

Abstract
Carotid body glomus cells release transmitters in response to hypoxia due to the increase of excitability resulting from inhibition of O2 -regulated K+ channels. However, the mechanisms involved in the detection of changes of O2 tension are unknown. We have studied the interaction between glomus cell O2 sensitivity and inhibition of the mitochondrial electron transport chain (ETC) in a carotid body thin slice preparation in which catecholamine release from intact single glomus cells can be monitored by amperometry. Inhibition of the mitochondrial ETC at proximal and distal complexes induces external Ca2+-dependent catecholamine secretion. At saturating concentration of the ETC inhibitors, the cellular response to hypoxia is maintained. However, rotenone, a complex I blocker, selectively occludes the responsiveness to hypoxia of glomus cells in a dose-dependent manner. The effect of rotenone is mimicked by 1-methyl-4-phenylpyridinium ion (MPP+), an agent that binds to the same site as rotenone, but not by complex I inhibitors acting on different sites. In addition, the effect of rotenone is not prevented by incubation of the cells with succinate, a substrate of complex II. These data strongly suggest that sensitivity to hypoxia of carotid body glomus cells is not linked in a simple way to mitochondrial electron flow and that a rotenone (and MPP+)-sensitive molecule critically participates in acute oxygen sensing in the carotid body.
Glomus cells are the major O2 -responsive elements in the carotid body. These are excitable neurosecretory cells that release transmitters in response to hypoxia to activate afferent sensory nerve fibres conveying the information to the respiratory centre (for recent review, see López-Barneo et al. 2001). It has been demonstrated that glomus cells sense O2 tension (PO2) through regulation of membrane ion channels and several types of K+ channels whose activity is inhibited by low PO2 have been described (López-Barneo et al. 1988; Delpiano & Hescheler, 1989; Peers, 1990; Stea & Nurse, 1991; Buckler, 1997). Reduction of K+ conductance by hypoxia causes cell depolarization and an increase of action potential firing frequency (López-López et al. 1989; Buckler & Vaughan-Jones, 1994; Wyatt & Peers, 1995; Montoro et al. 1996; Pérez-García et al. 2000), leading to extracellular Ca2+ influx through plasmalemmal voltage-gated channels (López-Barneo et al. 1993; Buckler & Vaughan-Jones, 1994; Ureña et al. 1994). Quantal catecholamine release induced by hypoxia in single glomus cells is absolutely dependent on extracellular Ca2+ influx (Ureña et al. 1994; Carpenter et al. 2000) and is mimicked by some K+-channel blockers (Pardal et al. 2000).
Despite the progress in the understanding of glomus cell electrophysiology and responsiveness to hypoxia, the molecular nature of the O2 sensor remains unknown. Because modulation of some K+ channels by PO2 is maintained in excised patches (Ganfornina & López-Barneo, 1991; Riesco-Fagundo et al. 2001) it was postulated that O2 sensing in glomus cells depends on membrane-delimited mechanisms (see López-Barneo, 1994; Lewis et al. 2002). On the other hand, several investigators have traditionally considered mitochondria as the site for glomus cell O2 sensing because, similarly to hypoxia, inhibitors of the electron transport chain (ETC) or mitochondrial uncouplers increase the afferent activity of the sinus nerve (see, for instance, Mills & Jöbsis, 1972; Mulligan et al. 1981). It has also been reported that cyanide and anoxia release Ca2+ from mitochondria in isolated rabbit glomus cells (Biscoe & Duchen, 1990; Duchen & Biscoe, 1992) and that mitochondrial uncouplers raise cytosolic Ca2+ and reduce background K+ permeability in rat carotid body cells (Buckler & Vaughan-Jones, 1998). Besides in the carotid body, mitochondria have also been postulated to participate in O2 sensing in other acutely responding systems, such as pulmonary vascular smooth muscle (Archer et al. 1993; Leach et al. 2001; Waypa et al. 2001) or chromaffin cells (Mojet et al. 1997; Inoue et al. 2002).
A major limitation for the study of the carotid body O2 sensors is the difficulty in obtaining a preparation with reproducible sensitivity to hypoxia, as this is easily altered by dispersion of the cells (see López-Barneo et al. 1998). We have recently developed the carotid body thin slice preparation in which the response of glomus cells to low PO2 can be studied in almost optimal physiological conditions (Pardal et al. 2000; Pardal & López-Barneo, 2002). Taking advantage of this technique we designed the present research to investigate whether sensitivity of intact glomus cells to hypoxia is altered by mitochondrial dysfunction. We report here that, as occurs with hypoxia, mitochondrial ETC inhibitors evoke an extracellular Ca2+-dependent secretory response from glomus cells. Sensitivity to lowering PO2 is not altered by blockade of mitochondrial electron flow, however responsiveness to hypoxia is selectively abolished by rotenone and 1-methyl-4-phenylpyridinium ion (MPP+). Thus the data suggest that a rotenone (and MPP+)-binding protein is part of the O2 sensor. A preliminary account of part of this work has previously been published (Pardal & López-Barneo, 2000).
METHODS
Preparation of carotid body thin slices
Wistar rats of ages between postnatal days 10 and 30 were deeply anaesthetized by injection of 100 mg kg−1 pentobarbital. Carotid bodies were dissected and placed on ice-cooled and O2-saturated modified Tyrode solution (mm: 148 NaCl, 2 KCl, 3 MgCl2, 10 Hepes, 10 glucose, pH 7.4). Slices were taken after inclusion of the carotid bodies in agarose as described in Pardal et al. (2000). Slices 150 μm thick were placed on 35 mm Petri dishes with culture medium and maintained at 37 °C in a 5 % CO2 incubator for 24–96 h before use. All experiments were performed according to the institutional animal care committee guidelines.
