

Abstract
In exercising skeletal muscle, vasoconstrictor responses to α-adrenoceptor activation are attenuated in part by nitric oxide (NO) produced by the neuronal isoform of NO synthase (nNOS), which is expressed constitutively in skeletal muscle cells. In skeletal muscle of pregnant animals, nNOS mRNA is upregulated, suggesting that muscle nNOS expression is modulated by the steroid hormone oestrogen. Whether oestrogen-induced changes in nNOS expression have measurable effects on vasoregulation in skeletal muscle is unknown. In this study, we hypothesized that oestrogen deficiency would reduce muscle nNOS expression, resulting in impaired modulation of sympathetic vasoconstriction in exercising skeletal muscle. Compared to gonadally intact rats, we found that ovariectomized (OVX) rats were characterized by greater sympathetic vasoconstriction in contracting hindlimb and reduced nNOS, but not eNOS, in skeletal muscle. In addition, NOS inhibition resulted in a greater enhancement of sympathetic vasoconstriction in contracting hindlimbs of intact compared to OVX rats. These effects of oestrogen deficiency were prevented by chronic treatment of OVX rats with 17β-oestradiol, but not with chronic progesterone or acute oestradiol. Further analysis revealed that skeletal muscle nNOS correlated directly with plasma 17β-oestradiol and inversely with the magnitude of sympathetic vasoconstrictor responses in contracting hindlimbs. These data indicate that NO-dependent attenuation of sympathetic vasoconstriction in contracting skeletal muscle is impaired in oestrogen-deficient female rats, and suggest that this impairment may be mediated by reduced skeletal muscle nNOS expression.
To sustain aerobic exercise, skeletal muscle blood flow must increase proportionately to match the increased metabolic demand of the contracting muscles. The mechanism by which muscle blood flow is closely coupled to aerobic metabolism is not well understood, but may be mediated in part by metabolites released from contracting skeletal muscle cells that diffuse to adjacent arterioles and cause vasodilatation (Lash, 1996; Delp & Laughlin, 1998). Skeletal muscle resistance arterioles are densely innervated by sympathetic vasoconstrictor nerves (Fuxe & Sedvall, 1965; Fleming et al. 1989), which display intermittent bursts of activity in quiescent muscle that are markedly increased in frequency and amplitude during exercise (Seals & Victor, 1991; Rowell & O'Leary, 1990). Despite this sympathetic activation, blood flow to contracting muscles increases during exercise, suggesting that muscle contraction may interfere with the normal ability of sympathetic nerves to cause vasoconstriction. Such interference is postulated to be due to muscle metabolites acting prejunctionally to reduce noradrenaline release from sympathetic nerve terminals (Burcher & Garlick, 1975; Verhaeghe et al. 1978) or postjunctionally to diminish the vasoconstrictor response to α-adrenoceptor activation (Remensnyder et al. 1962; Rowlands & Donald, 1968; Burcher & Garlick, 1973; Anderson & Faber, 1991).
One of the more recently identified vasoactive substances produced in contracting muscle that has been shown to modulate α-adrenergic vasoconstriction is the diffusible signalling molecule nitric oxide (NO) (Thomas et al. 1998; Thomas & Victor, 1998; Lau et al. 2000; Grange et al. 2001; Chavoshan et al. 2002). Both constitutive isoforms of nitric oxide synthase (NOS) are present in skeletal muscle, with endothelial NOS (eNOS) highly expressed in the vascular endothelium (Kobzik et al. 1995) and neuronal NOS (nNOS) highly expressed in the skeletal muscle cells (Nakane et al. 1993; Kobzik et al. 1994) where it localizes to the sarcolemma in association with the cytoskeletal protein dystrophin (Brenman et al. 1995; Chang et al. 1996). In healthy rodents and humans, the normal attenuation of α-adrenergic vasoconstriction in contracting skeletal muscle is impaired by concurrent pharmacological inhibition of eNOS and nNOS (Thomas et al. 1998; Thomas & Victor, 1998; Chavoshan et al. 2002). A similar impairment is observed when skeletal muscle nNOS, but not eNOS, is greatly reduced as in nNOS knockout mice (Thomas et al. 1998; Lau et al. 2000; Grange et al. 2001), or in mdx mice (Thomas et al. 1998) and boys with Duchenne muscular dystrophy (Sander et al. 2000) in which dystrophin deficiency results in a secondary reduction of muscle nNOS (Brenman et al. 1995; Chang et al. 1996). Impaired vasomodulation in the nNOS-deficient mouse muscles is not further exacerbated by pharmacological NOS inhibition, implying that the observed phenotype is due to lack of nNOS rather than eNOS (Thomas et al. 1998). Together, these previous studies indicate that substantial decreases in the activity or expression of skeletal muscle NOS, particularly the nNOS isoform, can have important functional consequences on vasoregulation in exercising muscle.
Relatively little is known about the factors that control NOS activity and expression in skeletal muscle. Although originally classified as constitutively expressed enzymes, both nNOS and eNOS expression in mature skeletal muscle can be modulated by factors such as contractile activity (Balon & Nadler, 1997; Reiser et al. 1997), innervation (Tews et al. 1997), and mechanical loading (Tidball et al. 1998). A potential role for oestrogen in the regulation of skeletal muscle NOS is suggested by the increased NOS catalytic activity and message for nNOS and eNOS observed in skeletal muscle of pregnant guinea-pigs, a condition in which serum oestrogen is elevated 20-fold above non-pregnant levels (Weiner et al. 1994a,b). Whether more modest changes in oestrogen levels have similar effects on skeletal muscle NOS activity or expression, and whether oestrogen-related changes in the NO system have a measurable impact on vasomodulation in skeletal muscle are not known.
We therefore investigated the effects of chronic oestrogen deficiency on NOS expression and vascular regulation in rat skeletal muscle. We hypothesized that oestrogen deficiency would result in impaired attenuation of sympathetic vasoconstriction in exercising muscle and that this impairment would be associated with dysfunction of the NO system caused mainly by reduced expression of muscle nNOS. We tested these hypotheses by using ovariectomy as a model of chronic oestrogen deficiency, performing in vivo experiments to measure sympathetic vasoconstriction in skeletal muscle and ex vivo analysis of constitutive NOS in skeletal muscle of ovariectomized rats with and without 17β-oestradiol or progesterone replacement.
Methods
All methods and protocols were approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center.
Experimental model
Bilateral ovariectomy or sham surgery was performed in female Sprague-Dawley rats at 9–10 weeks of age, anaesthetized with methohexital sodium (50 mg kg−1, i.p.). At the same time, 60-day timed-release pellets containing 17β-oestradiol (1.5 mg pellet−1) or progesterone (25 mg pellet−1) or placebo pellets (Innovative Research of America, Sarasota, FL, USA) were implanted subcutaneously at the nape of the neck. Experiments were performed 3–4 weeks post-surgery.
