

Abstract
Kainate receptors have recently been shown to be involved in synaptic transmission, to regulate transmitter release and to mediate synaptic plasticity in different regions of the CNS. However, very little is known about endogenous mechanisms that can control native kainate receptor signalling. In this study we have found that GluR5-containing kainate receptor-mediated actions can be modulated by activation of protein kinase C (PKC) but not protein kinase A (PKA). However, both PKA and PKC directly phosphorylate the GluR5 subunit of kainate receptors. Metabotropic glutamate (mGlu) receptors are well known to be involved in synaptic transmission, regulation of transmitter release and synaptic plasticity in a variety of brain regions. We now demonstrate that kainate receptor signalling is enhanced by activation of group I mGlu receptors, in a PKC-dependent manner. These data demonstrate for the first time that kainate receptor function can be modulated by activation of metabotropic glutamate receptors and have implications for understanding mechanisms of synaptic transmission, plasticity and disorders such as epilepsy.
Kainate receptors (KARs) are formed from heteromeric assemblies of GluR5, 6, 7 and KA1, 2 subunits (Cui & Mayer, 1999), are widespread throughout the CNS (Petralia et al. 1994) and are located both postsynaptically and presynaptically (see Chittajallu et al. 1999; Lerma, 2001 for reviews). Activation of KARs can contribute to postsynaptic EPSCs (Vignes et al. 1997; Castillo et al. 1997) and presynaptic regulation of transmitter release (Lauri et al. 2001; Schmitz et al. 2001). In addition, KARs are also involved in synaptic plasticity (Bortolotto et al. 1999; Contractor et al. 2001) and generation of seizure activity (Mulle et al. 1998). Therefore, probing molecular mechanisms that regulate KARs is vital in further understanding the key roles of KARs in the CNS.
Metabotropic glutamate receptors are composed of eight subtypes (mGlu1-8) classified into groups I, II and III. Group I receptors (mGlu1, 5) couple to an increase in phosphoinositide metabolism and PKC activity (Conn & Pin, 1997; Anwyl, 1999; De Blasi et al. 2001). The activation of group I receptors can result in a multitude of different consequences for neuronal function (Anwyl et al. 1999). An interesting aspect of mGlu receptors is that different subtypes can interact with one another and with other receptors to regulate intracellular signalling (Nicoletti et al. 1993; Schoepp et al. 1996; Cho et al. 2002).
Whilst it has been shown that NMDA receptor activation can regulate expressed KARs (Ghetti & Heinemann, 2000) it is not known whether other receptors can regulate native KARs. Here we show that the GluR5 subunit of KARs is phosphorylated by protein kinase A (PKA) and protein kinase C (PKC). In perirhinal cortex neurones activation of PKC resulted in an enhancement of KAR function and mGlu5 receptor activation enhanced KAR-mediated responses in a PKC-dependent manner. Finally, we demonstrate that synaptically evoked KAR EPSCs are enhanced by stimulation of mGlu5 receptors. These results establish that KARs can be regulated by metabotropic glutamate receptors that link to PKC signalling. This finding has important implications for understanding mechanisms controlling synaptic transmission, plasticity and epilepsy.
METHODS
Calcium imaging
Wistar rat pups (2–3 days old) were decapitated, in accordance with the UK Animals (Scientific Procedures) Act 1986. The perirhinal cortex was removed from the rest of the brain. Perirhinal cultures and imaging techniques were according to methods previously described (Cho et al. 2000). Briefly, cells were washed three times in HBS buffer (mm: NaCl, 119; KCl, 5; Hepes, 25; glucose, 33; CaCl2, 2; MgCl2, 2; TTX 500 nm; glycine, 1 μm; picrotoxin, 100 μm; pH 7.4; osmolarity 300–310 mosmol l−1) and loaded with 5 μm of the membrane-permeant Ca2+ indicator fluo-3 AM made up in 1 mg ml−1 bovine serum albumin/HBS at 37 °C for 30 min. Cells were then washed three times in HBS and incubated for 20 min in a 5 % CO2 atmosphere at 22 °C to allow de-esterification of the flurophore. Cells were viewed on a BioRad MRC600 confocal microscope with an argon ion laser, using green filter sets and perfused continuously with HBS buffer at ∼2 ml min−1. All cells in the field that responded with an increase in fluorescence to the application of (R,S)-2-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl propanoic acid (ATPA) were then used in the subsequent analysis with co-application of other agents. Integrations of five individual images were obtained every 10 s before, during and after agonist application. Fluorescence of individual cells in each preparation was measured using the public domain NIH program (http://rsb.info.nih.gov/nihimage/) and expressed relative to baseline. The mean peak fluorescence was calculated and expressed as mean ±s.e.m. Experiments were carried out in the presence of the NMDA receptor antagonist AP5 (50 μm; Tocris), the AMPA receptor antagonist GYKI 53655 (50 μm) and blockers of voltage-gated calcium channels (NiCl 20 μm, nifedepine 50 μm, ω-conotoxin MVIIC 1 μm; Tocris and Sigma). In some experiments 50 mm KCl was applied. Cyclopiazonic acid (CPA; 1 μm), which depletes calcium stores, was also used where indicated.
