

Abstract
At synapses, ATP is released and metabolised through ecto-nucleotidases forming adenosine, which modulates neurotransmitter release through inhibitory A1 or facilitatory A2A receptors, according to the amounts of extracellular adenosine. Neuromuscular junctions possess an ecto-AMP deaminase that can dissociate extracellular ATP catabolism from adenosine formation. In this study we have investigated the pattern of ATP release and its conversion into adenosine, to probe the role of ecto-AMP deaminase in controlling acetylcholine release from rat phrenic nerve terminals. Nerve-evoked ATP release was 28 ± 12 pmol (mg tissue)−1 at 1 Hz, 54 ± 3 pmol (mg tissue)−1 at 5 Hz and disproportionally higher at 50 Hz (324 ± 23 pmol (mg tissue)−1). Extracellular ATP (30 μm) was metabolised with a half time of 8 ± 2 min, being converted into ADP then into AMP. AMP was either dephosphorylated into adenosine by ecto-5′-nucleotidase (inhibited by ATP and blocked by 200 μmα,β-methylene ADP) or deaminated into IMP by ecto-AMP deaminase (inhibited by 200 μm deoxycoformycin, which increased adenosine formation). Dephosphorylation and deamination pathways also catabolised endogenously released adenine nucleotides, since the nerve-evoked extracellular AMP accumulation was increased by either α,β-methylene ADP (200 μm) or deoxycoformycin (200 μm). In the presence of nitrobenzylthioinosine (30 μm) to inhibit adenosine transport, deoxycoformycin (200 μm) facilitated nerve-evoked [3H]acetylcholine release by 77 ± 9 %, an effect prevented by the A2A receptor antagonist, ZM 241385 (10 nm). It is concluded that, while ecto-5′-nucleotidase is inhibited by released ATP, ecto-AMP deaminase activity transiently blunts adenosine formation, which would otherwise reach levels high enough to activate facilitatory A2A receptors on motor nerve terminals.
At the rat neuromuscular junction, adenosine acts as a neuromodulator either inhibiting (via A1 receptors) or facilitating (via A2A receptors) the release of acetylcholine (ACh) from motor nerve endings (Correia-de-Sáet al. 1991; reviewed by Ribeiro et al. 1996). The effect of endogenous extracellular adenosine depends on the pattern of nerve stimulation: at low stimulation frequencies there is a predominant tonic inhibition mediated by A1 receptors, whereas with increasing frequencies adenosine facilitates ACh release through tonic activation of A2A receptors (Correia-de-Sá et al. 1996). Interestingly, which adenosine receptor is predominantly activated is apparently determined by the differential contribution of the two main pathways leading to extracellular adenosine accumulation (Correia-de-Sá et al. 1996; Cunha et al. 1996a). Indeed, adenosine can either be released as such or can be formed upon the sequential extracellular dephosphorylation of released ATP (reviewed in Cunha, 2001a).
ATP contained in synaptic vesicles is released from stimulated nerve terminals as well as from the activated postsynaptic components (reviewed in Bodin & Burnstock, 2001). Once released, ATP can act as a fast neurotransmitter in some synapses or as a presynaptic neuromodulator (reviewed in Cunha & Ribeiro, 2000). Most commonly, released ATP is metabolised extracellularly into adenosine by a cascade of ecto-nucleotidases (reviewed in Cunha, 2001b). For the convenience of the reader, the pathways responsible for the extracellular catabolism of ATP and adenosine as well as the sites of action of various inhibitors are depicted in Fig. 1. With increasing frequencies of nerve stimulation, there is an increased contribution of ATP-derived adenosine (Cunha et al. 1996b). Since ATP is released in a frequency-dependent manner (Silinsky, 1975; Wieraszko et al. 1989; Cunha & Sebastião, 1993; Cunha et al. 1996b), it can reach levels high enough to inhibit ecto-5′-nucleotidase, the enzyme that forms adenosine from released adenine nucleotides (discussed in Cunha, 2001b). Under these conditions, there is a delayed burst-like formation of adenosine leading to high synaptic concentrations of the nucleoside similar to those required to activate facilitatory A2A receptors (Correia-de-Sá et al. 1996; Cunha et al. 1996a; reviewed by Cunha, 2001a,2001b). However, the relation between ATP release and adenosine receptor activation at the neuromuscular junction is complicated by the observation that AMP can also be extracellularly deaminated (Alertsen et al. 1958; Cunha & Sebastião, 1991) into the inactive metabolite IMP (e.g. Ribeiro & Sebastião, 1987), therefore bypassing adenosine formation. Understanding the effective contribution of this shunt-like deamination pathway is of central importance to predict when the neuromodulatory role of adenosine will shift from inhibition to facilitation with increasing frequencies of motoneuronal firing.
Figure 1. Schematic representation of the extracellular catabolism of ATP and adenosine.
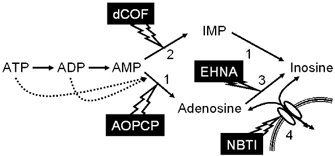
The solid arrows indicate the enzymatic activities identified. Dotted arrows represent inhibition of the ecto-5′-nucleotidase activity by ATP and ADP. The numbers in the figure represent: 1, ecto-5′-nucleotidase; 2, ecto-AMP deaminase; 3, ecto-adenosine deaminase; 4, adenosine transporter. The lightning-like arrows indicate the enzymatic activities that are the main targets of the indicated drugs (AOPCP, α,β-methylene ADP; dCOF, deoxycoformycin; ENHA, erythro-9-(2-hydroxy-3-nonyl)adenine; NBTI, S-(p-nitrobenzyl)-6-thioinosine). Note that dCOF also inhibits adenosine deaminase as well as adenosine transporters, EHNA inhibits adenosine deaminase, and AOPCP inhibits both the dephosphorylation of AMP as well as that of IMP.
In the present work, we studied the pattern of ATP release and its extracellular catabolism with particular emphasis on the relative contribution of ecto-5′-nucleotidase (forming adenosine) and of ecto-AMP deaminase (bypassing adenosine formation) pathways. We then used a pharmacological strategy to block ecto-AMP deaminase to probe the importance of this shunt-like pathway for the tonic adenosine modulation of ACh release.