Experimental preparation
Anaesthesia was initiated with ketamine (80 mg kg−1, i.p.) and maintained with α-chloralose (60 mg kg−1, i.v. supplemented with 10 mg kg−1 h−1, i.v.) for the duration of the study. Rats were mechanically ventilated with room air and supplemental O2. Core temperature was maintained at 37 °C with an external heat source. Rats were instrumented with jugular vein and carotid artery catheters, a Doppler flow probe (Crystal Biotech, Holliston, MA, USA) around the left femoral artery, and stimulating electrodes affixed to the left lumbar sympathetic chain and to the left sciatic nerve. The left hindlimb was connected to a force-displacement transducer (FT-10, Grass Instruments, Quincy, MA, USA) via the calcaneal tendon. Preparations were allowed to stabilize for at least 20 min before protocols were begun. At the beginning of each experiment blood was drawn and the plasma frozen at −80 °C for subsequent analysis of steroid hormones. At the end of the experiment rats were killed with an overdose of sodium pentobarbitone (150 mg kg−1, i.v.). In each rat, the presence or absence of ovarian tissue was confirmed visually and the uterus was excised and weighed.
Protocol 1. Effect of ovariectomy alone and in combination with chronic steroid hormone replacement on sympathetic vasoconstriction in rat hindlimb
Arterial pressure and femoral blood flow velocity responses to lumbar sympathetic nerve stimulation (1 ms pulses of 5 V at 1, 2.5, or 5 Hz) were measured in resting and contracting hindlimb. Contractions were produced by sciatic nerve stimulation at 2–3 × motor threshold voltage with 100 ms trains of pulses at a rate of 60 trains min−1.
Protocol 2. Effect of NOS inhibition on sympathetic vasoconstriction in hindlimb of ovariectomized rats
After completion of Protocol 1, rats were treated with the NOS inhibitor N-nitro-l-arginine methyl ester (l-NAME, Sigma; 5 mg kg−1, i.v.). Arterial pressure and femoral blood flow velocity responses to lumbar nerve stimulation were then measured in resting and contracting hindlimb.
Protocol 3. Effect of acute oestrogen replacement on sympathetic vasoconstriction in hindlimb of ovariectomized rats
Arterial pressure and femoral blood flow velocity responses to lumbar nerve stimulation were measured in resting and contracting hindlimb before and after infusion of 17β-oestradiol (100 μg kg−1, i.a.).
Protocol 4. Effect of restoration of basal NO after NOS inhibition on sympathetic vasoconstriction in rat hindlimb
Arterial pressure and femoral blood flow velocity responses to lumbar nerve stimulation were measured in resting and contracting hindlimb before and after infusion of l-NAME (5 mg kg−1, i.v.) alone or in combination with continuous infusion of sodium nitroprusside (NP; 25 μg ml−1 infused at rates of 5–20 μl min−1, i.v.) to restore arterial pressure and femoral blood flow velocity to pre-l-NAME levels.
Western blot analysis
Gastrocnemius muscle samples were frozen in liquid nitrogen and stored at −80 °C until analysed. Muscles were homogenized in 20 vol (w/v) of buffer containing 50 mm Tris HCl, pH 7.5, 1 μg ml−1 aprotinin, 2 μg ml−1 leupeptin, 20 μm tetrahydrobiopterin, 1 mm DTT, 1 μg ml−1 pepstatin A, 10 μg ml−1 soybean trypsin inhibitor, 1 mm benzamidine, 1 mm EDTA, 0.5 mm phenylmethylsulphonyl fluoride (PMSF), and 20 mm 3-[(3-cholamidopropyl)dimethylammonio]-1-propane sulphonate (CHAPS). Samples (100 μg) were resolved by SDS-PAGE on a 6 % gel and transferred to nitrocellulose. Membranes were incubated overnight at 4 °C with a rabbit polyclonal antibody raised against the N-terminus of nNOS (1:5000) or mouse monoclonal anti-eNOS (1:1000; Transduction Laboratories, Lexington, KY, USA). Membranes were washed and incubated for 1 h at room temperature with horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse antibodies. Immunoreactivity was detected by enhanced chemiluminescence and quantified by densitometry. Protein concentrations were determined using a Bio-Rad DC Protein Assay kit.
Steroid hormone assays
Plasma levels of 17β-oestradiol and progesterone were measured by competitive enzyme immunoassays according to kit instructions (Cayman Chemical, Ann Arbor, MI, USA). Samples were assayed in duplicate. Limits of detection were 12 pg ml−1 for 17β-oestradiol and 3 pg ml−1 for progesterone.
Data and statistical analysis
Haemodynamic and hindlimb force data were acquired and analysed using MacLab hardware and software (ADInstruments, Milford, MA, USA). Femoral vascular conductance (kHz mmHg−1) was calculated on-line as the quotient of mean Doppler shift and mean arterial pressure. Statistical analyses were performed by one-way or repeated-measures ANOVA followed by Bonferroni or Dunn's post hoc tests. Pearson product correlation coefficients were compared by Student's t test. A P value less than 0.05 was considered significant. All data are presented as means ± s.e.m.
Results
Forty-six female Sprague-Dawley rats (14 sham, 32 ovariectomized) were used for this study. Of the sham rats, 9 were used in protocols 1 and 2 (± l-NAME) and 5 were used in protocol 4 (l-NAME ± NP). Ovariectomized rats were implanted with placebo pellets (n = 10; OVX), or with pellets containing 17β-oestradiol (n = 10; OVX + E2) or progesterone (n = 7; OVX + P), and were used in protocols 1 and 2. Five additional placebo-treated OVX rats were used for protocol 3 (OVX + acute E2). General characteristics of the groups are provided in Table 1. As expected, at the time of study body weights were lighter and uterus weights were greater in the sham and OVX + E2 groups compared to the OVX and OVX + P groups. Plasma concentrations of 17β-oestradiol and progesterone were reduced in OVX rats and were restored to normal levels following treatment with the respective exogenous hormone. Haemodynamics in anaesthetized rats at rest and during unilateral hindlimb contraction are shown in Table 2. There were no significant pairwise differences in peak hindlimb force among sham (2.15 ± 0.03 kg), OVX (2.09 ± 0.07 kg), OVX + E2 (2.01 ± 0.10 kg), OVX + P (1.72 ± 0.03 kg), or OVX + acute E2 (1.82 ± 0.10 kg) groups, except for the comparison between sham and OVX + P rats (P < 0.05).
Table 1.