Phosphorylation
Glutathione-S-transferase (GST) and glutathione-S-transferase-C-terminal (GST-Ct)-GluR52b were purified by cation exchange chromatography. Five micrograms of fusion protein were used in each reaction. Standard procedures for PKC phosphorylation were used: reactions were performed at 30 °C for 30 min in phosphorylation buffer (40 mm TRIS-HCl, pH 7.5, 20 mm MgCl2, 200 μg ml−1l-α-phosphatidyl-l-serine, 40 μg ml−1 1, 2-dioleoyl-sn-glycerol) supplemented with reagents and purified PKC α (1.5 μg ml−1; Calbiochem) and ATP (100 μm). For PKA phosphorylation (Fig. 2) each reaction contained 20 units of PKA (Sigma) and was performed in 20 mm Hepes (pH 7.0), 10 mm MgCl2 and 250 μm ATP. All reactions included 5–10 μCi of [γ-32P]ATP and were stopped with SDS-PAGE sample buffer. The phosphorylated fusion proteins were resolved by SDS-PAGE and visualised by autoradiography.
Figure 2. Responses mediated by GluR5-containing KARs are modulated in a PKC-dependent manner.
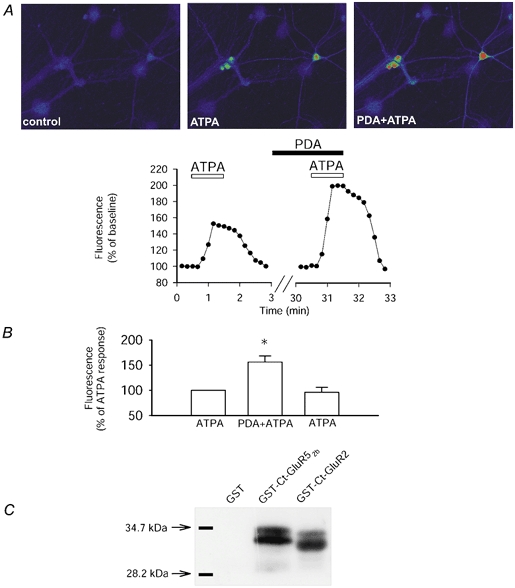
A, single example of a field of neurones (top) and the analysed change in fluorescence (bottom) showing that application of ATPA (2 μm), to selectively stimulate GluR5-containing KARs, results in an increase in fluorescence of the calcium indicator fluo-3 AM. The co-application of phorbol esters (PDA; 1 μm), to stimulate PKC, enhanced the GluR5-mediated calcium rise. B, pooled data showing that ATPA-induced calcium signalling is significantly increased by stimulation of PKC in a reversible manner (n = 18 cells, 3 dishes; P < 0.01). Following washout of PDA, the response to ATPA returns to baseline levels (96 ± 10 %). C, GluR52b is phosphorylated by PKC. A single representative example showing that GST (n = 10) is not phosphorylated by PKC. However, GST-Ct-GluR52b (n = 10) and GST-Ct-GluR2 (n = 2) are both phosphorylated by purified PKC.
