

Abstract
Defective regulation and/or reduced expression of the Na+-K+-2Cl− cotransporter NKCC1 may contribute to the severe secretory defect that is observed in cystic fibrosis, but data concerning the expression and function of NKCC1 in cystic fibrosis transmembrane conductance regulator (CFTR)-deficient cells are equivocal. We therefore investigated NKCC1 mRNA expression, Na+-K+-2Cl− cotransport activity and regulation by cAMP in crypts isolated from the proximal colon of CFTR-containing (CFTR (+/+)) and CFTR-deficient (CFTR (−/−)) mice. mRNA expression levels were determined by semiquantitative PCR, transport rates were measured fluorometrically in 2′,7′-bis(carboxyethyl)-5(6)-carboxyfluorescein acetomethylester (BCECF)-loaded crypts, cytoplasmic volume changes were assessed by confocal microscopy, and [Cl−]i changes were examined by N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide (MQAE) quenching. NKCC1 mRNA expression levels were not significantly reduced in CFTR (−/−) crypts compared to controls. Azosemide-sensitive NH4+ influx (used as a measure of Na+-K+-2Cl− cotransport) was 2.23 ± 0.72 vs. 1.56 ± 0.16 mm min−1, and increased by 63.6 % in (+/+) and 87.3 % in (−/−) crypts upon stimulation for 5 min with forskolin. After 20 min of stimulation with forskolin, the Na+-K+-2Cl− cotransport rates in (−/−) and (+/+) crypts were identical. Crypt cross-sectional area and [Cl−]i decreased only in (+/+) crypts upon stimulation. In conclusion, normal NKCC1 expression levels, somewhat reduced Na+-K+-2Cl− cotransport rates, but preserved activation by cAMP were found in colonic crypts from CFTR (−/−) mice, ruling out a severe dysfunction of the Na+-K+-2Cl− cotransporter in the CF intestine. Furthermore, these studies establish the existence of a direct, cell-volume- and [Cl−]i-independent activation of colonic NKCC1 by an increase in intracellular cAMP.
When Cl− secretion is activated in response to cAMP in intestinal epithelia, basolateral uptake must also increase to maintain cellular electrolyte composition. The importance of basolateral Cl− uptake as the rate-limiting step in the onset of apical Cl− secretion has been demonstrated in shark rectal glands (Lytle & Forbush, 1996). Colonic basolateral anion uptake is mediated predominantly by the bumetanide-sensitive Na+-K+-2Cl− cotransporter NKCC1 and by the alternative pathway of Na+-HCO3− cotransport coupled to basolateral anion exchange (Jacob et al. 2001).
In cystic fibrosis (CF), epithelial anion secretion is severely hampered, resulting in mucus accumulation in the pancreatic, biliary and epididymal ducts, and in the smaller airways and intestinal tract. Functional, molecular and immunohistochemical evidence exists for the presence of non-CF transmembrane conductance regulator (CFTR) anion conductances or transporters in the apical membrane of either intact intestine or intestinal cell lines (Hagos et al. 1999; Joo et al. 1999; Gaspar et al. 2000). Numerous attempts have been made by several laboratories to activate these alternative anion efflux pathways pharmacologically, but this has remained largely unsuccessful in the CFTR-deficient intestine (Seidler et al. 1997; Joo et al. 1998; Grubb & Boucher, 1999).
Based on experiments in CFTR-deficient and CFTR-transfected pancreatic tumour cells, Shumaker & Soleimani (1999) suggested that CFTR transcriptionally regulates NKCC1. They hypothesized that it is the CF-associated dysfunction of basolateral anion uptake, rather than reduced apical CFTR anion conductance, that may underlie the severe anion secretory defect observed in the pancreatic ducts of many CF patients (Soleimani & Ulrich, 2000). If this is true, even successful opening of alternative apical anion channels would not result in a substantial anion secretory response. In contrast, Haas et al. (1993) found that [3H]bumetanide binding increased upon addition of cAMP analogues in nasal epithelial cells from CF patients, although these cells did not display an increase in Cl− efflux, suggesting a direct activation of Na+-K+-2Cl− cotransport by cAMP.