Group characteristics
| Group | n | Initial body wt (g) | Final body wt (g) | Uterus wt (mg) | 17β-oestradiol (pg ml−1) | Progesterone (ng ml−1) |
|---|---|---|---|---|---|---|
| SHAM | 14 | 205 ± 4 | 242 ± 6 | 101 ± 9 | 70 ± 9 | 64 ± 5 |
| OVX | 15 | 198 ± 5 | 293 ± 6* | 29 ± 4* | 39 ± 6 | 31 ± 4* |
| OVX+E2 | 10 | 205 ± 5 | 210 ± 3*† | 128 ± 13† | 150 ± 20*† | 34 ± 4* |
| OVX+P | 7 | 188 ± 3 | 273 ± 8* | 20 ± 2* | 39 ± 5 | 66 ± 3† |
Data are means ±s.e.m. OVX, ovariectomized; OVX + E2, OVX + chronic 17β-oestradiol; OVX + P, OVX + chronic progesterone.
P < 0.05vs. sham
P < 0.05vs. OVX.
Table 2.
Haemodynamics at rest and during unilateral hindlimb contraction
| Group | MAP (mmHg) | FBF(kHz) | FVC (kHz (mmHg)−1 |
|---|---|---|---|
| Rest | |||
| SHAM | 84 ± 4 | 1.18 ± 0.13 | 0.014 ± 0.002 |
| OVX | 96 ± 7 | 1.59 ± 0.20 | 0.018 ± 0.003 |
| OVX + E2 | 97 ± 5 | 1.11 ± 0.15 | 0.012 ± 0.002 |
| OVX + P | 103 ± 5 | 0.86 ± 0.04† | 0.009 ± 0.001† |
| OVX + acute E2 | 101 ± 4 | 1.53 ± 0.24 | 0.016 ± 0.003 |
| Contraction | |||
| SHAM | 88 ± 6 | 4.37 ± 0.43 | 0.049 ± 0.003 |
| OVX | 99 ± 6 | 5.51 ± 0.47 | 0.057 ± 0.005 |
| OVX + E2 | 104 ± 4 | 4.18 ± 0.35 | 0.040 ± 0.003 |
| OVX + P | 112 ± 5* | 4.19 ± 0.41 | 0.037 ± 0.002† |
| OVX + acute E2 | 117 ± 3* | 5.53 ± 1.08 | 0.048 ± 0.010 |
Data are means ±s.e.m.. MAP, mean arterial pressure; FBF, femoral blood flow velocity; FVC, femoral vascular conductance. Sham (n = 9), OVX (n = 10), OVX + E2 (n = 10), OVX + P (n = 7), OVX + acute E2 (n = 5).
P < 0.05vs. sham
P < 0.05vs. OVX.
Modulation of sympathetic vasoconstriction is impaired in contracting skeletal muscle of OVX rats (Figs 1 and 2)
Figure 1. In vivo arterial blood pressure (BP) and femoral blood flow velocity responses to sympathetic nerve stimulation in resting and contracting rat hindlimb.

A, recordings from a sham rat showing that the frequency-dependent increases in BP and decreases in blood flow velocity elicited by sympathetic nerve stimulation in resting hindlimb (left) were attenuated in contracting hindlimb (right). B, recordings from an ovariectomized rat showing an impaired attenuation of sympathetic vasoconstriction in contracting hindlimb.
Figure 2. Effect of sympathetic nerve stimulation on femoral vascular conductance in resting and contracting hindlimb of sham and ovariectomized (OVX) rats with and without chronic hormone replacement.

Compared to responses in resting hindlimb, sympathetic vasoconstriction was attenuated in contracting hindlimb of sham (n = 9) and OVX (n = 10) rats, but the degree of attenuation was significantly less in OVX rats. This effect of ovariectomy was prevented with chronic 17β-oestradiol (OVX + E2; n = 10), but not with chronic progesterone (OVX + P; n = 7). *P < 0.05vs. sham.
In sham rats, sympathetic nerve stimulation evoked graded increases in arterial pressure and decreases in femoral blood flow velocity and vascular conductance in resting hindlimb. These sympathetically mediated decreases in blood flow and conductance were significantly attenuated in contracting hindlimb of sham rats. In OVX rats, sympathetic nerve stimulation elicited graded responses in resting hindlimb similar to those observed in sham rats. In contrast, the vasoconstrictor responses to sympathetic nerve stimulation were significantly greater in contracting hindlimb of OVX versus sham rats, indicating impaired modulation of sympathetic vasoconstriction.
Impaired vasomodulation in OVX rats is prevented by chronic treatment with 17β-oestradiol, but not progesterone (Fig. 2)
In resting hindlimb, sympathetic vasoconstrictor responses were similar in OVX + E2 rats compared to sham rats. Likewise, robust vasoconstriction was evoked at each frequency of sympathetic nerve stimulation in resting hindlimb of OVX + P rats, although the responses at 1 and 2.5 Hz were smaller than in sham rats. In contracting hindlimb, sympathetic vasoconstriction was attenuated to a similar degree in OVX + E2 compared to sham rats. In contrast, sympathetic vasoconstriction was attenuated to a lesser degree in OVX + P compared to sham rats.
Impaired vasomodulation in OVX rats is not reversed by acute 17β-oestradiol replacement (Fig. 3)
Figure 3. Effect of acute administration of 17β-oestradiol (E2) on sympathetic vasoconstriction in resting and contracting hindlimb of ovariectomized (OVX) rats.
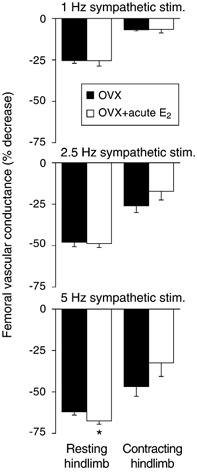
Acute E2 did not significantly attenuate vasoconstrictor responses to sympathetic nerve stimulation in resting or contracting hindlimb (n = 5). *P < 0.05vs. sham.
In OVX rats, acute infusion of 17β-oestradiol had no significant effects on arterial pressure (5 ± 3 mmHg), femoral blood flow velocity (-0.09 ± 0.11 kHz), or femoral vascular conductance (+0.001 ± 0.001 kHz mmHg−1). Acute 17β-oestradiol had no effect on the vasoconstrictor responses to sympathetic nerve stimulation in resting hindlimb, or on the degree of attenuation of sympathetic vasoconstriction in contracting hindlimb.
NOS inhibition impairs vasomodulation to a greater extent in oestrogen-replete rats than in oestrogen-deficient rats (Fig. 4)
Figure 4. Sympathetic vasoconstriction in resting and contracting hindlimb of sham and ovariectomized (OVX) rats after acute NOS inhibition with l-NAME.

Compared to responses in resting hindlimb, sympathetic vasoconstriction was attenuated similarly in contracting hindlimb of sham (n = 9), OVX (n = 9), OVX + E2 (n = 10), and OVX + P (n = 7) rats. *P < 0.05vs. sham.
