

Abstract
Rat and sheep cardiac myocytes become binucleate as they complete the ‘terminal differentiation’ process soon after birth and are not able to divide thereafter. Angiotensin II (Ang II) is known to stimulate hypertrophic changes in rodent cardiomyocytes under both in vivo and in vitro conditions via the AT1 receptor and intracellular extracellular regulated kinase (ERK) signalling cascade. We sought to develop culture methods for immature sheep cardiomyocytes in order to test the hypothesis that Ang II is a hypertrophic agent in the immature myocardium of the sheep. We isolated fetal sheep cardiomyocytes and cultured them for 96 h, added Ang II and phenylephrine (PE) for 48 h, and measured footprint area and proliferation (5-bromo-2′-deoxyuridine (BrdU) uptake) separately in mono- vs. binucleate myocytes. We found that neither Ang II nor PE changed the footprint area of mononucleated cells. PE stimulated an increase in footprint area of binucleate cells but Ang II did not. Ang II increased myocyte BrdU uptake compared to serum free conditions, but PE did not affect BrdU uptake. The MAP kinase kinase (MEK) inhibitor UO126 prevented BrdU uptake in Ang II-stimulated cells and prevented cell hypertrophy in PE-stimulated cells. This paper establishes culture methods for immature sheep cardiomyocytes and reports that: (1) Ang II is not a hypertrophic agent; (2) Ang II stimulates hyperplastic growth among mononucleate myocytes; (3) PE is a hypertrophic agent in binucleate myocytes; and (4) the ERK cascade is required for the proliferation effect of Ang II and the hypertrophic effect of PE.
Fetal cardiomyocytes are able to use both hyperplastic (cell division) and hypertrophic (cell enlargement) mechanisms to affect growth and maturation of the myocardium (Clubb & Bishop, 1984; Oparil et al. 1984). During early fetal life, growth is primarily hyperplastic but as myocytes mature during late prenatal or early postnatal life, they switch from a proliferative mode to the hypertrophic mode of growth. As working myocytes become mature and ‘terminally differentiated’ they appear to be unable to divide (Clubb & Bishop, 1984). Under these conditions, the working myocardium could increase its mass only by increasing myocyte size for the remaining life of the individual. However, the terminal differentiation concept may be oversimplified. Recent evidence from Anversa and co-workers challenges the assumption that cardiomyocytes escape the cell cycle irreversibly for life (Anversa & Kajstura, 1998; Kajstura et al. 1998; Beltrami et al. 2001; Quaini et al. 2002).
In rodents, the maturation process is marked by binucleation of cardiomyocytes over postnatal days 4 to 12 (Li et al. 1996; Kellerman et al. 1992; Soonpaa et al. 1996). Both fetal and early neonatal rats exposed to hypertension through carbon monoxide exposure clearly demonstrate cardiomyocyte hyperplasia before terminal differentiation (Clubb et al. 1986). Sheep undergo a similar process whereby the cardiac myocyte population goes from being 100 % mononucleated at around day 75 of gestation (term = 145 days of gestation) to 100 % binucleated soon after birth (K. L. Thornburg, A. Barbera, G. D. Giraud & J. G. Maylie, unpublished observation). The ratio of binucleated to mononucleated cells can be altered by environmental factors during fetal life. For example, an experimental systolic pressure load applied to the right ventricle in fetal animals stimulates cardiomyocyte hypertrophy, hyperplasia and binucleation simultaneously, increasing the percentage of myocytes with two nuclei and the total number of myocytes (Barbera et al. 2000). However, it is unclear what mechanical and chemical mechanisms regulate the maturation of myocytes in vivo.
Angiotensin II (Ang II) has been established by many investigators to stimulate hypertrophic changes in rodent cardiac myocytes both in vivo and in vitro. When exogenous Ang II is administered to rats in vivo, heart weight increases independently of increased afterload (Dostal & Baker, 1992; Geenen et al. 1993; Kim et al. 1995; Susic et al. 1996). The isolated whole heart also shows an increase in protein synthesis in response to Ang II (Schunkert et al. 1995). Ang II is well known as a stimulant of protein synthesis and expression of the early and late genes in association with a hypertrophic response in cardiomyocytes in vitro (Sadoshima & Izumo, 1993b; Miyata & Haneda, 1994; Booz & Baker, 1996; Takahashi et al. 1997; Gray et al. 1998; Liu et al. 1998). Studies have established that the AT1 receptor and not the AT2 receptor are responsible for these actions (Dostal & Baker, 1992; Sadoshima & Izumo, 1993b; Miyata & Haneda, 1994; Booz & Baker, 1996; Gray et al. 1998; Liu et al. 1998). By contrast, most investigators have shown little increase in proliferation of cardiomyocytes in response to Ang II (Sadoshima & Izumo, 1993b; Miyata & Haneda, 1994). However, Ang II has been shown to increase proliferation of mitotically capable cardiomyocytes overexpressing the AT1 receptor (Fukuda & Izumo, 1998), and Ang II does have mitotic actions on non-myocytes (Sadoshima & Izumo, 1993b; Schorb et al. 1993). It therefore remains unclear to what degree Ang II stimulates mitotically capable cardiomyocytes to proliferate in species other than rodents.
There is an extensive body of data showing that the local renin-angiotensin system (RAS) is important in regulating hypertrophic growth in the mammalian myocardium (Wollert & Drexler, 1999; Barlucchi et al. 2001). Periodic stretch upregulates the RAS in cardiac cells (Malhotra et al. 1999) and induces Ang II production (Sadoshima et al. 1992; Sadoshima & Izumo, 1993b). Blockade of angiotensin converting enzyme (ACE) and/or AT1 receptor block can attenuate pressure load hypertrophy (Linz et al. 1989; Baker et al. 1990; Scholkens et al. 1991; Jalil et al. 1991; Dussaillant et al. 1996; Ogawa et al. 1996; Grimm et al. 1998). Volume overload-induced hypertrophy can also be attenuated by Ang II blockade (Ruzicka et al. 1994; Everett et al. 1994; Brodsky et al. 1998). Thus, inhibition of the RAS has become an important clinical tool in amelioration of hypertension and the concomitant hypertrophic pathological alterations in the myocardium (Thurmann et al. 1998; Sleight, 2000).
While the case for Ang II-induced hypertrophy is well established in the rodent, it has not been shown conclusively to mediate hypertrophy in large adult mammals. In adult pigs, the angiotensinogen gene is upregulated in the setting of pressure or volume overload hypertrophy, but the upregulation is early and goes away after 24 h even while the pressure or volume overload stimulus remains (Modesti et al. 2000). In the adult dog, mitral regurgitation (volume overload) induces ACE and chymase-like activity (Dell'Italia et al. 1995), but ACE inhibitor therapy and AT1 blockade are unable to attenuate ventricular remodelling and hypertrophy (Dell'Italia et al. 1997; Perry et al. 2002). In the sheep fetus, Segar et al. (1997, 2001) have shown that development of right hypertrophy by pressure overload was not attenuated by blockade of the AT1 or AT2 receptor. This is consistent with the report showing that pressure load-induced hypertrophy in an isolated adult rat heart was not attenuated by AT1 blockade (Thienelt et al. 1997). When pressure overload was produced by Ang II infusion into fetal sheep, the left but not right ventricular mass increased (Segar et al. 2001). However, it is not known whether Ang II directly stimulates hypertrophy at the cellular level in the large mammal.