Experimental setup and measurement of secretion from single cells
For the experiments, a slice was transferred to the chamber (∼0.2 ml volume) placed on the stage of an upright microscope (Axioscope, Zeiss, Göttingen, Germany) and continuously perfused by gravity (flow 1–2 ml min−1) with a solution containing (mm): 117 NaCl, 4.5 KCl, 23 NaHCO3, 1 MgCl2, 2.5 CaCl2, 5 glucose and 5 sucrose. The ‘normoxic’ solution was bubbled with a gas mixture of 5 % CO2, 20 % O2 and 75 % N2 (PO2≈ 150 mmHg). The ‘hypoxic’ solution was bubbled with 5 % CO2 and 95 % N2 to reach a PO2 in the chamber of ∼20 mmHg. To attain lower PO2 values in the chamber, the use of oxygen scavengers is required. As these agents interfere with the amperometric measurements they were not used in our study. Equilibration of solutions in the chamber, determined with an O2 electrode, required ∼40 s. In the high K+ solutions, KCl replaced NaCl equimolarly. When external solution with 5 mm sodium succinate or methylsuccinate was used, the required amount of NaCl was removed. The pharmacological agents used were dissolved in distilled water, DMSO or ethanol as required. From these stock solutions they were added to the external solution at the desired concentration. At the concentrations used (lower than 1/1000 dilution), the vehicles (DMSO or ethanol) did not have any effect on glomus cell catecholamine secretion. Osmolality of solutions was ∼300 mosmol kg−1 and pH was 7.4. All the experiments were carried out at a temperature in the chamber of ∼36 °C. Secretory events were recorded with a 10 μm carbon-fibre electrode positioned near a cell under visual control (Ureña et al. 1994; Pardal et al. 2000). Amperometric currents were recorded with an EPC-8 patch-clamp amplifier (HEKA Electronics, Lambrecht/Pfaltz, Germany), filtered at 100 Hz and digitized at 250 Hz before storage on computer. Data acquisition and analysis were done with an ITC-16 interface (Instrutech Corporation, NY, USA) and PULSE/PULSEFIT software (HEKA Electronics). Samples were statistically compared with a Student's t test and their differences were considered to be significant if P < 0.05. Unless otherwise noted, the data are expressed as means ±s.e.m.
RESULTS
Electron transport chain inhibitors induce external Ca2+-dependent secretion from glomus cells
The secretory response to hypoxia (PO2≈ 20 mmHg) of glomus cells in carotid body slices is shown in Fig. 1A. As described before (Pardal et al. 2000; Pardal & López-Barneo, 2002), low PO2 induced spike-like quantal events corresponding to catecholamine release from individual vesicles. The cumulative secretion signal (lower trace in Fig. 1A) is a value of electric charge obtained by the sum of the time integral of successive amperometric events, which is proportional to the number of catecholamine molecules oxidized. To estimate the magnitude of the cell secretory responses to hypoxia or any other stimulus, we used the secretion rate (femtocoulombs (fC) min−1) calculated as the amount of charge transferred to the recording electrode during the 60 s after the solutions were equilibrated in the recording chamber. In our initial set of experiments we investigated if, as occurs with hypoxia, blockade of mitochondrial ETC also induced secretion from glomus cells. We studied the effect of inhibition at complex I with rotenone, at complex II with thenoyltrifluoroacetone (TTFA), at complex III with myxothiazol or antimycin A (respectively proximal and distal inhibitors of this complex) and at complex IV with cyanide. These agents were used in a broad range of concentrations, however as our experimental protocol required the application of saturating concentrations of the drugs, the lowest concentrations used were at least 5 to 10 times the reported K50 values (see Degli Esposti, 1998; Vaux et al. 2001). All the ETC inhibitors induced an exocytotic response from glomus cells in 1–3 min (Fig. 1B-F; see also Fig. 3). The difference in the secretagogue potency of applied ETC inhibitors was not very marked as they were used at concentrations above saturation. The fact that mitochondrial inhibition did not induce rapid cellular degranulation and preserved the responses to hypoxia or high potassium, indicates that ETC inhibitors did not produce major non-specific pharmacological alterations of the cells. The mean areas of individual quantal events (in fC, mean ±s.d.) triggered by the ETC inhibitors (rotenone: 40 ± 18, n = 245 spikes in 5 cells; myxothiazol: 42 ± 30, n = 102 spikes in 7 cells; antimycin A: 40 ± 22, n = 132 spikes in 5 cells; cyanide: 48 ± 33, n = 116 spikes in 7 cells) were not significantly different (P > 0.1) from the average values estimated with events evoked by hypoxia (43 ± 26, n = 576 spikes in 14 cells). Therefore, hypoxia and the ETC inhibitors appeared to induce the release of a common type of catecholaminergic vesicle. Secretion evoked by all the ETC inhibitors was completely abolished by blockade of membrane Ca2+ channels with Cd2+ (Fig. 1B-F). Only the secretory response induced by concentrations of cyanide in the millimolar range was partially maintained in the presence of 0.3 mm extracellular Cd2+, thus suggesting Ca2+ release from intracellular stores. These data indicate that, as described in cells exposed to hypoxia (Ureña et al. 1994; Pardal et al. 2000; Carpenter et al. 2000), activation of carotid body glomus cells by mitochondrial ETC inhibitors largely depends on extracellular Ca2+ influx through channels of the plasma membrane.
Figure 1. Secretory responses of glomus cells to hypoxia and to the inhibition of the mitochondrial electron transport.
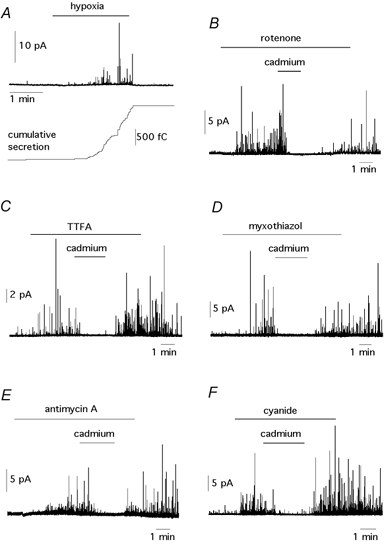
A, top, amperometric signal showing catecholamine release from a glomus cell exposed to low PO2 (≈ 20 mmHg). Each spike represents an exocytotic event; bottom, cumulative secretion signal (in femtocoulombs, fC) resulting from the time integral of the amperometric recording. B–F, catecholamine release induced by exposure to several electron transport inhibitors. The concentrations were: rotenone (5 μm), TTFA (0.3 μm), myxothiazol (1 μg ml−1), antimycin A (1 μg ml−1) and cyanide (100 μm). In all cases secretion was reversibly abolished by external application of 0.3 mm Cd2+.
Figure 3. Average secretion rate measured in cells in various experimental conditions.