METHODS
Experimental preparation
Rats (Wistar, 150–200 g) of either sex (Charles River, Barcelona, Spain) were kept at a constant temperature (21 ° C) and a regular light (06.30–19.30 h) dark (19.30–06.30 h) cycle with food and water provided ad libitum. The animals were decapitated and exsanguinated under halothane anaesthesia, according to the Portuguese and EU guidelines for handling and use of experimental animals. All experiments were conducted using left phrenic nerve-hemidiaphragm preparations (6-8 mm width) (see Correia-de-Sá et al. 1996). The preparations were mounted in 2 ml chambers, with direct oxygenation (95 % O2 and 5 % CO2), kept at 37 °C, and superfused (3 ml min−1) with gassed Tyrode solution containing (mm): NaCl 137.0, KCl 2.7, CaCl2 1.8, MgCl2 1.0, NaH2PO4 0.4, NaHCO3 11.9, and glucose 11.2. The nerve was then drawn into a suction electrode for stimulation. The tissue was allowed to equilibrate, under superfusion with gassed Tyrode solution, for 30 min.
Kinetic experiments
For kinetic experiments of purine catabolism, after the 30 min equilibration period, the bath was emptied and 2 ml of a 3–100 μm solution of the chosen initial substrate in Tyrode solution at 37 °C was added to the preparations at zero time. Samples of 75 μl were collected from the bath at different times up to 45 min for HPLC analysis of the variation of substrate disappearance and product formation (see Fig. 1) (Cunha & Sebastião, 1991; Salgado et al. 2000). When the modification of the extracellular catabolism of an initial substrate by an inhibitor was tested, the preparations were first incubated for at least 15 min with the modifiers before starting the kinetic experiment still in the presence of the modifier (see Cunha & Sebastião, 1991). In all experiments, the concentration of products at the different times of sample collection was corrected by subtracting the concentration of products in samples collected from the same preparation incubated without adding substrate. Only IMP, inosine and hypoxanthine were spontaneously released from the preparations in concentrations that did not exceed 1.4 μm.
[3H]Acetylcholine release experiments
The procedures used for labelling the preparations and measuring evoked [3H]acetylcholine ([3H]ACh) release have been previously described (Correia-de-Sá et al. 1991; 1996) and were used with minor modifications. Experiments were performed in the absence of cholinesterase inhibitors to prevent non-physiological extracellular accumulation of acetylcholine that might exaggerate cholinergic neuromodulation (cf. Correia-de-Sá & Ribeiro, 1994a; Oliveira et al. 2002). After a 30 min equilibration period, the superfusion was stopped and the nerve endings were labelled for 40 min with 1 μm[3H]choline (specific activity 2.5 μCi nmol−1) under electrical stimulation at 1 Hz. After the end of the labelling period, the preparations were again superfused (15 ml min−1) and the nerve stimulation stopped. From this time onwards, hemicholinium-3 (10 μm) was present to prevent uptake of choline. After a 60 min period of washout, bath samples (2 ml) were automatically collected every 3 min by emptying and refilling the organ bath with the solution in use, using a fraction collector (Gilson, FC 203B, France) coupled to a peristaltic pump (Gilson, Minipuls3, France) programmed device. Aliquots (0.5 ml) of the incubation medium were added to 3.5 ml of Packard Insta Gel II (USA) scintillation cocktail. Tritium content of the samples was measured by liquid scintillation (counting efficiency of 40 ± 2 %) after appropriate background subtraction, which did not exceed 5 % of the tritium content of the samples. The radioactivity was expressed as disintegrations per minute (DPM) per gram wet weight of the tissue determined at the end of the experiment. After the loading and washout periods, the preparations contained (5542 ± 248) × 103 DPM g−1 and the resting release was (132 ± 12) × 103 DPM g−1 in 3 min (n = 8). When the fractional release was calculated, this value was 2.38 ± 0.14 % of the radioactivity present in the tissue in the first collected sample.
Release of [3H]ACh was evoked by electrical stimulation of the phrenic nerve with 5 Hz frequency trains applied over 2.5 min. Supramaximal intensity rectangular pulses of 40 μs duration and a current strength of 8 mA were used. This was done to achieve synchronisation of phrenic motoneuron firing in order to reduce the number of silent units. Pulses were generated by a Grass S48 (USA) stimulator coupled to a stimulus isolation unit (Grass SIU5) operating in a constant current mode. The stimulation parameters were continuously monitored on an oscilloscope (Meguro, MO-1251A, Japan). Two stimulation periods were used: at 12 min (S1) and at 39 min (S2) after the end of washout (zero time). Electrical stimulation of the phrenic nerve increased only the release of [3H]ACh in a Ca2+- and tetrodotoxin-sensitive manner (Correia-de-Sáet al. 2000), while the output of [3H]choline remained unchanged during the stimulation periods (Wessler & Kilbinger, 1986). Therefore, the evoked release of [3H]ACh was calculated by subtracting the basal tritium outflow from the total tritium outflow during the stimulation period (cf. Correia-de-Sá et al. 1996).
Test drugs were added 15 min before S2 and were present up to the end of the experiments. The change in the ratio between the evoked [3H]ACh released during the two stimulation periods (S2/S1) relative to that observed in control situations (in the absence of test drugs) was taken as a measure of the effect of the tested drugs. When we evaluated the modifications of the effect of tested drugs by a modifier, this modifier was applied 15 min before starting sample collection and hence was present during S1 and S2. When present during S1 and S2, none of the tested modifiers significantly altered (P > 0.05) the S2/S1 ratio as compared to the S2/S1 ratio obtained in the absence of the modifiers (data not shown).
Release of ATP and of adenine nucleotides
For the ATP release experiments or to follow the nerve-evoked release of adenine nucleotides, after the 30 min equilibration period, the preparations were incubated as for the release of [3H]ACh, except that no [3H]choline was added to the Tyrode solution, although the 30 min prestimulation period (simulating loading) was maintained. The preparations were then superfused (3 ml min−1) for 60 min with gassed Tyrode solution containing hemicholinium-3 (10 μm) that was present from then on. After stopping superfusion, bath samples (1.2 ml) were collected every 2.5 min by emptying and refilling the organ bath with the solution in use. As for the release of [3H]ACh, the preparations were also stimulated twice using similar nerve stimulating conditions. When using the 5 Hz protocol, stimulation was delivered at a frequency of 5 Hz in two 2.5 min periods, at 7.5 min (S1) and at 32.5 min (S2) after starting sample collection (zero time). When using the 1 Hz protocol, stimulation was delivered at a frequency of 1 Hz in two 12.5 min periods, at 7.5 min (S1) and at 32.5 min (S2) after starting sample collection (zero time). When using the 50 Hz protocol, stimulation was delivered at a frequency of 50 Hz in two 15 s periods, at 7.5 min (S1) and at 32.5 min (S2) after starting sample collection (in this particular protocol, the bathing medium was changed immediately after stopping the application of the pulse). In these three protocols (where the number of pulses was kept constant), only the sample collected before stimulus application and the sample collected immediately after stimulation, were retained for analysis.