Ang II stimulates the three primary mitogen activated protein (MAP) kinase cascade pathways: p38 MAPK, JNK, and p42/44 MAPK/ERKs (Izumi et al. 2000; Wei et al. 2000). Of these, the intracellular ERK cascade is thought to be a key stimulant of hypertrophy in cardiomyocytes (Sadoshima & Izumo, 1993a; Fischer et al. 1998; Aoki et al. 2000; Bueno et al. 2000, 2001). Indeed stimulation of the ERK signalling cascade may be necessary to affect cell hypertrophy with exposure to phenylephrine, endothelin-1, leukaemia inhibitory factor, isoproterenol, sphingosylphosphorylcholine, and stretch (Lazou et al. 1998; Sekiguchi et al. 1999; Ueyama et al. 2000; Yue et al. 2000; Clerk et al. 2001; Wang & Proud, 2002). Stretch upregulates the renin-angiotensin system in cardiac cells (Malhotra et al. 1999) and also activates p21 Ras, which is upstream of ERK (Sadoshima & Izumo, 1993a). Ras activates Raf, which in turn activates MEK. Overexpression of MEK or expression of a constitutively active MEK induces hypertrophy in vivo or in vitro (Bueno et al. 2000; Ueyama et al. 2000). Expression of a dominant negative MEK blocks hypertrophy normally induced by phenylephrine, endothelin-1, leukaemia inhibitory factor, isoproterenol, and stretch (Ueyama et al. 2000). MEK phosphorylates the ERKs that affect the signalling programme.
The goals of this present study were: (1) to develop a primary cell culture method using cardiomyocytes from fetal sheep; (2) to determine the response of these cultured cells to the AT1 agonist Ang II; and (3) to elucidate the role of ERK signalling of Ang II actions in cultured sheep myoctes. We tested the hypothesis that Ang II would behave as a hypertrophic agent and not a hyperplastic agonist as predicted from immature rat myocyte findings. We reasoned that Ang II would induce hypertrophy of the more mature binucleated myocytes and not the immature mononucleated myocytes, based on our previous work with load-induced hypertrophy (Barbera et al. 2000). We further hypothesized that the p42/44 MAP kinase cascade would be essential to mediate the actions of Ang II.
METHODS
Animals
All procedures from which fetal cardiac cells were obtained conformed to the guidelines of the Oregon Health and Science University Institutional Animal Care and Use Committee protocol A050. Hearts from fetal sheep of 127–139 days gestation (term is 145 days) were collected from control animals of various experiments in the lab ongoing for other projects. Control animals included instrumented fetuses with saline infused into the right atrium, non-infused instrumented fetuses, and non-instrumented fetuses. Following the administration of a lethal dose to the ewe of commercial euthanasia solution (Delmarva Laboratories, Inc., VA, USA) containing pentobarbital sodium, the fetus was heparinized with 10 ml of heparin sulfate and given 3 ml of saturated KCl to arrest the heart in diastole. The heart was removed and taken for cell isolation.
Cell isolation
Our cell isolation procedure has been described previously (Barbera et al. 2000). Some modifications have been made for obtaining viable live cells for culture. The hearts were hung from a perfusion apparatus by cannulating the aorta and were perfused retrogradely by a series of oxygenated solutions at 39 °C through the coronary arteries. The protocol was 5–10 min perfusion with calcium-free Tyrode buffer; 10 min with Ca-free Tyrode solution containing collagenase (Worthington type II, 160 U ml−1) and protease (type XIV 13 mg in 80 ml; Sigma, St Louis, MO, USA); and 5–10 min with high potassium (KB) solution (mm: 74 glutamic acid, 30 KCl, 30 KH2PO4, 20 taurine, 3 MgSO4, 0.5 EGTA, 10 Hepes, and 10 glucose, adjusted to pH 7.37 using KOH). The right ventricle (RV) and left ventricle (LV) free walls were separately removed with scissors, cut into chunks, and gently agitated in KB solution to release the cells. Isolated cells were poured into separate tubes removing tissue chunks, resulting in a cell slurry containing > 95 % isolated individual cells (Fig. 1). Although cardiac myocytes seem to be released preferentially, endothelial cells, fibroblasts, and blood cells were also present. Some cells were stained to check cellular integrity. Some dissociated cells were fixed in 1% paraformaldehyde and dried on a glass slide. Monoclonal antibody (1:300 anti-myosin (Alexis Biochemicals, CA, USA, clone A4.1025) diluted in blocking buffer (PBS plus 1% bovine serum albumin (BSA) and 0.5% Triton X-100) was added and left overnight at 4 °C. Cells were washed in PBS and secondary antibody with nuclear stain was added. Secondary antibody was anti-mouse rhodamine red-X-conjugate (1:200; Jackson ImmunoResearch Laboratories, Inc, PA, USA), and the nuclear stain was Hoechst 33342 (1:10 000; Molecular Probes, Eugene, OR, USA). Secondary antibody was incubated for 1 h at room temperature. Cells were washed with PBS and covered with a coverslip mounted with ProLong anti-fade kit (Molecular Probes).
Figure 1. Dissociated cardiomyocytes from fetal sheep.
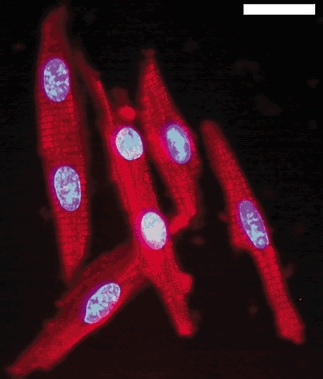
Cells were fixed after dissociation and viewed under × 400 power (Zeiss Axiophot). Myosin was stained with a secondary antibody conjugated to rhodamine red-X and the nuclei were stained with Hoechst 33342. Mononucleated and binucleated cells appear healthy and intact after dissociation. Bar = 20 μm.
Cell culture
LV and RV cells were kept separate throughout. Cells were left to rest for 30 min to 1 h at room temp in KB, while the concentration of cells was measured in a scientific counting chamber (VWR International, PA, USA). The total number of cells was estimated from the concentration and volume (ml) of slurry. Cells were gently pelletted at 1000 r.p.m. (≈200 g) for 3–5 min. KB solution was removed and cells were re-suspended to a concentration of 106 cells ml−1 in serum medium (MCDB105 medium (Sigma) supplemented with 2 mm KCl, 0.2 mm glycine, 5 mm creatine, 5 mm taurine, 2 mml-carnitine, 1 % insulin-transferrin-sodium selenite medium supplement (Sigma), 0.1 % antibiotic- antimycotic solution (× 100, Sigma), and 10 % (v/v) fetal bovine serum). The cell suspension was added to untreated tissue culture flasks for 2 h in the incubator (39 °C, 5 % CO2) to pre-plate fibroblasts and endothelial cells. Cells were gently poured from the first flask to a second flask and a second pre-plate interval followed for another 2 h. After pre-plating, unattached cells were moved to a tube and spun down again at 1000 r.p.m. The medium was removed and cells were resuspended in serum medium to the same concentration as above. This step helps remove any debris and possible contaminants. Lastly, cells were plated into 35 mm diameter tissue culture dishes holding sterile 22 mm × 22 mm coverslips pre-treated with laminin (1–5 μg ml−1) or plated into 6-well plates that were laminin treated. Cardiomyocytes were plated out at 500 000 per 35 mm diameter dish or 1–2 × 106 per well of a 6-well plate. Additional serum medium was added to make a final volume of 2.5 ml in each well or plate. After 24 h, the serum medium was replaced with serum-free medium (as serum medium but without serum). Cells were left for an additional 48 h, during which time the myocytes ‘plated out’ on the coverslips and wells. Fresh serum-free medium was added and left for a further 24 h before experiments began. Thus from the time the myocytes were first plated onto coverslips or wells, they were held in serum conditions for the first 24 h and then in serum-free conditions for an additional 72 h before experiments began.
