9 ± 1.6 mV after return to normoxia (Fig. 7A right). In some neurones, depolarisation due to hypoxia was accompanied by spike activity (Fig. 7B). Similarly, 2–5 mm halothane depolarised the membrane potential from −54.8 ± 2.6 to −50.4 ± 3.0 mV (n = 6; P < 0.05), followed by recovery to −54.9 ± 2.1 mV after washout. Moreover, 2–5 mm halothane occluded the depolarising effects of hypoxia (n = 4; see Fig. 7C), as expected if both agents acted via inhibition of the same K+ channels. In contrast, AA (5 μm) had the opposite effect, producing a long-lasting hyperpolarisation from −47.7 ± 3.2 to −56.3 ± 3.3 mV (n = 3; Fig. 7D), consistent with its enhancing effect on background K+ currents. As observed during voltage-clamp studies the effects of AA on membrane potential recovered only slowly after washout of the drug (Fig. 7D).
Figure 7. Effect of hypoxia, halothane and AA on the membrane potential and excitability in GPN neurones.
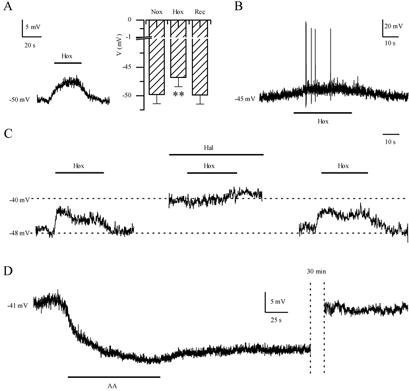
Representative examples of the hypoxia-induced membrane depolarisation and action potential firing in GPN neurones during current-clamp recordings are shown in A (left) and B, respectively. The histogram in A (right) shows that hypoxia (Hox) caused a significant (**P < 0.001) decrease of the resting potential to −46.8 ± 1.6 mV from −49.8 ± 1.6 mV (n = 18) during normoxia (Nox). C, representative example of the response to hypoxia before, during, and after application of halothane (Hal, 2–5 mm). Note that halothane caused membrane depolarisation and occlusion of the hypoxic response. D, representative example of the effect of AA (5 μm) on the resting potential. Note that AA caused a long-lasting hyperpolarisation and its effects were poorly reversible. In the example, recovery of the resting potential was reached ∼30 min after AA was removed from the extracellular solution.
DISCUSSION
The present study revealed a novel O2-sensing mechanism in peripheral neurones of the glossopharyngeal nerve (GPN) that innervate the rat carotid body (CB), the main arterial chemoreceptor in mammals (Gonzalez et al. 1994; Lopez-Barneo et al. 2001). These neurones were retrogradely labelled following injection of the lipophilic dye DiI in the CB, and were distributed along the GPN in two main groups, one of which was located near the branch point with the carotid sinus nerve (CSN). They also displayed positive immunoreactivity for vesicular ACh transporter (VAChT) and nNOS, and therefore correspond to neurones thought to underlie the basis for NO-mediated efferent inhibition of the CB (Wang et al. 1993, 1994a,b, 1995a,b; Höhler et al. 1994). Though we are unaware of data showing the presence of O2-sensing mechanisms in peripheral neurones, there are several reports of the presence of such mechanisms in central neurones, particularly those located in the ventrolateral medulla (Golanov & Reis, 1999; Mazza et al. 2000), hippocampus (Leblond & Krnjevic, 1989; Haddad & Jiang, 1997; Hammarström & Gage, 2000) and cerebellum (Plant et al. 2002). The O2-sensitive K+ current expressed in GPN neurones was unaffected by the classical voltage-dependent K+ channel blockers such as TEA and 4-AP, but it was reversibly blocked in a dose-dependent manner by quinidine. These data suggested the involvement of ‘leak’ or background K+ channels and this was validated in voltage-clamp studies where the hypoxia-sensitive ‘difference’ current IK,O2 reversed at the calculated K+ equilibrium potential in solutions of different extracellular K+ concentrations. Moreover, under symmetrical K+ conditions this current showed little or no voltage sensitivity, as expected of background K+ channels (Goldstein et al. 2001).