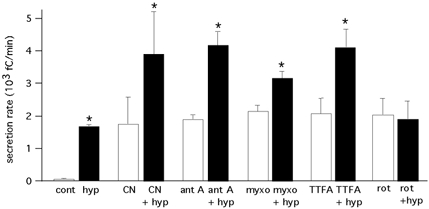
Secretion rate on the ordinate is expressed in fC min−1 (mean ±s.e.m.). Experimental conditions from left to right in the figure: control (cont; PO2≈ 150 mmHg; 75 ± 15 fC min−1; n = 17 cells) and hypoxia (PO2≈ 20 mmHg; 1710 ± 65 fC min−1; n = 17 cells); cyanide (CN; 100 μm; 1771 ± 842 fC min−1; n = 4 cells) and cyanide plus hypoxia (CN + hyp; 3932 ± 1339 fC min−1; n = 4 cells); antimycin A (ant A; 0.1–1 μg ml−1; 1910 ± 151 fC min−1; n = 13 cells) and antimycin A plus hypoxia (ant + hyp; 4201 ± 421 fC min−1; n = 7 cells); myxothiazol (myxo; 0.1–1 μg ml−1; 2167 ± 199 fC min−1; n = 6 cells) and myxothiazol plus hypoxia (myxo + hyp; 3188 ± 240 fC min−1; n = 6 cells); TTFA (0.1–0.3 μm; 2093 ± 488 fC min−1; n = 5 cells) andTTFA plus hypoxia (TTFA + hyp; 4134 ± 587 fC min−1; n = 5 cells); rotenone (rot; 0.1–5 μm; 2058 ± 550 fC min−1; n = 14 cells) and rotenone plus hypoxia (rot + hyp; 1915 ± 552 fC min−1; n = 12 cells). Asterisks indicate a statistically significant difference (P < 0.05) between each pair of samples.
Responsiveness of glomus cells to hypoxia is occluded by rotenone but unaffected by other electron transport chain inhibitors
The interaction between hypoxia and the mitochondrial electron flow was studied in cells exposed to low PO2 before and during application of ETC inhibitors acting at either proximal or distal mitochondrial complexes. The rationale behind these experiments was that if hypoxia exerts its effect through alteration of the mitochondrial electron flow, preincubation with saturating concentrations of ETC blockers would prevent any further effect of low PO2. In contrast, the effects of hypoxia and ETC inhibition would be additive, at least partially, if they were acting through separate pathways. Fig. 2A-D illustrates that when O2-responsive cells were treated with cyanide, antimycin A, myxothiazol or TTFA, the concomitant exposure to low PO2 elicited further increase in the secretory activity. In each case the amperometric recordings are shown on the left panel of the figure. The right panels show on an expanded time base, the cumulative secretion signals recorded during exposures to hypoxia (marked with an asterisk in the left panels) in the presence of the different ETC inhibitors. The rates of secretion immediately before hypoxia are illustrated diagrammatically by the slopes of the cumulative secretion signals (straight lines). The amperometric recordings show that responsiveness to hypoxia was preserved in cells treated with ETC inhibitors, even when a marked exocytotic activity was maintained after removal of the drugs. Similarly, the cumulative secretion traces clearly illustrate that in the presence of ETC inhibitors, hypoxia induced a reversible increase in the slope of the signals. An exception among the mitochondrial inhibitors tested in this set of experiments was rotenone, a flavoprotein inhibitor that blocks mitochondrial complex I. Figure 2E shows that, like other ETC inhibitors, rotenone elicited secretion from the cells; however, previous exposure to rotenone abolished any further increase of secretion by hypoxia. Therefore, rotenone occluded the response to hypoxia without mimicking it. In some cases, as in the example of Fig. 2E, the application of hypoxia in the presence of rotenone even reduced the secretory activity of the cells. In cells treated with rotenone the secretory response to depolarization with high potassium was unaltered (Fig. 2E).
Figure 2. Secretory responses of glomus cells exposed concomitantly to hypoxia (PO2≈ 20 mmHg) and to blockade of mitochondrial electron transport.
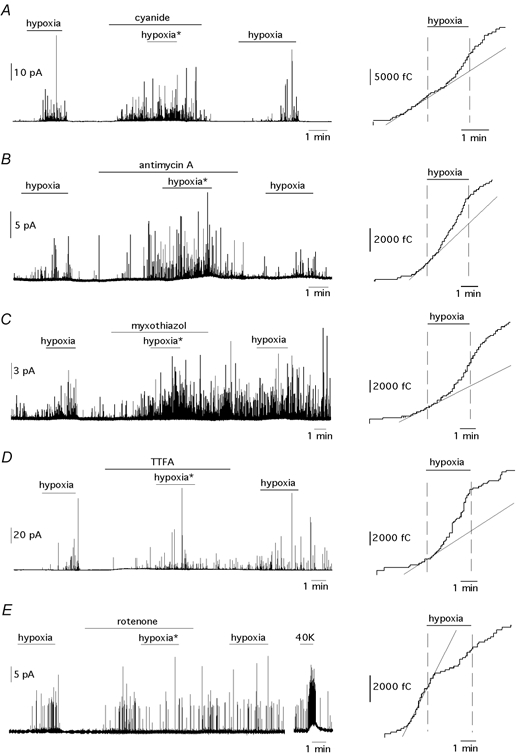
A–E, left panels, amperometric recordings. The concentration of drugs used in each experiment was: cyanide (A, 100 μm), antimycin A (B, 1 μg ml−1), myxothiazol (C, 1 μg ml−1), TTFA (D, 0.1 μm) and rotenone (E, 5 μm). A–E, right panels, cumulative secretion signals before, during and after exposure to hypoxia in the presence of the ETC inhibitors. The exposures to hypoxia are marked with an asterisk in the left panels. The straight lines represent the slopes (secretion rates) of the cumulative secretion signals immediately before exposure to hypoxia.
Average secretion rates measured in several cells exposed to hypoxia and the ETC inhibitors are given in the summary plot of Fig. 3. Inhibition at various sites along the ETC with saturating concentrations of mitochondrial inhibitors induced a secretory activity in glomus cells of a magnitude comparable to that evoked by low PO2 (∼20 mmHg). With the exception of rotenone, the effects of hypoxia and ETC inhibitors were additive, thus suggesting that they might act through separate signalling pathways. Selective occlusion of hypoxia sensitivity by rotenone was observed in all the cells studied (n = 28) with concentrations of the drug between 0.1 and 5 μm. The lowest concentration used in these experiments (0.1 μm) can produce full blockade of complex I (Degli Esposti, 1998; Vaux et al. 2001) or saturation of rotenone-binding sites (Higgins & Greenamyre, 1996).