Cell size experiments
Cardiomyocytes were exposed to Ang II in order to determine to what degree Ang II would cause hypertrophy. A dose-response curve for Ang II was generated using concentrations ranging from 10 nm to 5 μm. From these experiments, 100 nm Ang II was chosen; this dose corresponds to the optimal dose in the rodent myocyte model (Sadoshima et al. 1995; Aoki et al. 2000). Because PE is believed to cause hypertrophy through the α-adrenergic receptor mechanism, cardiomyocytes were exposed to PE as a positive control. A dose-response for PE was determined using concentrations of 0.2–20 μg ml−1 PE. A dose of 10 μg ml−1 PE was chosen for its maximal stimulation of cell hypertrophy. Further experiments were performed in the absence of BrdU, using × 1000 stocks of agonists to obtain final concentrations of 10 μg ml−1 (49.1 μm) PE and 100 nm Ang II. Cells were also stimulated with 10 μg ml−1 PE in the presence of 10 μm UO126 (a specific MEK inhibitor). After 48 h, cells were fixed for staining and measurement.
BrdU uptake experiments
Media were replaced with serum-free medium containing 10 μm> 5-bromo-2′-deoxyuridine (BrdU; Sigma). Experimental solutions were made by addition of × 1000 stocks of agonists and inhibitors to final concentrations of 10 μg ml−1 (49.1 μm) PE, 10 nm and 100 nm Ang II, and 10 μm UO126. Positive controls consisting of serum medium and 10 μm BrdU were also used. All experiments lasted for 48 h and then cells were fixed for staining.
Fixing cells to coverslips
The medium was removed and the coverslips were washed once with PBS in 35 mm plates. Cells were fixed in 70 % ethanol in 50 mm glycine, pH 2.0, for 30 min at −20 °C. Cells were washed again with PBS, stored at 4 °C and covered with PBS and 1 % sodium azide until staining.
BrdU immunofluorescence and counting
Cells were stained on the coverslips. Cells were treated with 5 μg ml−1 DNase I in a Tris buffer (66 mm Tris buffer, 0.66 mm MgCl2 and 1 mm 2-mercaptoethanol) for 30 min at 37 °C. Three washes of PBS + 1 mm EDTA were used to stop all DNase activity. Monoclonal antibodies (1:500 anti-myosin (Alexis Biochemicals, clone A4.1025) and 1:500 anti-BrdU (AbCam Ltd, Cambridge, UK, clone BU1/75), diluted in blocking buffer (PBS plus 1 % bovine serum albumin (BSA) and 0.5 % Triton X-100)) were added. Primary antibodies were left overnight at 4 °C. Cells were washed in PBS and secondary antibodies were added. Secondary antibodies were anti-mouse rhodamine red-X-conjugate (for detection of myosin) and anti-rat FITC-conjugate (for detection of BrdU) (1:200; Jackson ImmunoResearch Laboratories, Inc.) and were incubated for 2 h at room temperature. Cells were washed in PBS and inverted and mounted onto slides with ProLong anti fade kit (Molecular Probes). Under a fluorescence microscope (Zeiss Axiophot) at × 400, 300 myosin-positive cells were counted and the number of those cells positive for BrdU was recorded. This ensured that only myocytes and not any contaminating cells were counted. Data were kept as both numbers and as the percentages of the 300 cells counted.
Cell measurements
The Envision + System/HRP (DakoCytomation, Carpenteria, CA, USA) was used to stain myosin with diaminobenzidine to differentiate myocytes from contaminating cells. The kit protocols were followed using the primary antibody against myosin (1:500). Cells were counterstained with haemotoxylin and dehydrated, and coverslips were inverted and mounted in a xylene-based medium onto a glass slide. Optimas tissue analysis software (MediaCybernetics, Seattle, WA, USA) was used to capture images and measure the cell sizes under light microscopy at × 400 (Zeiss Axiophot). The threshold was set for each image so that the cells of interest were outlined while all other cells or non-cells were not. The software calculates area, length, width, and perimeter for each outlined cell. The areas of 50 mononucleated cells and 50 binucleated cells were saved in separate files from each experiment and the means for all values were calculated.
ERK stimulation
After following our cell culture protocol, fresh serum-free medium was added to each well and cells were left alone for 12 h before the experiment. Half the wells were pre-incubated for 20 min with 10 μm UO126. Both inhibited and non-inhibited wells were then stimulated with 100 nm Ang II for 0, 5, 10, or 20 min. In separate experiments, cells were stimulated with 10 μg ml−1 PE for 0, 5, 10, or 20 min along with separate wells stimulated with 100 nm Ang II for the same times. The stimulation was stopped on ice and the medium was removed. Cells were washed in ice-cold PBS and then scraped into Bos buffer (10 % glycerol, 1 % IGEPAL CA-630 (Sigma), 50 mm Tris HCl (pH7.4), 200 mm NaCl, 2 mm MgCl2, 0.5 mmβ-glycerolphosphate) with the protease inhibitors phenylmethylsulfonyl fluoride (PMSF) (1 mm), aprotinin (2 μg ml−1), leupeptin (1 μg ml−1), trypsin inhibitor (10 μg ml−1), and sodium orthovanadate (1 mm). Cell lysate was spun down and the supernatant removed and stored at −80 °C or immediately run on a gel.
Western blot analysis
Chemiluminescent reactions were used to detect levels of phospho-ERK stimulation using a phospho-specific antibody for ERK 1 and 2 normalized to total ERK 2. Protein concentrations were calculated from a Bradford reaction read on a 96-well plate. 20–40 μg of total protein from the lysate was run for each time point and experiment. The protein was run on a 12 % SDS-PAGE gel and then transferred to a polyvinyl-idene difluoride (PDVF) membrane. The membrane was blocked with 1 % BSA in PBS + 0.1 % Tween (PBST) for 1 h. The primary antibody was added (1:1000 phospho-p44/42 MAP kinase (Thr202/Tyr204); Cell Signaling Technology, Inc., Beverly, MA, USA) and left to shake overnight at 4 ° C. The PDVF membrane was washed three times and then secondary antibody (anti-rabbit HRP 1:10 000 in PBST) was added for 1 h at room temperature. The membrane was washed three times and then ECL Western blotting detection reagents (Amersham Pharmacia Biotech, Little Chalfont, UK) was added. A film was placed over the membrane, exposed, and then developed. The membrane was then stripped for 20 min at 55 ° C in a stripping solution containing β-mercaptoethanol. The membrane was washed thoroughly. Steps were repeated with primary antibody to ERK 2 (1:1000; clone sc-1647, Santa Cruz Biotechnology, Inc., CA, USA). Film images were scanned into TIFF files and analysed on multianalyst software (BioRad Laboratories, Hercules, CA, USA) and normalized to the 0 min time point of Ang II stimulation for each experiment.
Statistics
Multiple comparisons were tested using ANOVA with Tukey's post hoc test except for Western blot density, for which non-parametric ANOVA with Dunn's post hoc test was used. Student's t test was used to compare two groups. Comparisons were considered significant at P < 0.05.
RESULTS
One of the goals of this study was to develop a culture method for immature cardiomyocytes from sheep. Figure 1 shows an example of freshly isolated myocytes stained with anti-myosin antibody conjugated to rhodamine red-X and with Hoechst stain to visualize the nuclei. The cells were predominately cardiomyocytes, in good health and able to contract if placed in the appropriate solution. Freshly isolated myocytes were normally ≈60 % binucleated and ≈40 % mononucleated cells at age 135 days of gestation. These cells were maintained as a primary culture for up to eight weeks.
The effect of PE and Ang II on cardiac size was studied in vitro on cardiomyocytes from the LV and RV separately. Not surprisingly, binucleated cardiac cells were found to be larger than mononucleated cardiac cells. In cardiomyocytes from either ventricle, there was no change in mononucleated cardiomyocyte cell areas with any treatment (serum-free LV = 919.07 ± 72.28 μm2, mean ±s.e.m., RV = 1069.58 ± 89.59 μm2, Fig. 2). In LV cardiomyocytes, Ang II did not stimulate an increase in cell area (1461.30 ± 147.09 μm2) compared to cells grown in serum-free conditions (1613.28 ± 69.83 μm2, Fig. 2A). In RV cardiomyocytes the same pattern was true, Ang II did not cause hypertrophy (2342.06 ± 286.60 μm2) compared to serum-free conditions (2006.38 ± 172.19 μm2, Fig. 2B). As previously reported, binucleated cardiomyocytes from the RV tend to be larger than binucleated cardiomyocytes from the LV (Smolich et al. 1989; Barbera et al. 2000). In order to determine whether these immature cardiomyocytes were capable of hypertrophy under culture conditions, the cells were exposed to PE as a positive control. PE increased binucleated cardiomyocyte cell area for both LV (2116.46 ± 118.83 μm2) and RV (2456.99 ± 121.80 μm2) (Fig. 2).