Haemodynamics after systemic NOS inhibition with l-NAME are shown in Table 3. l-NAME had similar effects in all groups to increase arterial pressure and decrease femoral vascular conductance. In resting hindlimb, sympathetic vasoconstrictor responses were similar in all groups after l-NAME. In contracting hindlimb, sympathetic vasconstriction was attenuated in all groups, but in contrast to the situation before l-NAME, the degree of attenuation generally did not differ among the groups after l-NAME. Inspection of the data indicated that this was because l-NAME had a greater enhancing effect on sympathetic vasoconstrictor responses in contracting hindlimbs of sham and OVX + E2 rats compared with OVX or OVX + P rats (see Fig. 2).
Table 3.
Haemodynamics at rest and during unilateral hindlimb contraction after NOS inhibition with l-NAME
| Group | MAP (mmHg) | FBF (kHz) | FVC (kHz (mmHg)−) |
|---|---|---|---|
| Rest | |||
| SHAM | 134 ± 2 | 1.44 ± 0.17 | 0.011 ± 0.001 |
| OVX | 133 ± 5 | 1.97 ± 0.25 | 0.015 ± 0.002 |
| OVX + E2 | 127 ± 3 | 1.55 ± 0.24 | 0.013 ± 0.002 |
| OVX + P | 143 ± 3 | 1.03 ± 0.11* | 0.007 ± 0.001* |
| Contraction | |||
| SHAM | 129 ± 2 | 5.56 ± 0.46 | 0.043 ± 0.004 |
| OVX | 131 ± 4 | 7.22 ± 0.82 | 0.056 ± 0.007 |
| OVX + E2 | 127 ± 3 | 4.86 ± 0.40* | 0.039 ± 0.003 |
| OVX + P | 139 ± 2 | 4.71 ± 0.59* | 0.034 ± 0.004* |
Map, mean arterial pressure; FBF, femoral blood flow velocity; FVC, femoral vascular conductance. Sham (n = 9), OVX (n = 9). OVX + E2 (n = 10), OVX + P (n = 7).
P < 0.05vs. OVX.
Restoration of basal NO after NOS inhibition does not restore normal vasomodulation in oestrogen-replete rats (Fig. 5)
Figure 5. Sympathetic vasoconstriction in resting and contracting hindlimb of sham rats after acute NOS inhibition alone or in combination with an NO donor.

Sympathetic vasoconstriction was enhanced in contracting hindlimb after NOS inhibition with l-NAME (n = 5). In the same rats, this effect of l-NAME was not reversed by concomitant infusion of nitroprusside (NP) to restore basal NO. *P < 0.05vs. no drug.
In sham rats, the increases in arterial pressure and decreases in femoral vascular conductance produced by NOS inhibition were reversed by continuous infusion of the NO donor sodium nitroprusside (Table 4). Despite restoration of basal haemodynamics, nitroprusside infusion did not reverse the ability of l-NAME to enhance sympathetic vasoconstriction in contracting hindlimb. Peak force (2.01 ± 0.06 kg) was not significantly different after l-NAME alone (1.85 ± 0.09 kg) or after l-NAME + NP (1.88 ± 0.12 kg).
Table 4.
Haemodynamics at rest nd during unilateral hindlimb contraction after NOS inhibition alone or in combination with an NO donor
| Group | MAP (mmHg) | FBF (kHz) | FVC (kHz (mmHg)−1) |
|---|---|---|---|
| Rest | |||
| No drug | 94 ± 3 | 1.07 ± 0.27 | 0.011 ± 0.003 |
| l-NAME | 145 ± 1* | 0.83 ± 0.16 | 0.006 ± 0.001* |
| l-NAME + NP | 95 ± 4 | 0.93 ± 0.14 | 0.010 ± 0.002 |
| Contraction | |||
| No drug | 108 ± 6 | 5.29 ± 0.28 | 0.050 ± 0.005 |
| l-NAME | 135 ± 2* | 5.19 ± 0.94 | 0.039 ± 0.007 |
| l-MAME + NP | 111 ± 7 | 5.59 ± 0.78 | 0.052 ± 0.009 |
MAP, mean arterial pressure; FBF, femoral blood flow velocity; FVC, femoral vascular conductance; NP nitroprusside. n = 5.
P <0.05 vs. no drug.
Skeletal muscle nNOS is reduced in oestrogen-deficient rats and is restored by 17β -oestradiol, but not progesterone, replacement (Fig. 6)
Figure 6. Expression of nNOS in skeletal muscle homogenates from sham and ovariectomized (OVX) rats with and without hormone replacement.

A, Western blot showing nNOS immunoreactivity of gastrocnemius muscle homogenates (100 μg protein per lane). B, quantitative analysis of Western blots by densitometry. Compared to sham rats (n = 9), nNOS was reduced in muscles from OVX (n = 10) and OVX + progesterone (OVX + P; n = 7) rats, but not from OVX + 17β-oestradiol (OVX + E2; n = 10) rats. *P < 0.05vs. sham.
Immunoreactivity to nNOS in gastrocnemius muscle homogenates was reduced by 48 % in OVX compared to sham rats. Normal expression of skeletal muscle nNOS was restored by chronic treatment with 17β-oestradiol, but not by chronic treatment with progesterone. In contrast, immunoreactivity to eNOS was similar in muscle homogenates from sham (4612 ± 1060 arbitrary units; n = 9) and OVX (5895 ± 921 arbitrary units; n = 10) rats.
Skeletal muscle nNOS expression is highly correlated to plasma 17β-oestradiol and to vasomodulation in contracting skeletal muscle (Fig. 7)
Figure 7. Relationships between plasma oestradiol, skeletal muscle nNOS, and vasomodulation in contracting skeletal muscle.

A, plasma concentrations of 17β-oestradiol were correlated strongly with muscle levels of nNOS. B, muscle nNOS was correlated strongly with vasomodulation in contracting muscle expressed as the decrease in femoral vascular conductance (FVC) in response to 2.5 Hz sympathetic nerve stimulation. C, plasma 17β-oestradiol also was correlated with vasomodulation in contracting muscle. OVX, ovariectomized; E2, 17β-oestradiol; P, progesterone.
Skeletal muscle nNOS correlated directly with plasma levels of 17β-oestradiol and inversely with sympathetic vasoconstrictor responses in contracting hindlimb. Plasma 17β-oestradiol also correlated inversely with sympathetic vasoconstriction in contracting hindlimb. In contrast, there was no correlation between plasma progesterone and skeletal muscle nNOS (r = −0.004, P > 0.05) or sympathetic vasoconstriction in contracting hindlimb (r = −0.002, P > 0.05). There also was no correlation between eNOS in muscle homogenates and plasma 17β-oestradiol (r = −0.072, P > 0.05) or sympathetic vasoconstriction in contracting hindlimb (r = −0.110, P > 0.05).