Electrophysiology
Slices of perirhinal cortex were prepared from adult male DA rats (10 days of age, Bantin and Kingman, UK). Animals were anaesthetised with halothane and decapitated in accordance with the UK Animals (Scientific Procedures) Act 1986. The brain was rapidly removed and placed in ice-cold artificial cerebrospinal fluid (aCSF; bubbled with 95 % O2–5 % CO2) which comprised (mm): NaCl, 124; KCl, 3; NaHCO3, 26; NaH2PO4, 1.25; CaCl2, 2; MgSO4, 1; d-glucose, 10. A single slice was placed in a submerged recording chamber (28–30 °C, flow rate ∼2 ml min−1) when required. Whole-cell recordings were obtained from neurones in layer II-III. Pipette (4–7 MΩ) solutions (280 mosmol l−1, pH 7.2) comprised (mm): KMeSO4, 130; NaCl, 8; Mg-ATP, 4; Na-GTP, 0.3; EGTA, 0.5; Hepes 10; QX-314, 6. Neurones were voltage clamped at −70 mV. Data were analysed from one slice per rat (number = n). The change in holding current following agonist application was measured for each experiment and normalised. Application of (R, S)-3,5-dihydroxyphenylglycine (DHPG) produced a small inward current which was allowed to stabilise and was then used as the baseline for measuring effects of application of ATPA. Data pooled across slices are expressed as means ±s.e.m. Kainate EPSCs were isolated in the presence of AP5 and GYKI 53655. The kainate agonist (2S, 4R)-4-methylglutamate (SYM2081; 10 μm; Jones et al. 1997; DeVries, 2000) that produces rapid desensitisation of kainate responses was used to confirm that the EPSC evoked in response to five stimuli at 100 Hz was a kainate-mediated synaptic response (see DeVries, 2000).
RESULTS
Activation of kainate receptors triggers a rise in intracellular calcium
Bath application of 1 μm kainate increased intracellular calcium in cultured perirhinal cortex neurones (185 ± 7 % compared with baseline; P < 0.05, n = 35 cells, 5 dishes; Fig. 1) in the presence of NMDA and AMPA receptor antagonists, voltage-gated calcium channel blockers and cyclopiazonic acid to deplete intracellular calcium stores (see Methods for details). However, there was no increase in intracellular calcium with application of kainate in calcium-free aCSF (Fig. 1). Furthermore, activation of GluR5-containing KARs using the selective agonist ATPA also resulted in an increase in intracellular calcium (see Fig. 2A; 168 ± 7 % compared with baseline; P < 0.05, n = 18 cells, 3 dishes). KCl (50 mm) did not produce a rise in intracellular calcium, unless calcium channel blockers were absent from the bathing medium (data not shown). Together, these results suggest that the kainate- and ATPA-induced calcium signal relied on extracellular calcium and furthermore indicated that the calcium signal was not indirectly dependent on membrane depolarisation. Therefore, all subsequent experiments were carried out in the presence of the combination of pharmacological antagonists described above.
Figure 1. Kainate application results in an increase in intracellular calcium.
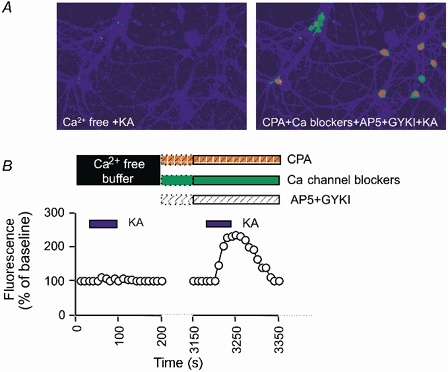
A, single example of a field of neurones and B, analysed pooled data showing that the kainate (KA; 1 μm)-induced increase in calcium is blocked by removing extracellular calcium. However, the calcium rise does not rely on entry via NMDA, AMPA or voltage-gated calcium channels, nor on release from intracellular calcium stores as evidenced by the KA-induced increase in calcium in the presence of the pharmacological cocktail (see Methods for details). In this and subsequent figures, the enhancement above baseline by kainate receptor agonists is normalised to 100 % and any subsequent changes expressed relative to the effects of agonist alone. The pharmacological cocktail was applied for 15 min (indicated by dotted lines) prior to the application of kainate.