Figure 2. Hypertrophy in culture.
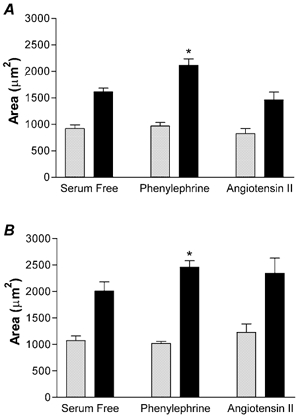
Area of LV (A) and RV (B) mononucleated (grey bars) and binucleated (black bars) cardiomyocytes from four separate fetuses. Cells were stimulated with 10 μg ml−1 PE or 100 nm AngII for 48 h. *P < 0.05 compared to serum-free binucleated, n = 4, paired ANOVA.
It has been reported that PE requires ERK stimulation to increase cardiomyocyte size (Ueyama et al. 2000; Wang & Proud, 2002). To determine whether ERK stimulation is required for PE-induced hypertrophy in immature sheep cardiomyocytes, we inhibited the ERK pathway using the MEK inhibitor UO126 in the presence of PE stimulation. In LV cardiomyocytes, PE stimulation increased average cell area (1977.89 ± 255.13 μm2) over serum-free conditions (1457.84 ± 206.20 μm2), but PE stimulation in the presence of UO126 had no effect on cell area (1426.22 ± 178.73 μm2) and UO126 alone did not affect cell area (1203.59 ± 137.71 μm2) compared to serum-free conditions (Fig. 3A). The same was true in RV cardiomyocytes; PE stimulation increased cell area (2666.25 ± 218.38 μm2) over serum-free conditions (1926.34 ± 310.46 μm2), but PE stimulation in the presence of UO126 had no effect on cell area (2051.89 ± 226.88 μm2) and UO126 alone did not affect cell area (2001.63 ± 318.47 μm2) compared to serum-free conditions (Fig. 3B).
Figure 3. Hypertrophy in the presence of ERK inhibition.
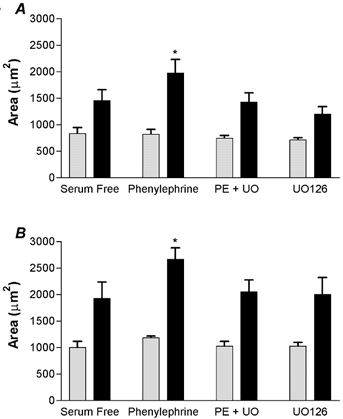
Area of LV (A) and RV (B) mononucleated (grey bars) and binucleated (black bars) cardiomyocytes from four separate fetuses. Inhibition of MEK with UO126 abolishes PE-induced hypertrophy and is not different from serum-free conditions. *P < 0.05 compared to serum-free binucleated myocytes, n = 4, paired ANOVA.
These experiments did not support the hypothesis that Ang II was an important stimulant of hypertrophy in these fetal cardiomyocytes, so we explored the ability of Ang II to stimulate hyperplasia by measuring BrdU incorporation. We first tested the ability of cardiomyocytes to incorporate BrdU in response to serum versus in serum-free conditions (Fig. 4). With regard to BrdU incorporation we saw no differences between LV and RV cells and the data were combined. Forty-eight hours of exposure to serum medium increased BrdU uptake (8.26 % ± 2.32, mean ±s.e.m., n = 9) compared to that in serum-free medium (1.11 % ± 0.23, n = 9, P < 0.05). These experiments showed that cardiomyocytes were readily able to proliferate in vitro, so we tested the ability of Ang II and PE to increase BrdU uptake. Figure 5 is a double immunofluorescence image of cardiomyocytes showing myosin staining red and incorporated BrdU staining green. Only myosin-rich cardiomyocytes were counted; contaminating cells such as fibroblasts did not stain for myosin and were thus not included in analyses. Figure 6 shows the incorporation of BrdU in response to PE and to two doses of Ang II. Phenylephrine did not stimulate BrdU incorporation (1.75 % ± 0.43, n = 8) while both doses of Ang II significantly increased BrdU incorporation (10 nm = 3.38 % ± 0.41, n = 8; 100 nm = 3.95 % ± 0.71, n = 7). The BrdU uptake stimulated by Ang II was dependent on the ERK signalling cascade (Fig. 7), as demonstrated by the inhibition of BrdU uptake in the presence of the MEK inhibitor UO126. Neither 100 nm Ang II plus UO126 nor UO126 alone stimulated BrdU uptake above the serum-free levels (P > 0.05, ANOVA).
Figure 4. BrdU uptake after 48 h in serum-free or serum medium conditions.
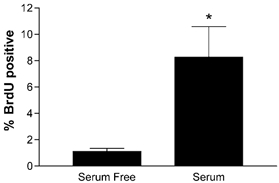
*P < 0.05, student's t test, n = 9.
Figure 5. Plated cardiomyocytes after stimulation with 100 nm Ang II in the presence of BrdU for 48 h.
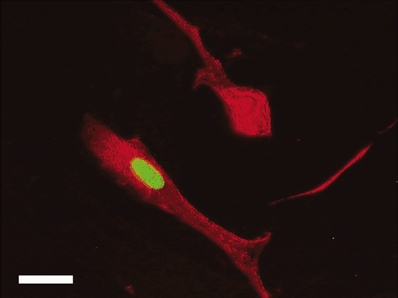
Note that one cell has incorporated BrdU into the nucleus (green). Myosin was stained with a secondary antibody conjugated to rhodamine red-X and nuclear BrdU was stained with a secondary antibody conjugated to FITC. Negative controls (without primary antibodies) did not stain for either myosin or BrdU (not shown). Bar = 20 μm.
Figure 6. BrdU uptake after 48 h in 10 μg ml−1 PE or two different doses of Ang II.
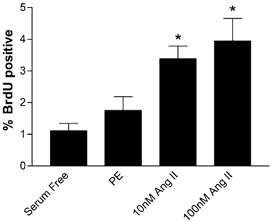
*P < 0.05, n = 7; compared to serum-free conditions, ANOVA.
Figure 7. BrdU uptake after 48 h with Ang II in the presence of the MEK inhibitor UO126.
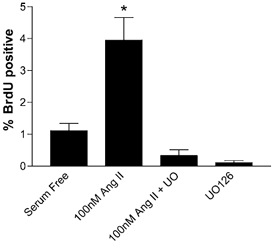
UO126 abolishes the increase in BrdU uptake that 100 nm Ang II induces in the absence of the MEK inhibitor. *P < 0.05 compared to serum-free conditions, ANOVA.
To further explore Ang II stimulation of the ERK cascade, we ran immunoblots of stimulated cell lysates with labels for anti-phospho-ERKs and anti-total ERK 2 (Fig. 8). Phospho-ERK stimulation with Ang II was significantly increased over baseline levels at 5 and 10 min, while PE-induced stimulation was significant at 5 min (Fig. 8C). The addition of UO126 abolished the Ang II-induced phospho-ERK (p-ERK) stimulation (Fig. 8A). There was no significant difference between Ang II and PE-induced stimulation of ERK (P > 0.05). It is noteworthy that the basal level of p-ERK was further decreased by the presence of UO126, which is consistent with the non-significant decrease in BrdU incorporation in the presence of UO126 compared to serum-free conditions (Fig. 7 and Fig. 8). This suggests that basal serum-free BrdU incorporation may also be dependent on ERK stimulation.