Rotenone-induced secretion and sensitivity of glomus cells to hypoxia
The effects of rotenone on the sensitivity to hypoxia in glomus cells were analysed in greater detail following the same experimental protocol as in Fig. 2E but using lower concentrations of the drug. Figure 4A-C illustrates that the potency of rotenone to evoke secretion decreased progressively at concentrations below 100 nm. Rotenone is a poorly reversible drug and washout from the cells was very slow in most experiments. The decrease of rotenone-evoked secretion was inversely related with its efficacy in occluding the responsiveness to hypoxia. This changed from full blockade of the cellular response to hypoxia at 100 nm (Fig. 4A) to lack of effect when the concentration of rotenone was reduced to 20 nm (Fig. 4C). With intermediate concentrations of rotenone (50 nm), blockade of responsiveness to hypoxia was only partial (Fig. 4B). Another observation suggesting a relationship between rotenone and hypoxia sensitivity was that cells insensitive to hypoxia were either unaffected or showed a minor response to rotenone (at concentrations between 0.1 and 5 μm; n = 8 cells), although they were healthy since they responded with a powerful burst of secretion to high [K+] (Fig. 5A). In contrast, O2- and rotenone-insensitive cells appeared to respond normally to myxothiazol, antimycin A or cyanide (Fig. 5B). It has been noticed before that glomus cells, either dispersed or in slices, can lose hypoxia responsiveness while maintaining normal ionic currents and secretory responses to other agents (López-Barneo et al. 1998; Pardal et al. 2000; see Discussion).
Figure 4. Dose-dependent occlusion of the responsiveness to hypoxia by rotenone.
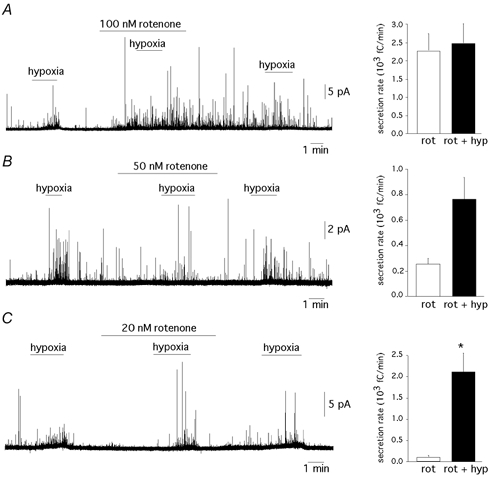
A–C, left panels, amperometric recordings illustrating the decrease in the potency of rotenone in blocking sensitivity to hypoxia as the concentration of the drug decreases from 100 to 20 nm. A–C, right panels, average secretion rates in the various experimental conditions were: A, rotenone 100 nm; 2300 ± 484 fC min−1; rotenone plus hypoxia, 2491 ± 566 fC min−1; n = 5 cells. P = 0.8. B, rotenone 50 nm; 264 ± 54 fC min−1; rotenone plus hypoxia, 772 ± 179 fC min−1; n = 3 cells. P = 0.05. C, rotenone 20 nm; 111 ± 50 fC min−1; rotenone plus hypoxia, 2130 ± 462 fC min−1; n = 3 cells. P < 0.05. Statistically significant difference (P < 0.05) indicated by an asterisk.
Figure 5. Cells insensitive to hypoxia respond weakly to rotenone.
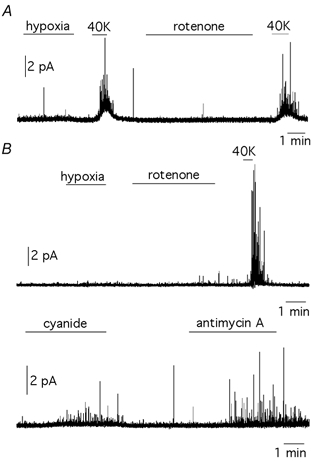
A, representative recording from a cell that did not respond to hypoxia (PO2≈ 20 mmHg) or rotenone (5 μm) but was stimulated to secrete by membrane depolarization with 40 mm potassium. B, top, representative recordings from another cell that did not respond to either hypoxia (PO2≈ 20 mmHg) or rotenone (5 μm) but produced a potent burst of secretory activity by membrane depolarization with 40 mm potassium. Bottom, in the same cell, secretion was induced by exposure to cyanide (0.1 mm) and antimycin A (1 μg ml−1).
To further investigate the relationships between hypoxia, occlusion of hypoxia responsiveness by rotenone and mitochondrial electron flow we tested to see if the effect of rotenone was prevented by the presence of succinate (5 mm), a substrate for complex II (see Leach et al. 2001). Similar experiments were also performed with methylsuccinate, a derivative with higher membrane permeability than succinate. Figure 6A shows that in O2-responsive cells, succinate did not prevent the abolition of responsiveness to hypoxia by rotenone (n = 3 cells). Similarly, in the presence of methylsuccinate both sensitivity to hypoxia (n = 7 cells, Fig. 6B) and its occlusion by rotenone (n = 6 cells, Fig. 6C) were observed. The amperometric trace in Fig. 6C (left) illustrates a full representative experiment in a cell with a clear response to hypoxia that was maintained in the presence of methylsuccinate but abolished by the addition of rotenone. The cumulative secretion signal is shown in the figure to facilitate its comparison with the results presented in Fig. 2. Therefore, occlusion of glomus cells’ responsiveness to hypoxia by rotenone is not prevented by feeding electrons to the mitochondrial ETC through complex II.
Figure 6. Hypoxia and mitochondrial electron flow.
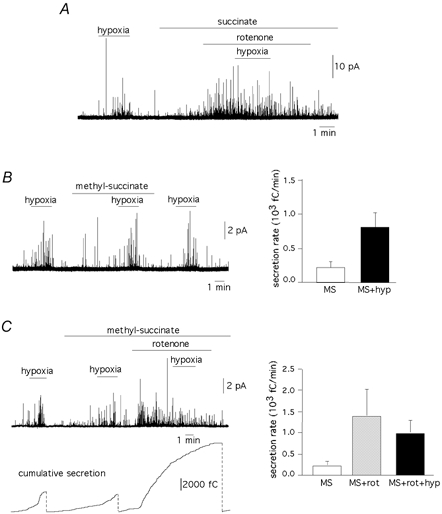
A, occlusion of responsiveness to hypoxia (PO2≈ 20 mmHg) by rotenone (0.1 μm) in the presence of 5 mm sodium succinate in the external solution. Similar qualitative results were obtained in two other cells tested. B, left, amperometric recording showing that methylsuccinate does not alter sensitivity to hypoxia; right, average secretion rates (fC min−1) in cells (n = 7) exposed to 5 mm methylsuccinate (230 ± 115) and methylsuccinate plus hypoxia (816 ± 229), P < 0.05. C, left, occlusion of responsiveness to hypoxia (PO2≈ 20 mmHg) by rotenone (1 μm) in the presence of 5 mm methylsuccinate in the external solution. The vertical dotted lines in the lower panel indicate the resetting of the integrator used to calculate the cumulative secretion signal; right, average secretion rates (fC min−1) in cells treated with 5 mm methylsuccinate (230 ± 115, n = 7 cells); methylsuccinate plus rotenone (0.1 to 1 μm) (1400 ± 648, n = 6 cells) and methylsuccinate plus rotenone plus hypoxia (1000 ± 309, n = 6). Statistical comparison between the two last pair of samples, P = 0.47.
