

Abstract
Damage to skeletal muscle occurs following excessive exercise, upon reperfusion following ischaemia and in disease states, such as muscular dystrophy. Key mechanisms by which damage is thought to occur include a loss of intracellular calcium homeostasis, loss of energy supply to the cell, increased activity of oxidising free radical-mediated reactions and activation of apoptosis pathways. An increased cellular content of heat shock proteins (HSPs) has been shown to protect skeletal muscle against some forms of damage, although the mechanistic basis of this protection is not clearly understood. The aim of this study was to establish a cell culture-based model of damage to C2C12 skeletal muscle cells using the calcium ionophore, A23187 and the mitochondrial uncoupler, 2,4-dinitrophenol (DNP) as damaging agents. Treatment of cells with 1 mm DNP for 60 min resulted in the release of 63.5 % of intracellular creatine kinase (CK) activity over the 3 h experimental period. Treatment of cells with 10 μm A23187 for 30 min resulted in the release of 47.9 % of CK activity. Exposure of myotubes to a period of hyperthermia resulted in a significant increase in their content of HSP25, HSP60, HSC70 (heat shock cognate) and HSP70. This increase in HSPs was associated with significant protection against both DNP-induced and A23187-induced damage to the myotubes. These results indicate that an increased content of HSPs may provide protection against the muscle damage that occurs by a pathological increase in intracellular calcium or uncoupling of the mitochondrial respiratory chain.
Damage to skeletal muscle occurs in various situations, such as following excessive or unaccustomed exercise, upon reperfusion following ischaemia and in disease states, including the muscular dystrophies. In these situations, the initial insult may be wide ranging, but the final route of damage is thought to occur via several key mechanisms (McArdle & Jackson, 1997). The pathways that have been extensively studied are: a loss of intracellular Ca2+ homeostasis, a loss of energy supply to the cell, or an increased activity of oxidising free radical-mediated reactions and, in recent years, activation of apoptosis pathways (McArdle & Jackson, 1997; McArdle et al. 1999).
Cells have endogenous mechanisms to protect against damage, facilitate recovery from non-lethal damage and adapt to prevent subsequent damage. One of the most extensively described of these is the stress response. All nucleated cells respond to a short period of (non-damaging) stress by increased synthesis of a family of proteins known as stress or heat shock proteins (HSPs; see Hightower, 1991; Feige et al. 1996; Gething, 1997; for comprehensive reviews). HSPs are named according to their molecular mass and include the small HSPs such as HSP25, HSP60, the HSP70 family of proteins and the larger HSPs such as HSP90 and HSP110. Some HSPs are constitutively expressed in the unstressed cell where they act as molecular chaperones, associating with newly synthesised proteins, facilitating folding and aiding translocation of newly synthesised proteins to intracellular sites such as the mitochondria (Hubbard & Sander, 1991; Gething, 1997; Fink, 1999). When cellular stress leads to the production of unfolded or misfolded proteins, the cellular content of HSPs is increased. Thus, the HSP content of cells is increased following heat, infection, incorporation of amino acid analogues and various forms of oxidative stress including exercise (Salo et al. 1991; Voellmy, 1996; Liu & Steinacker 2001; McArdle et al. 2001).
An increased cellular content of HSPs can provide cytoprotection against subsequent stresses. For example, in the heart, a period of hyperthermia provided substantial protection against damage induced by subsequent ischaemia and reperfusion, the calcium paradox or the oxygen paradox (Marber, 1994; Plumier & Currie, 1996). This protection was associated with a sixfold increase in the HSP70 content of the heart (Marber, 1994). Definitive evidence for the crucial role of HSP70 in this protection was presented by Marber et al. (1995) and Plumier et al. (1995). These studies independently demonstrated significant reductions in damage induced by ischaemia-reperfusion in hearts from transgenic mice overexpressing HSP70 in cardiac tissue.
A similar protective mechanism has also been reported in studies of skeletal muscle. Hyperthermia or exercise results in an increased content of HSPs in skeletal muscle (McArdle et al. 1997; Febbraio & Koukoulas, 2000; Khassaf et al. 2001; McArdle et al. 2001). In addition, a prior period of hyperthermia or a non-damaging period of ischaemia-reperfusion provides protection against subsequent ischaemia and reperfusion-induced skeletal muscle damage (Garramone et al. 1994; Lepore et al. 2001).
A variety of models have been developed to study muscle damage and adaptation. These include the use of cultured muscle cells, isolated single muscle fibres, bundles of fibres, intact isolated rodent muscles or damage to muscle in situ in animals and man (Faulkner et al. 1982, 1993; McArdle et al. 1992; Head, 1993; Pressmar et al. 1994; Faulkner & Brooks, 1997). Development of a reproducible model of skeletal muscle damage in cell culture would have a number of advantages over established in vitro, in vivo and in situ models. Most importantly, it could provide a model in which the specific response of muscle cells could be examined and to which approaches could be applied to examine the basic mechanisms underlying damage to skeletal muscle to help identify means of preventing damage.
The aim of this study was to examine the effect of prior heat stress on the susceptibility of skeletal muscle cells in culture to damage induced by the Ca2+ ionophore, A23187, and the mitochondrial uncoupler, 2,4-dinitrophenol (DNP), as damaging agents of relevance to processes known to occur in vivo. Our hypothesis was that a period of hyperthermia to myotubes in culture would lead to an increased content of HSPs and that this increased content would be associated with protection against damage induced by A23187 or DNP.