Discussion
In this study, we report that the attenuation of sympathetic vasoconstriction normally observed in contracting skeletal muscle is impaired in ovariectomized rats. This impairment is prevented by chronic treatment with 17β-oestradiol, but not progesterone, strongly implicating oestrogen deficiency as the underlying cause of the altered vasomodulation in the ovariectomized rats. We also provide evidence indicating that NO, particularly that derived from skeletal muscle nNOS, may mediate the effect of 17β-oestradiol to restore normal vascular regulation in the contracting muscle of ovariectomized rats. First, pharmacological NOS inhibition reproduces the ovariectomized phenotype in sham rats and in oestradiol-treated ovariectomized rats. Second, expression of nNOS, but not eNOS, is reduced in skeletal muscle of ovariectomized rats, an effect that is prevented by 17β-oestradiol replacement. Third, skeletal muscle nNOS, but not eNOS, is directly correlated with plasma 17β-oestradiol and inversely correlated with sympathetic vasoconstrictor responses in contracting muscle.
Oestrogen deficiency previously has been associated with the downregulation of nNOS in tissues such as brain, uterus, and neutrophils (Ceccatelli et al. 1996; Pelligrino et al. 1998; García-Durán et al. 1999; Zhang et al. 1999). Our data now extend these observations to skeletal muscle, a tissue in which an alternatively spliced isoform of nNOS is abundantly expressed (Silvagno et al. 1996). Although nNOS is constitutively expressed in mature skeletal muscle fibres, previous studies have shown that the level of protein expression is subject to modulation by factors such as contractile activity (Balon & Nadler, 1997; Reiser et al. 1997), innervation (Tews et al. 1997) and mechanical loading (Tidball et al. 1998). Regulation of nNOS expression by oestradiol was first suggested by Weiner et al. (1994a), who reported that NOS activity and nNOS mRNA were increased in skeletal muscle of near-term pregnant guinea-pigs and in non-pregnant gonadally intact guinea-pigs supplemented with 500 μg kg−1 day−1 of 17β-oestradiol. This dose of oestradiol, which is approximately 4-fold higher than the replacement dose we used in the present study, was chosen to mimic the 20-fold increase in serum oestrogen observed during pregnancy. A key finding of our study is that more moderate changes in oestrogen levels also appear to modulate skeletal muscle nNOS expression. In contrast to the findings of Weiner et al. (1994a), we did not observe an effect of 17β-oestradiol on eNOS expression in skeletal muscle. This could be due to differences in the muscles analysed (vastus vs. gastrocnemius), the endpoint detected (mRNA vs. protein), the dose of 17β-oestradiol used (500 vs. 120 μg kg−1 day−1), or species studied (guinea-pig vs. rat).
Our data further suggest that the changes in skeletal muscle nNOS expression observed in oestrogen-deficient compared with oestrogen-replete rats corresponded closely to measurable functional consequences. Thus, reduced skeletal muscle nNOS in the ovariectomized rats was associated with enhanced sympathetic vasoconstriction in contracting muscle. Oestradiol replacement restored normal nNOS expression and vasomodulation in skeletal muscle of ovariectomized rats, implicating nNOS as a key determinant of the responsiveness to sympathetic activation in contracting muscle. In contrast, changes in nNOS expression had no effect on sympathetic vasoconstrictor responses in resting skeletal muscle. This is probably due to differences in nNOS activity in resting and contracting muscle. Production of NO is low in resting muscle, but is increased substantially in contracting muscle presumably due to calcium- or mechanically induced activation of nNOS (Stamler & Meissner, 2001).
Increasing evidence indicates that some of the NO-dependent vascular effects of oestradiol are mediated by rapid, non-genomic mechanisms that enhance the catalytic activity of eNOS (Lantin-Hermoso et al. 1997; Shaul, 1999; Mendelsohn, 2000; Wyckoff et al. 2001). Oestradiol also may modulate nNOS activity acutely, although the effect appears to be biphasic such that nNOS activity is enhanced by low concentrations of oestrogen (0.1–100 nm) and attenuated by high concentrations (1–10 μm) (Hayashi et al. 1994). In our study, acute administration of oestradiol did not significantly improve the impaired modulation of sympathetic vasoconstriction in the contracting hindlimb of ovariectomized rats. In contrast, the finding that chronic oestradiol normalized sympathetic vasomodulation in ovariectomized rats indicates a predominant role for a delayed rather than rapid effect of oestradiol.
The upregulation of skeletal muscle nNOS that we observed in the ovariectomized rats treated chronically with oestradiol suggests a possible genomic mechanism of action of oestradiol. Transcriptional regulation by oestrogen is governed by its binding to the oestrogen receptors α and β, which are members of the steroid hormone superfamily of nuclear receptors (Nilsson et al. 2001). Oestrogen receptors are expressed at low levels in skeletal muscle, but their function is poorly understood (Dionne et al. 1979; Dahlberg, 1982). In skeletal myoblasts, oestrogen receptors modulate growth by transactivation of the immediate-early genes c-fos and egr-1 (Kahlert et al. 1997), perhaps by binding to full or half palindromic oestrogen response elements (EREs) in the promotor sequences of these genes. Although the nNOS promotor does not contain EREs, there is a consensus sequence for binding of the transcription factor Sp1 (Förstermann et al. 1998), which recently has been implicated in the oestrogen-induced transcription of the human eNOS gene (Kleinert et al. 1998).
Despite the strong association between chronic changes in oestradiol and skeletal muscle nNOS expression, we cannot definitively exclude other mechanisms by which oestradiol may impact vasomodulation in contracting muscle. For example, chronic oestradiol may attenuate vascular responses by suppressing expression of vascular α-adrenoceptors (Zhang & Davidge, 1999), reducing perivascular sympathetic innervation (Zoubina & Smith, 2001), or by modulating intracellular signalling pathways, such as decreasing protein kinase C activity (Kanashiro & Khalil, 2001) or calcium influx (Collins et al. 1993; Zhang et al. 1994). However, because altering oestrogen status did not have a measurable impact on sympathetic vasoconstriction in resting skeletal muscle in our study, it is unlikely that changes in vascular function per se underlie the differential modulation of vasoconstriction in contracting muscle of oestrogen-deficient versus oestrogen-replete rats. Oestrogen also is reported to increase fat oxidation by skeletal muscle (Campbell & Febbraio, 2001), which might alter vascular responsiveness by changing the metabolic profile of contracting muscle. Finally, oestrogen possesses antioxidant properties that may confer vasoprotection by scavenging oxygen free radicals such as the superoxide anion (Ayres et al. 1998; Barbacanne et al. 1999). We previously have shown in a rat model of heart failure that increased superoxide production in contracting muscle results in enhanced sympathetic vasoconstriction, presumably due to superoxide-mediated inactivation of NO (Thomas et al. 2001). Thus, by decreasing superoxide, oestrogen could increase the bioavailability of NO and improve the NO-dependent attenuation of sympathetic vasoconstriction in contracting muscle.