Effects of other complex I inhibitors
The action of rotenone appeared to depend on its interaction with specific targets rather than on general inhibition of electron flow in complex I because the effect of this agent was not mimicked by myxothiazol (Fig. 2 and Fig. 3), which also inhibits complex I (Degli Esposti, 1998). We also tested the effects of three other complex I inhibitors: diphenyleneiodonium (DPI), a flavoprotein inhibitor that acts on a site at complex I separated from the rotenone binding site; rhein, a competitive inhibitor of NADH; and 1-methyl-4-phenylpyridinium ion (MPP+), an agent that binds to the same site as rotenone (Higgins & Greenamyre, 1996; Degli Esposti, 1998). DPI used in a broad range of concentrations (0.2−5 μm, n = 5 cells) induced glomus cell secretion but, as other ETC inhibitors, did not prevent the stimulatory effect of hypoxia (Fig. 7A). A similar effect was observed for rhein when used at intermediate concentrations (10–25 μm, n = 7 cells; Fig. 7B). At higher concentrations, rhein produced inhibition of spontaneous and potassium-evoked secretion, thus suggesting that, as shown before for DPI in patch-clamped glomus cells (see Wyatt et al. 1994) this agent blocks extracellular Ca2+ entry through membrane channels. Therefore, responsiveness to hypoxia in the presence of fully saturating concentrations of rhein could not be studied in detail. Sensitivity of glomus cells to hypoxia was, however, occluded in the presence of MPP+ (50–150 μm, n = 7 cells).
Figure 7. Responsiveness to hypoxia and complex I inhibitors.
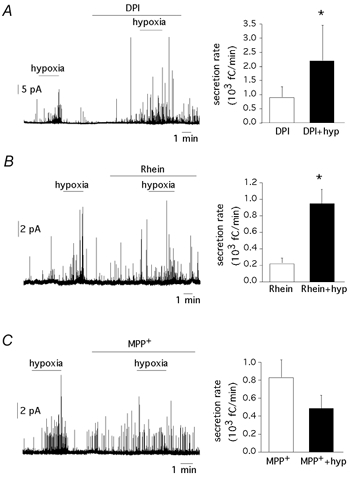
A, left, response to hypoxia in a cell treated with DPI (0.2 μm); right, increase in the secretory activity (fC min−1) induced by DPI (0.2–5 μm; 918 ± 389; n = 5 cells; P < 0.05) and by DPI plus hypoxia (2195 ± 1259; n = 5 cells). B, left, response to hypoxia in a cell treated with rhein (25 μm); right, increase in secretory activity (fC min−1) induced by rhein (10–25 μm; 228 ± 64; n = 5 cells) and by rhein plus hypoxia (957 ± 179; n = 5 cells; P < 0.05). C, left, abolition of the responsiveness to hypoxia by 1-methyl-4-phenylpyridinium ion (MPP+, 50 μm); right, secretory activity (fC min−1) induced by MPP+ (50–150 μm; 831 ± 198; n = 7 cells) and by hypoxia plus MPP+ (489 ± 150; n = 7 cells; P = 0.3). Statistically significant difference (P < 0.05) indicated by an asterisk.
DISCUSSION
The major observations in this study are: (1) inhibitors of proximal and distal complexes of the mitochondrial ETC elicit, as does hypoxia, a powerful external Ca2+-dependent secretory response in intact rat carotid body glomus cells; (2) cellular sensitivity to hypoxia is maintained after blockade of the mitochondrial ETC; and (3) rotenone and MPP+ selectively suppress responsiveness to hypoxia. These findings strongly suggest that mitochondrial electron flow is not directly involved in acute O2 sensing by glomus cells. However, a rotenone (and MPP+)-inhibitable molecule participates critically in this process.
The observations at the cellular level reported in this study explain the well-known stimulatory action of mitochondrial poisons on the carotid body chemoreceptors (Mills & Jöbsis, 1972; Mulligan et al. 1981). It had been reported before that cyanide increases cytosolic [Ca2+] in rabbit glomus cells due to release of the cation from mitochondria (Biscoe & Duchen, 1990); our results indicate that at moderate doses, cyanide-induced secretion is totally dependent on extracellular Ca2+. Similar dependence of external Ca2+ has been described for the effects of mitochondrial inhibitors on cytosolic [Ca2+] in rat glomus cells (Buckler & Vaughan-Jones, 1998) or the secretory activity in PC12 cells (Taylor et al. 2000). Taken together, these results could indicate that mitochondria can signal the cell membrane to regulate transmembrane Ca2+ influx. In support of this idea it has been reported that metabolic uncouplers reduce resting K+ permeability in rat glomus cells (Buckler & Vaughan-Jones, 1998) and that large concentrations of rotenone, antimycin A or DPI decrease the amplitude of voltage-dependent K+ currents in pulmonary arterial smooth muscle (Archer et al. 1993) and the carotid body (Wyatt et al. 1994; our own unpublished results) or model airway chemoreceptor (Searle et al. 2002) cells. Mitochondrial signalling could depend on either the production by these organelles of reactive oxygen species that modify the redox state of membrane channels (Archer et al. 1993; Chandel & Schumacker, 2000), or the level of cytosolic ATP concentration (Inoue et al. 2002). However, the effects of ETC inhibitors on membrane ionic conductances must be interpreted cautiously since at the concentrations used in most studies (several orders of magnitude higher than that required to inhibit mitochondrial ETC) some of them may behave as non-specific inhibitors of ion channels in glomus cells (Wyatt et al. 1994; our own unpublished results). An alternative explanation for the secretory response induced by mitochondrial inhibition is the existence in resting glomus cells of a maintained Ca2+ influx, which is normally counterbalanced by Ca2+ uptake in mitochondria located adjacent to the cell membrane. As described by Montero et al. (2000) in chromaffin cells, inhibition of submembranous mitochondria and the loss of their Ca2+-buffering capacity, would then raise cytosolic Ca2+ levels and trigger catecholamine release.