In general, background or resting K+-selective channels play a key role in setting the resting membrane potential and input resistance of the cell (Lesage & Lazdunski, 2000; Goldstein et al. 2001; Patel & Honoré, 2001b). They are therefore important determinants of the magnitude and kinetics of synaptic inputs in neurones and help shape neuronal excitability. More recently, these channels have additionally been recognised as critical sites for neuromodulation by endogenous ligands, as well as targets for clinically important volatile anaesthetics (Goldstein et al. 2001; Patel & Honoré, 2001a,b; Talley & Bayliss, 2002). Moreover, they can be regulated by important biophysical and biochemical parameters including pH, temperature and O2 tension (Buckler et al. 2000; Patel & Honoré, 2001b; Plant et al. 2002). So far, two members of the background 2P domain K+ channel family, the acid-sensitive TASK-1 (Buckler et al. 2000; Plant et al. 2002) and TASK-3 (Hartness et al. 2001) channels, have been shown to possess O2 sensitivity. These two background channels are, however, inhibited by AA and activated by halothane, effects that are opposite to the O2-sensitive current (IK,O2) found in GPN neurones in the present study. Moreover, the relatively weak pH sensitivity of IK,O2 contrasts with that of TASK-1 and TASK-3 channels (Duprat et al. 1997; Leonoudakis et al. 1998; Buckler et al. 2000; Sirois et al. 2000; Hartness et al. 2001). For example, IK,O2 displayed only ∼50 % inhibition at pH 5.5, compared to the almost total inhibition of TASK-1 channels expressed in either heterologous systems (Lopes et al. 2000, Sirois et al. 2000; Duprat et al. 1997; Leonoudakis et al. 1998; Kim et al. 1999) or native cerebellar granule neurones (Plant et al. 2002).
Similar to the O2-sensitive channels in GPN neurones, two other members of the background channel family, THIK-1 (Kcnk13) and TWIK-2 (Kcnk6), are inhibited by halothane and activated by AA. However, when rat TWIK-2 was expressed in Xenopus oocytes (Chavez et al. 1999) or COS cells (Patel et al. 2000), the currents were reversibly inhibited by barium and quinidine in physiological K+ solutions, but the inhibition was not observed in a symmetrical K+ gradient (Chavez et al. 1999; Patel et al. 2000), even with high barium concentrations (10 mm, Patel et al. 2000). These properties contrast with those of IK,O2 expressed in GPN neurones and in addition, TWIK-2 currents display an inward rectification and inactivation at positive test potentials (Chavez et al. 1999; Patel et al. 2000) not seen in IK,O2. Therefore, TWIK-2 is unlikely to be the O2-sensitive conductance expressed in GPN neurones. Rather, the properties of this current, including its relatively weak pH sensitivity, more closely resembled those of the only functional member of the THIK family (THIK-1) described to date (Rajan et al. 2001).
The O2-sensitive current was undetectable during voltage- and current-clamp recordings in dialysed GPN neurones studied with conventional whole-cell recording. These data suggest that the O2-sensitive K+ channel requires intact cytosolic components in order to be functional, as previously observed for the TASK-1-like O2-sensitive K+ channels expressed in CB body chemoreceptors (Buckler et al. 2000). In the case of GPN neurones, in preliminary experiments we applied AA and halothane while recording membrane potential in physiological solutions to test whether the O2-sensitive conductance was still functional in dialysed preparations (n = 4, not shown). Interestingly, the effects of AA and halothane did not differ from those observed during perforated-patch recordings (see Fig. 7C and D), suggesting that the relevant background conductance was functional in dialysed cells but it lacked O2 sensitivity. Further studies are needed in order to understand the cytosolic regulation of the background channels in GPN neurones.