METHODS
Preparation of myoblasts and myotubes
C2C12 cells derived from a mouse cell line originally established by Yaffe & Saxel (1977) were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 0.45 % glucose with 2 mm glutamine, 50 i.u. penicillin, 50 μg ml−1 streptomycin and 12 % (v/v) fetal calf serum (FCS). Cells were grown at 37 °C in 5 % (v/v) CO2 in a humidified environment. Myoblasts were subcultured using 0.25 % (w/v) trypsin and 0.025 % (wv) EDTA in Dulbecco's PBS (D-PBS; Sigma Chemical Co. Ltd, UK). Myoblasts were passaged when 70–80 % confluence was reached.
To induce myotube formation, the medium was replaced with DMEM containing 2 % horse serum (HS) with 0.45 % (wv) glucose and 2 mm glutamine prior to the myoblasts reaching confluence. The change of 12 % FCS to 2 % HS enhances the formation of myotubes by encouraging fusion of myoblasts. Development of myotubes was assessed by light microscopy and by analysis of the total cellular activity of the muscle specific enzyme, creatine kinase (CK). The HSP content of myoblasts and myotubes during development was analysed by Western blotting as described below.
Basic model of damage to myotubes in culture
Myoblasts were grown as an adherent monolayer in sterile 6-well tissue culture plates. Seven days after the addition of HS, the medium was removed and myotubes were washed in sterile D-PBS for 30 min to remove excess medium and serum. Cells were incubated in 1 ml of D-PBS at 37 °C for 30 min. The D-PBS was removed and replaced for 1 × 30 min with D-PBS containing the calcium ionophore A23187 or for 2 × 30 min with D-PBS containing DNP. Control, untreated cells were incubated in D-PBS alone. This medium was then removed and replaced with 1 ml D-PBS every 30 min for a further 60–90 min. All media were analysed for CK activity and soluble protein content. At the end of the experiment, cells were harvested in 1 ml D-PBS. The resulting suspension was sonicated and analysed for CK activity and protein content. Release of CK activity and soluble protein from cells into the medium was expressed as a percentage of the total CK activity.
The effect of prior hyperthermia on HSP content and damage to myotubes in culture
Seven days after the addition of HS, myotubes were cultured in 6-well tissue culture plates. The temperature of the test cells was raised to 42 °C for 30 min. Control cells were maintained at 37 °C. At 4, 8, 12, 18 and 24 h following hyperthermia, the cells were washed with excess D-PBS and harvested into 1 ml D-PBS. The cells were pelleted and resuspended in homogenisation buffer containing 1 % SDS and a range of protease inhibitors. Cells were sonicated and analysed for HSP content as described below.
Alternatively, myotubes at 4, 8, 12, 18 and 24 h following hyperthermia were subjected to damage induced by treatment with 1 mm DNP or 10 μm A23187 as described above. Damage was assessed by release of CK activity into surrounding medium.
Methods of analysis of cellular damage and death
The protein content of the D-PBS and myotubes was measured using the bicinchoninic acid (BCA) protein assay (Sigma), based on the method of Smith et al. (1985).
CK activity of cells and medium was determined using a modification of the spectrophotometric method described by Jones et al. (1983). A 50 μl sample was added to 200 μl of a cocktail of assay reagents in a 96-well microtitre plate and the change in absorbance in each well was measured for 10 min at a wavelength of 340 nm using a microplate reader (Benchmark, Biorad, UK). Activity of CK was calculated by the conversion of NADP+ to NADPH where one unit of CK activity is equivalent to the conversion of 1 μmol of creatine phosphate substrate per minute at 20 °C.
An alternative method to assess plasma membrane permeability of myotubes was also applied. Cells were stained by incubation in 1 ml of D-PBS containing 2 μl of a Live/Dead cell viability kit (Molecular Probes, OR, USA) for 30 min in the dark. The medium was then removed and cells were fixed with 4 % gluteraldehyde in D-PBS. The cells were viewed immediately using a fluorescent microscope (Polyvar, Germany). The kit comprises two reagents. The first is the cell permeant green fluorescent SYTO 10, which is an indicator of cells that have esterase activity as well as an intact membrane to retain the esterase products. The second component is ethidium homodimer-1, which is a red fluorescent nucleic acid stain that is only able to pass through compromised membranes of dead cells where it shows as a red nuclear stain. The dye concentrations and their relative affinities are balanced such that a cell population exposed simultaneously to both dyes becomes differentially stained.
Analysis of HSP content of myotubes by SDS-PAGE and Western blotting
Cells were harvested into a 1 % solution of SDS containing (mm): 1 iodoacetamide, 1 benzethonium chloride, 5.7 phenylmethyl sulphonyl fluoride and 5 EGTA. Cells were then sonicated and centrifuged at 2000 g for 5 min. Protein content of the supernatant was determined using the BCA protein assay (Sigma). A 50 μg sample of total cellular protein was boiled for 5 min in a water bath in an equal volume of Laemmli buffer (National Diagnostics, Atlanta, GA, USA). The cooled sample was then applied to a 12 % polyacrylamide gel with a 4 % stacking gel (Protogel, National Diagnostics). Electrophoresis was carried out at a constant current of 40 mA. The separated proteins were then transferred from the gel onto a nitrocellulose membrane by Western blotting using a Multiphore II discontinuous blotting system (Pharmacia, Uppsala, Sweden). A constant current of 0.8 mA cm−2 was applied to the system for 90 min. Following electroblotting, the nitrocellulose membrane was analysed for HSP content using a panel of antibodies raised against HSP25 (Stressgen, Victoria, Canada), HSP60 (Stressgen), HSC70 (Sigma) and HSP70 (Amersham International Laboratories, Amersham, UK). Bands were visualised using the ECL film and chemiluminescent detection kit (Amersham). Membranes were exposed to film for 3–4 different exposure times to ensure that saturation of film had not occurred. Samples from each experiment were applied to the same gel. The intensity of staining for individual HSPs was quantified by densitometry and the content of HSPs was expressed as a percentage of experimental control values to allow comparisons between Western blots. Previous data have demonstrated that this results in an error of less than ± 10 %.