Although hypoxia and mitochondrial ETC inhibitors induce similar external Ca2+-dependent secretory responses from glomus cells in carotid body slices, our results indicate that activation of these cells by low PO2 is not directly dependent on mitochondrial electron flow. Mitochondria have been classically postulated to participate in carotid body O2 sensing due to the similarity of the stimulatory effects of hypoxia and mitochondrial poisoning (Mulligan et al. 1981). They have also been ascribed a role in acute O2 sensing in chromaffin cells (Mojet et al. 1997; Inoue et al. 2002) and pulmonary arterial smooth muscle (Archer et al. 1993). Using this last preparation it has recently been hypothesized that O2 tension regulates superoxide production at the semiubiquinone site of mitochondrial complex III and that an increase of radical production during exposure to low PO2 is the signal that mediates the acute phase of hypoxic pulmonary vasoconstriction (HPV) (Leach et al. 2001; Waypa et al. 2001). A major argument in favour of this model is the differential effect of blockers of the ETC acting upstream and downstream of the semiubiquinone site. Antimycin A, a downstream blocker that leaves complex III reduced, does not alter HPV whereas this response is blocked by upstream ETC inhibitors, such as rotenone and myxothyazol, that maintain complex III permanently oxidized. In addition, occlusion of HPV by rotenone is reversed by stimulation of electron transport via complex II with succinate (Leach et al. 2001). In contrast with these data, we observe in carotid body cells that several blockers acting upstream of the semiubiquinone site at complexes I (DPI, rhein and myxothiazol), II (TTFA) and III (myxothiazol) are able to trigger a powerful secretory response but they do not prevent further increase of secretion by hypoxia. Moreover, the occlusion of hypoxia responsiveness by rotenone is unaltered after incubation of the cells with succinate, which feeds electrons to the ETC through complex II. The effect of rotenone is mimicked by MPP+, a drug that binds to the same site as rotenone (Higgins & Greenamyre, 1996). In parallel with the present study focused on intact glomus cells in carotid body slices, we have observed in preliminary experiments that at submicromolar concentrations rotenone, but not antimycin A, reduces the amplitude of the macroscopic K+ current and blocks the inhibition of this same current by hypoxia. Altogether, these observations strongly suggest that although a rotenone-sensitive molecule appears to be necessary, mitochondrial electron transport per se is not directly involved in acute O2 sensing by carotid body glomus cells. This conclusion, however, is compatible with the data reported for pulmonary smooth muscle (Leach et al. 2001; Waypa et al. 2001) as it is generally agreed that distinct cell types may use different strategies to sense O2. For example, knocking out the NADPH oxidase gene abolishes O2 sensitivity in neuroepithelial cells of the lung but leaves unaltered the responses to hypoxia in pulmonary smooth muscle or carotid body cells (for review and discussion, see López-Barneo et al. 2001). In addition, sensitivity of macroscopic outward currents to hypoxia in model airway chemoreceptor cells is maintained in the presence of high concentrations of rotenone and antimycin A (Searle et al. 2002). Cross et al. (1990), have reported the abolition of the hypoxia-induced increase in carotid body sinus nerve chemoreceptor discharge by DPI (that inhibits both mitochondrial complex I and NADPH oxidase). These results can, however, be explained by the non-specific blocking effect of this agent on Ca2+ channels at the glomus cell-afferent fibre synapse (Wyatt et al. 1994).
Besides the effects of rotenone and MPP+ on the fast response of glomus cells to acute hypoxia described here, there are several reports suggesting that rotenone-binding proteins might be involved in chronic cellular responses to low PO2. Erythropoietin gene induction by hypoxia, although unaffected by downstream mitochondrial inhibitors like azide or cyanide, is blocked by rotenone (Necas & Thorling, 1972; Goldberg et al. 1988; Pugh et al. 1991). Similarly, activation and cell accumulation of hypoxia-inducible factor 1 α in low PO2 is prevented by rotenone (Chandel et al. 1998; Semenza, 1999) and MPP+ (Agani et al. 2000). These effects of rotenone are, however, unclear because they are only observed at concentrations higher than that required to inhibit oxygen consumption by the cells (Vaux et al. 2001).
The nature and location of the rotenone (and MPP+)-inhibitable molecule involved in glomus cell O2 sensing is unknown. It could be a membrane K+ channel subunit with a flavoprotein modified by rotenoids that changes its conformation in response to lowering PO2. This hypothesis, although speculative, is in accord with the existence of cytosolic aggregates of pre-assembled complex I proteins of unknown function (Schulte, 2001), which in glomus cells could be associated with membrane ion channels. However, we cannot discount a more elaborate arrangement constituted by mitochondria with complex I proteins regulated by changes of O2 tension in close contact with membrane K+ channels. As described in other systems (Kennedy et al. 1999; Montero et al. 2000), peripheral mitochondria may form functional units with membrane proteins to delimit submembraneous cytosolic microdomains. These ideas are compatible with previous observations describing the modulation of membrane K+ channels by O2 tension in excised membrane patches (Ganfornina & López-Barneo, 1991; Riesco-Fagundo et al. 2001; Lewis et al. 2002) as these are known to contain mitochondria and other intracellular organelles (Sather et al. 1992; Rustenbeck et al. 1999). The existence of a separate sensor that confers O2 sensitivity to the K+ channels helps to explain why in some conditions, in which the sensor is possibly detached from the channels, glomus cells lose the responsiveness to hypoxia while maintaining normal electrophysiological properties (López-Barneo et al. 1998). This scheme of glomus cell O2 sensing could also account for the existence of O2-insensitive cells with low responsiveness to rotenone. Detachment of mitochondria from the cell membrane would render cells O2-insensitive and make mitochondrial signalling to the membrane more difficult. These cells would then require complete blockade of the ETC (with cyanide or antimycin A) to induce significant secretion.
In conclusion, we report that, as with hypoxia, mitochondrial inhibitors activate external Ca2+-dependent catecholamine secretion from carotid body glomus cells. However, our results indicate that mitochondrial electron flow is not linked in a simple way to acute regulation of glomus cell activity by changes of O2 tension. Hypoxia and mitochondrial inhibitors, acting through separate pathways, converge to raise cytosolic [Ca2+], which triggers secretion. We have identified rotenone (and MPP+) as highly selective and specific inhibitors of the responsiveness to hypoxia. Therefore, these drugs and their derivatives could be used as tools to pursue investigation of the molecular characterization and location of the O2 sensing mechanism in glomus cells.
Acknowledgments
We wish to thank Dr J. Toledo-Aral for comments on the manuscript. Research was supported by grants from the Spanish Ministry of Science and Technology (1FD97-1614) and Fundaciones La Caixa and Ramón Areces. P.O.-S. was supported by a postdoctoral fellowship of the Fondo de Investigación Sanitaria, and R.P. and M.G.-F. by fellowships of the FPI and FPU programmes of the Spanish Ministry of Education. J.L.-B. received the ‘Ayuda a la Investigación 2000′ of the Juan March Foundation.