Figure 8. ERK stimulation.
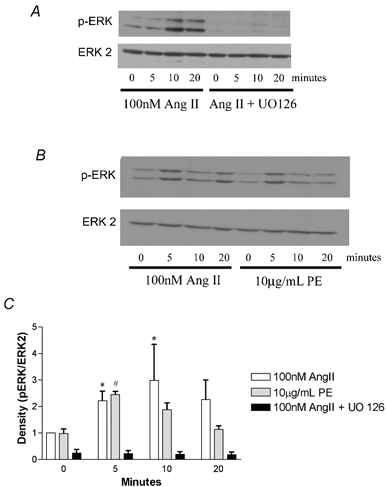
A, shows a typical Western blot (n = 4). Results shown in the left 4 columns were from cells stimulated with 100 nm Ang II in the absence of UO126 for the indicated times. The right 4 columns are from cells pre-treated for 20 min with UO126 before stimulation with Ang II. B, a typical Western blot of Ang II and PE stimulation (n = 4). The left 4 columns are from cells that were stimulated with 100 nm Ang II and the right 4 columns from cells treated with 10 μg ml−1 PE. All membranes were probed for phospho-ERKs, then stripped and probed for ERK 2. C, average band density of Western blots normalized to baseline Ang II stimulation (0 min). Density is plotted in normalized units of p-ERK/ERK 2 levels. At 5 and 10 min, phospho-ERK levels were increased over basal unstimulated levels with Ang II (*P < 0.05, non-parametric ANOVA). At 5 min PE significantly increased phospho-ERK levels over 0 min of PE stimulation (#P < 0.05, non-parametric ANOVA).
DISCUSSION
This study yielded four primary findings: first, and contrary to our hypothesis, Ang II did not behave as a hypertrophic agent for fetal sheep cardiomyocytes in culture. Second, the pro-hypertrophic action of phenylephrine was dependent on the state of cardiomyocyte maturity as indicated by the number of nuclei. Third, Ang II stimulated hyperplastic growth in cardiomyocytes under similar culture conditions. Fourth, the ERK signalling cascade was necessary for Ang II-induced hyperplasia and PE-induced hypertrophy, but ERK stimulation does not appear to be sufficient for either hypertrophy or hyperplasia.
A dose of 100 nm Ang II has been routinely used by other investigators to induce hypertrophy in neonatal rat cardiomyocytes in vitro (Sadoshima & Izumo, 1993b), but the same dose did not stimulate proliferation in those cells (Sadoshima et al. 1997). In fetal sheep cardiomyocytes, Ang II had no effect on the size of cells from either the LV or RV at the stage of maturity that we studied (Fig. 2). It is possible that, with further maturation, Ang II would stimulate binucleate cardiomyocytes to undergo hypertrophy in neonatal or adult sheep. However, our results are consistent with previous findings that Ang II is not important for fetal sheep in generation of cardiomyocyte hypertrophy during fetal life (Segar et al. 1997).
During ovine fetal development, mitotically competent mononucleated cells co-exist with terminally differentiated binucleated cells (Barbera et al. 2000). Our work establishes for the first time a difference in the response of sheep cardiomyocytes to a hypertrophic stimulus that is dependent on the state of myocyte maturation. We report here that binucleated cells increased in size in the presence of PE while mononucleated cells were unresponsive (Fig. 2 and Fig. 3). Clubb et al. (1986) were the first to suggest that binucleation was a marker for the transformation from hyperplastic growth to hypertrophic growth. In future studies we plan to determine whether the actions of other hypertrophic agents are limited to increasing the size of binucleated cardiomyocytes.
The fact that Ang II can stimulate hypertrophy is well established in neonatal rat cardiomyocytes and we were surprised to discover in our system that sheep cardiomyocytes are not enlarged under the influence of Ang II. However, the finding that Ang II can induce proliferation is not novel. Ang II has been shown to cause proliferation in cardiac fibroblasts (Sadoshima & Izumo, 1993b; Schorb et al. 1993), vascular smooth muscle cells (Watanabe et al. 2001; Mueller et al. 2002), and other cell types as well (Muscella et al. 2002; Rossi et al. 2002). However, cardiomyocytes have not been shown to proliferate in culture in response to Ang II (Sadoshima & Izumo, 1993b; Miyata & Haneda, 1994) unless transformed (Fukuda & Izumo, 1998). We first tested the ability of fetal sheep cardiomyocytes to proliferate in culture. We stimulated cardiomyocytes with either a serum-free or serum-enriched medium for 48 h in the presence of BrdU (Fig. 4). We obtained the same positive response for BrdU uptake in sheep cardiomyocytes as that of neonatal rat cardiomyocytes exposed to fetal calf serum for 48 h in Izumo's laboratory (Sadoshima et al. 1997). However, in their experiments the neonatal rat cells failed to increase BrdU uptake in response to Ang II over serum-free levels during the window of time before terminal differentiation in culture (Sadoshima et al. 1997). In our system, a 100 nm dose of Ang II increased BrdU uptake by nearly 4-fold above serum-free levels and about half as much as with serum-enriched media (Fig. 6). In one study, Ang II was shown to increase LV mass and not RV mass, while PE increased mass in neither ventricle (Segar et al. 2001). Our data are consistent with the explanation that, in their experiments, the LV increased mass by increasing cell numbers. However, it is not clear why RV mass did not increase. We found no difference in LV and RV response to Ang II in vitro and we found profound RV hypertrophy with increased mechanical systolic load (Barbera et al. 2000). Nor is it clear why PE was unable to induce an increase in ventricular mass in the experiments of Segar et al. (2001).
BrdU uptake was counted in all cardiomyocytes as we were not able to distinguish mononucleated from binucleated cells that were BrdU negative. It could be argued then that the increase in BrdU uptake was leading only to binucleation and not proliferation. BrdU-positive binucleated cells were seen, but the vast majority of BrdU-positive myocytes were mononucleated (≈90 % of all BrdU-positive myocytes). We expect that only mononucleated cells are capable of proliferation and that the more mature binucleated cells are not. If true, we expect that all of the BrdU-positive binucleated cells that were detected were the result of mononucleated cells undergoing binucleation. If binucleated cells are able to divide in the fetal myocardium they do not fit the definition of being ‘terminally’ differentiated. However, division in binuclear cells is highly unlikely because Ang II treatment did not affect the proportion of binucleated BrdU-positive cells (data not shown) and, thus, evidently did not lead to increased DNA synthesis in these cells. While the constant proportion of binucleation is not proof that binucleated cells cannot divide, it seems more likely and consistent with our data that the mononucleated cells in the fetal myocardium are capable of proliferation in response to Ang II, while binucleated cells are unable to proliferate but are able to increase cell size in response to PE. The fact that Ang II is not capable of inducing enlargement of even a binucleated cell at this stage of gestation, suggests a species difference between sheep and rats. It may indeed indicate that there is a general difference in myocardial growth regulation between larger mammals and rodents (Dell'Italia et al. 1997; Segar et al. 1997; Modesti et al. 2000; Perry et al. 2002).
ERKs are involved in diverse cellular responses including proliferation and hypertrophy. We set out to determine the roles that ERKs were playing in Ang II signalling in immature ovine cardiomyocytes. Thus, we studied the effect of MEK inhibition on BrdU uptake. Inhibition with 10 μm UO126 completely inhibited all phospho-ERK stimulation even reducing basal p-ERK stimulation from the serum-free medium conditions (Fig. 8). BrdU uptake and ERK stimulation by Ang II were completely inhibited in the presence of the MEK inhibitor (Fig. 7 and Fig. 8). We conclude that ERK stimulation is necessary for Ang II action in sheep cardiomyocytes.