Aliquots of 250 μl of each sample were used for the luminometric assay of ATP conducted with the luciferin-luciferase assay (see Cunha et al. 1996b). To measure AMP by reverse-phase HPLC, we used 200 μl aliquots from collected samples (see Cunha & Sebastião, 1993). Nerve-evoked release of ATP (or adenine nucleotides) was calculated by subtracting the basal release, measured in the sample collected before stimulation, from the total release of ATP (or adenine nucleotides) determined after stimulus application. When testing the ability of a modifier to affect ATP or adenine nucleotide release, this modifier was added to the bath 15 min before starting sample collection and was present throughout the protocol.
Reagents
ATP, ADP, AMP, IMP, adenosine, inosine, α,β-methylene ADP (AOPCP), S-(p-nitrobenzyl)-6-thioinosine (NBTI), hemicholinium-3, tetrodotoxin, luciferin and luciferase (ATP assay mix) were from Sigma Ibérica, 1,3-dipropyl-8-cyclopenthylxanthine (DPCPX) was from Research Biochemicals Inc. (Sigma Ibérica), erythro-9-(2-hydroxy-3-nonyl)adenine (EHNA) was from Burroughs Wellcome, dipyridamole was from Boehringer Ingelheim (Germany), 4-(2-[7-amino-2-(2-furyl) (1,2,4)triazolo(2,3-a)(1,3,5)triazin-5-ylamino]ethyl)phenol (ZM 241385) was from Tocris Cookson (UK), [methyl-3H]choline chloride (80 Ci mmol−1) was from Amersham (Pharmacia, Portugal) and deoxycoformycin was from Parke-Davies.
Dipyridamole, EHNA, NBTI, deoxycoformycin and ZM241385 were made up to a 5 mm stock solution in dimethylsulfoxide and DPCPX was made up into 5 mm stock in 99 % dimethylsulfoxide and 1 % NaOH (1 m). These stock solutions were aliquoted and stored at −20 °C and aqueous dilution of these solutions was made daily.
Statistics
The values are presented as means ±s.e.m. To test the significance of the effect of drugs versus control, Student's paired t test was used. When making comparisons from a different set of experiments with control, one way analysis of variance (ANOVA) followed by Dunnett's test was used. P < 0.05 was considered to represent a significant difference.
RESULTS
ATP release
Stimulation of the phrenic nerve at a frequency of 5 Hz for 2.5 min led to an increased accumulation of ATP in the bath effluent from an average basal value of 21 ± 3 pmol (mg tissue)−1 to a total value of 75 ± 5 pmol (mg tissue)−1 (n = 6). As illustrated in Fig. 2, this evoked release of ATP was frequency dependent, increasing sharply upon high frequency stimulation (50 Hz). Nerve-evoked release of ATP was Ca2+ dependent, since omission of Ca2+ in the Tyrode solution essentially abolished the ATP outflow (n = 4) and was dependent on neuronal activity since it was nearly abolished by 1 μm tetrodotoxin (n = 3).
Figure 2. The amount of ATP released upon stimulating the phrenic nerve depends on the stimulus frequency.
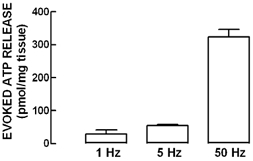
The ordinates represent the evoked release of ATP, quantified in the effluent with the luciferin-luciferase assay, upon application of electrical stimulation through the phrenic nerve trunk using different frequencies of stimulation (1–50 Hz) while keeping the number of pulses constant. Nerve-evoked release of ATP was calculated by subtracting the basal release from the total release of ATP determined after stimulus application. The data are means ±s.e.m. of 4–6 experiments.
Extracellular catabolism of ATP and formation of adenosine
We then investigated the fate of extracellular ATP in phrenic nerve-hemidiaphragm preparations. As illustrated in Fig. 3, extracellular ATP (30 μm) was catabolised with a half-degradation time of 8 ± 2 min (n = 8). The ATP metabolites detected in the bath were ADP, whose concentration reached a maximum of 5.4 ± 0.1 μm at 30 min, AMP, whose concentration reached a maximum of 3.2 ± 0.7 μm at 45 min, adenosine, whose maximum concentration (5.7 ± 1.5 μm) was reached at 45 min, and IMP, whose maximum concentration (2.0 ± 0.6 μm) was reached at 45 min. Surprisingly, the accumulation of IMP was preferential during the first 5 min; beyond this period, the speed of IMP accumulation tapered while adenosine concentration progressively increased. This pattern of extracellular catabolism of ATP was similar to that described in other neuromuscular junctions (e.g. Manery & Dryden, 1979; Cunha & Sebastião, 1991; but see Vizi et al. 2000) and differed from that found in the central nervous system, where no deamination metabolites of adenine nucleotides (like IMP) were found (reviewed in Cunha, 2001b). Therefore, we investigated in more detail the pattern of extracellular catabolism of AMP, using selective inhibitors of both dephosphorylation and deamination pathways.
Figure 3. Time course of extracellular ATP metabolism.
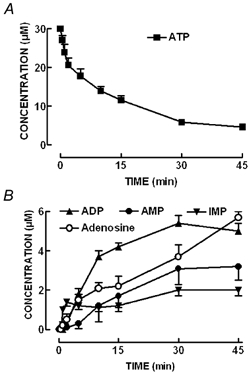
ATP (30 μm) was added at zero time to the preparation and samples were collected from the bath at the times indicated on the abscissa. Each collected sample was analysed by HPLC to separate and quantify A, ATP (▪) and its metabolites and B, ADP (▴), AMP (•), IMP (▾) and adenosine (○). Inosine was also detected but is not presented for the sake of clarity. To quantify the spontaneous release of purines, samples were also collected from the bath at the same time indicated on the abscissa adding a solution without ATP at zero time. The data are means ±s.e.m. of 8 experiments.
As shown in Fig. 4A, extracellular AMP (30 μm) was catabolised with a half-degradation time of 23 ± 5 min (n = 12). The concentration of AMP metabolites detected in the bath rose almost linearly with time up to 45 min. The metabolites identified were adenosine, which reached a maximum concentration of 14.3 ± 1.8 μm; IMP, which reached a maximum concentration of 5.3 ± 0.9 μm; and inosine, which reached a maximum concentration of 14.5 ± 1.1 μm. As expected, dephosphorylation of AMP (30 μm) leading to adenosine formation was more effective than AMP deamination forming IMP during the whole assay (inosine was not considered in this comparison since it can be formed both from the dephosphorylation of IMP and from the intra- and/or extracellular deamination of adenosine, see Fig. 1 and text below).
Figure 4. Extracellular catabolism of AMP in the absence and in the presence of the ecto-5′-nucleotidase inhibitor, α,β-methylene ADP (AOPCP, 200 μm), and/or of the AMP deaminase inhibitor, deoxycoformycin (dCOF, 200 μm).
