

Abstract
The hyperpolarization-activated current (Ih) is important in the control of resting membrane potential, in the regulation of network firing pattern and in the modulation of presynaptic transmitter release in central neurons. Recent studies on native and cloned Ih channels have demonstrated that the Ih channel is commonly modulated by cAMP through a positive shift in its voltage dependence without a change in its maximum current. The present study demonstrates that activation of κ-opioid receptors enhances Ih by increasing its maximum current in brainstem neurons in the nucleus raphe magnus. Agents that interfere with the release of intracellular calcium from calcium stores altered the maximum Ih and significantly attenuated the κ-receptor-mediated enhancement of Ih. These results suggest that κ-opioid receptors enhance the maximum Ih by mobilizing intracellular calcium from calcium stores. This provides a physiological function for κ-receptor-stimulated calcium release and may suggest another Ih-regulating mechanism by intracellular calcium in central neurons.
The hyperpolarization-activated current (Ih) is a non-selective, inward cation current that is slowly activated at membrane potentials more negative than the resting membrane potential. Although it was first described in both cardiac cells and neurons more than 20 years ago (Brown et al. 1979; DiFrancesco, 1981; Halliwell & Adams, 1982), only in recent years has there been rapid progress in our understanding of its modulation and regulatory functions.
It has now been clearly shown that Ih is increased by intracellular cAMP through a positive shift in its voltage dependence without changing its maximum in various types of central and peripheral neurons (Bobker & Williams, 1989; McCormick & Pape, 1990; Pape, 1992; Banks et al. 1993; Erickson et al. 1993; Ingram & Williams, 1994; Pedarzani & Storm, 1995; Luthi & McCormick, 1999) and in cloned Ih channels (Gauss et al. 1998; Ludwig et al. 1998; Santoro et al. 1998). A variety of functions of Ih have been described in central neurons (McCormick & Pape, 1990; Kaupp & Seifert, 2001). A membrane depolarizing current activated near the resting membrane potential, Ih is an important contributor to the control of resting membrane potential and neuronal excitability (Rainnie et al. 1994; Saitow & Konishi, 2000; Lupica et al. 2001; Williams et al. 2002). In thalamocortical relay neurons, Ih plays a key role in the burst firing pattern and controls the synchronized rhythmic activity in the thalamocortical system during sleep (Pape & McCormick, 1989; Steriade et al. 1993; von Krosigk et al. 1993; Luthi & McCormick, 1999). Recently, activation of presynaptic Ih has been shown to enhance synaptic transmission through a cAMP-dependent mechanism (Beaumont & Zucker, 2000; Saitow & Konishi, 2000; Southan et al. 2000; Lupica et al. 2001) and mediate hippocampal mossy fibre long-term potentiation (Mellor et al. 2002).
Opioid peptides are among various neurotransmitters that modulate Ih in neurons. It is interesting that many neurotransmitters, including opioids, either enhance (Colino & Halliwell, 1993) or inhibit the maximum amplitude of Ih (Jiang et al. 1993; Cathala & Paupardin-Tritsch, 1997; Parkis & Berger, 1997). However, the mechanism underlying the modulation of the maximum Ih by these neurotransmitters is unknown. The present study investigated the modulation of Ih by κ-opioid receptors and its underlying intracellular mechanisms in brainstem neurons in the nucleus raphe magnus (NRM), which are critically involved in mediating opioid analgesia (Fields et al. 1991; Pan et al. 1997).
METHODS
All procedures involving the use of animals conformed to the guidelines set by the University of Texas-MD Anderson Cancer Center Animal Care and Use Committee.
Male, neonatal (9–14 days old) Wistar rats were used for brain slice preparations and in vitro recordings. Animals were anaesthetized with halothane and then killed by decapitation. The brain was removed and cut in a vibratome in cold physiological saline at 4 °C to obtain brainstem slices (200 μm thick) containing the nucleus raphe magnus (NRM). A single slice was transferred and submerged in a shallow recording chamber with preheated (35 °C) physiological saline flowing through. The physiological saline contained (mm): NaCl 126, KCl 2.5, NaH2PO4 1.2, MgCl2 1.2, CaCl2 2.4, glucose 11 and NaHCO3 25; saturated with 95 % O2−5 % CO2. Calcium-free external solution was prepared by replacing CaCl2 with an equal concentration of MgCl2. Neonatal rats were used for better visualization of neurons in brain slices with an infrared Nomarski microscope. It has been demonstrated that the physiological and pharmacological properties of neurons from these young rats are indistinguishable from those of adult rats (Pan et al. 1997).
Visualized whole-cell patch-clamp recordings were made from identified neurons with a glass pipette (resistance, 3–5 MΩ) filled with a solution containing (mm): potassium gluconate 126, NaCl 10, MgCl2 1, EGTA 11, Hepes 10, ATP 2 and GTP 0.25; pH was adjusted to 7.3 with KOH. In some experiments, BAPTA (30 mm) or heparin (500 μm) was included in the internal solution with a reduced concentration of potassium gluconate to reach a final osmolarity of 280–290 mosmol l−1. An AxoPatch 1-D amplifier and AxoGraph software (Axon Instruments, Inc.) were used for data acquisition and on-/off-line data analyses. The filter and sampling frequencies for acquisition were 2 kHz. A seal resistance of 2 GΩ or above and an access resistance of 15 MΩ or less were considered acceptable. Series resistance was optimally compensated. All drugs were applied through the perfusing solution.
In all experiments, NRM cells were first classified into either a primary cell type or secondary cell type according to the criteria described in our previous study (Pan et al. 1990). Only primary cells that were hyperpolarized by κ-receptor agonists (Pan et al. 1997) were included in the results of this study. The amplitude of Ih was determined by the difference in the current values at the beginning and the end of a 1 s hyperpolarizing voltage step. Numerical data were statistically analysed using Student's paired t test or ANOVA and are presented as means ±s.e.m. All drugs used were purchased from Research Biochemicals International and Sigma-Aldrich.
RESULTS
Properties of Ih
Visualized whole-cell patch clamp recordings were made from neurons of the nucleus raphe magnus (NRM) in brainstem slices in vitro. All results were obtained from a class of cells (termed primary cells) that express κ-opioid receptors (Pan et al. 1997). In the majority of primary cells under voltage clamp, voltage steps from a holding potential of −60 mV to more negative potentials activated a slowly activating, inward sag current (Ih) (Fig. 1A). In normal extracellular potassium concentration ([K+]o) of 2.5 mm, the average amplitude of the Ih at −120 mV was 18.6 ± 1.3 pA (n = 38). That is in strong contrast to the Ih amplitude in the other cell type (termed secondary cells), which displayed a much larger Ih at the same membrane potential (58 ± 8 pA, n = 18, Fig. 1B).
Figure 1. Properties of Ih in primary cells of the nucleus raphe magnus (NRM).