While the present data point to the possible involvement of the recently identified THIK (tandem pore-domain halothane-inhibited K+ channels) subfamily of background K+ channels (Girard et al. 2001; Rajan et al. 2001) in mediating the O2 sensitivity of GPN neurones, other candidates are not excluded. Conceivably, the O2-sensitive current might be carried by a new member of the 2P domain family not yet described, or by a new variant of one of the already described background channels (Goldstein et al. 2001). Therefore, molecular identification of the O2-sensitive K+ conductance will be necessary to clarify which member of the 2P domain family is indeed responsible for conferring O2 sensitivity to GPN neurones. Nevertheless, it is interesting that the pharmacological profile of IK,O2 resembles that of THIK channels. Of the two closely related members of this family, i.e. THIK-1 and THIK-2 (58 % identity at the amino acid level; Rajan et al. 2001), THIK-1 was broadly expressed in several tissues and was functional when heterologously expressed in Xenopus oocytes (Rajan et al. 2001). In contrast, the expression of THIK-2 (Kcnk12) was more restricted, though it was especially abundant in brain (Rajan et al. 2001). Moreover, it could not be functionally expressed in Xenopus oocytes and therefore its physiological function awaits characterisation (Girard et al. 2001; Rajan et al. 2001). Since the physiological function of THIK channels is unknown, our studies raise, for the first time, the possibility of an O2-sensing function for this background channel family.
Physiological relevance
This study describes a novel O2-sensitive background K+ conductance in GPN neurones. Since background channels play a key role in setting the resting membrane potential and controlling neuronal excitability, modulation of IK,O2 may help control neuronal function during ischaemic stress. In GPN neurones these O2-sensitive channels may also play a hitherto unrecognised role in the control of respiration, since these neurones are thought to provide NO-mediated efferent inhibition of CB chemoreceptors (Wang et al. 1995b), which are excited by hypoxia. Since GPN neurones are also excited by hypoxia, via inhibition of background channels, the possibility is raised that this excitation leads to increase in intracellular Ca2+ levels, activation of nNOS, followed by synthesis and release of NO. The resulting inhibitory effects of NO on carotid body function may provide a means of negative feedback modulation of chemoreceptor activity during hypoxia. In this way the O2-sensitive background conductance expressed in GPN neurones may contribute to the control of respiration via regulation of carotid body function during hypoxia and conceivably, following exposure to volatile anaesthetics, e.g. halothane.
Acknowledgments
We thank Cathy Vollmer for expert technical assistance, and Dr James Mwanjewe for his generous gift of the nNOS antibody. This work was supported by a grant from the Canadian Institutes for Health Research to C.A.N. (MOP-57909). In addition, V.A.C was supported by a Dr Sydney Segal Research Grant from The SIDS Foundation of Canada and I.M.F. was supported by a Wellcome Trust Prize International Travelling Research Fellowship (Ref. no. 16154).
REFERENCES
- Archer SL, Huang J, Henry T, Peterson D, Weir EK. A redox based oxygen sensor in rat pulmonary vasculature. Circ Res. 1993;73:1100–1112. doi: 10.1161/01.res.73.6.1100. [DOI] [PubMed] [Google Scholar]
- Archer SL, Souil E, Dinh-Xuan AT, Schremmer B, Mercier JC, El Yaagoubi A, Nguyen-Huu L, Reeve HL, Hampl V. Molecular identification of the role of voltage-gated K+ channels, Kv1. 5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest. 1998;101:2319–2330. doi: 10.1172/JCI333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashmole I, Goodwin PA, Stanfield PR. TASK-5, a novel member of the tandem pore K+ channel family. Pflugers Arch. 2001;442:828–833. doi: 10.1007/s004240100620. [DOI] [PubMed] [Google Scholar]