Most CF patients and cells from these patients have some residual CFTR expression, which could potentially explain the discrepant results reported by Shumaker & Soleimani (1999) and Haas et al. (1993). The only native epithelia with zero CFTR expression are those of the CFTR null mutant mice (Clarke et al. 1992; Ratcliff et al. 1993). These CFTR (−/−) mice display ATP-activated anion conductances in their bronchial, biliary and pancreatic systems, but not in the intestine (Clarke et al. 1992). To measure the expression, transport rates and cAMP-dependent regulation of the Na+-K+-2Cl− cotransporter in the same cellular system, we isolated colonic crypts from the proximal colon of CFTR (−/−) mice and their normal (CFTR (+/+)) littermates, determined NKCC1 mRNA expression levels and assessed Na+-K+-2Cl− activity fluorometrically in 2′,7′-biscarboxyethyl-5(6)-carboxyfluorescein acetomethylester (BCECF AM)-loaded crypts in the absence and presence of forskolin. To assess potential cAMP-evoked changes in cell volume and [Cl−]i unrelated to CFTR channel opening, we determined volume changes in CFTR (+/+) and (−/−) crypts using confocal microscopy, and cAMP-induced changes of [Cl−]i using the anion-sensitive dye N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide (MQAE).
METHODS
Materials
Azosemide was supplied by Sanofi-Synthelabo (Berlin, Germany), bovine serum albumin (BSA; cell culture grade) from Paesel and Lorei (Frankfurt, Germany) and BCECF AM, calcein AM and MQAE were from Molecular Probes (Leiden, The Netherlands). All other chemicals were either obtained from Sigma (Deisenhofen, Germany) or from Merck (Darmstadt, Germany) at tissue culture grade or the highest grade available.
Animal breeding and genotyping
The CF null mice (Cftrtm1Cam) were established in the laboratories of R. Ratcliff, W. H. Colledge and M. Evans and had been characterized previously (Ratcliff et al. 1993); the NKCC1-knockout mice were established in the laboratories of G. Shull (Flagella et al. 1999). Both strains were bred in the animal facility of the Department of Biochemistry in Tübingen. The CF null mice were genotyped as described previously (Seidler et al. 1997). For genotype analysis of the NKCC1-knockout mice, genomic DNA was prepared from tail samples of 1- to 2-week-old mice using the QIAamp tissue kit (Qiagen, Hilden, Germany) and added to 25 μl of a PCR mixture: 2 U Qiagen Taq DNA polymerase; 2.5 μl Qiagen PCR buffer B; 2.5 mm Qiagen MgCl−; 20 mm of each dNTP; 20 μm of forward and reverse primers. For detection of the wild-type allele, the oligonucleotides (5′-GGAACATTCCATACTTATGATAGATG-3′) and (5′-CTCACCTTTGCTTCCCACTCCATTCC-3′) were used as primers and amplified a 105 base pair (bp) product. The primers (5′-GGAACATTCCATACTTATGATAGATG-3′) and (5′-GACAATAGCAGGCATGCTGG-3′) amplified a 156 bp product of the mutant allele. The PCR was performed using the following temperature parameters: 40 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s Heterozygous breeding pairs were maintained on a NMRI (Naval Medical Research Institute) background by periodically backcrossing to avoid impaired viability due to inbreeding. Animal experiments followed approved protocols at the University of Tübingen and the local authorities for the regulation of animal welfare (Regierungspräsidium).