A, a current trace showing the Ih during a 1 s voltage step (indicated above) under voltage clamp. The holding potential was −60 mV. B, a similar current trace but from a secondary cell for comparison. Note the larger Ih amplitude than the primary cell shown in A. C, a graph of Ih amplitudes against voltage across the range for activation (n = 38). D, the activation curve of Ih tails (n = 13). Tail current amplitudes were normalized to the current amplitude at −130 mV as the maximum and fitted with the Boltzmann equation. The estimated half-maximum voltage (V1/2) was −86.8 ± 0.4 mV. E, a graph showing the average Ih amplitudes in control (○) and in the presence of BaCl2 (200 μm, •) in an extracellular potassium concentration of 6.5 mm ([K+]o) (n = 6). F, a graph showing the average Ih amplitudes in control (○) and in the presence of CsCl (2 mm, •) in 6.5 mm[K+]o (n = 8).
The activation of Ih started at potentials more negative than the holding potential and the Ih amplitude increased at more negative potentials (Fig. 1C). The time course of activation of Ih was typically fitted by a single exponential and the activation rate accelerated as the membrane potential became more negative with a time constant of 248 ± 25 ms at −120 mV (n = 8). The voltage dependence of Ih was further characterized by the activation curve of Ih tails fitted with the Boltzmann equation (Fig. 1D). The maximum tail current was reached at around −120 mV and the estimated half-maximal voltage (V1/2) was −86.8 ± 0.4 mV (n = 13, Fig. 1D). When the [K+]o was increased to 6.5 mm, the Ih amplitude was significantly increased (40.2 ± 8.8 pA at −120 mV, n = 11), suggesting that the Ih channel is permeable to potassium. The presence of BaCl2 (200 μm) did not affect Ih (at −120 mV: control, 41.5 ± 14.1 pA; BaCl2, 44.3 ± 10.8 pA; P > 0.05, n = 6, Fig. 1E), nor did it change the amplitude of the Ih tail at −120 mV (control, 27.4 ± 6.5 pA; BaCl2, 26.7 ± 6.5 pA; P > 0.05, n = 4). However, the presence of CsCl (2 mm) completely blocked Ih in all cells tested (n = 8, Fig. 1F). These properties of Ih, including slow activation, voltage dependence, insensitivity to barium and blockade by caesium, are consistent with the previously reported properties of Ih in various types of neurons from other brain sites (McCormick & Pape, 1990; Uchimura et al. 1990; Khakh & Henderson, 1998; Zhu et al. 1999).
It has been reported that ZD7288 (1–100 μm) selectively blocks Ih in a variety of neurons (Harris & Constanti, 1995; Khakh & Henderson, 1998; Takigawa et al. 1998; Larkman & Kelly, 2001). In the present study, ZD7288 at 25–50 μm completely blocked Ih at all potentials (at −120 mV: control, 19.9 ± 3.9 pA; ZD7288, 1.6 ± 0.6 pA; P < 0.01, n = 16, Fig. 2A and B). Consistent with previous reports, lower concentrations of ZD7288 (< 20 μm) were also effective at blocking the Ih, though the onset of action was significantly slower. These results further confirm the identity of the Ih described here in these brainstem neurons.
Figure 2. ZD7288 blocks Ih.

A, current traces during a voltage step (−60 to −120 mV) in the absence (upper panel) and presence (lower panel) of ZD7288 (25 μm). B, Ih amplitudes at −120 mV from a group of primary cells (n = 16) in control and in the presence of ZD7288. **P < 0.01 vs. control.
κ-Receptor agonist enhances the maximum Ih
We have previously reported that these cells are hyperpolarized by κ-receptor agonists through activation of inwardly rectifying potassium channels, or GIRK channels (Pan et al. 1997). In addition to the GIRK channel-mediated hyperpolarization, it was found in the present study that the κ-receptor agonist U69593 also enhanced the Ih, particularly its maximum amplitude. In cells that displayed measurable Ih (> 5 pA, n = 31), U69593 (300 nm) induced an outward current of 26.3 ± 3.9 pA from the holding potential (−60 mV) with an average reversal potential of −93.8 ± 1.5 mV, suggesting GIRK channel activation. In the same cells (n = 31), U69593 concomitantly increased the Ih amplitude (Fig. 3A and B). The increase in the maximum Ih estimated at −120 mV varied from cell to cell and the average increase was 223 ± 16 % (16.6 ± 1.0 pA in control and 35.2 ± 2.2 pA in U69593, P < 0.01, n = 31). The effect of U69593 was reversible by wash out or by addition of naloxone (1 μm, n = 3, Fig. 3A). The selective κ-receptor antagonist norBNI (100 nm) completely blocked both the U69593-induced hyperpolarization, as shown in our previous study (Pan et al. 1997), and the U69593-induced Ih enhancement in this study (17.6 ± 4.9 pA in norBNI and 17.2 ± 5.6 pA in norBNI + U69593 at −120 mV, n = 5), suggesting a specific κ-receptor-mediated effect. In addition to the amplitude increase, U69593 accelerated the Ih activation rate with an activation time constant of 133 ± 21 ms, a significant effect when compared to 248 ± 25 ms in control (P < 0.01, n = 8). Similar Ih enhancement by U69593 was also observed in higher [K+]o (6.5 mm, Fig. 3B).
Figure 3. κ-opioid receptor agonist U69593 enhances the Ih.

A, current traces during a voltage step to −120 mV in control, in the presence of U69593 (300 nm) and in the presence of U69593 + naloxone (1 μm). Note the increase in Ih in the presence of U69593. B, a graph showing the average Ih amplitudes in control (open symbols) and in U69593 (300 nm, filled symbols) in 2.5 mm (circles, n = 31) and 6.5 mm (squares, n = 5) [K+]o. C, current traces similar to those in A and aligned at the holding potential from another cell showing a U69593-dependent increase in both the Ih tail and GIRK conductance. D, a graph showing the Ih tail amplitudes in control (○) and in the presence of U69593 (•), n = 7. E, activation curves of Ih tails in control (○) and in the presence of U69593 (•), n = 7. The estimated V1/2 was −85.7 mV and −86.9 mV in control and in the presence of U69593, respectively.
The κ-receptor agonist also enhanced the maximum amplitude of the Ih tails across the activation range with an average of 155 ± 14 % increase at −120 mV (control, 26.1 ± 7.4 pA; U69593, 38.5 ± 9.2 pA; P < 0.05, n = 7, Fig. 3C and D). The Ih augmentation by U69593 was voltage dependent being more effective at negative membrane potentials (Fig. 3B and D). Analysis of the Ih activation curves in control and in the presence of U69593 revealed that the κ-receptor agonist produced only a small change in V1/2 that was not statistically significant (control, −85.7 ± 0.4 mV; U69593, −86.9 ± 0.6 mV; P > 0.05, n = 7, Fig. 3E). The small change in V1/2 was probably due to an underestimate (at −120 mV) of the maximum Ih that had increased in the presence of U69593. These results suggest that in addition to membrane hyperpolarization through GIRK channel activation, κ-receptor activation enhances the maximum amplitude of the Ih without a significant change in its voltage dependence in these cells.