Statistical analysis
Data are presented as means ±s.e.m. Data were analysed using one-way analysis of variance and Bonferroni modified t test where appropriate. *P < 0.05 for comparisons stated in figure legends.
RESULTS
Development of myoblasts and myotubes in culture
Figure 1A–C shows the mean change in HSP25, HSP60 and HSC70 content of myoblasts and myotubes during fusion and differentiation following addition of 2 % HS and Fig. 1D shows representative Western blots. Data are expressed as a percentage of HSP content of myoblasts, prior to addition of HS. The content of HSPs per unit protein fell to approximately 50–70 % of the value for myoblasts by 7 days following fusion. HSP70 was not detected in samples from any time point following the addition of HS.
Figure 1.
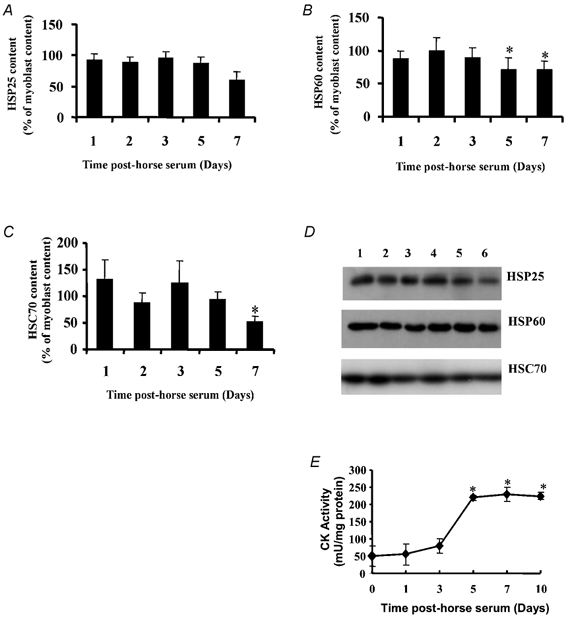
HSP25 (A), HSP60 (B) and HSC70 (C) content of C2C12 cells at 1, 2, 3, 5 and 7 days following treatment with HS. Data are expressed as a percentage of HSP content of myoblasts. D, representative Western blot showing HSP content of C2C12 cells prior to (lane 1) and at 1, 2, 3, 5 and 7 days following treatment with HS (lanes 2–6). E, specific CK activity in C2C12 cells prior to (0 time point) and at specific time points following treatment with HS. *P < 0.05 cf. myoblast content.
Figure 1E shows the activity of CK per unit of cellular protein in myoblasts and myotubes following induction of differentiation. Cultured myoblasts contained a low level of CK activity. The CK activity of myotubes rose steadily during differentiation to reach a plateau at 5–7 days following the addition of HS.
These data therefore indicated that the development of myotubes in culture reaches a plateau at 5–7 days following addition of 2 % HS. The use of myotubes at this time point was therefore considered to be the most appropriate and sensitive time where release of CK activity could be used as a marker of damage to myotubes.
Development of a model for the study of damage to myotubes in culture
Figure 2 shows the effect of treatment of myotubes with 250 μm, 1 mm or 10 mm DNP on release of CK activity from myotubes in culture. Data are expressed as the proportion of the total CK activity released per 30 min (Fig. 2A) and cumulative release of CK activity (Fig. 2B). The percentage of total CK activity that was released throughout the time course of the experiment is given in Table 1. Treatment of cells with 10 mm DNP resulted in loss of a significant proportion of the CK activity over the initial stages of the experiment, markedly reducing the cellular CK activity remaining in the myotubes and therefore available for further loss in the later stages of the experiment (Fig. 2A). Treatment of myotubes with 1 mm DNP resulted in the release of a total of 63.5 ± 2.6 % of CK activity. Table 1 also shows the proportion of soluble protein that was released form the cells following treatment with DNP. This demonstrates that the proportion of protein released was generally comparable with the percentage of CK activity that was released following treatment.
Figure 2.
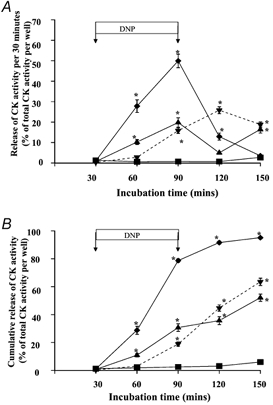
Time course of release of CK activity expressed as percentage of total CK activity in the well per 30 min (A) and cumulative release of activity (B), from control myotubes (▪) and myotubes treated with 0.25 (▴), 1 (▾) and 10 mm (♦) DNP. *P < 0.05 compared with untreated myotubes.
Table 1.
Total release of CK activity and protein from myotubes following treatment with DNP or A23187
| Agent | Concentration | CK activity | Protein released |
|---|---|---|---|
| (% of total activity) | (% of total protein) | ||
| Control | — | 6.0±1.2 | 11.5 ± 0.8 |
| DNP | 0.25 mm | 52.1±2.6* | 35.5±11.2* |
| 1 mm | 63.5±2.6* | 55.5±6.74* | |
| 10 mm | 97.5±0.4* | 78.0±3.0* | |
| A23187 | 2μm | 3.1±0.4 | 16.3±1.9 |
| 10 μm | 47.9±3.5* | 56.3±1.7* | |
| 20 μm | 92.7±1.5* | 65.2±4.7* | |
| 50μm | 87.9±1.2* | 80.7±4.0* |
Significantly different from control values.