Phenylephrine-induced hypertrophy is also reported to require ERK stimulation in rodents (Ueyama et al. 2000; Yue et al. 2000). We have shown that ERK stimulation is required for the induction of hypertrophy by PE in binucleate cells (Fig. 3). It is curious that ERK stimulation is required for both PE-induced hypertrophy and Ang II-induced proliferation, two mutually exclusive processes. However, since both agonists stimulate ERK and are only capable of hypertrophy or hyperplasia, but not both, the outcome of ERK stimulation must depend upon the maturation state of the myocyte. If this is true, it is likely that the cascades themselves are significantly ‘remodelled’ with maturation. It is also possible that PE, together with ERK, stimulates a separate signalling cascade that leads to hypertrophy, while Ang II does not, and that Ang II stimulates a separate cascade together with ERK that is required for hyperplastic growth that is not stimulated by PE. The focus of this study was limited to ERK signalling. Further investigation of PE and Ang II signalling in the maturing cardiomyocyte will be required to distinguish between these possibilities.
In summary, this paper reports on the establishment of culture methods for immature sheep cardiomyocytes. It also reports on several novel findings regarding Ang II and its effects on cardiomyocytes. It is shown here that Ang II stimulates the ERK cascade in fetal sheep cardiomyocytes without stimulating hypertrophy. Ang II is able to stimulate hyperplasia in fetal sheep cardiomyocytes and the stimulation of the ERK cascade is required for the increase in the proliferative index of BrdU uptake. It is also shown that the ability of the cardiomyocyte to enlarge in response to PE is dependent on its state of maturation. Binucleation is considered to be a marker for the transformation from hyperplastic growth to hypertrophic growth. Our studies support this hypothesis by showing that, in the immature myocardium containing a mix of immature mononucleated cells and differentiated binucleated cells, it is possible for both hyperplasia and hypertrophy to take place but in a maturation state-dependent manner. This may reflect the situation in vivo, as augmented afterload conditions will induce both hyperplasia and hypertrophy in the fetal right ventricle (Barbera et al. 2000).
Acknowledgments
The authors thank Patricia Renwick, Robert Webber, Krista Wehrley, Sonnet Jonker and Kirstin Labudda for help with the completion of these experiments and Dr Peter Rotwein for expert advice. This work was supported by NIH grant 2PO1 HD34430. N.C.S. is supported by a pre-doctoral fellowship, grant 0215264Z, from the AHA.
References
- Anversa P, Kajstura J. Ventricular myocytes are not terminally differentiated in the adult mammalian heart. Circ Res. 1998;83:1–14. doi: 10.1161/01.res.83.1.1. [DOI] [PubMed] [Google Scholar]
- Aoki H, Richmond M, Izumo S, Sadoshima J. Specific role of the extracellular signal-regulated kinase pathway in angiotensin II-induced cardiac hypertrophy in vitro. Biochem J. 2000;347:275–284. [PMC free article] [PubMed] [Google Scholar]
- Baker KM, Chernin MI, Wixson SK, Aceto JF. Renin-angiotensin system involvement in pressure-overload cardiac hypertrophy in rats. Am J Physiol. 1990;259:H324–332. doi: 10.1152/ajpheart.1990.259.2.H324. [DOI] [PubMed] [Google Scholar]
- Barbera A, Giraud GD, Reller MD, Maylie J, Morton MJ, Thornburg KL. Right ventricular systolic pressure load alters myocyte maturation in fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1157–1164. doi: 10.1152/ajpregu.2000.279.4.R1157. [DOI] [PubMed] [Google Scholar]
- Barlucchi L, Leri A, Dostal DE, Fiordaliso F, Tada H, Hintze TH, Kajstura J, Nadal-Ginard B, Anversa P. Canine ventricular myocytes possess a renin-angiotensin system that is upregulated with heart failure. Circ Res. 2001;88:298–304. doi: 10.1161/01.res.88.3.298. [DOI] [PubMed] [Google Scholar]
- Beltrami AP, Urbanek K, Kajstura J, Yan SM, Finato N, Bussani R, Nadal-Ginard B, Silvestri F, Leri A, Beltrami CA, Anversa P. Evidence that human cardiac myocytes divide after myocardial infarction. N Engl J Med. 2001;344:1750–1757. doi: 10.1056/NEJM200106073442303. [DOI] [PubMed] [Google Scholar]
- Booz GW, Baker KM. Role of type 1 and type 2 angiotensin receptors in angiotensin II-induced cardiomyocyte hypertrophy. Hypertension. 1996;28:635–640. doi: 10.1161/01.hyp.28.4.635. [DOI] [PubMed] [Google Scholar]
- Brodsky S, Gurbanov K, Abassi Z, Hoffman A, Ruffolo RR, Feuerstein GZ, Winaver J. Effects of eprosartan on renal function and cardiac hypertrophy in rats with experimental heart failure. Hypertension. 1998;32:746–752. doi: 10.1161/01.hyp.32.4.746. [DOI] [PubMed] [Google Scholar]
- Bueno OF, DeWindt LJ, Lim HW, Tymitz KM, Witt SA, Kimball TR, Molkentin JD. The dual-specificity phosphatase MKP-1 limits the cardiac hypertrophic response in vitro and in vivo. Circ Res. 2001;88:88–96. doi: 10.1161/01.res.88.1.88. [DOI] [PubMed] [Google Scholar]
- Bueno OF, DeWindt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, Hewett TE, Jones SP, Lefer DJ, Peng CF, Kitsis RN, Molkentin JD. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000;19:6341–6350. doi: 10.1093/emboj/19.23.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerk A, Pham FH, Fuller SJ, Sahai E, Aktories K, Marais R, Marshall C, Sugden PH. Regulation of mitogen-activated protein kinases in cardiac myocytes through the small G protein Rac1. Mol Cell Biol. 2001;21:1173–1184. doi: 10.1128/MCB.21.4.1173-1184.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clubb FJ, Bishop SP. Formation of binucleated myocardial cells in the neonatal rat. An index for growth hypertrophy. Lab Invest. 1984;50:571–577. [PubMed] [Google Scholar]
- Clubb FJ, Penney DG, Baylerian MS, Bishop SP. Cardiomegaly due to myocyte hyperplasia in perinatal rats exposed to 200 ppm carbon monoxide. J Mol Cell Cardiol. 1986;18:477–486. doi: 10.1016/s0022-2828(86)80913-0. [DOI] [PubMed] [Google Scholar]
- Dell'Italia LJ, Balcells E, Meng QC, Su X, Schultz D, Bishop SP, Machida N, Straeter-Knowlen IM, Hankes GH, Dillon R, Cartee RE, Oparil S. Volume-overload cardiac hypertrophy is unaffected by ACE inhibitor treatment in dogs. Am J Physiol. 1997;273:H961–970. doi: 10.1152/ajpheart.1997.273.2.H961. [DOI] [PubMed] [Google Scholar]
- Dell'Italia LJ, Meng QC, Balcells E, Straeter-Knowlen IM, Hankes GH, Dillon R, Cartee RE, Orr R, Bishop SP, Oparil S. Increased ACE and chymase-like activity in cardiac tissue of dogs with chronic mitral regurgitation. Am J Physiol. 1995;269:H2065–2073. doi: 10.1152/ajpheart.1995.269.6.H2065. [DOI] [PubMed] [Google Scholar]