REFERENCES
- Agani FH, Pichiule P, Chavez JC, Lamanna JC. The role of mitochondria in the regulation of hypoxia-inducible factor 1 expression during hypoxia. J Biol Chem. 2000;275:35863–35867. doi: 10.1074/jbc.M005643200. [DOI] [PubMed] [Google Scholar]
- Archer SL, Huang J, Henry T, Peterson D, Weir EK. A redox-based O2 sensor in rat pulmonary vasculature. Circ Res. 1993;73:1100–1112. doi: 10.1161/01.res.73.6.1100. [DOI] [PubMed] [Google Scholar]
- Biscoe TJ, Duchen MR. Responses of type I cells dissociated from the rabbit carotid body to hypoxia. J Physiol. 1990;428:39–59. doi: 10.1113/jphysiol.1990.sp018199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ. A novel oxygen-sensitive potassium current in rat carotid body type I cells. J Physiol. 1997;498:649–662. doi: 10.1113/jphysiol.1997.sp021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. J Physiol. 1994;476:423–428. doi: 10.1113/jphysiol.1994.sp020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of mitochondrial uncouplers on intracellular calcium, pH and membrane potential in rat carotid body type I cells. J Physiol. 1998;513:819–833. doi: 10.1111/j.1469-7793.1998.819ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter E, Hatton CJ, Peers C. Effects of hypoxia and dithionite on catecholamine release from isolated type I cells of the rat carotid body. J Physiol. 2000;523:719–729. doi: 10.1111/j.1469-7793.2000.00719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, Schumacker PT. Cellular oxygen sensing by mitochondria: old questions, new insight. J Appl Physiol. 2000;88:1880–1889. doi: 10.1152/jappl.2000.88.5.1880. [DOI] [PubMed] [Google Scholar]
- Cross AR, Henderson AL, Jones OTG, Delpiano MA, Hentschel J, Acker H. Involvement of an NAD(P)H oxidase as a PO2 sensor protein in the rat carotid body. Biochem J. 1990;272:743–747. doi: 10.1042/bj2720743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degli Esposti M. Inhibitors of the NADH-ubiquinone reductase: an overview. Biochim Biophys Acta. 1998;1364:222–235. doi: 10.1016/s0005-2728(98)00029-2. [DOI] [PubMed] [Google Scholar]
- Delpiano MA, Hescheler J. Evidence for a PO2-sensitive K+ channel in the type-I cell of the rabbit carotid body. FEBS Lett. 1989;249:195–198. doi: 10.1016/0014-5793(89)80623-4. [DOI] [PubMed] [Google Scholar]
- Duchen MR, Biscoe TJ. Relative mitochondrial membrane potential and [Ca2+]i in type I cells isolated from the rabbit carotid body. J Physiol. 1992;450:33–61. doi: 10.1113/jphysiol.1992.sp019115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganfornina MD, López-Barneo J. Single K+ channels in membrane patches of arterial chemoreceptor cells are modulated by O2 tension. Proc Natl Acad Sci U S A. 1991;88:2927–2930. doi: 10.1073/pnas.88.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MA, Dunning SP, Bunn HF. Regulation of the erythropoietin gene: evidence that the oxygen sensor is a heme protein. Science. 1988;242:1412–1415. doi: 10.1126/science.2849206. [DOI] [PubMed] [Google Scholar]
- Higgins DS, Greenamyre JT. [3H]dihydrorotenone binding to NADH: Ubiquinone reductase (complex I) of the electron transport chain: an autoradiographic study. J Neurosci. 1996;16:3807–3816. doi: 10.1523/JNEUROSCI.16-12-03807.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Fujishiro N, Imanaga I, Sakamoto Y. Role of ATP decrease in secretion induced by mitochondrial dysfunction in guinea-pig adrenal chromaffin cells. J Physiol. 2002;539:145–155. doi: 10.1113/jphysiol.2001.012936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy HJ, Pouli AE, Ainscow EK, Jouaville LS, Rizzuto R, Rutter GA. Glucose generates subplasma membrane ATP microdomains in single islet β-cells. J Biol Chem. 1999;274:13281–13291. doi: 10.1074/jbc.274.19.13281. [DOI] [PubMed] [Google Scholar]
- Leach RM, Hill HM, Snetkov VA, Robertson TP, Ward JPT. Divergent roles of glycolysis and the mitochondrial electron transport chain in hypoxic pulmonary vasoconstriction of the rat: identity of the hypoxic sensor. J Physiol. 2001;536:211–224. doi: 10.1111/j.1469-7793.2001.00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis A, Peers C, Ashford ML, Kemp PJ. Hypoxia inhibits human recombinant large conductance, Ca2+-activated K+ (maxi-K) channels by a mechanism which is membrane-delimited and Ca2+-sensitive. J Physiol. 2002;540:771–780. doi: 10.1113/jphysiol.2001.013888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Barneo J. Oxygen-sensitive ion channels: how ubiquitous are they? Trends Neurosci. 1994;17:133–135. doi: 10.1016/0166-2236(94)90084-1. [DOI] [PubMed] [Google Scholar]
- López-Barneo J, Benot AR, Ureña J. Oxygen sensing and the electrophysiology of arterial chemoreceptor cells. News Physiol Sci. 1993;8:191–195. [Google Scholar]
- López-Barneo J, López-López JR, Ureña J, González C. Chemotransduction in the carotid body: K+ current modulated by PO2 in type I chemoreceptor cells. Science. 1988;241:580–582. doi: 10.1126/science.2456613. [DOI] [PubMed] [Google Scholar]
- López-Barneo J, Montoro R, Ortega-Sáenz P, Ureña J. Oxygen-regulated ion channels: functional roles and mechanisms. In: López-Barneo J, Weir EK, editors. Oxygen Regulation of Ion Channels and Gene Expression. Armonk NY: Futura Publishing Company; 1998. pp. 127–144. [Google Scholar]
- López-Barneo J, Pardal R, Ortega-Sáenz P. Cellular mechanisms of oxygen sensing. Annu Rev Physiol. 2001;63:259–287. doi: 10.1146/annurev.physiol.63.1.259. [DOI] [PubMed] [Google Scholar]
- López-López JR, González C, Ureña J, López-Barneo J. Low PO2 selectively inhibits K+ channel activity in chemoreceptor cells of the mammalian carotid body. J Gen Physiol. 1989;93:1001–1015. doi: 10.1085/jgp.93.5.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills E, Jöbsis FF. Mitochondrial respiratory chain of carotid body and chemoreceptor response to changes in oxygen tension. J Neurophysiol. 1972;35:405–428. doi: 10.1152/jn.1972.35.4.405. [DOI] [PubMed] [Google Scholar]