Prior to treatment with DNP, approximately 5 % of cells were permeable to the ethidium homodimer-1 component of the Live/Dead viability kit (Fig. 3). Treatment of cells with 1 mm DNP resulted in no significant increase in the percentage of cells that was permeable to the vital stain over the time course of the experiment (Fig. 3). Microscopic examination of myotubes indicated that the treatment with 1 mm DNP also resulted in an apparent reduction in cell volume (Fig. 3).
Figure 3.
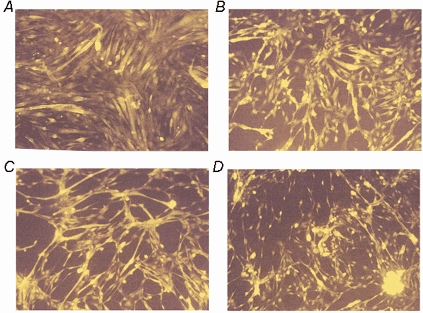
Live/Dead staining of control myotubes (A), and myotubes at 90 (B), 120 (C) and 150 min (D) following treatment with 1 mm DNP.
The loss of cytosolic components compared with the lack of increase in plasma membrane integrity may have occurred because: (1) the cells were able to release intracellular proteins without loss of the ability of the plasma membrane to exclude the vital stain or (2) only viable cells remained adhered to the tissue culture plate. To address these possibilities, the contents of any media removed from cells were collected onto a microscope slide using a cytospin (Eppendorf, Hamburg, Germany). The slides were viewed using a fluorescent microscope and the number of nuclei in each well was estimated. The total number of nuclei recovered from the media was 4.4 ± 0.4 % of the total cells in each well. Thus, the release of CK activity, which was measured in the media, is primarily due to the cells releasing cytoplasmic CK without loss of membrane integrity.
The effect of treatment of myotubes with different concentrations of A23187 on release of CK activity is shown in Fig. 4. Data are expressed as the proportion of CK activity released per 30 min (Fig. 4A) and cumulative release of CK activity (Fig. 4B). The total percentage of CK activity which was released throughout the time course of the experiment is given in Table 1. No significant increase in release of CK activity was seen when myotubes were treated with 2 μm A23187 compared with that of control myotubes. Treatment of myotubes with 10 μm A23187 resulted in the total loss of 45.8 ± 3.0 % of total cellular CK activity at 1 h following treatment. At the same time point, the plasma membrane of approximately 10 % of the cells had become permeable to the ethidium homodimer-1 vital stain (data not shown in detail). Values for release of CK activity and permeability to the vital stain did not change significantly over the remainder of the experiment.
Figure 4.
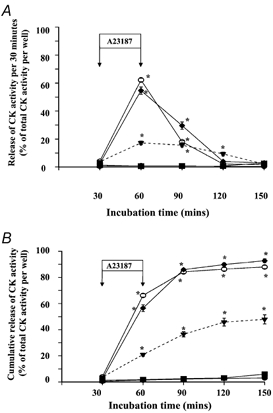
Time course of release of CK activity expressed as a percentage of total CK activity in the well per 30 min (A) and cumulative release of activity (B), from control myotubes(▪) and myotubes treated with 2 (•), 10 (▾), 20 (♦) and 50 μm (○) A23187. Data from control myotubes and myotubes treated with 2 μm A23187 overlap. *P < 0.05 compared with untreated myotubes.
HSP content of myotubes following hyperthermia
Figure 5A–C shows summary data of the time course of HSP25, HSP60 and HSC70 content of myotubes following hyperthermia. Data are expressed as a percentage of levels in untreated cells which were harvested at the same time point. HSP70 was not detected in untreated cells, thus representative Western blots of HSP70 and the other HSPs are shown in Fig. 1D. In general, the HSP content of heat-shocked myotubes had reached peak levels by 12–18 h following hyperthermia. The HSP70, HSC70 and HSP60 content remained elevated at 24 h following hyperthermia.
Figure 5.
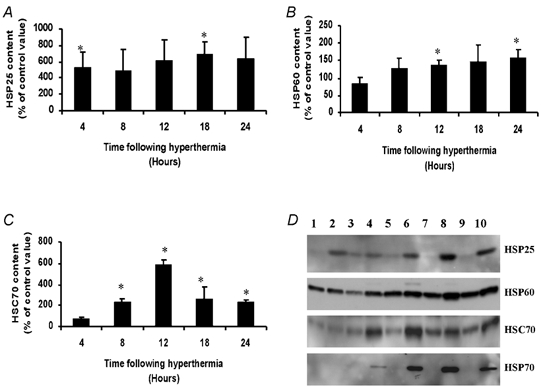
HSP25 (A), HSP60 (B) and HSC70 (C) content of myotubes at 4, 8, 12, 18 and 24 h following hyperthermia. Data are expressed as a percentage of non-heated cells which were harvested at the same time point. D, representative Western blots showing HSP content of myotubes at 4 (lane 2), 8 (lane 4), 12 (lane 6), 18 (lane 8) and 24 h (lane 10) following hyperthermia and control cells harvested at the same time point (lanes 1, 3, 5, 7, 9). *P < 0.05 cf. control (non-heated) value.