Although eNOS expression in skeletal muscle homogenates was not altered by changes in oestrogen status in our study, we cannot completely discount eNOS as a potential source of vasomodulatory NO in contracting muscle. Our attempts to distinguish effects of nNOS from those of eNOS in our rat model using NOS inhibitors with reportedly greater selectivity for nNOS than eNOS (7-nitroindazole, 1-(2-trifluoromethylphenyl) imidazole, 1400W) (Boucher et al. 1999) have been unsuccessful to date because large systemic doses of these compounds produced significant changes in basal haemodynamics that confounded interpretation of the experiments (W. Zhao & G. D. Thomas, unpublished observations). However, ex vivo experiments on skeletal muscle from nNOS or eNOS knockout mice have indicated that nNOS is the predominant source of contraction-induced increases in NO (Hirschfield et al. 2000; Lau et al. 2000), whereas eNOS is the principal source of tonically produced NO that maintains basal vascular tone (Grange et al. 2001). Our data showing that infusion of nitroprusside to restore basal NO did not reverse the impaired vasomodulation in NOS-inhibited rats suggest that newly synthesized, rather than tonically produced, NO is required for the attenuation of sympathetic vasoconstriction in contracting muscle.
Compared to the large number of studies that have examined the effects of chronic changes in oestrogen levels on vascular function, relatively few studies have focused on the role of progesterone. In our study, sympathetic vasoconstriction was attenuated in resting muscle of ovariectomized rats treated chronically with progesterone compared with responses in untreated ovariectomized rats or sham rats. These data are consistent with previous studies using isolated arteries in which progesterone was reported to diminish the vasoconstrictor response to exogenous noradrenaline (Dogterom & De Jong, 1974; Mukerji et al. 2000). Despite this effect of progesterone on resting muscle, sympathetic vasoconstrictor responses in contracting muscle were greater in progesterone-treated ovariectomized rats than in sham rats, excluding a role for progesterone as a key factor mediating the vasomodulatory effect of muscle contraction.
In summary, these data indicate that oestrogen deficiency alters vasoregulation in contracting skeletal muscle by impairing the normal attenuation of sympathetic vasoconstriction. Such defective modulation of sympathetic vasoconstrictor responses in oestrogen-deficient states may be mediated by reduced skeletal muscle nNOS, which is an important source of NO in contracting muscle. In previous studies, we have observed attenuated α-adrenergic vasoconstriction in contracting skeletal muscle of both male and female mice as well as human subjects (Thomas et al. 1998; Sander et al. 2000; Chavoshan et al. 2002). Because oestradiol levels in males are low, within the range reported for females following menopause or ovariectomy, we speculate that there may be sex-related differences in some of the mechanisms that modulate sympathetic vasoconstriction in exercising skeletal muscle. In this regard, it will be interesting to compare the role of NO in the attenuation of sympathetic vasoconstriction in contracting muscle of male and female rats.
Acknowledgments
This work was supported by NIH grant HL64784 (G.D.T.). P.J.F. was supported by NIH training grant HL07360 and individual NRSA HL69648.
References
- Anderson KM, Faber JE. Differential sensitivity of arteriolar α1- and α2-adrenoceptor constriction to metabolic inhibition during rat skeletal muscle contraction. Circ Res. 1991;69:174–184. doi: 10.1161/01.res.69.1.174. [DOI] [PubMed] [Google Scholar]
- Ayres S, Abplanalp W, Liu JH, Subbiah MT. Mechanisms involved in the protective effect of estradiol-17β on lipid peroxidation and DNA damage. Am J Physiol. 1998;274:E1002–1008. doi: 10.1152/ajpendo.1998.274.6.E1002. [DOI] [PubMed] [Google Scholar]
- Balon TW, Nadler JL. Evidence that nitric oxide increases glucose transport in skeletal muscle. J Appl Physiol. 1997;82:359–363. doi: 10.1152/jappl.1997.82.1.359. [DOI] [PubMed] [Google Scholar]
- Barbacanne MA, Rami J, Michel JB, Souchard JP, Philippe M, Besombes JP, Bayard F, Arnal JF. Estradiol increases rat aorta endothelium-derived relaxing factor (EDRF) activity without changes in endothelial NO synthase gene expression: possible role of decreased endothelium-derived superoxide anion production. Cardiovasc Res. 1999;41:672–681. doi: 10.1016/s0008-6363(98)00254-5. [DOI] [PubMed] [Google Scholar]
- Boucher JL, Moali C, Tenu JP. Nitric oxide biosynthesis, nitric oxide synthase inhibitors and arginase competition for l-arginine utilization. Cell Mol Life Sci. 1999;55:1015–1028. doi: 10.1007/s000180050352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Xia H, Aldape K, Bredt DS. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995;82:743–752. doi: 10.1016/0092-8674(95)90471-9. [DOI] [PubMed] [Google Scholar]
- Burcher E, Garlick D. Antagonism of vasoconstrictor responses by exercise in the gracilis muscle of the dog. J Pharmacol Exp Ther. 1973;187:78–85. [PubMed] [Google Scholar]
- Burcher E, Garlick D. Effects of exercise metabolites on adrenergic vasoconstriction in the gracilis muscle of the dog. J Pharmacol Exp Ther. 1975;192:149–156. [PubMed] [Google Scholar]
- Campbell SE, Febbraio MA. Effect of ovarian hormones on mitochondrial enzyme activity in the fat oxidation pathway of skeletal muscle. Am J Physiol Endocrinol Metab. 2001;281:E803–808. doi: 10.1152/ajpendo.2001.281.4.E803. [DOI] [PubMed] [Google Scholar]
- Ceccatelli S, Grandison L, Scott REM, Pfaff DW, Kow L-M. Estradiol regulation of nitric oxide synthase mRNAs in rat hypothalamus. Neuroendocrinology. 1996;64:357–363. doi: 10.1159/000127139. [DOI] [PubMed] [Google Scholar]