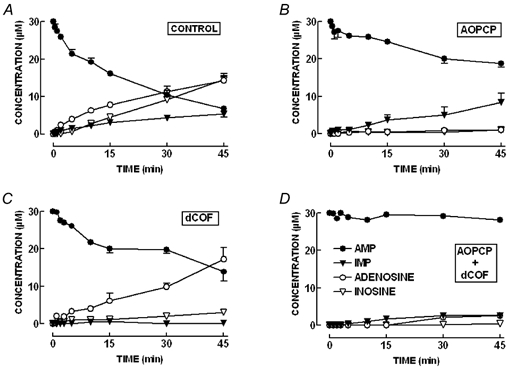
A, the pattern of extracellular AMP metabolism in the rat innervated hemidiaphragm preparation. AMP (30 μm) was added at zero time to the preparation. Bath samples were collected at the times indicated on the abscissa, for HPLC analysis to separate and quantify AMP (•) and its metabolites, IMP (▾), adenosine (○) and inosine (▿). Samples were also collected from the bath at the same time indicated on the abscissa but adding a solution without AMP at zero time to quantify the spontaneous release of purines at the different time points. The data are means ±s.e.m. of 12 experiments. B, a similar time course of extracellular catabolism of AMP, but now in the presence of AOPCP (200 μm). There is an impairment of the extracellular conversion of AMP into adenosine whereas there is a relative increase in the conversion of AMP into IMP. The data are means ±s.e.m. of 4 experiments. C, a similar time course of catabolism of extracellular AMP, but in the presence of dCOF (200 μm). It is evident that there is an impairment of the extracellular conversion of AMP into IMP and an increase in the effectiveness of the conversion of AMP into adenosine. The data are means ±s.e.m. of 4 experiments. D, the near-complete blockade of the extracellular catabolism of AMP in the simultaneous presence of AOPCP (200 μm) and dCOF (200 μm). The data are means ±s.e.m. of 4 experiments. All the experiments were performed in the presence of NBTI (30 μm) to block adenosine transport.
As shown in Fig. 4B, the ecto-5′-nucleotidase inhibitor, α,β-methylene ADP (200 μm) (Naito & Lowenstein, 1985), essentially prevented the formation of adenosine from exogenously added AMP. Under these conditions, there was a decrease in the extracellular catabolism of AMP since only 37.3 ± 1.0 % of the initial amount of AMP added at time zero was extracellularly metabolised after 45 min (n = 4). In the presence of α,β-methylene ADP (200 μm), the AMP-derived IMP formation was slightly (P > 0.05) enhanced, since IMP reached a maximal concentration of 8.4 ± 2.4 μm at 45 min. This is probably due to the blockade of IMP dephosphorylation to inosine, since both AMP and IMP are dephosphorylated by ecto-5′-nucleotidase, i.e. the extracellular catabolism of IMP is nearly blocked by 200 μmα,β-methylene ADP (data not shown). The ability of α,β-methylene ADP to inhibit ecto-5′-nucleotidase is due to the known capacity of adenine nucleotides, namely ATP and ADP, to inhibit ecto-5′-nucleotidase activity (Naito & Lowenstein, 1985; Cunha, 2001b). This inhibition can be evaluated by quantifying the ratio ([AMP]+[IMP])/([adenosine]+[inosine]), which is an inverse measure of the activity of ecto-5′-nucleotidase (Cunha & Sebastião, 1991; Cunha, 2001b). When adding as an initial substrate 30 μm of either ATP, ADP or AMP, this ratio at 45 min was 1.10 ± 0.21 (n = 8) for ATP, 1.00 ± 0.14 (n = 6) for ADP and 0.45 ± 0.06 (n = 12) for AMP, confirming that ATP and ADP inhibited ecto-5′-nucleotidase in phrenic nerve hemidiaphragm preparations. The inhibition of ecto-5′-nucleotidase by ATP can be exaggerated by incubating the preparations with a higher concentration (100 μm) of ATP. In such conditions, the hydrolysis of ATP is delayed, as indicated by a significant (P < 0.05) increase in the nucleotides/nucleosides ratio to 2.89 ± 0.46 (n = 3). In contrast, this ratio was not significantly altered (P > 0.05) when the initial concentration of AMP was increased to 100 μm (0.57 ± 0.09, n = 3).
The ecto-AMP deaminase inhibitor, deoxycoformycin (200 μm) (Agarwal & Parks, 1977), essentially prevented the formation of IMP from exogenously added AMP (Fig. 4C). Under these conditions, there was a decrease in the extracellular catabolism of AMP that was now metabolised with a half-degradation time of 39 ± 6 min (n = 4). In the presence of deoxycoformycin (200 μm), the AMP-derived adenosine formation was slightly (P > 0.05) enhanced, since adenosine now reached a maximal concentration of 19.0 ± 4.0 μm at 45 min. This tendency for the extracellular concentration of adenosine to increase results both from the greater availability of AMP as a substrate of ecto-5′-nucleotidase, and from the ability of deoxycorformycin to inhibit the removal of extracellular adenosine, namely through adenosine transporters (Rogler-Brown & Parks, 1980) and through adenosine deaminase (Agarwal & Parks, 1977). In fact, in experiments designed to follow the time course of extracellular adenosine removal, it was confirmed that the conversion of adenosine into inosine was attenuated by deoxycoformycin (n = 6, data not shown). The removal of extracellular adenosine was inhibited by an inhibitor of adenosine transport, S-(p-nitrobenzyl)-6-thioinosine (NBTI, 5 μm, 36 ± 5 % inhibition, n = 5), and further inhibited by an inhibitor of adenosine deaminase, erythro-9-(2-hydroxy-3-nonyl)adenine (EHNA, 25 μm, 26 ± 3 % inhibition, n = 5), indicating that both adenosine uptake and ecto-adenosine deaminase might be involved in the clearance of extracellular adenosine at the rat neuromuscular junction as predicted from functional studies (Correia-de-Sá & Ribeiro, 1996).
Finally, as shown in Fig. 4D, the simultaneous presence of α,β-methylene ADP (200 μm) and of deoxycoformycin (200 μm) virtually prevented the extracellular catabolism of AMP.