Effect of hyperthermia on damage to myotubes induced by A23187 and DNP
The effect of prior hyperthermia on release of CK activity from myotubes treated with 1 mm DNP or 10 μm A23187 is shown in Fig. 6A and B respectively. Treatment of cells with 1 mm DNP resulted in the release of 63.5 ± 2.6 % of the total cellular CK activity into the medium. This was significantly reduced at all time points following hyperthermia. Treatment of cells with 10 μm A23187 resulted in the release of 47.9 ± 3.5 % of total cellular CK activity into the medium. Prior hyperthermia resulted in a significant reduction in the release of CK activity from myotubes treated with A23187, at 18 and 24 h following hyperthermia.
Figure 6.
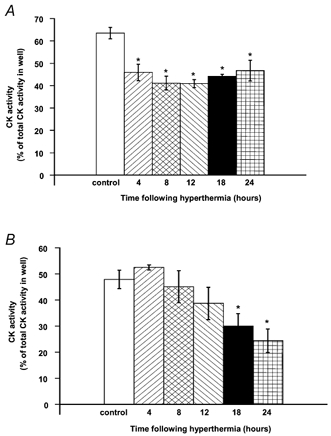
Effect of prior hyperthermia on release of CK activity from myotubes following treatment with 1 mm DNP (A) or 10 μm A23187 (B). *P < 0.05 cf. control (non-heated) value.
DISCUSSION
HSP25, HSP60 and HSC70 were detected in both actively dividing and non-dividing myoblasts (data not shown in detail). This detection was maintained for several days during differentiation of myoblasts to form myotubes and then fell significantly once differentiation was complete (Fig. 1). In the unstressed cell, HSPs act as molecular chaperones, facilitating folding and translocation of newly synthesised proteins (Hubbard & Sander, 1991; Gething, 1997). During initial differentiation, myoblasts undergo substantial remodelling to form mature myotubes with the increased expression of muscle specific proteins. HSPs are required during cellular remodelling and the relatively high HSP content during this remodelling phase appears to reflect this. The lack of expression of HSP70 during differentiation suggests that HSP70 does not play a major role in this remodelling.
Myoblasts contained negligible amounts of CK activity, but the CK activity in myotubes increased steadily following induction to differentiate (Fig. 1E). Release of creatine kinase (CK) activity from mature skeletal muscle has long been used as an index of damage to muscle in both in vivo and in vitro studies. The CK activity of myotubes reached maximum and stable levels at 5–7 days following change of the medium to HS, suggesting that CK activity could be used as a marker of membrane damage to myotubes at this time point.
Treatment of myotubes with 1 mm DNP or with 10 μm A23187 resulted in the reproducible release of an average of 64 and 48 % of CK activity respectively over the experimental period (Fig. 2 and Fig. 4). This provided a reproducible model of damage initiated by a mitochondrial uncoupler or a rapid increase in intracellular calcium content.
Treatment of intact muscles with DNP has previously been shown to result in a decline in muscle ATP concentration (West-Jordan et al. 1990, 1991) and it was suggested that this led to a gradual failure of energy-dependent pumps and thus a slow rise in free calcium concentration within the cells. This was reflected in a delayed release of CK activity from the muscles (Jackson et al. 1984). The data presented here are compatible with these results from studies of intact muscles. In contrast, treatment of cells with A23187 resulted in the rapid efflux of CK activity (Fig. 4). Treatment of isolated muscles with A23187 also results in an immediate influx of external Ca2+ (McArdle et al. 1992) with the resultant rapid activation of Ca2+-dependent degenerative processes and, in a similar manner to the myotubes in culture, a rapid efflux of creatine kinase (McArdle et al. 1992).
Comparison of the pattern of efflux of CK activity with analysis of plasma membrane integrity using the Live/Dead stain demonstrated that the plasma membranes of only 5–10 % of cells were permeable to the ethidium homodimer-1 when 50–60 % of CK activity and cellular protein (data not shown) had been lost following treatment with either DNP or A23187. These differences appear temporal with release of cytosolic components preceding the loss of the ability to exclude the vital stain by 1–2 h. The molecular mass of the ethidium homodimer-1 component of the Live/Dead viability kit is 857, which is considerably less that that of creatine kinase (82 000). This suggests that this temporal difference is not due to a ‘size’-dependent process. One possible explanation is that the release of cytoplasmic contents (which would include CK) is an early attempt by the cell to reduce cell swelling and prevent lysis. The mechanism by which muscle damage leads to loss of CK activity is unclear. However, data from our group and others suggest that efflux of CK activity does not occur through non-specific leaks in the plasma membrane. Diedrichs et al. (1979) demonstrated that damage to muscle fibres resulted in a gradual increase in muscle cell volume and that cell volume begins to fall when CK activity is seen to be released from the damaged muscle. These authors suggest that the muscle cell attempts to maintain viability by extruding cytoplasm via an exocytotic type of mechanism and, in doing so, extrudes cytoplasmic components such as CK and other proteins. Previous data from our group have demonstrated that the pattern of proteins released from muscle cells during damage induced by excessive contractile activity or treatment with A23187 is different to that induced by treatment with a detergent and is selective for only some of the total cytosolic protein fraction from the muscle (Jackson et al. 1991). Data also suggested that this did not reflect a complete loss of cell viability or demonstrate a ‘size’-related release of proteins. This lack of size-dependent release may be due to the different location and intracellular binding of some proteins. Data from the current study suggest that the total percentage and time course of soluble protein and CK efflux from A23187-treated cells are similar, suggesting that an active cytosolic extrusion process may be occurring. Further data from our group demonstrated that release of CK activity can occur in several systems without a clear loss of membrane integrity (West-Jordan et al. 1990, 1991 and authors' unpublished observations) and the current observations are in keeping with this. These data suggest that the sole use of CK activity as an index of muscle damage may be misleading whereby early detection of increased release of CK activity may be indicative of an attempt by cells to survive. Further studies of the time course of release of CK activity in comparison with structural and functional deficits are necessary.