Ih enhancement is independent of GIRK channel activation
Since the κ-receptor agonist produced Ih enhancement that was accompanied by GIRK channel activation, it is interesting and necessary to investigate whether the Ih enhancement is a separate effect of the κ-receptor, independent of the GIRK channel activation, or whether its effect on Ih is secondary to GIRK activation. Therefore, the following experiments were performed after pharmacological blockade of either GIRK channels or Ih channels.
Barium has been shown to block GIRK channels in central neurons (Watts et al. 1996; Svoboda & Lupica, 1998; Cathala & Paupardin-Tritsch, 1999). In the presence of 200 μm BaCl2, the outward current induced by U69593 (300 nm) was blocked in all cells tested (control, 58.9 ± 10.1 pA; BaCl2, 2.3 ± 1.1 pA; P < 0.01, n = 10). In the majority of these cells (8/10) after blockade of GIRK channels by BaCl2, U69593 still enhanced the Ih amplitude (at −120 mV: BaCl2, 11.3 ± 3.0 pA; BaCl2+ U69593, 26.3 ± 4.0 pA; P < 0.01, n = 8). U69593 did not affect the Ih in the remaining two cells. The amplitude of Ih tails was also enhanced by U69593 over its entire activation range (196 ± 38 % increase at −120 mV, P < 0.05, n = 9, Fig. 4A) after the U69593-induced GIRK current had been blocked by BaCl2 in the same cells (before BaCl2, 55.8 ± 7.3 pA; after BaCl2, 2.2 ± 0.9 pA; P < 0.01, n = 9, Fig. 4B). These results indicate that κ-receptor activation produces a comparable Ih enhancement after blockade of GIRK channels.
Figure 4. The κ-receptor-mediated Ih enhancement is independent of GIRK channel activation.
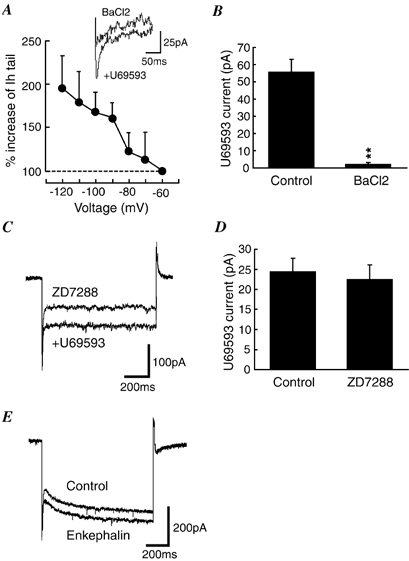
A, a graph showing the percentage increase in Ih tails induced by U69593 (300 nm) in the presence of BaCl2 (200 μm, n = 9). The inset shows current traces of Ih tails at −120 mV in the presence of either BaCl2 or BaCl2+ U69593. B, U69593-induced outward currents in control and in the presence of BaCl2 from the same cells as in A (n = 9). Holding potential was −60 mV. **P < 0.01 vs. control. C, current traces during a voltage step to −120 mV in the presence of either ZD7288 (1 μm) or ZD7288 + U69593 in a brain slice pre-treated with ZD7288 (1 μm). Note the absence of Ih in both conditions. D, U69593 (300 nm)-induced outward currents in normal cells (control, n = 39) and in ZD7288-treated cells (n = 14). E, current traces during a voltage step to −120 mV before (control) and during application of met-enkephalin (10 μm) in a secondary cell. Note the increase in GIRK conductance but not in the Ih.
Next, the κ-receptor-mediated effect was further investigated after blockade of the Ih channel by ZD7288. It was found that although ZD7288 at concentrations of 25–50 μm completely blocked the Ih, it also significantly reduced the U69593-induced outward current by various amounts, making the results inconclusive at those ZD7288 concentrations. Experiments were then conducted in brain slices pre-incubated with 1 μm ZD7288. Subsequent recordings were made with 1 μm ZD7288 in the perfusing solution. In these ZD7288-treated slices, no detectable Ih was observed in any of the cells tested (n = 14). In the presence of U69593 (300 nm), there was still no detectable Ih in these cells (n = 14, Fig. 4C) although U69593 produced a comparable GIRK current in the same cells (ZD7288-treated slices, 22.6 ± 3.5 pA, n = 14; all untreated slices, 24.5 ± 3.2 pA, n = 39; P > 0.05, Fig. 4D).
We have recently shown that nociceptin/orphanin FQ (N/OFQ) hyperpolarizes these NRM primary cells by activating GIRK channels (Pan et al. 2000). In the present study, while activating GIRK channels, N/OFQ (100 nm) did not significantly change the Ih amplitude in the same cells (at −120 mV: control, 15.0 ± 1.3 pA; N/OFQ, 19.1 ± 3.4 pA; P > 0.05, n = 15). In addition, as we reported previously (Pan et al. 1990, 1997), the other NRM cell type, secondary cells, are hyperpolarized by μ-receptor agonists through activation of GIRK channels. In the current study, met-enkephalin (ME, 10 μm) acting on μ-receptors (Pan et al. 1990) caused an outward current of 25 ± 3 pA in secondary cells (n = 18), but it did not alter the basal Ih amplitude in these cells (at −120 mV: control, 58 ± 8 pA; ME, 57 ± 9 pA; P > 0.05, n = 18, Fig. 4E). These findings demonstrate that membrane hyperpolarization by GIRK channel activation does not necessarily lead to an increase in Ih in these neurons under our experimental conditions.
Overall, the above results obtained using barium, ZD7288, N/OFQ and ME suggest that κ-receptor-mediated Ih enhancement is independent of κ-receptor-mediated activation of GIRK channels and may be mediated through a separate signal transduction mechanism coupled to κ-receptors.
Actions of cAMP analogues
Since it has often been reported that cAMP modulates Ih channels, an aim of the present study was to investigate whether cAMP was also involved in modulation of the Ih in these brainstem neurons. The cAMP analogue 8-bromoadenosine 3′,5′-cyclic monophosphate (8-Br-cAMP) (1 mm) caused a small increase in the Ih at sub-maximal potentials, but did not change the maximum current amplitude (at −120 mV in 6.5 mm[K+]o: control, 33.3 ± 6.1 pA; 8-Br-cAMP, 33.2 ± 5.9 pA; P > 0.05, n = 5, Fig. 5A). The V1/2 value estimated from activation curves of the Ih tails was −94.6 mV in control and −89.7 mV in the presence of 8-Br-cAMP at this higher level of [K+]o (P < 0.05, n = 5, Fig. 5B). Thus, the cAMP analogue increased the Ih by a positive shift of 4.9 mV in its voltage dependence. Similar effects were observed with the adenylyl cyclase activator forskolin (10 μm, n = 5, data not shown). These results are in agreement with those previously reported in native Ih channels in other neurons (McCormick & Pape, 1990; Tokimasa & Akasu, 1990; Ingram & Williams, 1994) and similar to those in the cloned HCN1 channel, which shows a small shift in V1/2 by cAMP (2–13 mV)(Kaupp & Seifert, 2001). Nevertheless, with no effect on the maximum Ih amplitude, the action of cAMP is apparently distinct from that of the κ-receptor agonist, which produced a large enhancement of the maximum Ih amplitude.