Phosphorylation by PKC modulates GluR5-KAR signalling
Phosphorylation of intracellular targets can modulate receptor function. Therefore, we investigated whether phosphorylation has any physiological consequences for KAR signalling. Stimulation of protein kinase C (PKC) by phorbol esters significantly enhanced (P < 0.01) the GluR5-induced calcium rise (Fig. 2A and B; ATPA plus phorbol 12-myristate 13-acetate (PDA) gave a 56 ± 12 % increase compared with ATPA alone, n = 18). Activation of PKC could affect kainate receptors either indirectly or directly through altering phosphorylation of the receptor itself. Therefore, we examined whether the above effects on GluR5 function could potentially be due to direct phosphorylation of the GluR5 subunit by PKC. As shown in Fig. 2C, PKC can phosphorylate the C-terminal of GluR52b in an in vitro assay and it has recently been shown that PKC phosphorylates GluR52b at S880 and/or S886 (Hirbec et al. 2003). As a control we also demonstrated that PKC can phosphorylate the C-terminal of GluR2 but has no effect on GST alone (Fig. 2C). In both the GluR52b and GluR2 lanes there is a double band which is most probably due to protein degradation, reflecting the labile nature of GST-fusion proteins. These results raise the possibility that PKC-dependent phosphorylation of GluR5 may be responsible for enhancing the physiological GluR5-mediated calcium signal.
Phosphorylation by PKA does not modulate GluR5-KAR signalling
Previous evidence has shown that PKA-dependent phosphorylation can alter properties of expressed GluR6 receptors (Raymond et al. 1993; Wang et al. 1993). The next set of experiments was performed to examine whether protein kinase A (PKA) also affects GluR5-KARs in a similar manner to the effects mediated by PKC. However, GluR5-stimulated calcium mobilisation was unaffected by increasing cAMP levels (Fig. 3A; ATPA plus 8-bromo cAMP: 2 ± 13 % increase compared with ATPA, P > 0.05). To test whether this lack of effect was due to endogenous basal PKA activity we utilised the PKA inhibitor KT5720. However, PKA inhibition did not affect ATPA-induced calcium signalling (Fig. 3B; ATPA plus PKA inhibitor KT5720: 3 ± 4 % decrease compared with ATPA; P > 0.05. n = 27 cells, 3 dishes). This lack of effect is not due to poor penetration of these compounds since we have previously shown these compounds to be effective under similar conditions (Cho et al. 2002). Despite the finding that PKA activation did not affect GluR5-containing KAR signalling, the catalytic subunit of PKA did nevertheless phosphorylate the C-terminal of GluR52b (Fig. 3C).
Figure 3. Responses mediated by GluR5-containing KARs are unaffected by activation or inhibition of PKA.
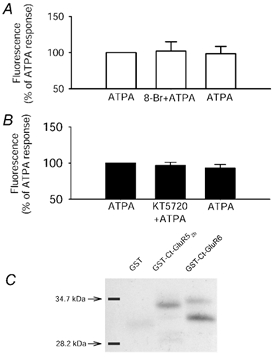
A and B, ATPA-stimulated calcium rise is not affected by application of 8-bromo cAMP (1 μm; A) (ATPA plus 8-bromo cAMP: 2 ± 13 % increase compared with ATPA, P > 0.05. ATPA after washout of 8-bromo cAMP: 2 ± 10 % decrease, n = 27 neurones, 3 dishes) nor by the PKA inhibitor KT5720 (1 μm; B) (ATPA plus PKA inhibitor KT5720: 3 ± 4 % decrease compared with ATPA; P > 0.05. ATPA after KT5720 washout: 7 ± 5 % decrease; n = 27 cells, 3 dishes n = 21 neurones, 3 dishes). C, a single example showing that the catalytic subunit of PKA phosphorylates the C-terminal of GluR52b (n = 3) and GluR6 (n = 3).
Metabotropic glutamate receptor activation enhances GluR5-KAR signalling
We next examined whether activation of glutamate receptors that positively couple to PKC might also enhance KAR function. Therefore, the group I mGlu receptor agonist DHPG was co-applied with ATPA. DHPG (in the presence of cyclopiazonic acid) did not produce an increase in intracellular calcium by itself (not shown), but resulted in a significant enhancement (P < 0.05) of GluR5-stimulated calcium signalling (Fig. 4A; ATPA plus DHPG: 35 ± 5 % increase compared with ATPA alone). Crucially, the enhancement by group I mGlu receptor stimulation was blocked by the PKC inhibitor BIM (Fig. 4A; ATPA plus DHPG plus BIM: 6 ± 3 % increase compared with ATPA alone, P > 0.05, n = 59 cells, 9 dishes).