Data presented show that myotubes have a rapid stress response following 30 min of hyperthermia. This increase in the cellular content of HSPs peaked at approximately 12–18 h following hyperthermia and remained elevated at 24 h following hyperthermia. This is compatible with the time course of production of HSPs in other rodent tissues such as the rat heart (Marber, 1994) and mouse skeletal muscle in vivo following hyperthermia or a period of non-damaging isometric exercise (McArdle & Jackson, 1996; McArdle et al. 2001). Studies using isolated cardiac cells and tissue have demonstrated that an increased content of HSPs was associated with cytoprotection against a (normally) damaging insult (Yellon et al. 1992; Yellon & Latchman 1992; Heads et al. 1994; Mestril & Dillman, 1995; Plumier & Currie, 1996). Data presented here support a role for HSPs in providing cytoprotection against calcium-induced damage to myotubes in culture. Prior treatment of myotubes by a period of hyperthermia resulted in significant reduction in the release of CK activity when myotubes were subsequently treated with the calcium ionophore A23187 at the time of maximal content of HSPs. Comparable data for the rat heart were presented by Marber (1994), who demonstrated that prior increase in the tissue content of HSPs was associated with a significant reduction in damage following the calcium paradox in isolated hearts. Data also demonstrate that an increased content of HSPs was associated with rapid protection against DNP-induced damage to myotubes. HSP70 expression increased dramatically following hyperthermia, in contrast to the lack of HSP70 expression during differentiation of muscle. This suggests that HSP70 plays a role in providing protection against acute periods of damage, but plays little role in the remodelling that occurs during differentiation of muscle cells. The time post-hyperthermia, which was necessary to provide protection against damage induced by treatment with A23187, was longer than the time course for protection against DNP-induced damage. It is likely that the protection is due to increases in multiple HSPs and so does not solely occur at the peak time of one HSP. In addition, some HSPs provide a more specific protection, for instance, HSP25 is thought to be involved in the stabilisation of microfilaments (McArdle & Jackson, 2002). This suggests that different HSPs or different patterns of HSPs may be responsible for protection against A23187- and DNP-induced muscle damage.
The difference in the time course of protection of heat stress against damage induced by DNP or A23187 suggests that different patterns of protein expression may be involved in providing this protection. Indeed, differential protective effects exerted by specific HSPs have previously been demonstrated (Dillmann, 1999). Work in the heart has suggested that a period of hyperthermia resulted in increased activity of antioxidant defence proteins that are not members of the HSP family (Yamashita et al. 1998) although this is currently controversial (Xi et al. 2001; Arnaud et al. 2002). The effect of hyperthermia on the activity of antioxidant defence proteins has not been studied in skeletal muscle although experiments in the heart have demonstrated an increase in some proteins, particularly in manganese superoxide dismutase in the heart following hyperthermia (Yamashita et al. 1998). Zuo et al. (2000) have shown that exposure of diaphragm muscle to hyperthermia (42 °C) for 30 min resulted in a significant increase in the production of the superoxide anion radical. Further studies are therefore necessary in skeletal muscle to examine the relationship between hyperthermia, HSP expression, antioxidant defence protein expression and cytoprotection.
The possibility that prior hyperthermia might protect against muscle damage induced by a pathological rise in Ca2+ concentration or a fall in ATP in vivo has widespread implications (McArdle & Jackson, 1997). A loss of Ca2+ homeostasis has been implicated in muscle damage in a variety of situations, including exercise-induced muscle damage. A fall in ATP concentration is an important contributor to the muscle damage which occurs during ischaemia and reperfusion, and this is mimicked by treatment with the mitochondrial uncoupler, DNP (West-Jordan et al. 1990, 1991). Muscle damage following an ischaemic episode occurs in skeletal muscle in a variety of situations, including prolonged use of a tourniquet in limb surgery, aneurism repair and microsurgical transfer of skeletal muscle (Lundberg et al. 2002). The current study has demonstrated that a prior heat shock is associated with an induction of HSP synthesis and provides considerable protection against this form of injury. These data suggest that further knowledge of the mechanisms of induction and time course of expression of HSPs in human skeletal muscle may be of potential benefit in protection against these damaging processes.
In summary, this study has established reproducible models of chemical damage to myotubes in culture using DNP or A23187 as damaging agents and has provided further insight into the mechanisms by which these agents cause muscle damage. The cytoprotective effect of heat stress against both Ca2+-mediated damage, and damage induced by a fall in ATP levels, which was observed in this system, may have wide ranging implications. These data suggest that a prior induction of the stress response may (at least partially) protect skeletal muscle against damage in these disparate situations.