Relative contribution of the ecto-AMP deaminase pathway for adenosine formation
To demonstrate that the two alternative pathways of extracellular metabolism of AMP also contributed to the removal of endogenously released adenine nucleotides, we tested the effect of α,β-methylene ADP (200 μm) and/or deoxycoformycin (200 μm) on the accumulation of adenine nucleotides triggered by stimulating the phrenic nerve. We quantified the released adenine nucleotides as AMP accumulating in the extracellular milieu, since we have pharmacological tools to interfere with the extracellular catabolism of AMP, but not with that of ATP. As illustrated in Fig. 5, we could not detect a nerve-evoked accumulation of endogenous AMP in the absence of any inhibitor of extracellular AMP catabolism. This suggests that the rate of extracellular catabolism of endogenously formed AMP is larger than that of exogenously added AMP, in accordance with the hypothesis that the ecto-nucleotidase pathway displays channelling properties (reviewed in Cunha, 2001b). However, when we blocked either the ecto-AMP deaminase activity, with deoxycoformycin (200 μm, n = 4), or the ecto-5′-nucleotidase, with α,β-methylene ADP (200 μm, n = 5), we now observed a net evoked accumulation of endogenous extracellular AMP. Interestingly, we found that these two inhibitors of extracellular AMP catabolism increased the total accumulation of extracellular AMP in a manner similar to the basal accumulation of extracellular AMP (Fig. 5). This indicates that both ecto-AMP deaminase and ecto-5′-nucleotidase control both the basal and the total accumulation of endogenous extracellular AMP on nerve stimulation (Fig. 5). Simultaneous application of the two inhibitors did not cause an accumulation of the evoked endogenous extracellular AMP (1.15 ± 0.11 nmol (g tissue)−1, n = 6) that was significantly (P > 0.05) higher than that caused by 200 μmα,β-methylene ADP (1.09 ± 0.33 nmol (g tissue)−1, n = 5), though it was greater than that caused by 200 μm deoxycoformycin (0.58 ± 0.14 nmol (g tissue)−1, n = 4). These observations suggest that endogenous extracellular AMP is catabolised more efficiently by ecto-5′-nucleotidase than by ecto-AMP deaminase. These results fully agree with our findings measuring the extracellular catabolism of exogenously added AMP (see Fig. 4A), where dephosphorylation of AMP leading to adenosine formation was preferential as compared with the generation of its inactive metabolite, IMP (e.g. Ribeiro & Sebastião, 1987), via the ecto-AMP deaminase pathway.
Figure 5. Extracellular accumulation of released adenine nucleotides is controlled by the activities of both ecto-5′-nucleotidase and ecto-AMP deaminase.
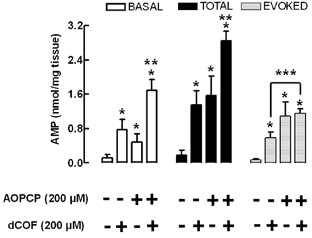
AMP was quantified by HPLC in bath samples collected after 2.5 min incubation with the preparations during rest (basal) or after electrical stimulation (total) of the phrenic nerve trunk with 5 Hz trains. The ecto-5′-nucleotidase inhibitor, α,β-methylene ADP (AOPCP, 200 μm), and/or the ecto-AMP deaminase inhibitor, deoxycoformycin (dCOF, 200 μm), were added to the incubation medium 15 min before sampling. The absence (-) or presence (+) of each drug is indicated below each bar. T he evoked release of adenine nucleotides (evoked) was calculated by subtracting the basal release from the total release of adenine nucleotides. The data are means ±s.e.m. of 3–8 experiments. *P < 0.05 when compared with 0 nmol (mg tissue)−1; **P < 0.05 when compared with the effect of either AOPCP or dCOF alone; ***P < 0.05 between the indicated bars.
Contribution of the ecto-AMP deaminase pathway for adenosine modulation of ACh release
We have previously documented the physiological relevance of ecto-5′-nucleotidase on the evoked acetylcholine (ACh) release from phrenic nerve terminals (Correia-de-Sá et al. 1996; Cunha et al. 1996a), which is under dual control of inhibitory A1 receptors and facilitatory A2A receptors depending on the levels of extracellular adenosine (Correia-de-Sá & Ribeiro, 1996). We have reported that prevention of ATP-derived adenosine formation with α,β-methylene ADP (200 μm) caused an inhibition of the evoked release of [3H]ACh and, in addition, we pharmacologically confirmed that this was due to removal of tonic activation of facilitatory A2A receptors (Cunha et al. 1996a). These findings indicated that adenosine originating from catabolism of adenine nucleotides preferentially activates facilitatory A2A receptors in motor nerve terminals. As we have now found that ecto-AMP deaminase also contributed to the removal of endogenous extracellular AMP, we tested the functional impact of the blockade of ecto-AMP deaminase on the nerve-evoked release of [3H]ACh. The experiments were performed in the presence of a supramaximal concentration of an inhibitor of adenosine transport, NBTI (30 μm) (see Correia-de-Sá & Ribeiro, 1996), since deoxycoformycin has previously been shown to inhibit adenosine transporters (e.g. Rogler-Brown & Parks, 1980). In these conditions, deoxycoformycin (200 μm) facilitated the evoked [3H]ACh release by 77 ± 9 % (n = 7). Deoxycoformycin-induced facilitation of [3H]ACh release was not related to the modulation of [3H]ACh release via adenosine A1 receptors, since the facilitatory effect of deoxycorformycin was essentially maintained (58 ± 8 % facilitation, n = 4) in the presence of the selective A1 receptor antagonist, 1,3-dipropyl-8-cyclopenthylxanthine (DPCPX, 2.5 nm) (Fig. 6). Instead, facilitation of [3H]ACh caused by deoxycorformycin (200 μm) was due to the activation of adenosine A2A receptors, since the selective A2A receptor antagonist, ZM 241385 (10 nm), completely prevented this facilitatory effect. In fact, upon blockade of A2A receptors with ZM 241385 (10 nm), deoxycorformycin (200 μm) now inhibited the evoked release of [3H]ACh by 33 ± 8 % (n = 4) (see Fig. 6). This was predicted since the blockade of facilitatory A2A receptors might exaggerate inhibition of ACh release operated by the activation of inhibitory A1 receptors. These findings are in good agreement with the previously reported A1/A2A inter-receptor equilibrium controlling ACh release evoked by 5 Hz trains (Pereira et al. 2000), which tightly depends on the extracellular levels of adenosine (Correia-de-Sá & Ribeiro, 1996; Correia-de-Sá et al. 1996).
Figure 6. Blockade of ecto-AMP deaminase with deoxycoformycin (200 μm) leads to the facilitation of evoked acetylcholine (ACh) release through the activation of facilitatory A2A adenosine receptors.