- Dostal DE, Baker KM. Angiotensin II stimulation of left ventricular hypertrophy in adult rat heart. Mediation by the AT1 receptor. Am J Hypertens. 1992;5:276–280. doi: 10.1093/ajh/5.5.276. [DOI] [PubMed] [Google Scholar]
- Dussaillant GR, Gonzalez H, Cespedes C, Jalil JE. Regression of left ventricular hypertrophy in experimental renovascular hypertension: diastolic dysfunction depends more on myocardial collagen than it does on myocardial mass. J Hypertens. 1996;14:1117–1123. doi: 10.1097/00004872-199609000-00012. [DOI] [PubMed] [Google Scholar]
- Everett AD, Tufro-McReddie A, Fisher A, Gomez RA. Angiotensin receptor regulates cardiac hypertrophy and transforming growth factor-beta 1 expression. Hypertension. 1994;23:587–592. doi: 10.1161/01.hyp.23.5.587. [DOI] [PubMed] [Google Scholar]
- Fischer TA, Singh K, O'Hara DS, Kaye DM, Kelly RA. Role of AT1 and AT2 receptors in regulation of MAPKs and MKP-1 by ANG II in adult cardiac myocytes. Am J Physiol. 1998;275:H906–916. doi: 10.1152/ajpheart.1998.275.3.H906. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Izumo S. Angiotensin II potentiates DNA synthesis in AT-1 transformed cardiomyocytes. J Mol Cell Cardiol. 1998;30:2069–2080. doi: 10.1006/jmcc.1998.0770. [DOI] [PubMed] [Google Scholar]
- Geenen DL, Malhotra A, Scheuer J. Angiotensin II increases cardiac protein synthesis in adult rat heart. Am J Physiol. 1993;265:H238–243. doi: 10.1152/ajpheart.1993.265.1.H238. [DOI] [PubMed] [Google Scholar]
- Gray MO, Long CS, Kalinyak JE, Li HT, Karliner JS. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovasc Res. 1998;40:352–363. doi: 10.1016/s0008-6363(98)00121-7. [DOI] [PubMed] [Google Scholar]
- Grimm D, Kromer EP, Bocker W, Bruckschlegel G, Holmer SR, Riegger GA, Schunkert H. Regulation of extracellular matrix proteins in pressure-overload cardiac hypertrophy: effects of angiotensin converting enzyme inhibition. J Hypertens. 1998;16:1345–1355. doi: 10.1097/00004872-199816090-00016. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Kim S, Zhan Y, Namba M, Yasumoto H, Iwao H. Important role of angiotensin II-mediated c-Jun NH(2)-terminal kinase activation in cardiac hypertrophy in hypertensive rats. Hypertension. 2000;36:511–516. doi: 10.1161/01.hyp.36.4.511. [DOI] [PubMed] [Google Scholar]
- Jalil JE, Janicki JS, Pick R, Weber KT. Coronary vascular remodeling and myocardial fibrosis in the rat with renovascular hypertension. Response to captopril. Am J Hypertens. 1991;4:51–55. doi: 10.1093/ajh/4.1.51. [DOI] [PubMed] [Google Scholar]
- Kajstura J, Leri A, Finato N, Di Loreto C, Beltrami CA, Anversa P. Myocyte proliferation in end-stage cardiac failure in humans. Proc Natl Acad Sci U S A. 1998;95:8801–8805. doi: 10.1073/pnas.95.15.8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellerman S, Moore JA, Zierhut W, Zimmer HG, Campbell J, Gerdes AM. Nuclear DNA content and nucleation patterns in rat cardiac myocytes from different models of cardiac hypertrophy. J Mol Cell Cardiol. 1992;24:497–505. doi: 10.1016/0022-2828(92)91839-w. [DOI] [PubMed] [Google Scholar]
- Kim NN, Villarreal FJ, Printz MP, Lee AA, Dillmann WH. Trophic effects of angiotensin II on neonatal rat cardiac myocytes are mediated by cardiac fibroblasts. Am J Physiol. 1995;269:E426–437. doi: 10.1152/ajpendo.1995.269.3.E426. [DOI] [PubMed] [Google Scholar]
- Lazou A, Sugden PH, Clerk A. Activation of mitogen-activated protein kinases (p38-MAPKs, SAPKs/JNKs and ERKs) by the G-protein-coupled receptor agonist phenylephrine in the perfused rat heart. Biochem J. 1998;332:459–465. doi: 10.1042/bj3320459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol. 1996;28:1737–1746. doi: 10.1006/jmcc.1996.0163. [DOI] [PubMed] [Google Scholar]
- Linz W, Scholkens BA, Ganten D. Converting enzyme inhibition specifically prevents the development and induces regression of cardiac hypertrophy in rats. Clin Exp Hypertens A. 1989;11:1325–1350. doi: 10.3109/10641968909038172. [DOI] [PubMed] [Google Scholar]
- Liu Y, Leri A, Li B, Wang X, Cheng W, Kajstura J, Anversa P. Angiotensin II stimulation in vitro induces hypertrophy of normal and postinfarcted ventricular myocytes. Circ Res. 1998;82:1145–1159. doi: 10.1161/01.res.82.11.1145. [DOI] [PubMed] [Google Scholar]
- Malhotra R, Sadoshima J, Brosius FC, Izumo S. Mechanical stretch and angiotensin II differentially upregulate the renin-angiotensin system in cardiac myocytes in vitro. Circ Res. 1999;85:137–146. doi: 10.1161/01.res.85.2.137. [DOI] [PubMed] [Google Scholar]
- Miyata S, Haneda T. Hypertrophic growth of cultured neonatal rat heart cells mediated by type 1 angiotensin II receptor. Am J Physiol. 1994;266:H2443–2451. doi: 10.1152/ajpheart.1994.266.6.H2443. [DOI] [PubMed] [Google Scholar]
- Modesti PA, Vanni S, Bertolozzi I, Cecioni I, Polidori G, Paniccia R, Bandinelli B, Perna A, Liguori P, Boddi M, Galanti G, Serneri GG. Early sequence of cardiac adaptations and growth factor formation in pressure- and volume-overload hypertrophy. Am J Physiol Heart Circ Physiol. 2000;279:H976–985. doi: 10.1152/ajpheart.2000.279.3.H976. [DOI] [PubMed] [Google Scholar]
- Mueller C, Baudler S, Welzel H, Bohm M, Nickenig G. Identification of a novel redox-sensitive gene, Id3, which mediates angiotensin II-induced cell growth. Circulation. 2002;105:2423–2428. doi: 10.1161/01.cir.0000016047.19488.91. [DOI] [PubMed] [Google Scholar]
- Muscella A, Greco S, Elia MG, Storelli C, Marsigliante S. Angiotensin II stimulation of Na+/K+ATPase activity and cell growth by calcium-independent pathway in MCF-7 breast cancer cells. J Endocrinol. 2002;173:315–323. doi: 10.1677/joe.0.1730315. [DOI] [PubMed] [Google Scholar]
- Ogawa T, Linz W, Stevenson M, Bruneau BG, Kuroski DeBold ML, Chen JH, Eid H, Scholkens BA, DeBold AJ. Evidence for load-dependent and load-independent determinants of cardiac natriuretic peptide production. Circulation. 1996;93:2059–2067. doi: 10.1161/01.cir.93.11.2059. [DOI] [PubMed] [Google Scholar]
- Oparil S, Bishop SP, Clubb FJ. Myocardial cell hypertrophy or hyperplasia. Hypertension. 1984;6:III38–43. doi: 10.1161/01.hyp.6.6_pt_2.iii38. [DOI] [PubMed] [Google Scholar]
- Perry GJ, Wei CC, Hankes GH, Dillon SR, Rynders P, Mukherjee R, Spinale FG, Dell'Italia LJ. Angiotensin II receptor blockade does not improve left ventricular function and remodeling in subacute mitral regurgitation in the dog. J Am Coll Cardiol. 2002;39:1374–1379. doi: 10.1016/s0735-1097(02)01763-1. [DOI] [PubMed] [Google Scholar]
- Quaini F, Urbanek K, Beltrami AP, Finato N, Beltrami CA, Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Chimerism of the transplanted heart. N Engl J Med. 2002;346:5–15. doi: 10.1056/NEJMoa012081. [DOI] [PubMed] [Google Scholar]