Figure 4. Responses mediated by GluR5-containing KARs are enhanced by mGluR stimulation.
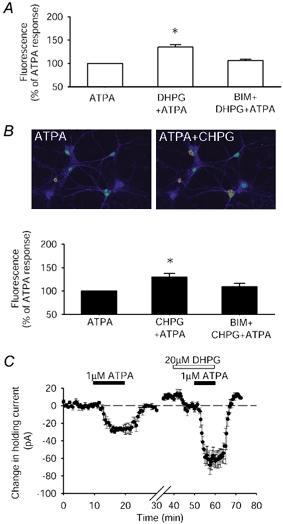
A, DHPG, 20 μm (n = 54 cells, 9 dishes), increases the GluR5-mediated calcium signal (P < 0.05). This enhancement is prevented by the PKC inhibitor BIM. B, single example of a field of cells and pooled data showing that the mGlu5 agonist CHPG, 500 μm (n = 39 cells, 5 dishes), enhanced GluR5 calcium signalling, in a PKC-dependent manner (P < 0.001). These experiments were performed in the presence of cyclopiazonic acid (CPA) to prevent calcium mobilisation from intracellular stores by group I mGlu receptor stimulation. C, the inward current resulting from activation of GluR5-containing KARs by ATPA in perirhinal cortex neurones is enhanced (P < 0.05) by co-activation of group I mGlu receptors by DHPG (ATPA: 26 ± 3 pA; ATPA and DHPG: 57 ± 11 pA, n = 4).
To determine which of the group I receptor subtypes (mGlu5 or mGlu1) was responsible for enhancing KAR signalling we firstly used the mGlu5 agonist (R,S)-2-chloro-5-hydroxyphenylglycine (CHPG). Like DHPG this also enhanced (P < 0.001) the GluR5-mediated calcium signal in a PKC-dependent manner (Fig. 4B; ATPA plus CHPG: 30 ± 8 % increase compared with ATPA; ATPA plus CHPG plus BIM: 9 ± 7 % increase compared with ATPA, P > 0.05; n = 39 cells, 5 dishes). In contrast, however, activation of mGlu1 (by DHPG in the presence of the mGlu5 receptor antagonist 2-methyl-6-(phenylethynyl) pyridine hydrochloride (MPEP)) did not enhance the GluR5-stimulated calcium signal (ATPA plus DHPG in MPEP: 5 ± 6 % decrease compared with ATPA, P > 0.05, n = 32 neurones from 4 dishes; data not shown).
Interaction between mGlu5 and GluR5-KARs in intact cortex
To examine whether an interaction between mGlu5 and GluR5 also occurred in intact neuronal preparations, whole-cell recordings were made from neurones in layer II-III of perirhinal cortex slices. Application of the GluR5 agonist ATPA resulted in an inward current (Fig. 4C; 26 ± 3 pA, n = 4) that returned to baseline following washout of ATPA. Co-application of the group I mGlu receptor agonist DHPG resulted in a significant enhancement (P < 0.05, n = 4) of the ATPA-induced inward current (57 ± 11 pA). These results show that the interaction between group I mGlu receptors and GluR5-containing KARs is not confined to calcium signalling but can regulate KAR-mediated electrophysiological properties in intact cortical preparations.
We next investigated whether mGlu receptor activation can modulate synaptically evoked KAR EPSCs. In the presence of AP5 and GYKI 53655 (to block NMDA and AMPA receptor transmission) a short burst of high frequency stimulation evoked a slow inward current (Fig. 5), reminiscent of previously described KAR EPSCs (Vignes & Collingridge, 1997). That this was a KAR EPSC was demonstrated by using the selective agonist SYM 2081 that activates and produces a rapid desensitization of KAR-mediated responses (Jones et al. 1997; DeVries, 2000). Application of the mGlu5 agonist CHPG produced a significant enhancement (37 ± 9 %, n = 4, P < 0.05) of the amplitude of the KAR EPSC (measured 10 min after start of CHPG application) that returned to baseline following washout of CHPG. These results demonstrate that mGlu5 receptor stimulation can enhance synaptically evoked KAR EPSCs in the cortex.