Figure 5. Cyclic AMP analogue increases the Ih through a positive shift in its voltage dependence.

A, a graph showing the average Ih amplitudes in control (○) and in the presence of 8-Br-cAMP (1 mm, •) in 6.5 mm[K+]o, n = 5. B, activation curves of Ih tails before (control, ○) and during application of 8-Br-cAMP (1 mm, •) in 6.5 mm[K+]o. The V1/2 was −94.6 mV in control and −89.7 mV in the presence of 8-Br-cAMP.
The κ-receptor-mediated enhancement of maximum Ih requires release of intracellular calcium
Since it appeared that cAMP was not involved in the mechanism underlying κ-receptor-mediated Ih enhancement, the focus then moved to the role of intracellular calcium (Cai2+) in the Ih modulation by κ-receptors. Caffeine induces calcium release into cytoplasm from Cai2+ stores (Henzi & MacDermott, 1992). In this study, caffeine (10 mm) caused a significant increase in the Ih amplitude including its maximum estimated at −120 mV in the majority of cells tested (18/24, 75 %, Fig. 6A). Compared to controls, the average increase in Ih amplitude by caffeine at −120 mV was 257 ± 35 % (control, 11.8 ± 1.5 pA; caffeine, 23.6 ± 1.8 pA; P < 0.01, n = 18, Fig. 6B and C). The effect of caffeine was similar to that of U69593 (223 % increase by U69593, Fig. 3B). If the κ-receptor-mediated Ih augmentation also involves the release of Ca2+, caffeine should occlude, at least partially, the κ-receptor-mediated effect on the Ih. This hypothesis was then tested by examining the effect of U69593 in the presence of caffeine. Under this condition, the average Ih amplitude at −120 mV was 27.2 ± 4.0 pA in the presence of caffeine and 35.2 ± 8 pA in the presence of caffeine + U69593 (n = 6). Thus, the Ih-enhancing effect of U69593 was significantly reduced by caffeine (130 ± 25 % increase at −120 mV, P < 0.05, n = 6, compared to 223 ± 16 % increase by U69593 in control, Fig. 6C).
Figure 6. Caffeine and thapsigargin reduce the Ih-enhancing effect of κ-receptors.

A, current traces during a voltage step to −120 mV in control and in the presence of caffeine (10 mm). Note the similar Ih enhancement by caffeine and by U69593 in Fig. 3A. B, a graph showing the average Ih amplitudes in control (○) and in the presence of caffeine (•), n = 18. C, percentage increases of the Ih at −120 mV by caffeine (10 mm) compared to controls (P < 0.01, n = 18) and by U69593 (300 nm) in control (P < 0.01, n = 31), and in the presence of caffeine (*P < 0.05, n = 6, compared to the effect of U69593 in control). D, percentage changes of the Ih at −120 mV by thapsigargin compared to controls (n = 7), and by U69593 in control (as in C) and in the presence of thapsigargin (3 μm, *P < 0.05, compared to the effect of U69593 in control, n = 6). Con, control; Thaps, thapsigargin.
Because caffeine increased the basal Ih amplitude before addition of U65593, it was possible that the reduction of the effect of U69593 in caffeine was simply due to Ih saturation after caffeine plus U69593. To determine whether this was the case, thapsigargin was used to further confirm the role of Cai2+ in the κ-receptor-mediated effect. Thapsigargin has been shown to deplete and prevent refilling of inositol 1,4,5-trisphosphate (IP3)-sensitive calcium stores (Jin et al. 1994; Hauser et al. 1996). In contrast to the effect of caffeine, thapsigargin (3 μm) decreased the Ih amplitude (at −120 mV: control, 17.0 ± 2.6 pA; thapsigargin, 11.1 ± 2.7 pA, n = 7, Fig. 6D). However, similar to the results with caffeine, the effect of U69593 was also significantly attenuated in the presence of thapsigargin (control, 223 ± 6 % increase, n = 31; thapsigargin, 141 ± 23 % increase, n = 6; P < 0.05, Fig. 6D). These results suggest the involvement of Cai2+ release in the mechanism underlying κ-receptor-mediated Ih enhancement.
If the κ-receptor-mediated enhancement of the Ih in these cells requires the release of Cai2+, chelation of Cai2+ released from calcium stores would be expected to block or reduce the κ-receptor-mediated effect. Therefore, the fast-acting Cai2+ chelator BAPTA was used in the following experiments. BAPTA (30 mm) was included in the recording pipette and the cell was held under voltage clamp for at least 5 min before data collection. In this condition, Ih tended to increase (31.7 ± 7.6 pA at −120 mV, n = 13, Fig. 7A). Although the reason for this is not clear, it may indirectly indicate the involvement of Cai2+ in the modulation of Ih. With the BAPTA-filled pipette, U69593 induced only a 124 ± 13 % increase in the Ih amplitude at −120 mV (33.7 ± 8.5 pA, n = 13, Fig. 7B and C), a significant reduction when compared to the effect of U69593 in control conditions (226 ± 16 %, P < 0.01, n = 31, Fig. 7C). While the Ih-enhancing effect of U69593 was significantly reduced by BAPTA, the κ-receptor agonist produced an outward current with an average amplitude of 25.3 ± 2.8 pA (n = 13), comparable to the U69593-induced current in normal conditions (26.3 ± 3.9 pA, P > 0.05, n = 30, Fig. 7D). This suggests that the GIRK channels and their coupling to κ-receptors are not affected by intracellular BAPTA.
Figure 7. BAPTA blocks the Ih-enhancing effect of κ-receptors.

The data were obtained from experiments with BAPTA (30 mm) included in recording pipettes. A, current traces during a voltage step to −120 mV in the absence and presence of U69593 (300 nm). B, a graph showing the Ih amplitudes in the absence (○) and presence of U69593 (•), n = 13. C, percentage increases of the Ih at −120 mV induced by U69593 in normal conditions (as in Fig. 6C and D) and with BAPTA-filled recording pipettes (n = 13). **P < 0.01, compared to the effect of U69593 in normal conditions. D, U69593-induced outward currents in normal conditions (n = 30) and with BAPTA-filled recording pipettes in the same cells as in C (n = 13).