- Rossi F, Ferraresi A, Romagni P, Silvestroni L, Santiemma V. Angiotensin II stimulates contraction and growth of testicular peritubular myoid cells in vitro. Endocrinology. 2002;143:3096–3104. doi: 10.1210/endo.143.8.8955. [DOI] [PubMed] [Google Scholar]
- Ruzicka M, Yuan B, Leenen FH. Effects of enalapril versus losartan on regression of volume overload-induced cardiac hypertrophy in rats. Circulation. 1994;90:484–491. doi: 10.1161/01.cir.90.1.484. [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Aoki H, Izumo S. Angiotensin II and serum differentially regulate expression of cyclins, activity of cyclin-dependent kinases, and phosphorylation of retinoblastoma gene product in neonatal cardiac myocytes. Circ Res. 1997;80:228–241. doi: 10.1161/01.res.80.2.228. [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Izumo S. Mechanical stretch rapidly activates multiple signal transduction pathways in cardiac myocytes: potential involvement of an autocrine/paracrine mechanism. EMBO J. 1993a;12:1681–1692. doi: 10.1002/j.1460-2075.1993.tb05813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoshima J, Izumo S. Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res. 1993b;73:413–423. doi: 10.1161/01.res.73.3.413. [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Qiu Z, Morgan JP, Izumo S. Angiotensin II and other hypertrophic stimuli mediated by G protein-coupled receptors activate tyrosine kinase, mitogen-activated protein kinase, and 90-kD S6 kinase in cardiac myocytes. The critical role of Ca(2+)-dependent signaling. Circ Res. 1995;76:1–15. doi: 10.1161/01.res.76.1.1. [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Takahashi T, Jahn L, Izumo S. Roles of mechano-sensitive ion channels, cytoskeleton, and contractile activity in stretch-induced immediate-early gene expression and hypertrophy of cardiac myocytes. Proc Natl Acad Sci U S A. 1992;89:9905–9909. doi: 10.1073/pnas.89.20.9905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholkens BA, Linz W, Martorana PA. Experimental cardiovascular benefits of angiotensin-converting enzyme inhibitors: beyond blood pressure reduction. J Cardiovasc Pharmacol. 1991;18(suppl. 2):S26–30. [PubMed] [Google Scholar]
- Schorb W, Booz GW, Dostal DE, Conrad KM, Chang KC, Baker KM. Angiotensin II is mitogenic in neonatal rat cardiac fibroblasts. Circ Res. 1993;72:1245–1254. doi: 10.1161/01.res.72.6.1245. [DOI] [PubMed] [Google Scholar]
- Schunkert H, Sadoshima J, Cornelius T, Kagaya Y, Weinberg EO, Izumo S, Riegger G, Lorell BH. Angiotensin II-induced growth responses in isolated adult rat hearts. Evidence for load-independent induction of cardiac protein synthesis by angiotensin II. Circ Res. 1995;76:489–497. doi: 10.1161/01.res.76.3.489. [DOI] [PubMed] [Google Scholar]
- Segar JL, Dalshaug GB, Bedell KA, Smith OM, Scholz TD. Angiotensin II in cardiac pressure-overload hypertrophy in fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2001;281:R2037–2047. doi: 10.1152/ajpregu.2001.281.6.R2037. [DOI] [PubMed] [Google Scholar]
- Segar JL, Scholz TD, Bedell KA, Smith OM, Huss DJ, Guillery EN. Angiotensin AT1 receptor blockade fails to attenuate pressure-overload cardiac hypertrophy in fetal sheep. Am J Physiol. 1997;273:R1501–1508. doi: 10.1152/ajpregu.1997.273.4.R1501. [DOI] [PubMed] [Google Scholar]
- Sekiguchi K, Yokoyama T, Kurabayashi M, Okajima F, Nagai R. Sphingosylphosphorylcholine induces a hypertrophic growth response through the mitogen-activated protein kinase signaling cascade in rat neonatal cardiac myocytes. Circ Res. 1999;85:1000–1008. doi: 10.1161/01.res.85.11.1000. [DOI] [PubMed] [Google Scholar]
- Sleight P. The HOPE Study (Heart Outcomes Prevention Evaluation) J Renin Angiotensin Aldosterone Syst. 2000;1:18–20. doi: 10.3317/jraas.2000.002. [DOI] [PubMed] [Google Scholar]
- Smolich JJ, Walker AM, Campbell GR, Adamson TM. Left and right ventricular myocardial morphometry in fetal, neonatal, and adult sheep. Am J Physiol. 1989;257:H1–9. doi: 10.1152/ajpheart.1989.257.1.H1. [DOI] [PubMed] [Google Scholar]
- Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol. 1996;271:H2183–2189. doi: 10.1152/ajpheart.1996.271.5.H2183. [DOI] [PubMed] [Google Scholar]
- Susic D, Nunez E, Frohlich ED, Prakash O. Angiotensin II increases left ventricular mass without affecting myosin isoform mRNAs. Hypertension. 1996;28:265–268. doi: 10.1161/01.hyp.28.2.265. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Azuma M, Taira K, Baba A, Yamamoto I, Schaffer SW, Azuma J. Effect of taurine on angiotensin II-induced hypertrophy of neonatal rat cardiac cells. J Cardiovasc Pharmacol. 1997;30:725–730. doi: 10.1097/00005344-199712000-00004. [DOI] [PubMed] [Google Scholar]
- Thienelt CD, Weinberg EO, Bartunek J, Lorell BH. Load-induced growth responses in isolated adult rat hearts. Role of the AT1 receptor. Circulation. 1997;95:2677–2683. doi: 10.1161/01.cir.95.12.2677. [DOI] [PubMed] [Google Scholar]
- Thurmann PA, Kenedi P, Schmidt A, Harder S, Rietbrock N. Influence of the angiotensin II antagonist valsartan on left ventricular hypertrophy in patients with essential hypertension. Circulation. 1998;98:2037–2042. doi: 10.1161/01.cir.98.19.2037. [DOI] [PubMed] [Google Scholar]
- Ueyama T, Kawashima S, Sakoda T, Rikitake Y, Ishida T, Kawai M, Yamashita T, Ishido S, Hotta H, Yokoyama M. Requirement of activation of the extracellular signal-regulated kinase cascade in myocardial cell hypertrophy. J Mol Cell Cardiol. 2000;32:947–960. doi: 10.1006/jmcc.2000.1135. [DOI] [PubMed] [Google Scholar]
- Wang L, Proud CG. Ras/Erk signaling is essential for activation of protein synthesis by Gq protein-coupled receptor agonists in adult cardiomyocytes. Circ Res. 2002;91:821–829. doi: 10.1161/01.res.0000041029.97988.e9. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Pakala R, Katagiri T, Benedict CR. Serotonin potentiates angiotensin II-induced vascular smooth muscle cell proliferation. Atherosclerosis. 2001;159:269–279. doi: 10.1016/s0021-9150(01)00505-6. [DOI] [PubMed] [Google Scholar]
- Wei C, Cardarelli MG, Downing SW, Mclaughlin JS. The effect of angiotensin II on mitogen-activated protein kinase in human cardiomyocytes. J Renin Angiotensin Aldosterone Syst. 2000;1:379–384. doi: 10.3317/jraas.2000.070. [DOI] [PubMed] [Google Scholar]
- Wollert KC, Drexler H. The renin-angiotensin system and experimental heart failure. Cardiovasc Res. 1999;43:838–849. doi: 10.1016/s0008-6363(99)00145-5. [DOI] [PubMed] [Google Scholar]
- Yue TL, Gu JL, Wang C, Reith AD, Lee JC, Mirabile RC, Kreutz R, Wang Y, Maleeff B, Parsons AA, Ohlstein EH. Extracellular signal-regulated kinase plays an essential role in hypertrophic agonists, endothelin-1 and phenylephrine-induced cardiomyocyte hypertrophy. J Biol Chem. 2000;275:37895–37901. doi: 10.1074/jbc.M007037200. [DOI] [PubMed] [Google Scholar]