- Buckler KJ. A novel oxygen-sensitive potassium current in rat carotid body type I cells. J Physiol. 1997;498:649–662. doi: 10.1113/jphysiol.1997.sp021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Williams BA, Honoré E. An oxygen-, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemoreceptor cells. J Physiol. 2000;525:135–142. doi: 10.1111/j.1469-7793.2000.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez RA, Gray AT, Zhao BB, Kindler CH, Mazurek MJ, Mehta Y, Forsayeth JR, Yost CS. TWIK-2, a new weak inward rectifying member of the tandem pore domain potassium channel family. J Biol Chem. 1999;274:7887–7892. doi: 10.1074/jbc.274.12.7887. [DOI] [PubMed] [Google Scholar]
- Decher N, Maier M, Dittrich W, Gassenhuber J, Bruggemann A, Busch AE, Steinmeyer K. Characterisation of TASK-4, a novel member of the pH-sensitive, two-pore domain potassium channel family. FEBS Lett. 2001;492:84–89. doi: 10.1016/s0014-5793(01)02222-0. [DOI] [PubMed] [Google Scholar]
- Duprat F, Lesage F, Fink M, Reyes R, Heurteaux C, Lazdunski M. TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J. 1997;16:5464–5471. doi: 10.1093/emboj/16.17.5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XW, Wang D, Nurse CA, Dinauer MC, Cutz E. NADPH oxidase is an O2 sensor in airway chemoreceptors: Evidence from K+ current modulation in wild-type and oxidase-deficient mice. Proc Natl Acad Sci U S A. 2000;97:4374–4379. doi: 10.1073/pnas.97.8.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung ML, Ye JS, Fung PC. Acute hypoxia elevates nitric oxide generation in rat carotid body in vitro. Pflugers Arch. 2001;442:903–909. doi: 10.1007/s004240100610. [DOI] [PubMed] [Google Scholar]
- Girard C, Duprat F, Terrenoire C, Tinel N, Fosset M, Romey G, Lazdunski M, Lesage F. Genomic and functional characteristics of novel human pancreatic 2P domain K+ channels. Biochem Biophys Res Commun. 2001;282:249–256. doi: 10.1006/bbrc.2001.4562. [DOI] [PubMed] [Google Scholar]
- Golanov EV, Reis DJ. A role for KATP-channels in mediating the elevations of cerebral blood flow and arterial pressure by hypoxic stimulation of oxygen-sensitive neurons of rostral ventrolateral medulla. Brain Res. 1999;827:10–14. doi: 10.1016/s0006-8993(99)01256-1. [DOI] [PubMed] [Google Scholar]
- Goldstein SA, Bockenhauer D, O'Kelly I, Zilberberg N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci. 2001;2:175–184. doi: 10.1038/35058574. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A, Rigual R. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiol Rev. 1994;74:829–898. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- Grimes PA, Lahiri S, Stone R, Mokashi A, Chug D. Nitric oxide synthase occurs in neurons and nerve fibers of the carotid body. Adv Exp Med Biol. 1994;360:221–224. doi: 10.1007/978-1-4615-2572-1_33. [DOI] [PubMed] [Google Scholar]
- Haddad GG, Jiang C. O2-sensing mechanisms in excitable cells: role of plasma membrane K+ channels. Annu Rev Physiol. 1997;59:23–42. doi: 10.1146/annurev.physiol.59.1.23. [DOI] [PubMed] [Google Scholar]
- Haddad GG, Jiang C. Mechanisms of anoxia-induced depolarization in brainstem neurons: in vitro current and voltage-clamp studies in the adult rat. Brain Res. 1993;625:261–268. doi: 10.1016/0006-8993(93)91067-3. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hammarström AK, Gage PW. Oxygen-sensing persistent sodium channels in rat hippocampus. J Physiol. 2000;529:107–118. doi: 10.1111/j.1469-7793.2000.00107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartness ME, Lewis A, Searle GJ, O'Kelly I, Peers C, Kemp PJ. Combined antisense and pharmacological approaches implicate hTASK as an airway O2-sensing K+ channel. J Biol Chem. 2001;276:26499–26508. doi: 10.1074/jbc.M010357200. [DOI] [PubMed] [Google Scholar]