Acknowledgments
The authors would like to thank the Wellcome Trust (grant number 43364/Z) for generous financial support of this work
REFERENCES
- Arnaud C, Joyeux M, Garrel C, Godin-Ribuot D, Demenge P, Ribuot C. Free-radical production triggered by hyperthermia contributes to heat stress-induced cardioprotection in isolated rat hearts. Br J Pharmacol. 2002;135:1776–1782. doi: 10.1038/sj.bjp.0704619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diedrichs F, Muhlhaus K, Trautschold I. On the mechanism of lactate dehydrogenase release from skeletal muscle in relation to the control of cell volume. Enzyme. 1979;24:404–415. doi: 10.1159/000458695. [DOI] [PubMed] [Google Scholar]
- Dillmann WH. Heat shock proteins and protection against ischaemic injury. Infect Dis Obstet Gynecol. 1999;7:55–57. doi: 10.1155/S1064744999000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JA, Brooks SV. Muscle damage induced by contraction: in an in situ single skeletal muscle model. In: Salmons S, editor. Muscle Damage. Oxford: Oxford University Press; 1997. pp. 28–40. [Google Scholar]
- Faulkner JA, Brooks SV, Opiteck JA. Injury to skeletal muscle fibres during contractions: conditions of occurrence and prevention. Phys Ther. 1993;73:911–921. doi: 10.1093/ptj/73.12.911. [DOI] [PubMed] [Google Scholar]
- Faulkner JA, Clafin DR, McKully KK, Jones DA. Contractile properties of bundles of fibre segments from skeletal muscles. Am J Physiol. 1982;243:C66–73. doi: 10.1152/ajpcell.1982.243.1.C66. [DOI] [PubMed] [Google Scholar]
- Febbraio MA, Koukoulas I. HSP72 gene expression progressively increases in human skeletal muscle during prolonged, exhaustive exercise. J Appl Physiol. 2000;89:1055–1060. doi: 10.1152/jappl.2000.89.3.1055. [DOI] [PubMed] [Google Scholar]
- Feige U, Morimoto RI, Yahara I, Polla BS. Stress-Inducible Cellular Responses. Basel: Birkhauser; 1996. [Google Scholar]
- Fink AL. Chaperone-mediated protein folding. Physiol Rev. 1999;79:425–449. doi: 10.1152/physrev.1999.79.2.425. [DOI] [PubMed] [Google Scholar]
- Garramone RR, Winters RM, Das DK, Deckers PJ. Reduction of skeletal muscle injury through stress conditioning using the heat shock response. Plast Reconstr Surg. 1994;93:1242–1247. doi: 10.1097/00006534-199405000-00021. [DOI] [PubMed] [Google Scholar]
- Gething MJ. Guidebook to Molecular Chaperones and Protein-Folding Catalysts. Oxford: Oxford University Press; 1997. [Google Scholar]
- Head SI. Membrane potential, resting calcium and calcium transients in isolated muscle fibres from normal and dystrophic mice. J Physiol. 1993;469:11–19. doi: 10.1113/jphysiol.1993.sp019801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heads RJ, Latchman DS, Yellon DM. Stable high level expression of a transfected human HSP70 gene protects a heart-derived muscle cell line against thermal stress. J Mol Cell Cardiol. 1994;26:695–699. doi: 10.1006/jmcc.1994.1084. [DOI] [PubMed] [Google Scholar]
- Hightower L. Heat shock, stress proteins, chaperones and proteotoxicity. Cell. 1991;66:191–197. doi: 10.1016/0092-8674(91)90611-2. [DOI] [PubMed] [Google Scholar]
- Hubbard T, Sander C. The role of heat shock and chaperone proteins in protein folding: possible molecular mechanisms. Protein Eng. 1991;4:711–717. doi: 10.1093/protein/4.7.711. [DOI] [PubMed] [Google Scholar]
- Jackson MJ, Jones DA, Edwards RHT. Experimental skeletal muscle damage: the nature of the calcium-activated degenerative processes. Eur J Clin Invest. 1984;14:369–374. doi: 10.1111/j.1365-2362.1984.tb01197.x. [DOI] [PubMed] [Google Scholar]
- Jackson MJ, Page S, Edwards RHT. The nature of the proteins lost from isolated rat skeletal muscle during experimental damage. Clin Chim Acta. 1991;197:1–8. doi: 10.1016/0009-8981(91)90342-a. [DOI] [PubMed] [Google Scholar]
- Jones DA, Jackson MJ, Edwards RHT. Release of intracellular enzymes from an isolated mammalian skeletal muscle preparation. Clin Sci (Lond) 1983;65:193–201. doi: 10.1042/cs0650193. [DOI] [PubMed] [Google Scholar]
- Khassaf M, Child RB, McArdle A, Brodie DA, Esanu C, Griffiths RD, Jackson MJ. Time course of responses of human skeletal muscle to oxidative stress induced by nondamaging exercise. J Appl Physiol. 2001;90:1031–1035. doi: 10.1152/jappl.2001.90.3.1031. [DOI] [PubMed] [Google Scholar]
- Lepore DA, Hurley JV, Stewart AG, Morrison WA, Anderson RL. Prior heat stress improves survival of ischemic-reperfused skeletal muscle in vivo. Muscle Nerve. 2001;23:1847–1855. doi: 10.1002/1097-4598(200012)23:12<1847::aid-mus8>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Liu Y, Steinacker JM. Changes in skeletal muscle heat shock proteins: pathological significance. Front Biosci. 2001;6:D12–25. doi: 10.2741/liu. [DOI] [PubMed] [Google Scholar]
- Lundberg J, Elander A, Rakotonirainy O, Zetterlund T, Fogdestam I, Soussi B. Energy metabolism during microsurgical transfer of human skeletal muscle assessed by high-pressure liquid chromatography and by 31P-nuclear magnetic resonance. Scand J Plast Reconstr Surg Hand Surg. 2002;36:141–148. doi: 10.1080/028443102753718014. [DOI] [PubMed] [Google Scholar]