Preparation of colonic crypts and surface cells
Crypts were prepared from the proximal colon because our previous Ussing-chamber data on the relevance of NKCC1 and other anion transporters for colonic anion secretion were obtained from this segment of the colon. Mice were anaesthetized with a rising concentration of CO2 and killed by cervical dislocation. A 3 cm segment of proximal colon was excised, washed with ice-cold buffer A gassed with 100 % O2 (mm: 120 NaCl, 14 2-Hepes, 7 Tris, 3 KH2PO4, 2 K2HPO4, 1.2 MgSO4, 1.2 calcium gluconate, 20 glucose; pH 7.4), everted and filled with Ca2+-free buffer B: (mm: 127 NaCl, 5 KCl, 1 MgCl2, 5 sodium pyruvate, 10 Hepes, 5 EDTA, with 1 % BSA; pH 7.4). The segment was placed into a tube filled with solution B and vibrated at 10 Hz at room temperature. At defined intervals the solution was replaced and epithelial fragments harvested by low-speed centrifugation. Early fractions contained surface cells, later fractions crypts, both easily identified under a microscope and their viability tested by trypan blue exclusion.
Semiquantitative RT-PCR
Isolation of total and purified poly (A+) RNA was carried out according to the method of Chomczyinski & Sacchi (1987), as described in Rossmann et al. (1999). Homologous primers for murine NKCC1 were designed from published sequence information (NKCC1: forward primer 5′-GGGCTAATGAACAACTTTCAGG-3′, reverse primer 5′-GGAGAACCTCTCATTACAAGGG-3′). An 18 s rRNA fragment was amplified as an internal control with the use of primers supplied by Ambion (Austin, TX, USA). Semiquantitative RT-PCR was performed as described previously (Rossmann et al. 1999, 2000), but using different annealing temperatures (56 °C for NKCC1, and 58 °C for 18 s rRNA). The relationship between the studied gene and 18 s rRNA was calculated and regarded as the relative expression level.
Ussing-chamber experiments
Mice were anaesthetized with ether and killed by cervical dislocation. Proximal mouse colon was muscle-stripped with the aid of a microscope and the mucosa was mounted between two specially designed oval lucite half-chambers with an exposed area of 0.62 cm2 and placed in an Ussing apparatus. Both half-chambers were perfused with a buffer containing (mm): 140.5 Na+, 4.5 K+, 2 Ca2+, 1.3 Mg2+, 116 Cl−, 1.3 SO42-, 20 HCO3−, 1.5 HPO42-. The serosal perfusate also contained 11.9 mm dextrose, 10 mm pyruvate and 0.01 mm indomethacin, and the luminal perfusate contained mannitol instead of dextrose. The osmolarity of both solutions was 308 mosmol l−. Before placing the tissue into the chamber, the series resistance of the solutions and the chamber were assessed and a fluid resistance compensation performed before each experiment. Subsequently, the epithelium was continuously short-circuited using the EVC4000 voltage-clamp apparatus (World Precision Instruments) and the short-circuit current (ISC) was recorded.
Assessment of NKCC1 activity in murine colonic crypts
NKCC1 activity was measured fluorometrically in the presence of Ba2+ (except for the experiments to investigate the difference between flux rates in the absence and presence of Ba2+) and in the absence of HCO3−, as described by Heitzmann et al. (2000). This technique is based on the fact that NKCC1 can transport NH4+ instead of K+, with a resultant change in intracellular pH (pHi). Crypt cell pHi was measured as described previously (Bachmann et al. 1998; Rossmann et al. 1999) after loading crypts for 30 min at room temperature with 5 μm BCECF AM in buffer A. Crypts were fixed between a glass coverslip and a 0.3 μm polycarbonate membrane (Osmonics, Minnetonka, MN, USA) on the stage of an inverted microscope (Nikon Diaphot TMD, Nikon, Düsseldorf, Germany) and superfused following the appropriate protocol with buffer A and buffer C (40 mm NaCl was replaced by 40 mm NH4Cl). Crypts were alternately excited at 440 ± 10 nm and 490 ± 10 nm at a rate of 100 s−1. Data acquisition and processing was performed using the software provided by the manufacturer (Photon Technology International, Lawrenceville, NJ, USA). Emitted light from an individual crypt was collected through a 510 nm dichroic mirror, a 530 nm long-pass filter and an adjustable diaphragm, and was recorded by a photomultiplier. At the end of each experiment, the 490: 440 nm ratio was calibrated to pHi using the high K+-nigericin method (mm: 100 potassium gluconate, 40 KCl, 7 Hepes, 1.2 calcium gluconate, 1.2 MgSO4, 20 glucose, 0.01 nigericin; two buffers with pH values of 6.6 and 7.4 were prepared for two-point calibration). Background fluorescence was found to be negligible. CO2/HCO3−-free buffers were used to minimize pHi recovery by HCO3−-dependent transport mechanisms, and 1 mm Ba2+ was used to minimize NH4+ influx via K+ channels (Heitzmann et al. 2000).