Figure 5. KAR-mediated EPSCs in perirhinal cortex are enhanced by stimulation of mGlu5.
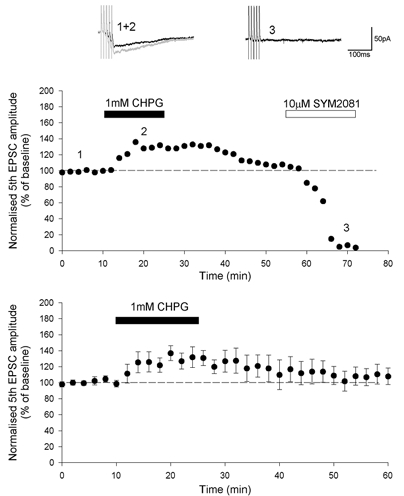
KAR EPSCs are evoked by high frequency stimulation (5 stimuli, 100 Hz) in the presence of AP5 and GYKI 53655. Application of SYM2081 produced a complete block of KAR EPSCs (DeVries, 2000). Synaptic traces are averages of four consecutive responses from the single experiment shown, taken at the time points indicated. The amplitude of KAR EPSCs is enhanced by activation of mGlu5 by the application of CHPG. The bottom graph illustrates pooled data demonstrating the enhancement of KAR EPSCs by CHPG (maximum enhancement 37 ± 9 %, P < 0.05, n = 4).
DISCUSSION
The results presented in this study demonstrate that cellular signalling mediated by GluR5-containing KARs can be enhanced by PKC or by activation of mGlu5 receptors. Furthermore, we show that the GluR5 subunit can be phosphorylated directly by PKC. This raises the possibility that the enhancement of KAR signalling by PKC and mGlu5 receptor stimulation relies on phosphorylation of a serine and/or threonine substrate on GluR5.
PKC and mGlu5 regulation of GluR5-containing kainate receptors
A simple scenario for the enhancement of GluR5-KAR signalling by mGlu5 is through an mGlu-mediated increase in PKC. Indeed the finding that the enhancement of GluR5 signalling was blocked by PKC inhibition supports this hypothesis. PKC regulation of KAR-mediated signalling has previously been reported (Rodriguez-Moreno & Lerma, 1998; Melyan et al. 2002). However, the precise mechanisms underlying the enhancement of GluR5-containing KAR function are yet to be determined. There are several possible mechanisms by which PKC or mGlu5 might regulate KAR signalling. For example, PKC-dependent phosphorylation of GluR5 may directly affect channel conductance or could modulate G-protein interaction with GluR5, in line with previous suggestions (Rodriguez-Moreno & Lerma, 1998; Melyan et al. 2002). Alternatively, phosphorylation of KAR subunits may alter KAR desensitisation for example, by an interaction with SAP90 (Garcia et al. 1998). We do not consider that mGlu5 enhancement of KAR function requires an increase in intracellular calcium since this was observed in the presence of CPA; conditions under which there was no detectable mGlu5-dependent increase in intracellular calcium. Therefore, the present mechanism may differ from that in which NMDA receptor regulation of KAR function was described to be calcium-dependent in hippocampal cultures (Ghetti & Heinemann, 2000).
Neither PKA nor mGlu1 receptors affect GluR5 function
Interestingly, the interaction between KARs and mGlu receptors appeared to be specific for mGlu5 and did not involve mGlu1 receptors. The reason for this specificity is not clear but parallels recent observations that group II mGlu receptors can interact with mGlu5 but not mGlu1 (Cho et al. 2002). Despite our finding that PKA can phosphorylate GluR5, the activation of PKA had no effect on GluR5-mediated calcium signalling. This result suggests that, under the conditions of our experiments, the PKA phosphorylation site does not play a critical role in modulation of GluR5-containing KAR-mediated calcium signalling.