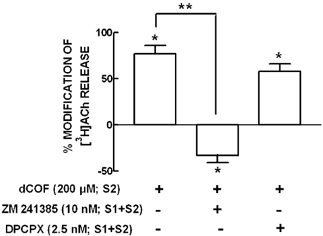
The evoked release of [3H]ACh was elicited by two stimulation periods of the phrenic nerve (S1 and S2, separated by 27 min) with a frequency of 5 Hz and 750 supramaximal intensity pulses of 40 μs duration. Deoxycoformycin (dCOF, 200 μm) was added from 15 min before S2 onwards. When the effect of dCOF was tested in the presence of the selective adenosine A2A receptor antagonist, ZM 241385 (10 nm), or the selective adenosine A1 receptor antagonist, DPCPX (2.5 nm), these antagonists were added 15 min before starting sample collection (present during S1 and S2). The absence (-) or presence (+) of each drug during S2 or during S1 and S2 is indicated below each bar. The ordinates are percentage changes in the S2/S1 ratios as compared with the S2/S1 ratios in control experiments. Zero percent represents identity between the two ratios; positive and negative values represent facilitation or inhibition of the evoked release of [3H]ACh, respectively. The results are means ±s.e.m. of 4–7 experiments. *P < 0.05 when compared with controls; **P < 0.05 when compared with the effect of deoxycoformycin in the absence of adenosine receptor antagonists. Note that the inhibitor of adenosine transporters, nitrobenzylthioinosine, was present in a supramaximal concentration (30 μm) in all superfusion solutions.
DISCUSSION
The present results confirm that adenine nucleotides, namely ATP, are released to the extracellular milieu upon stimulation of motor nerve endings and that they are metabolised extracellularly by a series of ecto-nucleotidases (see Fig. 1 for a schematic representation). The activity of the ecto-nucleotidase pathway appears critical to define the pattern of formation of extracellular ATP-derived adenosine to allow the activation of either inhibitory A1 or facilitatory A2A adenosine receptors. In particular, the neuromuscular junction is equipped with an ecto-AMP deaminase pathway that metabolises adenine nucleotides bypassing adenosine formation, and this activity is critical to divert part of the released adenine nucleotides from adenosine formation. Thus, while the ecto-5′-nucleotidase pathway modulates the rate of adenosine formation from released nucleotides, alternative AMP deamination controls the amount of AMP available to be converted into adenosine. The combined effect of both pathways might serve to maintain the local concentration of adenosine at lower levels allowing the predominant activation of inhibitory A1 receptors to operate at moderate frequencies of nerve stimulation (e.g. 5 Hz trains) (see Correia-de-Sá & Ribeiro, 1996).
Several studies using neuromuscular junctions from different species and different stimulation patterns have concluded that nerve stimulation triggers the release of ATP (Silinsky, 1975; Smith, 1991; Silinsky & Redman, 1996; Vizi et al. 2000). There is some debate as to whether this released ATP originates from nerve terminals (e.g. Silinsky, 1975), from activated muscle fibres (e.g. Smith, 1991; Vizi et al. 2000) or from peri-synaptic Schwann cells (discussed in Fields & Stevens, 2000). But, irrespective of the source of ATP released upon stimulating motor nerve terminals, the relevant question from the functional point of view is what might be the role of extracellular ATP in the control of neuromuscular transmission. Although some reports support possible direct effects of ATP as such (Giniatullin & Sokolova, 1998; Salgado et al. 2000; Deuchars et al. 2001; Galkin et al. 2001), the most likely role for extracellular ATP is to act as a source of adenosine that fulfils a key neuromodulatory role in the control of neuromuscular transmission through activation of inhibitory A1 or facilitatory A2A receptors (Ribeiro & Walker, 1975; Correia-de-Sáet al. 1991, 1996; Cunha et al. 1996a; Silinsky et al. 1999). This contention obviously places greater emphasis on the understanding of the enzymes responsible for the extracellular metabolism of ATP into adenosine, generically named ecto-nucleotidases.
The extracellular ATP hydrolysis at neuromuscular junctions shares many similarities with that described in many different preparations, being mainly catalysed by an ATP and/or ADP metabolising enzyme(s) and an ecto-5′-nucleotidase designed to form adenosine (reviewed in Zimmermann & Braun, 1999; Cunha, 2001b). The main difference found at neuromuscular junctions is the presence of an ecto-AMP deaminase activity that converts AMP into IMP (see Fig. 1), which has been reported to be present at various neuromuscular junctions (Manery & Dryden, 1979; Cunha & Sebastião, 1991). Skeletal muscle fibres are one of the mammalian tissues with higher activity of AMP deamination (Ogasawara et al. 1974). Most of this enzymatic activity is associated with muscle fibres, but it is also present in the vicinity of neuronal and circulatory elements (Thompson et al. 1992), although it is not known if AMP deaminase might also be located presynaptically. We have now confirmed that the AMP deaminase involved in the extracellular catabolism of adenine nucleotides at the innervated hemidiaphragm is indeed an ecto-nucleotidase (see also Alertsen et al. 1958). The expected role of this ecto-AMP deaminase activity might be to divert released adenine nucleotides from extracellular adenosine formation, since AMP is inactivated into IMP (and inosine) rather than dephosphorylated into adenosine. In fact, the extracellular accumulation of endogenous adenine nucleotides is increased by deoxycoformycin. Likewise, the kinetic experiments comparing the pattern of extracellular adenosine formation upon addition of AMP as initial substrate show a more than 50 % increase in the amounts of extracellular adenosine formed in the presence of the ecto-AMP deaminase inhibitor, deoxycoformycin, compared with its absence. This indicates that there is a large drain of adenine nucleotides into the ecto-AMP deaminase shunt-like pathway since its blockade increased the extracellular levels of endogenous adenosine.
We had previously shown that increasing the levels of extracellular adenosine, either by maximal inhibition of adenosine transport or of adenosine deaminase, leads to a facilitation of the evoked release of acetylcholine from phrenic motor nerve endings (Correia-de-Sá & Ribeiro, 1996). Accordingly, we now observed that the blockade of ecto-AMP deaminase also leads to a facilitation of the evoked release of acetylcholine through the activation of facilitatory A2A, rather than inhibitory A1, adenosine receptors. The experiments were carried out in the presence of a supramaximal concentration of the adenosine transport inhibitor, NBTI, to rule out the possibility that deoxycoformycin might be facilitating acetylcholine release because of its ability to inhibit adenosine transporters (Rogler-Brown & Parks, 1980). Deoxycorformycin is also an inhibitor of adenosine deaminase, and it has previously been suggested that an ecto-adenosine deaminase activity might be controlling the removal of extracellular adenosine at the rat neuromuscular junction (Correia-de-Sá & Ribeiro, 1996). However, a previous study has shown that the effect of adenosine deaminase inhibition was not further enhanced in the presence of a supramaximal concentration of an adenosine transport blocker (Correia-de-Sá & Ribeiro, 1996). This reinforces our contention that the effect of deoxycoformycin on ACh release results from the inhibition of ecto-AMP deaminase, which has a profound effect on the extracellular levels of adenosine tonically controlling the evoked release of acetylcholine. Indeed, if the levels of adenine nucleotides released during stimulation were fully converted into adenosine, the extracellular levels of adenosine would be high enough to predominantly activate facilitatory A2A receptors, systematically overtaking the A1 receptor activation required to restrain superfluous transmitter release at lower frequencies of nerve stimulation. In addition, fine-tuning control of cholinergic and peptidergic neuromodulatory systems, which are regulated by the balanced activation of adenosine A1 and A2A receptors (Correia-de-Sá & Ribeiro, 1994a,1994b; Oliveira et al. 2002), would also be disrupted. This ecto-AMP deaminase activity might be critically required at neuromuscular junctions since the amount of adenine nucleotides released at these synapses is between one and two orders of magnitude greater than the levels of extracellular adenine nucleotides that are released from central nervous system preparations (cf. Silinsky, 1975; Potter & White, 1980; Wieraszko et al. 1989; Smith, 1991; Cunha & Sebastião, 1993; Cunha et al. 1996b).