- Mojet MH, Mills E, Duchen MR. Hypoxia-induced catecholamine secretion in isolated newborn rat adrenal chromaffin cells is mimicked by inhibition of mitochondrial respiration. J Physiol. 1997;504:175–189. doi: 10.1111/j.1469-7793.1997.175bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero M, Alonso MT, Carnicero E, Cuchillo-Ibañez I, Albillos A, García AG, García-Sancho J, Alvarez J. Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat Cell Biol. 2000;2:57–61. doi: 10.1038/35000001. [DOI] [PubMed] [Google Scholar]
- Montoro RJ, Ureña J, Fernández-Chacón R, Alvarez de Toledo G, López-Barneo J. Oxygen sensing by ion channels and chemotransduction in single glomus cells. J Gen Physiol. 1996;107:133–143. doi: 10.1085/jgp.107.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan E, Lahiri S, Storey BT. Carotid body O2 chemoreception and mitochondrial oxydative phosphorylation. J Appl Physiol. 1981;51:438–446. doi: 10.1152/jappl.1981.51.2.438. [DOI] [PubMed] [Google Scholar]
- Necas E, Thorling EB. Unresponsiveness of erythropoietin-producing cells to cyanide. Am J Physiol. 1972;222:1187–1190. doi: 10.1152/ajplegacy.1972.222.5.1187. [DOI] [PubMed] [Google Scholar]
- Pardal R, López-Barneo J. Differential effects of mitochondrial inhibitors on the hypoxia sensitivity of glomus cells in slices of rat carotid body. Eur J Neurosci. 2000;12(suppl. 11):499. [Google Scholar]
- Pardal R, López-Barneo J. Low glucose-sensing cells in the carotid body. Nat Neurosci. 2002;5:197–198. doi: 10.1038/nn812. [DOI] [PubMed] [Google Scholar]
- Pardal R, Ludewig U, García-Hirschfeld J, López-Barneo J. Secretory responses of intact glomus cells in thin slices of rat carotid body to hypoxia and tetraethylammonium. Proc Natl Acad Sci U S A. 2000;97:2361–2366. doi: 10.1073/pnas.030522297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peers C. Hypoxic suppresion of K+ currents in type I carotid body cells: selective effect on the Ca2+ -activated K+ current. Neurosci Lett. 1990;119:253–256. doi: 10.1016/0304-3940(90)90846-2. [DOI] [PubMed] [Google Scholar]
- Pérez-García MT, López-López JR, Riesco AM, Hoppe UC, Marban E, Gonzalez C, Johns DC. Viral gene transfer of dominant-negative Kv4 construct suppresses an O2 -sensitive K+ current in chemoreceptor cells. J Neurosci. 2000;20:5689–5695. doi: 10.1523/JNEUROSCI.20-15-05689.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh CW, Tan CC, Jones RW, Ratcliffe PJ. Functional analysis of an oxygen-regulated transcriptional enhancer lying 3′ to the mouse erythropoietin gene. Proc Natl Acad Sci U S A. 1991;88:10553–10557. doi: 10.1073/pnas.88.23.10553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesco-Fagundo AM, Pérez-García MT, González C, López-López JR. O2 modulates large conductance Ca2+-dependent K+ channels of rat chemoreceptor cells by a membrane-restricted and CO-sensitive mechanism. Circ Res. 2001;89:430–436. doi: 10.1161/hh1701.095632. [DOI] [PubMed] [Google Scholar]
- Rustenbeck I, Dickel C, Herrmann C, Grimmsmann T. Mitochondria present in excised patches from pancreatic B-cells may form microcompartments with ATP-dependent potassium channels. Biosci Rep. 1999;19:89–98. doi: 10.1023/a:1020106409700. [DOI] [PubMed] [Google Scholar]
- Sather W, Dieudonne S, MacDonald J, Ascher P. Activation and desensitization of N-methyl-D-aspartate receptors in nucleated outside-out patches from mouse neurons. J Physiol. 1992;450:643–672. doi: 10.1113/jphysiol.1992.sp019148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte U. Biogenesis of respiratory complex I. J Bioenerg Biomembr. 2001;33:205–212. doi: 10.1023/a:1010730919074. [DOI] [PubMed] [Google Scholar]
- Searle GJ, Hartness ME, Hoareau R, Peers C, Kemp PJ. Lack of contribution of mitochondrial electron transport to acute O2 sensing in model airway chemoreceptors. Biochem Biophys Res Commun. 2002;291:332–337. doi: 10.1006/bbrc.2002.6428. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- Stea A, Nurse CA. Whole-cell and perforated-patch recordings from O2- sensitive rat carotid body cells grown in short- and long-term culture. Pflugers Arch. 1991;418:93–101. doi: 10.1007/BF00370457. [DOI] [PubMed] [Google Scholar]
- Taylor SC, Shaw SM, Peers C. Mitochondrial inhibitors evoke catecholamine release from pheochromocytoma cells. Biochem Biophys Res Commun. 2000;273:17–21. doi: 10.1006/bbrc.2000.2894. [DOI] [PubMed] [Google Scholar]
- Ureña J, Fernández-Chacón R, Benot AR, Alvarez de Toledo G, López-Barneo J. Hypoxia induces voltage-dependent Ca2+ entry and quantal dopamine secretion in carotid body glomus cells. Proc Natl Acad Sci U S A. 1994;91:10208–10211. doi: 10.1073/pnas.91.21.10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux EC, Metzen E, Yeates KM, Ratcliffe PJ. Regulation of hypoxia inducible factor is preserved in the absence of a functioning mitochondrial respiratory chain. Blood. 2001;98:296–302. doi: 10.1182/blood.v98.2.296. [DOI] [PubMed] [Google Scholar]
- Waypa GB, Chandel NS, Schumacker PT. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ Res. 2001;88:1259–1266. doi: 10.1161/hh1201.091960. [DOI] [PubMed] [Google Scholar]
- Wyatt CN, Peers C. Ca2+-activated K+ channels in isolated type I cells of the neonatal rat carotid body. J Physiol. 1995;483:559–565. doi: 10.1113/jphysiol.1995.sp020606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt CN, Weir EK, Peers C. Diphenylene iodonium blocks K+ and Ca2+ channels in type I cells isolated from newborn rats. Neurosci Lett. 1994;172:63–66. doi: 10.1016/0304-3940(94)90663-7. [DOI] [PubMed] [Google Scholar]