Possible physiological roles for interaction between kainate and mGlu receptors
The results presented here show an interaction between mGlu5 and GluR5-containing kainate receptors. Since mGlu receptors play a critical role in synaptic plasticity in perirhinal cortex (Cho & Bashir, 2002; Brown & Bashir, 2002) this phenomenon may also be important in plastic mechanisms in perirhinal cortex. Furthermore, since KARs have been described to have a range of pre- and post-synaptic actions in other brain regions (see Chittajallu et al. 1999; Lerma, 2001) and have been implicated in synaptic plasticity (Bortolotto et al. 1999; Contractor et al. 2001) the results of the present study add a new dimension to the ways in which both mGlu and KARs may orchestrate synaptic transmission, synaptic plasticity and forms of epileptiform activity (Mulle et al. 1998).
Acknowledgments
This work was supported by the BBSRC, the Wellcome Trust and the MRC. KAR clones were gifts from Drs Hollman and Heineman.
REFERENCES
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Bortolotto ZA, Clarke VRJ, Delany CM, Parry MC, Smolders I, Vignes M, Hoo KH, Brinton B, Fantaske R, Ogden A, Gates M, Ornstein PL, Lodge D, Bleakman D, Collingridge GL. Kainate receptors are involved in synaptic plasticity. Nature. 1999;402:297–301. doi: 10.1038/46290. [DOI] [PubMed] [Google Scholar]
- Brown MW, Bashir ZI. Evidence concerning how neurons of the perirhinal cortex may effect familiarity discrimination. Philos Trans R Soc Lond B Biol Sci. 2002;357:1083–1095. doi: 10.1098/rstb.2002.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Malenka RC, Nicoll RA. Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature. 1997;388:182–186. doi: 10.1038/40645. [DOI] [PubMed] [Google Scholar]
- Chittajallu R, Braithwaite SP, Clarke VR, Henley JM. Kainate receptors: subunits, synaptic localisation and function. Trends Pharmacol Sci. 1999;20:26–35. doi: 10.1016/s0165-6147(98)01286-3. [DOI] [PubMed] [Google Scholar]
- Cho K, Bashir ZI. Cooperation between mGlu receptors: a depressing mechanism? Trends Neurosci. 2002;25:405–411. doi: 10.1016/s0166-2236(02)02228-2. [DOI] [PubMed] [Google Scholar]
- Cho K, Kemp N, Noel J, Aggleton JP, Brown MW, Bashir ZI. A novel form of long-term depression in the perirhinal cortex. Nature Neurosci. 2000;3:150–156. doi: 10.1038/72093. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Contractor A, Swanson G, Heinemann SF. Kainate receptors are involved in short- and long-term plasticity at mossy fibre synapses in the hippocampus. Neuron. 2001;29:209–216. doi: 10.1016/s0896-6273(01)00191-x. [DOI] [PubMed] [Google Scholar]
- Cui C, Mayer ML. Heteromeric kainate receptors formed by the coassembly of GluR5, GluR6, and GluR7. J Neurosci. 1999;19:8281–8291. doi: 10.1523/JNEUROSCI.19-19-08281.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBlasi A, Conn PJ, Pin J-P, Nicoletti F. Molecular determinants of metabotropic glutamate receptor signalling. TIPS. 2001;22:114–120. doi: 10.1016/s0165-6147(00)01635-7. [DOI] [PubMed] [Google Scholar]
- DeVries SH. Bipolar cells use kainate and AMPA receptors to filter visual information into separate channels. Neuron. 2000;28:847–856. doi: 10.1016/s0896-6273(00)00158-6. [DOI] [PubMed] [Google Scholar]
- Garcia EP, Mehta S, Blair LA, Wells DG, Shang J, Fukushima T, Fallon JR, Garner C, Marshall J. SAP90 binds and clusters kainate receptors causing incomplete desensitisation. Neuron. 1998;21:727–739. doi: 10.1016/s0896-6273(00)80590-5. [DOI] [PubMed] [Google Scholar]