In conclusion, the present results illustrate the functional importance of ecto-AMP deaminase, an ecto-nucleotidase activity only found at neuromuscular synapses, for the controlled formation of extracellular adenosine and proper functioning of the adenosine neuromodulatory system at the neuromuscular junction. It remains to be explored if ecto-AMP deaminase also controls the formation of adenosine involved in the regulation of functional hyperaemia (e.g. Proctor & Dubling, 1982) or of the insulin sensitivity of muscle cells (e.g. Espinal et al. 1983).
Acknowledgments
Supported by Fundação para a Ciência e a Tecnologia (SAU/14014/1998 and POCTI/36545/FCB/2000). L.O. is in receipt of an FCT Young Researcher studentship.
REFERENCES
- Agarwal RP, Parks RE., Jr Potent inhibition of muscle 5′-AMP deaminase by nucleoside antibiotics coformycin and deoxycoformycin. Biochem Pharmacol. 1977;26:633–636. doi: 10.1016/0006-2952(77)90046-6. [DOI] [PubMed] [Google Scholar]
- Alertsen AAR, Walaas O, Walaas E. Enzymic conversion of mononucleotides by rat diaphragm in vitro. Acta Physiol Scand. 1958;43:122–134. doi: 10.1111/j.1748-1716.1958.tb01582.x. [DOI] [PubMed] [Google Scholar]
- Bodin P, Burnstock G. Purinergic signalling: ATP release. Neurochem Res. 2001;26:959–969. doi: 10.1023/a:1012388618693. [DOI] [PubMed] [Google Scholar]
- Correia-de-Sá P, Ribeiro JA. Tonic adenosine A2A receptor activation modulates nicotinic autoreceptor function at the rat neuromuscular junction. Eur J Pharmacol. 1994a;271:349–355. doi: 10.1016/0014-2999(94)90793-5. [DOI] [PubMed] [Google Scholar]
- Correia-de-Sá P, Ribeiro JA. Potentiation by tonic A2a -adenosine receptor activation of CGRP-facilitated [3H]-ACh release from rat motor nerve endings. Br J Pharmacol. 1994b;111:582–588. doi: 10.1111/j.1476-5381.1994.tb14777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia-de-Sá P, Ribeiro JA. Adenosine uptake and deamination regulate tonic A2a -receptor facilitation of evoked [3H]-ACh release from the motor nerve terminals. Neuroscience. 1996;73:85–92. doi: 10.1016/0306-4522(96)00028-0. [DOI] [PubMed] [Google Scholar]
- Correia-de-Sá P, Sebastião AM, Ribeiro JA. Inhibitory and excitatory effects of adenosine receptor agonists on evoked transmitter release from phrenic nerve endings of the rat. Br J Pharmacol. 1991;103:1614–1620. doi: 10.1111/j.1476-5381.1991.tb09836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia-de-Sá P, Timóteo MA, Ribeiro JA. Presynaptic A1 inhibitory/A2A facilitatory adenosine receptor activation balance depends on motor nerve stimulation paradigm at the rat hemidiaphragm. J Neurophysiol. 1996;76:3910–3919. doi: 10.1152/jn.1996.76.6.3910. [DOI] [PubMed] [Google Scholar]
- Correia-de-Sá P, Timóteo MA, Ribeiro JA. A2A adenosine receptor facilitation of neuromuscular transmission: influence of stimulus paradigm on calcium mobilization. J Neurochem. 2000;74:2462–2469. doi: 10.1046/j.1471-4159.2000.0742462.x. [DOI] [PubMed] [Google Scholar]
- Cunha RA. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: different roles, different sources and different receptors. Neurochem Int. 2001a;38:107–125. doi: 10.1016/s0197-0186(00)00034-6. [DOI] [PubMed] [Google Scholar]
- Cunha RA. Regulation of the ecto-nucleotidase pathway in rat hippocampal nerve terminals. Neurochem Res. 2001b;26:979–991. doi: 10.1023/a:1012392719601. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Correia-de-Sá P, Sebastião AM, Ribeiro JA. Preferential activation of excitatory adenosine receptors at rat hippocampal and neuromuscular synapses by adenosine formed from released adenine nucleotides. Br J Pharmacol. 1996a;119:253–260. doi: 10.1111/j.1476-5381.1996.tb15979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RA, Ribeiro JA. ATP as a presynaptic modulator. Life Sci. 2000;68:119–137. doi: 10.1016/s0024-3205(00)00923-1. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Sebastião AM. Extracellular metabolism of adenine nucleotides and adenosine in the innervated skeletal muscle of the frog. Eur J Pharmacol. 1991;197:83–92. doi: 10.1016/0014-2999(91)90368-z. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Sebastião AM. Adenosine and adenine nucleotides are independently released from both the nerve terminals and the muscle fibres upon electrical stimulation of the innervated skeletal muscle of the frog. Pflugers Arch. 1993;424:503–510. doi: 10.1007/BF00374914. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Vizi ES, Ribeiro JA, Sebastião AM. Preferential release of ATP and its extracellular catabolism as a source of adenosine upon high- but not low-frequency stimulation of rat hippocampal slices. J Neurochem. 1996b;67:2180–2187. doi: 10.1046/j.1471-4159.1996.67052180.x. [DOI] [PubMed] [Google Scholar]
- Deuchars SA, Atkinson L, Brooke RE, Musa H, Milligan CJ, Batten TFC, Buckley NJ, Parson SH, Deuchars J. Neuronal P2X7 receptors are targeted to presynaptic terminals in the central and peripheral nervous systems. J Neurosci. 2001;21:7143–7152. doi: 10.1523/JNEUROSCI.21-18-07143.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinal J, Challiss RA, Newsholme EA. Effect of adenosine deaminase and an adenosine analogue on insulin sensitivity in soleus muscle of the rat. FEBS Lett. 1983;158:103–106. doi: 10.1016/0014-5793(83)80685-1. [DOI] [PubMed] [Google Scholar]
- Fields RD, Stevens B. ATP: an extracellular signalling molecule between neurons and glia. Trends Neurosci. 2000;23:625–633. doi: 10.1016/s0166-2236(00)01674-x. [DOI] [PubMed] [Google Scholar]