- Chang WJ, Iannaccone ST, Lau KS, Masters BSS, McCabe TJ, McMillan K, Padre RC, Spencer MJ, Tidball JG, Stull JT. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc Natl Acad Sci U S A. 1996;93:9142–9147. doi: 10.1073/pnas.93.17.9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavoshan B, Sander M, Sybert TE, Hansen J, Victor RG, Thomas GD. Nitric oxide-dependent modulation of sympathetic neural control of oxygenation in exercising human skeletal muscle. J Physiol. 2002;540:377–386. doi: 10.1113/jphysiol.2001.013153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins P, Rosano GM, Jiang C, Lindsay D, Sarrel PM, Poole-Wilson PA. Cardiovascular protection by oestrogen - a calcium antagonist effect? Lancet. 1993;341:1264–1265. doi: 10.1016/0140-6736(93)91158-i. [DOI] [PubMed] [Google Scholar]
- Delp MD, Laughlin MH. Regulation of skeletal muscle perfusion during exercise. Acta Physiol Scand. 1998;162:411–419. doi: 10.1046/j.1365-201X.1998.0324e.x. [DOI] [PubMed] [Google Scholar]
- Dionne FT, Lesage RL, Dube JY, Tremblay RR. Estrogen binding proteins in rat skeletal and perineal muscles: in vitro and in vivo studies. J Steroid Biochem. 1979;11:1073–1080. doi: 10.1016/0022-4731(79)90156-0. [DOI] [PubMed] [Google Scholar]
- Dogterom J, De Jong W. Diminished pressor response to noradrenaline of the perfused tail artery of pregnant rats. Eur J Pharmacol. 1974;25:267–269. doi: 10.1016/0014-2999(74)90062-4. [DOI] [PubMed] [Google Scholar]
- Fleming BP, Gibbins IL, Morris JL, Gannnon BJ. Noradrenergic and peptidergic innervation of the extrinsic vessels and microcirculation of the rat cremaster muscle. Microvasc Res. 1989;38:255–268. doi: 10.1016/0026-2862(89)90004-6. [DOI] [PubMed] [Google Scholar]
- Förstermann U, Boissel J-P, Kleinert H. Expressional control of the ‘constitutive’ isoforms of nitric oxide synthase (NOS I and NOS III) FASEB J. 1998;12:773–790. [PubMed] [Google Scholar]
- Fuxe K, Sedvall G. The distribution of adrenergic nerve fibres to the blood vessels in skeletal muscle. Acta Physiol Scand. 1965;64:75–86. doi: 10.1111/j.1748-1716.1965.tb04155.x. [DOI] [PubMed] [Google Scholar]
- García-Durán M, De Frutos T, Díaz-Recasens J, García-Gálvez G, Jiménez A, Montón M, Farré J, Sánchez De Miguel L, González-Fernández F, Del Mar Arriero M, Rico L, García R, Casado S, López-Farré A. Estrogen stimulates neuronal nitric oxide synthase protein expression in human neutrophils. Circ Res. 1999;85:1020–1026. doi: 10.1161/01.res.85.11.1020. [DOI] [PubMed] [Google Scholar]
- Grange RW, Isotani E, Lau KS, Kamm KE, Huang PL, Stull JT. Nitric oxide contributes to vascular smooth muscle relaxation in contracting fast-twitch muscles. Physiol Genomics. 2001;5:35–44. doi: 10.1152/physiolgenomics.2001.5.1.35. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Ishikawa T, Yamada K, Kuzuya M, Naito M, Hidaka H, Iguchi A. Biphasic effect of estrogen on neuronal constitutive nitric oxide synthase via Ca2+-calmodulin dependent mechanism. Biochem Biophys Res Commun. 1994;203:1013–1019. doi: 10.1006/bbrc.1994.2283. [DOI] [PubMed] [Google Scholar]
- Hirschfield W, Moody MR, O'Brien WE, Gregg AR, Bryan R M, Jr, Reid MB. Nitric oxide release and contractile properties of skeletal muscles from mice deficient in type III NOS. Am J Physiol. 2000;278:R95–100. doi: 10.1152/ajpregu.2000.278.1.R95. [DOI] [PubMed] [Google Scholar]
- Kahlert S, Grohe C, Karas RH, Lobbert K, Neyses L, Vetter H. Effects of estrogen on skeletal myoblast growth. Biochem Biophys Res Commun. 1997;232:373–378. doi: 10.1006/bbrc.1997.6223. [DOI] [PubMed] [Google Scholar]
- Kanashiro CA, Khalil RA. Gender-related distinctions in protein kinase C activity in rat vascular smooth muscle. Am J Physiol Cell Physiol. 2001;280:C34–45. doi: 10.1152/ajpcell.2001.280.1.C34. [DOI] [PubMed] [Google Scholar]
- Kleinert H, Wallerath T, Euchenhofer C, Ihrig-Biedert I, Li H, Förstermann U. Estrogens increase transcription of the human endothelial NO synthase gene. Analysis of the transcription factors involved. Hypertension. 1998;31:582–588. doi: 10.1161/01.hyp.31.2.582. [DOI] [PubMed] [Google Scholar]
- Kobzik L, Reid MB, Bredt DS, Stamler JS. Nitric oxide in skeletal muscle. Nature. 1994;372:546–548. doi: 10.1038/372546a0. [DOI] [PubMed] [Google Scholar]
- Kobzik L, Stringer B, Balligand J-L, Reid MB, Stamler JS. Endothelial type nitric oxide synthase in skeletal muscle fibers: mitochondrial relationships. Biochem Biophys Res Commun. 1995;211:375–381. doi: 10.1006/bbrc.1995.1824. [DOI] [PubMed] [Google Scholar]
- Lantin-Hermoso RL, Rosenfeld CR, Yuhanna IS, German Z, Chen Z, Shaul PW. Estrogen acutely stimulates nitric oxide synthase activity in fetal pulmonary artery endothelium. Am J Physiol. 1997;273:L119–126. doi: 10.1152/ajplung.1997.273.1.L119. [DOI] [PubMed] [Google Scholar]
- Lash JM. Regulation of skeletal muscle blood flow during contractions. Proc Soc Exp Biol Med. 1996;211:218–235. doi: 10.3181/00379727-211-43965. [DOI] [PubMed] [Google Scholar]
- Lau KS, Grange RW, Isotani E, Sarelius IH, Kamm KE, Huang PL, Stull JT. nNOS and eNOS modulate cGMP formation and vascular response in contracting fast-twitch skeletal muscle. Physiol Genomics. 2000;2:21–27. doi: 10.1152/physiolgenomics.2000.2.1.21. [DOI] [PubMed] [Google Scholar]
- Mendelsohn ME. Nongenomic, estrogen receptor-mediated activation of endothelial nitric oxide synthase. How does it work? What does it mean? Circ Res. 2000;87:677–682. doi: 10.1161/01.res.87.11.956. [DOI] [PubMed] [Google Scholar]
- Mukerji MS, Leathard HL, Huddart H. The effect of progesterone on spontaneous and agonist-evoked contractions of the rat aorta and portal vein. J Pharm Pharmacol. 2000;52:843–849. doi: 10.1211/0022357001774525. [DOI] [PubMed] [Google Scholar]