It has been shown that κ-opioid receptors couple to the IP3 pathway (Ueda et al. 1995; Smart & Lambert, 1996). It is possible that the κ-receptor agonist enhances the Ih by inducing Ca2+ release from IP3-sensitive stores through activation of the IP3 pathway. To test that hypothesis, further experiments were carried out using the recording pipette containing heparin (500 μm), which inhibits Ca2+ release from IP3-sensitive stores (Kiselyov et al. 1998; Kapur et al. 2001). Ih was unaffected by the presence of heparin (16.7 ± 2.3 pA at −120 mV, n = 17, Fig. 8A). However, the Ih-enhancing effect of U69593 was blocked by internal application of heparin (120 ± 13 % of control Ih at −120 mV, P > 0.05, n = 7, Fig. 8A and C). The amplitude of Ih at −120 mV was 17.6 ± 3.8 pA in control and 19.0 ± 2.8 pA in the presence of U69593 (300 nm, n = 7). The difference between the effects of U69593 with normal and heparin-containing pipettes was also statistically confirmed (P < 0.01, Fig. 8C). While the Ih augmentation by U6953 was blocked, the U69593-induced GIRK current was not affected by heparin in the same cells (35.7 ± 2.9 pA, n = 7, Fig. 8C). Thus, κ-receptor agonists appear to augment the Ih through Ca2+ release from IP3-sensitive stores.
Figure 8. Heparin and Ca2+-free external solution block the Ih-enhancing effect of κ-receptors.

A, current traces during a voltage step (−60 to −120 mV) in control and in the presence of U69593 (300 nm), with heparin (500 μm) included in the recording pipette. B, similar current traces to those in A, but obtained from another cell in Ca2+-free external solution. C, percentage changes induced by U69593 (300 nm) in the Ih amplitude at −120 mV (□) and in the GIRK currents (▪) under normal conditions (n = 31), with heparin-filled recording pipettes (n = 7), and in Ca2+-free external solution (n = 7). **P < 0.01, compared to the effect of U69593 in normal conditions. Con, control.
Finally, to further examine the dependence of Ca2+ release from intracellular stores and the effect of U69593 on extracellular Ca2+ (Cao2+) in these cells, the κ-receptor-mediated effect was tested in Ca2+-free external solution (replaced by MgCl2). After at least 8 min in this Ca2+-free solution, the Ih amplitude at −120 mV was 20.4 ± 5.2 pA in control and 22.6 ± 6.2 pA in the presence of 300 nm U69593 (n = 7, Fig. 8B). Thus, U69593 was no longer effective at enhancing the maximum Ih amplitude in the absence of Cao2+ (117 ± 16 % of control at −120 mV, P > 0.05, n = 7, Fig. 8C). When removal of Cao2+ eliminated the effect of U69593 on the Ih amplitude (P < 0.01 by comparing its effects in normal and Ca2+-free solutions), the U69593-dependent activation of GIRK current was not significantly changed in the same cells (38.3 ± 8.5 pA, n = 7, Fig. 8C). It is interesting that after cells were reintroduced to standard physiological saline solution with Ca2+, U69593 was able to enhance the maximum Ih amplitude again with an average increase of 214 ± 21 % at −120 mV (control, 15.7 ± 4.8 pA; U69593, 34.0 ± 11.5 pA; n = 3). These findings suggest that the κ-receptor-induced Ca2+ release and augmentation of the Ih are largely dependent on extracellular Ca2+.
DISCUSSION
The present study demonstrates that in addition to GIRK channel activation, κ-opioid receptors enhance the maximum amplitude of Ih in NRM neurons. The current data also show that this κ-receptor-mediated effect can be significantly attenuated or blocked by various compounds that interfere with Ca2+ release, suggesting that this κ-receptor-mediated enhancement of maximum Ih amplitude involves mobilization of intracellular calcium released from calcium stores, particularly IP3-sensitive stores. Thus, the κ-receptor-mediated calcium-dependent modulation of the maximum Ih amplitude may represent an intracellular mechanism for neurotransmitter regulation of the Ih channel and its associated cellular functions in central neurons.
Calcium-dependent modulation of the Ih
The current results provide evidence for the involvement of intracellular calcium released from calcium stores in the κ-receptor-mediated enhancement of maximum Ih amplitude. Besides the large number of studies on cAMP-dependent modulation of Ih channels, other studies have suggested a calcium dependence of Ih channels. In heart cells, an increase in intracellular calcium concentration enhances the Ih amplitude at all membrane potentials and the Ih enhancement is independent of protein kinase or calmodulin activities, indicating a direct regulation of the Ih channel by calcium (Hagiwara & Irisawa, 1989). A calcium-dependent, cAMP-independent potentiation of the Ih has also been reported after activation of muscarinic receptors in hippocampal neurons (Colino & Halliwell, 1993). In thalamocortical neurons, elevation of intracellular calcium concentration initiates the cAMP-mediated Ih enhancement (Luthi & McCormick, 1999), whereas the results from a separate study suggest the lack of a direct regulation of Ih by intracellular calcium in thalamic neurons (Budde et al. 1997). Furthermore, it was recently demonstrated that an increase in calcium concentration in hippocampal mossy fibre terminals enhances the Ih through calcium/calmodulin-sensitive adenylyl cyclase and cAMP (Mellor et al. 2002).
An apparent difference in the modulation of Ih by κ-receptors described here and by cAMP is that κ-receptor agonists augment the Ih by increasing its maximum current amplitude whereas cAMP increases Ih by shifting its voltage dependence to more depolarized potentials. This suggests that cAMP is not involved in the Ih enhancement by κ-receptors. Consistent with this, previously reported modulation of the maximum Ih by various neurotransmitters is independent of the cAMP pathway (Colino & Halliwell, 1993; Jiang et al. 1993; Cathala & Paupardin-Tritsch, 1997; Parkis & Berger, 1997). The current results suggest that intracellular calcium may mediate the effects of these neurotransmitters. It is not clear from this study whether calcium modulates the Ih directly or indirectly through calcium-dependent mechanisms other than the cAMP pathway. In view of the observation that the increase in maximum conductance of Ih by intracellular calcium is independent of protein kinases and calmodulins in cardiac cells (Hagiwara & Irisawa, 1989), it is possible that calcium directly regulates the maximum conductance through a calcium modulation site in Ih channels. The results of the present study may indicate a yet unidentified mechanism in the modulation of Ih channels by intracellular calcium, either directly or indirectly.