- Galkin AV, Giniatullin RA, Mukhtarov MR, Svandová I, Grishin SN, Vyskocil F. ATP but not adenosine inhibits nonquantal acetylcholine release at the mouse neuromuscular junction. Eur J Neurosci. 2001;13:2047–2053. doi: 10.1046/j.0953-816x.2001.01582.x. [DOI] [PubMed] [Google Scholar]
- Giniatullin RA, Sokolova EM. ATP and adenosine inhibit transmitter release at the frog neuromuscular junction through distinct presynaptic receptors. Br J Pharmacol. 1998;124:839–844. doi: 10.1038/sj.bjp.0701881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manery JF, Dryden EE. Ecto-enzymes concerned with nucleotide metabolism. In: Baer HP, Drummond GI, editors. Physiological and Regulatory Functions of Adenosine and Adenine Nucleotides. New York: Raven Press; 1979. pp. 323–339. [Google Scholar]
- Naito Y, Lowenstein JM. 5′-Nucleotidase from rat heart membranes. Inhibition by adenine nucleotides and related compounds. Biochem J. 1985;226:645–651. doi: 10.1042/bj2260645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara N, Goto H, Watanabe T, Kawamura Y, Yoshino M. Multiple forms of AMP deaminase in various rat tissues. FEBS Lett. 1974;44:63–66. doi: 10.1016/0014-5793(74)80306-6. [DOI] [PubMed] [Google Scholar]
- Oliveira L, Timoteo MA, Correia-de-Sá P. Modulation by adenosine of both muscarinic M1-facilitation and M2-inhibition of [3H]acetylcholine release from the rat motor nerve terminals. Eur J Neurosci. 2002;15:1728–1736. doi: 10.1046/j.1460-9568.2002.02020.x. [DOI] [PubMed] [Google Scholar]
- Pereira MF, Cunha RA, Ribeiro JA. Tonic adenosine neuromodulation is preserved in motor nerve endings of aged rats. Neurochem Int. 2000;36:563–566. doi: 10.1016/s0197-0186(99)00164-3. [DOI] [PubMed] [Google Scholar]
- Potter P, White TD. Release of adenosine 5′-triphosphate from synaptosomes from different regions of rat brain. Neuroscience. 1980;5:1351–1356. doi: 10.1016/0306-4522(80)90207-9. [DOI] [PubMed] [Google Scholar]
- Proctor KG, Dubling BR. Adenosine and free-flow functional hyperemia in striated muscle. Am J Physiol. 1982;242:H688–697. doi: 10.1152/ajpheart.1982.242.4.H688. [DOI] [PubMed] [Google Scholar]
- Ribeiro JA, Cunha RA, Correia-de-Sá P, Sebastião AM. Purinergic regulation of acetylcholine release. Prog Brain Res. 1996;109:231–241. doi: 10.1016/s0079-6123(08)62107-x. [DOI] [PubMed] [Google Scholar]
- Ribeiro JA, Sebastião AM. On the role, inactivation and origin of endogenous adenosine at the frog neuromuscular junction. Br J Pharmacol. 1987;384:571–585. doi: 10.1113/jphysiol.1987.sp016470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro JA, Walker J. The effects of adenosine triphosphate and adenosine diphosphate on transmission at the rat and frog neuromuscular junctions. Br J Pharmacol. 1975;54:213–218. doi: 10.1111/j.1476-5381.1975.tb06931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogler-Brown T, Parks RE., Jr Tight binding inhibitors - VIII. Studies of the interactions of 2′-deoxycoformycin and adenosine transport inhibitors with the erythrocytic nucleoside transport system. Biochem Pharmacol. 1980;29:2491–2497. doi: 10.1016/0006-2952(80)90354-8. [DOI] [PubMed] [Google Scholar]
- Salgado AI, Cunha RA, Ribeiro JA. Facilitation by P2 receptor activation of acetylcholine release from rat motor nerve terminals: interaction with presynaptic nicotinic receptors. Brain Res. 2000;877:245–250. doi: 10.1016/s0006-8993(00)02679-2. [DOI] [PubMed] [Google Scholar]
- Silinsky EM. On the association between transmitter secretion and the release of adenine nucleotides from mammalian motor nerve terminals. J Physiol. 1975;247:145–162. doi: 10.1113/jphysiol.1975.sp010925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silinsky EM, Hirsh JK, Searl TJ, Redman RS, Watanabe M. Quantal ATP release from motor nerve endings and its role in neurally mediated depression. Prog Brain Res. 1999;120:145–158. doi: 10.1016/s0079-6123(08)63552-9. [DOI] [PubMed] [Google Scholar]
- Silinsky EM, Redman RS. Synchronous release of ATP and neurotransmitter within milliseconds of a motor nerve impulse in the frog. J Physiol. 1996;492:815–822. doi: 10.1113/jphysiol.1996.sp021348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DO. Sources of adenosine released during neuromuscular transmission in the rat. J Physiol. 1991;432:343–354. doi: 10.1113/jphysiol.1991.sp018388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JL, Sabina RL, Ogasawara N, Riley DA. AMP deaminase histochemical activity and immunofluorescent isozyme localization in rat skeletal muscle. J Histochem Cytochem. 1992;40:931–946. doi: 10.1177/40.7.1607642. [DOI] [PubMed] [Google Scholar]
- Vizi ES, Nitahara K, Sato K, Sperlágh B. Stimulation-dependent release, breakdown, and action of endogenous ATP in mouse hemidiaphragm preparation: the possible role of ATP in neuromuscular transmission. J Auton Nerv Syst. 2000;81:278–284. doi: 10.1016/s0165-1838(00)00129-6. [DOI] [PubMed] [Google Scholar]
- Wessler I, Kilbinger H. Release of [3H]-acetylcholine from a modified rat phrenic nerve-hemidiaphragm preparation. Naunyn-Schmiedebergs Arch Pharmacol. 1986;334:357–364. doi: 10.1007/BF00569370. [DOI] [PubMed] [Google Scholar]
- Wieraszko A, Goldsmith G, Seyfried TN. Stimulation-dependent release of adenosine triphosphate from hippocampal slices. Brain Res. 1989;485:244–250. doi: 10.1016/0006-8993(89)90567-2. [DOI] [PubMed] [Google Scholar]
- Zimmermann H, Braun N. Ecto-nucleotidases - molecular structures, catalytic properties, and functional roles in the nervous system. Prog Brain Res. 1999;120:371–385. [PubMed] [Google Scholar]