- Nakane M, Schmidt HH, Pollock JS, Forstermann U, Murad F. Cloned human brain nitric oxide synthase is highly expressed in skeletal muscle. FEBS Lett. 1993;316:175–180. doi: 10.1016/0014-5793(93)81210-q. [DOI] [PubMed] [Google Scholar]
- Nilsson S, Mäkelä S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- Pelligrino DA, Santizo R, Baughman VL, Wang Q. Cerebral vasodilating capacity during forebrain ischemia: effects of chronic estrogen depletion and repletion and the role of neuronal nitric oxide synthase. Neuroreport. 1998;9:3285–3291. doi: 10.1097/00001756-199810050-00026. [DOI] [PubMed] [Google Scholar]
- Reiser PJ, Kline WO, Vaghy PL. Induction of neuronal type nitric oxide synthase in skeletal muscle by chronic electrical stimulation in vivo. J Appl Physiol. 1997;82:1250–1255. doi: 10.1152/jappl.1997.82.4.1250. [DOI] [PubMed] [Google Scholar]
- Remensnyder JP, Mitchell JH, Sarnoff SJ. Functional sympatholysis during muscular activity. Circ Res. 1962;11:370–380. doi: 10.1161/01.res.11.3.370. [DOI] [PubMed] [Google Scholar]
- Rowell LB, O'Leary DS. Reflex control of the circulation during exercise: chemoreflexes and mechanoreflexes. J Appl Physiol. 1990;69:407–418. doi: 10.1152/jappl.1990.69.2.407. [DOI] [PubMed] [Google Scholar]
- Rowlands DJ, Donald DE. Sympathetic vasoconstrictive responses during exercise- or drug-induced vasodilation. Circ Res. 1968;23:45–60. doi: 10.1161/01.res.23.1.45. [DOI] [PubMed] [Google Scholar]
- Sander M, Chavoshan B, Harris SA, Iannoccone ST, Stull JT, Thomas GD, Victor RG. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 2000;97:13818–13823. doi: 10.1073/pnas.250379497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals DR, Victor RG. Regulation of muscle sympathetic nerve activity during exercise in humans. Exerc Sport Sci Rev. 1991;19:313–349. [PubMed] [Google Scholar]
- Shaul PW. Rapid activation of endothelial nitric oxide synthase by estrogen. Steroids. 1999;64:28–34. doi: 10.1016/s0039-128x(98)00105-6. [DOI] [PubMed] [Google Scholar]
- Silvagno F, Xia H, Bredt DS. Neuronal nitric-oxide synthase-μ, an alternatively spliced isoform expressed in differentiated skeletal muscle. J Biol Chem. 1996;271:11204–11208. doi: 10.1074/jbc.271.19.11204. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81:209–237. doi: 10.1152/physrev.2001.81.1.209. [DOI] [PubMed] [Google Scholar]
- Tews DS, Goebel HH, Schneider I, Gunkel A, Stennert E, Neiss WF. Expressionn of different isoforms of nitric oxide synthase in experimentally denervated and reinnervated skeletal muscle. J Neuropathol Exp Neurol. 1997;56:1283–1289. doi: 10.1097/00005072-199712000-00003. [DOI] [PubMed] [Google Scholar]
- Thomas GD, Sander M, Lau KS, Huang PL, Stull JT, Victor RG. Impaired metabolic modulation of α-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc Natl Acad Sci U S A. 1998;95:15090–15095. doi: 10.1073/pnas.95.25.15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GD, Victor RG. Nitric oxide mediates contraction-induced attenuation of sympathetic vasoconstriction in rat skeletal muscle. J Physiol. 1998;506:817–826. doi: 10.1111/j.1469-7793.1998.817bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GD, Zhang W, Victor RG. Impaired modulation of sympathetic vasoconstriction in contracting skeletal muscle of rats with chronic myocardial infarctions. Role of oxidative stress. Circ Res. 2001;88:816–823. doi: 10.1161/hh0801.089341. [DOI] [PubMed] [Google Scholar]
- Tidball JG, Lavergne E, Lau KS, Spencer MJ, Stull JT, Wehling M. Mechanical loading regulates NOS expression and activity in developing and adult skeletal muscle. Am J Physiol. 1998;275:C260–266. doi: 10.1152/ajpcell.1998.275.1.C260. [DOI] [PubMed] [Google Scholar]
- Verhaeghe RH, Lorenz RR, Mcgrath MA, Shepherd JT, Vanhoutte PM. Metabolic modulation of neurotransmitter release-adenosine, adenine nucleotides, potassium, hyperosmolarity, and hydrogen ion. Fed Proc. 1978;37:208–211. [PubMed] [Google Scholar]
- Weiner CP, Knowles RG, Moncada S. Induction of nitric oxide synthases early in pregnancy. Am J Obstet Gynecol. 1994a;171:838–843. doi: 10.1016/0002-9378(94)90108-2. [DOI] [PubMed] [Google Scholar]
- Weiner CP, Lizasoain I, Baylis SA, Knowles RG, Charles IG, Moncada S. Induction of calcium-dependent nitric oxide synthases by sex hormones. Proc Natl Acad Sci U S A. 1994b;91:5212–5216. doi: 10.1073/pnas.91.11.5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff MH, Chambliss KL, Mineo C, Yuhanna IS, Mendelsohn ME, Mumby SM, Shaul PW. Plasma membrane estrogen receptors are coupled to endothelial nitric-oxide synthase through Gαi. J Biol Chem. 2001;276:27071–27076. doi: 10.1074/jbc.M100312200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Davidge ST. Effect of estrogen replacement on vasoconstrictor responses in rat mesenteric arteries. Hypertension. 1999;34:1117–1122. doi: 10.1161/01.hyp.34.5.1117. [DOI] [PubMed] [Google Scholar]
- Zhang J, Massmann GA, Mirabile CP, Figueroa JP. Nonpregnant sheep uterine type I and type III nitric oxide synthase is differentially regulated by estrogen. Biol Reprod. 1999;60:1198–1203. doi: 10.1095/biolreprod60.5.1198. [DOI] [PubMed] [Google Scholar]
- Zhang F, Ram JL, Standley PR, Sowers JR. 17β-estradiol attenuates voltage-dependent Ca++ currents in A7r5 vascular smooth muscle cell line. Am J Physiol. 1994;266:C975–980. doi: 10.1152/ajpcell.1994.266.4.C975. [DOI] [PubMed] [Google Scholar]
- Zoubina EV, Smith PG. Sympathetic hyperinnervation of the uterus in the estrogen receptor alpha knock-out mouse. Neuroscience. 2001;103:237–244. doi: 10.1016/s0306-4522(00)00549-2. [DOI] [PubMed] [Google Scholar]