- Henrich M, Hoffmann K, Konig P, Gruss M, Fischbach T, Godecke A, Hempelmann G, Kummer W. Sensory neurons respond to hypoxia with NO production associated with mitochondria. Mol Cell Neurosci. 2002;20:307–322. doi: 10.1006/mcne.2002.1111. [DOI] [PubMed] [Google Scholar]
- Hohler B, Mayer B, Kummer W. Nitric oxide synthase in the rat carotid body and carotid sinus. Cell Tissue Res. 1994;276:559–564. doi: 10.1007/BF00343953. [DOI] [PubMed] [Google Scholar]
- Kim Y, Bang H, Kim D. TBAK-1 and TASK-1, two-pore K+ channel subunits: kinetic properties and expression in rat heart. Am J Physiol. 1999;277:H1669–1678. doi: 10.1152/ajpheart.1999.277.5.H1669. [DOI] [PubMed] [Google Scholar]
- Kim Y, Bang H, Kim D. TASK-3, a new member of the tandem pore K+ channel family. J Biol Chem. 2000;275:9340–9347. doi: 10.1074/jbc.275.13.9340. [DOI] [PubMed] [Google Scholar]
- Leblond J, Krnjevic K. Hypoxic changes in hippocampal neurons. J Neurophysiol. 1989;62:1–14. doi: 10.1152/jn.1989.62.1.1. [DOI] [PubMed] [Google Scholar]
- Leonoudakis D, Gray AT, Winegar BD, Kindler CH, Harada M, Taylor DM, Chavez RA, Forsayeth JR, Yost CS. An open rectifier potassium channel with two pore domains in tandem cloned from rat cerebellum. J Neurosci. 1998;18:868–877. doi: 10.1523/JNEUROSCI.18-03-00868.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage F, Lazdunski M. Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol. 2000;279:F793–801. doi: 10.1152/ajprenal.2000.279.5.F793. [DOI] [PubMed] [Google Scholar]
- Lopes CM, Gallagher PG, Buck ME, Butler MH, Goldstein SA. Proton block and voltage gating are potassium-dependent in the cardiac leak channel Kcnk3. J Biol Chem. 2000;275:16969–16978. doi: 10.1074/jbc.M001948200. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Pardal R, Ortega-Saenz P. Cellular mechanism of oxygen sensing. Annu Rev Physiol. 2001;63:259–287. doi: 10.1146/annurev.physiol.63.1.259. [DOI] [PubMed] [Google Scholar]
- Mazza E, Jr, Edelman NH, Neubauer JA. Hypoxic excitation in neurons cultured from the rostral ventrolateral medulla of the neonatal rat. J Appl Physiol. 2000;88:2319–2329. doi: 10.1152/jappl.2000.88.6.2319. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E. Anesthetic-sensitive 2P domain K+ channels. Anesthesiology. 2001a;95:1013–1021. doi: 10.1097/00000542-200110000-00034. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E. Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci. 2001b;24:339–346. doi: 10.1016/s0166-2236(00)01810-5. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Maingret F, Magnone V, Fosset M, Lazdunski M, Honoré E. TWIK-2, an inactivating 2P domain K+ channel. J Biol Chem. 2000;275:28722–28730. doi: 10.1074/jbc.M003755200. [DOI] [PubMed] [Google Scholar]
- Peers C. Oxygen-sensitive ion channels. Trends Pharmacol Sci. 1997;18:405–408. doi: 10.1016/s0165-6147(97)01120-6. [DOI] [PubMed] [Google Scholar]
- Plant LD, Kemp PJ, Peers C, Henderson Z, Pearson HA. Hypoxic depolarisation of cerebellar granule neurons by specific inhibition of TASK-1. Stroke. 2002;33:2324–2328. doi: 10.1161/01.str.0000027440.68031.b0. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. NO and CO as second messengers in oxygen sensing in the carotid body. Respir Physiol. 1999;115:161–168. doi: 10.1016/s0034-5687(99)00019-5. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. Oxygen sensing by the carotid body chemoreceptors. J Appl Physiol. 2000;88:2287–2295. doi: 10.1152/jappl.2000.88.6.2287. [DOI] [PubMed] [Google Scholar]