- McArdle A, Edwards RHT, Jackson MJ. Accumulation of calcium by normal and dystrophin-deficient mdx mouse. Clin Sci (Lond) 1992;80:367–371. [Google Scholar]
- McArdle A, Jackson MJ. Heat shock expression in skeletal muscle. Biochem Soc Trans. 1996;24:485S. doi: 10.1042/bst024485s. [DOI] [PubMed] [Google Scholar]
- McArdle A, Jackson MJ. Intracellular mechanisms involved in skeletal muscle damage. In: Salmons S, editor. Muscle Damage. Oxford: Oxford University Press; 1997. pp. 90–106. [Google Scholar]
- McArdle A, Jackson MJ. Stress proteins and exercise induced muscle damage. In: Locke M, Noble E, editors. Stress Proteins and Tissue Damage. Boca Raton: CRC Press; 2002. pp. 137–150. [Google Scholar]
- McArdle A, McArdle C, Jackson MJ. Stress proteins and protection of skeletal muscle against contraction-induced skeletal muscle damage in anaesthetised mice. J Physiol. 1997;499.P:9P. [Google Scholar]
- McArdle A, Pattwell D, Vasilaki A, Griffiths RD, Jackson MJ. Contractile activity-induced oxidative stress: cellular origin and adaptive responses. Am J Physiol Cell Physiol. 2001;280:C621–627. doi: 10.1152/ajpcell.2001.280.3.C621. [DOI] [PubMed] [Google Scholar]
- McArdle A, van der Meulen JH, Catapano M, Symons MCR, Faulkner JA, Jackson MJ. Contraction-induced injury to the extensor digitorum longus muscle of rats: Effects on muscle free radical injury. Free Rad Biol Med. 1999;26:1085–1091. doi: 10.1016/s0891-5849(98)00317-7. [DOI] [PubMed] [Google Scholar]
- Marber MS. Stress proteins and myocardial protection. Clin Sci (Lond) 1994;86:375–381. doi: 10.1042/cs0860375. [DOI] [PubMed] [Google Scholar]
- Marber MS, Mestril R, Chi SH, Sayen MR, Yellon DM, Dillmann WH. Overexpression of the rat inducible 70-kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J Clin Invest. 1995;95:1446–1456. doi: 10.1172/JCI117815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestril R, Dillman WH. Heat shock proteins and protection against myocardial ischemia. J Mol Cell Cardiol. 1995;27:45–52. doi: 10.1016/s0022-2828(08)80006-5. [DOI] [PubMed] [Google Scholar]
- Plumier JC, Currie RW. Heat shock-induced myocardial protection against ischemic injury: a role for HSP70? Cell Stress Chaperones. 1996;1:13–17. doi: 10.1379/1466-1268(1996)001<0013:hsimpa>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pressmar J, Brinkmeier H, Seewald MJ, Naumann T, Rudel R. Intracellular Ca2+ concentrations are not elevated in resting cultured muscle from Duchenne (DMD) patients and in MDX mouse muscle fibres. Pflugers Arch. 1994;426:499–505. doi: 10.1007/BF00378527. [DOI] [PubMed] [Google Scholar]
- Salo DC, Donovan CM, Davies KJ. HSP70 and other possible heat shock or oxidative stress proteins are induced in skeletal muscle, heart, and liver during exercise. Free Rad Biol Med. 1991;11:239–46. doi: 10.1016/0891-5849(91)90119-n. [DOI] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klein DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Voellmy R. Sensing stress and responding to stress. In: Feige U, Morimoto RI, Yahara I, Polla BS, editors. Stress-Inducible Cellular Responses. Basel: Birkhauser; 1996. pp. 121–137. [Google Scholar]
- West-Jordan J, Martin PA, Abraham RJ, Edwards RHT, Jackson MJ. Energy dependence of cytosolic enzyme efflux from rat skeletal muscle. Clin Chim Acta. 1990;189:63–72. doi: 10.1016/0009-8981(90)90088-a. [DOI] [PubMed] [Google Scholar]
- West-Jordan J, Martin PA, Abraham RJ, Edwards RHT, Jackson MJ. Energy metabolism during damaging contractile activity in isolated skeletal muscle: a 13P-NMR study. Clin Chim Acta. 1991;203:119–134. doi: 10.1016/0009-8981(91)90284-j. [DOI] [PubMed] [Google Scholar]
- Xi L, Tekin D, Bhargava P, Kukreja RC. Whole body hyperthermia and preconditioning of the heart: basic concepts, complexity and potential mechanisms. Int J Hyperthermia. 2001;17:439–455. doi: 10.1080/02656730110064342. [DOI] [PubMed] [Google Scholar]
- Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1977;284:555–556. doi: 10.1038/270725a0. [DOI] [PubMed] [Google Scholar]
- Yamashita N, Hoshida S, Taniguchi N, Kuzuya T, Hori M. Whole-body hyperthermia provides biphasic cardioprotection against ischaemia/reperfusion injury in the rat. Circulation. 1998;98:1414–1421. doi: 10.1161/01.cir.98.14.1414. [DOI] [PubMed] [Google Scholar]
- Yellon DM, Latchman DS. Stress proteins and myocardial protection. J Mol Cell Cardiol. 1992;24:113–124. doi: 10.1016/0022-2828(92)93148-d. [DOI] [PubMed] [Google Scholar]
- Yellon D, Pasini E, Cargnoni A, Marber M, Latchman D, Ferrari R. The protective role of heat stress in the ischaemic and reperfused rabbit myocardium. J Mol Cell Cardiol. 1992;24:895–907. doi: 10.1016/0022-2828(92)91102-b. [DOI] [PubMed] [Google Scholar]
- Zuo L, Christofi FL, Wright VP, Liu CY, Merola AJ, Berliner LJ, Clanton TL. Intra- and extracellular measurement of reactive oxygen species produced during heat stress in diaphragm muscle. Am J Physiol Cell Physiol. 2000;279:C1058–1066. doi: 10.1152/ajpcell.2000.279.4.C1058. [DOI] [PubMed] [Google Scholar]