- Ghetti A, Heinemann SF. NMDA-dependent modulation of hippocampal kainate receptors by calcineurin and Ca/calmodulin-dependent protein kinase. J Neurosci. 2000;20:2766–2773. doi: 10.1523/JNEUROSCI.20-08-02766.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirbec H, Francis JC, Lauri SE, Braithwaite SP, Dev KK, Couthino V, Meyer G, Isaac JTRI, Collingridge GL, Henley JM. PDZ proteins bind and dynamically regulate GluR5 and GluR6-containing kainate receptors. Neuron. 2003;37:625–638. doi: 10.1016/s0896-6273(02)01191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Wilding TJ, Huettner JE, Costa A-M. Desensitisation of kainate receptors by kainate, glutamate and diasteromers of 4-methylglutamate. Neuropharmacology. 1997;36:853–863. doi: 10.1016/s0028-3908(97)00066-x. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Bortolotto ZA, Bleakman D, Ornstein PL, Lodge D, Isaac TR, Collingridge GL. A critical role of a facilitatory presynaptic kainate receptor in mossy fiber LTP. Neuron. 2001;32:697–709. doi: 10.1016/s0896-6273(01)00511-6. [DOI] [PubMed] [Google Scholar]
- Lerma J, Paternain AV, Rodriguez-Moreno A, Lopez-Garcia JC. Molecular physiology of kainate receptors. Physiol Rev. 2001;81:971–998. doi: 10.1152/physrev.2001.81.3.971. [DOI] [PubMed] [Google Scholar]
- Melyan Z, Wheal HV, Lancaster B. Metabotropic-mediated kainate receptor regulation of ISAHP and excitability in pyramidal cells. Neuron. 2002;34:107–114. doi: 10.1016/s0896-6273(02)00624-4. [DOI] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Perez-Otano I, Dickinson-Anson H, Castillo PE, Bureu I, Maron C, Gage FH, Mann JE, Bettler B, Heinemann SF. Altered synaptic physiology and reduced susceptibility to kainate induced seizures in GluR6-deficient mice. Nature. 1998;392:601–605. doi: 10.1038/33408. [DOI] [PubMed] [Google Scholar]
- Nicoletti F, Casabona G, Genazzani AA, L'esiscopo MR, Shinozaki H. (2S, 1′ R, 2′ R, 3′ R)-2-(2, 3-dicarboxycyclopropyl)glycine enhances quisqualate-stimulated inositol phospholipid hydrolysis in hippocampal slices. Eur J Pharmacol. 1993;245:297–298. doi: 10.1016/0922-4106(93)90111-l. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Wenthold RJ. Histological and ultrastuctural localisation of the kainate receptor subunits, KA2 and GluR6/7, in the rat nervous system using selective antipeptide antibodies. J Comp Neurol. 1994;349:85–110. doi: 10.1002/cne.903490107. [DOI] [PubMed] [Google Scholar]
- Raymond LA, Blackstone CD, Huganir RL. Phosphorylation and modulation of recombinant GluR6 glutamate receptors by cAMP-dependent protein kinase. Nature. 1993;361:637–641. doi: 10.1038/361637a0. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Morena A, Lerma J. Kainate receptor modulation of GABA release involves a metabotropic function. Neuron. 1998;20:1211–1218. doi: 10.1016/s0896-6273(00)80501-2. [DOI] [PubMed] [Google Scholar]
- Schmitz D, Mellor J, Nicoll RA. Synaptic activation of presynaptic kainate receptors on hippocampal mossy fiber synapses. Neuron. 2001;27:327–338. doi: 10.1016/s0896-6273(00)00040-4. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Salhoff CR, Wright RA, Johnson BG, Burnett JP, Mayne NG, Belagage R, Wu S, Monn JA. The novel metabotropic glutamate receptor agonist 2R, 4R-APDC potentiates stimulation of phosphoinositide hydrolysis in the rat hippocampus by 3, 5-dihydroxyphenylglycine: evidence for a synergistic interaction between group I and group II receptors. Neuropharmacol. 1996;35:1661–1672. doi: 10.1016/s0028-3908(96)00121-9. [DOI] [PubMed] [Google Scholar]
- Vignes M, Collingridge GL. The synaptic activation of kainate receptors. Nature. 1997;388:179–182. doi: 10.1038/40639. [DOI] [PubMed] [Google Scholar]
- Wang LY, Taverna FA, Huang HP, Macdonald JF, Hampson DR. Phosphorylation and modulation of a kainate receptor (GluR6) by cAMP dependent protain kinase. Science. 1993;259:1173–1175. doi: 10.1126/science.8382377. [DOI] [PubMed] [Google Scholar]