Mobilization of intracellular calcium by κ-opioid receptors
Ample biochemical evidence shows that in addition to their negative coupling to the cAMP signal transduction pathway, opioid receptors, including κ-receptors, also couple to and stimulate the phospholipase C pathway, whose activation leads to production of IP3 and calcium release from IP3-sensitive calcium stores (Smart et al. 1994; Ueda et al. 1995; Smart & Lambert, 1996). Consequently, κ and other opioid receptors increase IP3 concentration and mobilize intracellular calcium from IP3-sensitive stores in a calcium influx-dependent or -independent manner in various cell types (Jin et al. 1994; Tang et al. 1994; Fields et al. 1995; Allouche et al. 1996; Gurwell et al. 1996). These reports and the current results obtained using caffeine, thapsigargin, BAPTA and heparin strongly suggest that κ-receptors enhance the maximum Ih through mobilization of intracellular calcium, involving calcium release primarily from IP3-sensitive stores. Consistent with this notion, inositol phospholipids have been implicated in the calcium-dependent, cAMP-independent increase of the Ih by muscarinic receptors (Colino & Halliwell, 1993; Allouche et al. 1996).
In addition, the current data obtained in Ca2+-free solution indicate that the κ-receptor-mediated enhancement of Ih is largely dependent on external Ca2+ concentration or on Ca2+ influx. Similar dependence on Ca2+ influx has been reported in muscarinic receptor-mediated Ca2+ release from IP3-sensitive stores (Allouche et al. 1996). It is intriguing to consider possible mechanisms for the dependence of the κ-receptor-mediated effect on Ca2+ influx. Under the present experimental conditions, it is unlikely that mobilization of intracellular Ca2+ by κ-receptors is due to Ca2+ influx through voltage-sensitive or ligand-gated calcium channels. It has recently been demonstrated that depletion of Ca2+ stores by agonist-stimulated IP3 production activates store-operated channels (SOCs) through interaction between IP3-bound IP3 receptors and SOCs, causing Ca2+ influx (Kiselyov et al. 1998). In view of the current evidence for κ-receptor-stimulated IP3 production and Ca2+ release from IP3-sensitive stores to increase the Ih, it appears likely that such a mechanism mediated by IP3 receptors and SOCs is involved in the κ-receptor-mediated augmentation of the Ih, and that Ca2+ influx through SOCs is necessary for proper Ca2+ release and refill of Ca2+ stores induced by κ-receptors. Despite extensive biochemical data demonstrating opioid receptor-stimulated mobilization of intracellular calcium, evidence has been lacking regarding the physiological function of this signalling pathway for opioid receptors. The current findings demonstrate an example of the roles of this opioid receptor coupling in regulating neuronal activity.
Modulation of maximum Ih and activation of GIRK channels
There have been several reports of receptor-mediated inhibition of the maximum Ih and concomitant membrane hyperpolarization via activation of GIRK channels in the same cell. Although some attributed the reduced Ih to an indirect effect secondary to the GIRK channel activation in substantia nigra neurons (Watts et al. 1996; Cathala & Paupardin-Tritsch, 1999), other studies have shown that inhibition of the maximum Ih is independent of GIRK channel activation and is mediated by a separate, unknown mechanism (Rainnie et al. 1994; Svoboda & Lupica, 1998). In the present study, several lines of evidence suggest that κ-receptor-mediated enhancement of the maximum Ih is a direct effect independent of GIRK channel activation. Firstly, after blockade of GIRK channels by barium, the κ-receptor agonist still increased the maximum Ih. Secondly, after the Ih was blocked by ZD7288, κ-receptor activation induced a similar outward current without increasing the Ih, indicating that these two actions of κ-receptors are separate and independent of each other in these cells. Finally, N/OFQ and μ-receptor agonists activated GIRK channels in primary and secondary cells, respectively, but neither of them affected the Ih, making it unlikely that GIRK channel activation leads to the increase in Ih in our recording conditions.
Physiological implications
Given the general role of Ih in control of neuronal excitability and the important modulating role of these NRM neurons in opioid analgesia (Fields et al. 1991; Pan et al. 1997), the κ-receptor-mediated enhancement of the maximum Ih in these cells could have physiological significance in κ-opioid receptor-mediated acute and chronic actions. At the single cell level, it is proposed that concomitant enhancement of the Ih by κ-receptors could normally serve as a stopping mechanism to prevent extreme or over-inhibition due to GIRK channel activation by κ-receptors. It is also possible that in chronic conditions with elevated intracellular calcium levels, the activity of these cells driven by other excitatory inputs may increase due to enhanced Ih. At the system level, it is interesting to note that brain levels of intracellular calcium are significantly higher in morphine-tolerant rats (Welch & Bass, 1995). Moreover, κ-receptor agonists attenuate morphine tolerance and potentiate morphine analgesia in opioid-tolerant but not opioid-naive animals (Bhargava, 1994; Pan, 1998). Given the analgesic action of these brainstem cells on spinal pain transmission pathways (Pan et al. 1997), the κ-receptor-mediated and intracellular calcium-promoted increase in the firing activity of these cells in chronic opioid conditions may be one of the mechanisms for the anti-tolerance action of κ-receptors.
Acknowledgments
The author would like to thank Dr Howard Fields for his support at the preliminary stage of this study. This work was supported by a grant from the National Institute on Drug Abuse, NIH.