- Rajan S, Wischmeyer E, Karschin C, Preisig-Muller R, Grzeschik KH, Daut J, Karschin A, Derst C. THIK-1 and THIK-2, a novel subfamily of tandem pore domain K+ channels. J Biol Chem. 2001;276:7302–7311. doi: 10.1074/jbc.M008985200. [DOI] [PubMed] [Google Scholar]
- Reyes R, Duprat F, Lesage F, Fink M, Salinas M, Farman N, Lazdunski M. Cloning and expression of a novel pH-sensitive two pore domain K+ channel from human kidney. J Biol Chem. 1998;273:30863–30869. doi: 10.1074/jbc.273.47.30863. [DOI] [PubMed] [Google Scholar]
- Rigoni F, Deana R. Ruthenium red inhibits the mitochondrial Ca2+ uptake in intact bovine spermatozoa and increases the cytosolic Ca2+ concentration. FEBS Lett. 1986;198:103–108. doi: 10.1016/0014-5793(86)81193-0. [DOI] [PubMed] [Google Scholar]
- Sirois JE, Lei Q, Talley EM, Lynch C III, Bayliss DA. The TASK-1 two-pore domain K+ channel is a molecular substrate for neuronal effects of inhalation anesthetics. J Neurosci. 2000;20:6347–6354. doi: 10.1523/JNEUROSCI.20-17-06347.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stea A, Nurse CA. Whole-cell currents in two subpopulations of cultured rat petrosal neurons with different tetrodotoxin sensitivities. Neuroscience. 1992;47:727–736. doi: 10.1016/0306-4522(92)90180-a. [DOI] [PubMed] [Google Scholar]
- Talley EM, Bayliss DA. Modulation of TASK-1 (Kcnk3) and TASK-3 (Kcnk9) potassium channels. Volatile anesthetics and neurotransmitters share a molecular site of action. J Biol Chem. 2002;277:17733–17742. doi: 10.1074/jbc.M200502200. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Nurse CA. Anoxia differentially modulates multiple K+ currents and depolarises neonatal rat adrenal chromaffin cells. J Physiol. 1998;512:421–434. doi: 10.1111/j.1469-7793.1998.421be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Saenz DeMiera E, Lau DH, Zhadina M, Pountney D, Coetzee WA, Rudy B. Kt3. 2 and kt3.3, two novel human two-pore K+ channels closely related to TASK-1. J Neurophysiol. 2001;86:130–142. doi: 10.1152/jn.2001.86.1.130. [DOI] [PubMed] [Google Scholar]
- Wang ZZ, Bredt DS, Fidone SJ, Stensaas LJ. Neurons synthesising nitric oxide innervate the mammalian carotid body. J Comp Neurol. 1993;336:419–432. doi: 10.1002/cne.903360308. [DOI] [PubMed] [Google Scholar]
- Wang ZZ, Dinger BG, Stensaas LJ, Fidone SJ. The role of nitric oxide in carotid chemoreception. Biol Signals. 1995a;4:109–116. doi: 10.1159/000109430. [DOI] [PubMed] [Google Scholar]
- Wang ZZ, Stensaas LJ, Bredt DS, Dinger B, Fidone SJ. Localisation and actions of nitric oxide in the cat carotid body. Neuroscience. 1994a;60:275–286. doi: 10.1016/0306-4522(94)90221-6. [DOI] [PubMed] [Google Scholar]
- Wang ZZ, Stensaas LJ, Bredt DS, Dinger BG, Fidone SJ. Mechanisms of carotid body inhibition. Adv Exp Med Biol. 1994b;360:229–235. doi: 10.1007/978-1-4615-2572-1_35. [DOI] [PubMed] [Google Scholar]
- Wang ZZ, Stensaas LJ, Dinger BG, Fidone SJ. Nitric oxide mediates chemoreceptor inhibition in the cat carotid body. Neuroscience. 1995b;65:217–229. doi: 10.1016/0306-4522(94)00437-a. [DOI] [PubMed] [Google Scholar]
- Youngson C, Nurse C, Yeger H, Cutz E. Oxygen sensing by airway chemoreceptors. Nature. 1993;365:153–155. doi: 10.1038/365153a0. [DOI] [PubMed] [Google Scholar]
- Zhong H, Zhang M, Nurse CA. Synapse formation and hypoxic signalling in co-cultures of rat petrosal neurones and carotid body type 1 cells. J Physiol. 1997;503:599–612. doi: 10.1111/j.1469-7793.1997.599bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Bers DM. Time course of action of antagonists of mitochondrial Ca2+ uptake in intact ventricular myocytes. Pflugers Archiv. 2002;445:132–138. doi: 10.1007/s00424-002-0909-7. [DOI] [PubMed] [Google Scholar]