REFERENCES
- Allouche S, Polastron J, Jauzac P. The delta-opioid receptor regulates activity of ryanodine receptors in the human neuroblastoma cell line SK-N-BE. J Neurochem. 1996;67:2461–2470. doi: 10.1046/j.1471-4159.1996.67062461.x. [DOI] [PubMed] [Google Scholar]
- Banks MI, Pearce RA, Smith PH. Hyperpolarization-activated cation current (Ih) in neurons of the medial nucleus of the trapezoid body: voltage-clamp analysis and enhancement by norepinephrine and cAMP suggest a modulatory mechanism in the auditory brain stem. J Neurophysiol. 1993;70:1420–1432. doi: 10.1152/jn.1993.70.4.1420. [DOI] [PubMed] [Google Scholar]
- Beaumont V, Zucker RS. Enhancement of synaptic transmission by cyclic AMP modulation of presynaptic Ih channels. Nat Neurosci. 2000;3:133–141. doi: 10.1038/72072. [DOI] [PubMed] [Google Scholar]
- Bhargava HN. Diversity of agents that modify opioid tolerance, physical dependence, abstinence syndrome and self-administrative behavior. Pharmacol Rev. 1994;46:293–324. [PubMed] [Google Scholar]
- Bobker DH, Williams JT. Serotonin augments the cationic current Ih in central neurons. Neuron. 1989;2:1535–1540. doi: 10.1016/0896-6273(89)90041-x. [DOI] [PubMed] [Google Scholar]
- Brown HF, DiFrancesco D, Noble SJ. How does adrenaline accelerate the heart? Nature. 1979;280:235–236. doi: 10.1038/280235a0. [DOI] [PubMed] [Google Scholar]
- Cathala L, Paupardin-Tritsch D. Neurotensin inhibition of the hyperpolarization-activated cation current (Ih) in the rat substantia nigra pars compacta implicates the protein kinase C pathway. J Physiol. 1997;503:87–97. doi: 10.1111/j.1469-7793.1997.087bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathala L, Paupardin-Tritsch D. Effect of catecholamines on the hyperpolarization-activated cationic Ih and the inwardly rectifying potassium I(Kir) currents in the rat substantia nigra pars compacta. Eur J Neurosci. 1999;11:398–406. doi: 10.1046/j.1460-9568.1999.00452.x. [DOI] [PubMed] [Google Scholar]
- Colino A, Halliwell JV. Carbachol potentiates Q current and activates a calcium-dependent non-specific conductance in rat hippocampus in vitro. Eur J Neurosci. 1993;5:1198–1209. doi: 10.1111/j.1460-9568.1993.tb00974.x. [DOI] [PubMed] [Google Scholar]
- Difrancesco D. A new interpretation of the pace-maker current in calf Purkinje fibres. J Physiol. 1981;314:359–376. doi: 10.1113/jphysiol.1981.sp013713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson KR, Ronnekleiv OK, Kelly MJ. Electrophysiology of guinea-pig supraoptic neurones: role of a hyperpolarization-activated cation current in phasic firing. J Physiol. 1993;460:407–425. doi: 10.1113/jphysiol.1993.sp019478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields A, Gafni M, Oron Y, Sarne Y. Multiple effects of opiates on intracellular calcium level and on calcium uptake in three neuronal cell lines. Brain Res. 1995;687:94–102. doi: 10.1016/0006-8993(95)00475-6. [DOI] [PubMed] [Google Scholar]
- Fields H, Heinricher M, Mason P. Neurotransmitters in nociceptive modulatory circuits. Ann Rev Neurosci. 1991;14:219–245. doi: 10.1146/annurev.ne.14.030191.001251. [DOI] [PubMed] [Google Scholar]
- Gauss R, Seifert R, Kaupp UB. Molecular identification of a hyperpolarization-activated channel in sea urchin sperm. Nature. 1998;393:583–587. doi: 10.1038/31248. [DOI] [PubMed] [Google Scholar]
- Gurwell JA, Duncan MJ, Maderspach K, Stiene-Martin A, Elde RP, Hauser KF. kappa-opioid receptor expression defines a phenotypically distinct subpopulation of astroglia: relationship to Ca2+ mobilization, development, and the antiproliferative effect of opioids. Brain Res. 1996;737:175–187. doi: 10.1016/0006-8993(96)00728-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara N, Irisawa H. Modulation by intracellular Ca2+ of the hyperpolarization-activated inward current in rabbit single sino-atrial node cells. J Physiol. 1989;409:121–141. doi: 10.1113/jphysiol.1989.sp017488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell JV, Adams PR. Voltage-clamp analysis of muscarinic excitation in hippocampal neurons. Brain Res. 1982;250:71–92. doi: 10.1016/0006-8993(82)90954-4. [DOI] [PubMed] [Google Scholar]
- Harris NC, Constanti A. Mechanism of block by ZD 7288 of the hyperpolarization-activated inward rectifying current in guinea pig substantia nigra neurons in vitro. J Neurophysiol. 1995;74:2366–2378. doi: 10.1152/jn.1995.74.6.2366. [DOI] [PubMed] [Google Scholar]
- Hauser KF, Stiene-Martin A, Mattson MP, Elde RP, Ryan SE, Godleske CC. mu-opioid receptor-induced Ca2+ mobilization and astroglial development: morphine inhibits DNA synthesis and stimulates cellular hypertrophy through a Ca(2+)-dependent mechanism. Brain Res. 1996;720:191–203. doi: 10.1016/0006-8993(96)00103-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzi V, MacDermott AB. Characteristics and function of Ca(2+)- and inositol 1,4,5-trisphosphate-releasable stores of Ca2+ in neurons. Neuroscience. 1992;46:251–273. doi: 10.1016/0306-4522(92)90049-8. [DOI] [PubMed] [Google Scholar]
- Ingram SL, Williams JT. Opioid inhibition of Ih via adenylyl cyclase. Neuron. 1994;13:179–186. doi: 10.1016/0896-6273(94)90468-5. [DOI] [PubMed] [Google Scholar]
- Jiang ZG, Pessia M, North RA. Dopamine and baclofen inhibit the hyperpolarization-activated cation current in rat ventral tegmental neurones. J Physiol. 1993;462:753–764. doi: 10.1113/jphysiol.1993.sp019580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Lee NM, Loh HH, Thayer SA. Opioids mobilize calcium from inositol 1,4,5-trisphosphate-sensitive stores in NG108–15 cells. J Neurosci. 1994;14:1920–1929. doi: 10.1523/JNEUROSCI.14-04-01920.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur A, Yeckel M, Johnston D. Hippocampal mossy fibre activity evokes Ca2+ release in CA3 pyramidal neurons via a metabotropic glutamate receptor pathway. Neuroscience. 2001;107:59–69. doi: 10.1016/s0306-4522(01)00293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupp UB, Seifert R. Molecular diversity of pacemaker ion channels. Annu Rev Physiol. 2001;63:235–257. doi: 10.1146/annurev.physiol.63.1.235. [DOI] [PubMed] [Google Scholar]
- Khakh BS, Henderson G. Hyperpolarization-activated cationic currents (Ih) in neurones of the trigeminal mesencephalic nucleus of the rat. J Physiol. 1998;510:695–704. doi: 10.1111/j.1469-7793.1998.00695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselyov K, Xu X, Mozhayeva G, Kuo T, Pessah I, Mignery G, Zhu X, Birnbaumer L, Muallem S. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature. 1998;396:478–482. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- Larkman PM, Kelly JS. Modulation of the hyperpolarisation-activated current, Ih, in rat facial motoneurones in vitro by ZD-7288. Neuropharmacology. 2001;40:1058–1072. doi: 10.1016/s0028-3908(01)00024-7. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M. A family of hyperpolarization-activated mammalian cation channels. Nature. 1998;393:587–591. doi: 10.1038/31255. [DOI] [PubMed] [Google Scholar]
- Lupica CR, Bell JA, Hoffman AF, Watson PL. Contribution of the hyperpolarization-activated current (I(h)) to membrane potential and GABA release in hippocampal interneurons. J Neurophysiol. 2001;86:261–268. doi: 10.1152/jn.2001.86.1.261. [DOI] [PubMed] [Google Scholar]
- Luthi A, McCormick DA. Modulation of a pacemaker current through Ca(2+)-induced stimulation of cAMP production. Nat Neurosci. 1999;2:634–641. doi: 10.1038/10189. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Pape HC. Noradrenergic and serotonergic modulation of a hyperpolarization-activated cation current in thalamic relay neurones. J Physiol. 1990;431:319–342. doi: 10.1113/jphysiol.1990.sp018332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor J, Nicoll RA, Schmitz D. Mediation of hippocampal mossy fibre long-term potentiation by presynaptic Ih channels. Science. 2002;295:143–147. doi: 10.1126/science.1064285. [DOI] [PubMed] [Google Scholar]
- Pan ZZ. mu-opposing actions of the kappa-opioid receptor. Trends Pharmacol Sci. 1998;19:94–98. doi: 10.1016/s0165-6147(98)01169-9. [DOI] [PubMed] [Google Scholar]
- Pan ZZ, Hirakawa N, Fields H. A cellular mechanism for the bidirectional pain-modulating actions of orphanin FQ/nociceptin. Neuron. 2000;26:515–522. doi: 10.1016/s0896-6273(00)81183-6. [DOI] [PubMed] [Google Scholar]
- Pan ZZ, Tershner S, Fields H. Cellular mechanism for anti-analgesic action of agonists of the κ-opioid receptor. Nature. 1997;389:382–385. doi: 10.1038/38730. [DOI] [PubMed] [Google Scholar]
- Pan ZZ, Williams J, Osborne P. Opioid actions on single raphe magnus neurons from rat and guinea pig in vitro. J Physiol. 1990;427:519–532. doi: 10.1113/jphysiol.1990.sp018185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC. Adenosine promotes burst activity in guinea-pig geniculocortical neurones through two different ionic mechanisms. J Physiol. 1992;447:729–753. doi: 10.1113/jphysiol.1992.sp019026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC, McCormick DA. Noradrenaline and serotonin selectively modulate thalamic burst firing by enhancing a hyperpolarization-activated cation current. Nature. 1989;340:715–718. doi: 10.1038/340715a0. [DOI] [PubMed] [Google Scholar]
- Parkis MA, Berger AJ. Clonidine reduces hyperpolarization-activated inward current (Ih) in rat hypoglossal motoneurons. Brain Res. 1997;769:108–118. doi: 10.1016/s0006-8993(97)00677-x. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF. Protein kinase A-independent modulation of ion channels in the brain by cyclic AMP. Proc Natl Acad Sci U S A. 1995;92:11716–11720. doi: 10.1073/pnas.92.25.11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainnie DG, Grunze HC, McCarley RW, Greene RW. Adenosine inhibition of mesopontine cholinergic neurons: implications for EEG arousal. Science. 1994;263:689–692. doi: 10.1126/science.8303279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitow F, Konishi S. Excitability increase induced by beta-adrenergic receptor-mediated activation of hyperpolarization-activated cation channels in rat cerebellar basket cells. J Neurophysiol. 2000;84:2026–2034. doi: 10.1152/jn.2000.84.4.2026. [DOI] [PubMed] [Google Scholar]
- Santoro B, Liu DT, Yao H, Bartsch D, Kandel ER, Siegelbaum SA, Tibbs GR. Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell. 1998;93:717–729. doi: 10.1016/s0092-8674(00)81434-8. [DOI] [PubMed] [Google Scholar]
- Smart D, Lambert DG. The stimulatory effects of opioids and their possible role in the development of tolerance. Trends Pharmacol Sci. 1996;17:264–269. doi: 10.1016/0165-6147(96)10023-7. [DOI] [PubMed] [Google Scholar]
- Smart D, Smith G, Lambert DG. mu-opioid receptor stimulation of inositol (1,4,5)trisphosphate formation via a pertussis toxin-sensitive G protein. J Neurochem. 1994;62:1009–1014. doi: 10.1046/j.1471-4159.1994.62031009.x. [DOI] [PubMed] [Google Scholar]
- Southan AP, Morris NP, Stephens GJ, Robertson B. Hyperpolarization-activated currents in presynaptic terminals of mouse cerebellar basket cells. J Physiol. 2000;526:91–97. doi: 10.1111/j.1469-7793.2000.t01-1-00091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Svoboda KR, Lupica CR. Opioid inhibition of hippocampal interneurons via modulation of potassium and hyperpolarization-activated cation (Ih) currents. J Neurosci. 1998;18:7084–7098. doi: 10.1523/JNEUROSCI.18-18-07084.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takigawa T, Alzheimer C, Quasthoff S, Grafe P. A special blocker reveals the presence and function of the hyperpolarization-activated cation current IH in peripheral mammalian nerve fibres. Neuroscience. 1998;82:631–634. doi: 10.1016/s0306-4522(97)00383-7. [DOI] [PubMed] [Google Scholar]
- Tang T, Kiang JG, Cox BM. Opioids acting through delta receptors elicit a transient increase in the intracellular free calcium concentration in dorsal root ganglion-neuroblastoma hybrid ND8–47 cells. J Pharmacol Exp Ther. 1994;270:40–46. [PubMed] [Google Scholar]
- Tokimasa T, Akasu T. Cyclic AMP regulates an inward rectifying sodium-potassium current in dissociated bull-frog sympathetic neurones. J Physiol. 1990;420:409–429. doi: 10.1113/jphysiol.1990.sp017920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchimura N, Cherubini E, North RA. Cation current activated by hyperpolarization in a subset of rat nucleus accumbens neurons. J Neurophysiol. 1990;64:1847–1850. doi: 10.1152/jn.1990.64.6.1847. [DOI] [PubMed] [Google Scholar]
- Ueda H, Miyamae T, Fukushima N, Takeshima H, Fukuda K, Sasaki Y, Misu Y. Opioid mu- and kappa-receptor mediate phospholipase C activation through Gi1 in Xenopus oocytes. Brain Res Mol Brain Res. 1995;32:166–170. doi: 10.1016/0169-328x(95)00077-6. [DOI] [PubMed] [Google Scholar]
- vonKrosigk M, Bal T, McCormick DA. Cellular mechanisms of a synchronized oscillation in the thalamus. Science. 1993;261:361–364. doi: 10.1126/science.8392750. [DOI] [PubMed] [Google Scholar]
- Watts AE, Williams JT, Henderson G. Baclofen inhibition of the hyperpolarization-activated cation current, Ih, in rat substantia nigra zona compacta neurons may be secondary to potassium current activation. J Neurophysiol. 1996;76:2262–2270. doi: 10.1152/jn.1996.76.4.2262. [DOI] [PubMed] [Google Scholar]
- Welch SP, Bass PP. Modulation of free intracellular calcium levels [(Ca2+)i] in brain and spinal cord of morphine-tolerant rats and mice. Pharmacol Biochem Behav. 1995;51:57–63. doi: 10.1016/0091-3057(94)00356-n. [DOI] [PubMed] [Google Scholar]
- Williams SR, Christensen SR, Stuart GJ, Hausser M. Membrane potential bistability is controlled by the hyperpolarization-activated current IH in rat cerebellar Purkinje neurons in vitro. J Physiol. 2002;539:469–483. doi: 10.1113/jphysiol.2001.013136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ, Uhlrich DJ, Lytton WW. Properties of a hyperpolarization-activated cation current in interneurons in the rat lateral geniculate nucleus. Neuroscience. 1999;92:445–457. doi: 10.1016/s0306-4522(98)00759-3. [DOI] [PubMed] [Google Scholar]