Determination of the intrinsic intracellular buffering power
Determination of the intracellular buffering power (βi) was performed as described by Boyarsky et al. (1988) using increasing or decreasing concentrations of NH4Cl (TMA-Cl instead of NaCl, and 0.5-80 mm TMA-Cl was replaced by NH4Cl in buffer A) and measuring the resulting pHi changes. βi was calculated over a wide pHi range (6.6-7.8), resulting in buffering curves that were not different between CFTR (+/+) and CFTR (−/−) crypts (Fig. 1).
Figure 1. Intrinsic buffering capacity of colonic crypts from CFTR (+/+) and CFTR (−/−) mice.
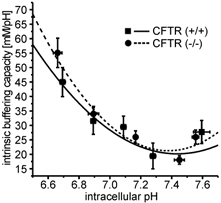
To calculate proton-base flux rates, βi was determined in murine colonic crypts from CFTR (+/+) (▪-) and CFTR (−/−) mice (•–) between pHi 6.6 and 7.8, as described in Methods. The buffering curves with a βi of 45.1 mm (pH unit)−1 and 49.5 mm (pH unit)−1 at pHi 6.7, 24.7 mm (pH unit)−1 and 26.0 mm (pH unit)−1 at pHi 7.1 and 26.7 mm (pH unit)−1 and 24.3 mm (pH unit)−1 at pHi 7.6 in CFTR (+/+) and CFTR (−/−) mice, respectively, resemble that found in other cell types. Data points were fitted with a second-order polynomial function. (n = 13 crypts from 9 mice for normal mice, n = 11 crypts from 6 mice for knockout animals).
Volume measurements
Confocal images of the crypt cross-sectional area were used as a measure for the crypt volume, as described by Heitzmann et al. (2000). Single crypts were selected with the aid of a binocular microscope, transferred into a perfusion chamber on the stage of a Zeiss LSM 410 confocal microscope, fixed with two holding pipettes, loaded with the fluorescent dye calcein AM (10 μm for 15 min) and then perfused with buffer A containing agonists and inhibitors where appropriate. Internal validation was performed by an osmotic challenge at the end of the experiment: omitting 100 mm NaCl or adding 100 mm mannitol. Cross-sectional images were obtained by z-scanning with an excitation wavelength of 488 nm and a pinhole of 33 nm every 5 s. Cross-sectional areas were determined using the Metafluor software (Universal Imaging, Downingtown, PA, USA), with the initial value normalized to 100 %.
[Cl−]i measurements
Changes in [Cl−]i were measured using the fluorescent dye MQAE as a qualitative tool for measuring concentration changes, not absolute Cl− concentrations. The same experimental set-up as for the intracellular pH measurements was used (see above). Crypts were loaded with 10 mm MQAE for 1 h and then excited with 370 ± 10 nm at a rate of 100 s. The fluorescence signal was measured at an emission wavelength of 460 nm, with the initial value normalized to 100 %.
Statistics and calculation of proton fluxes
Results are given as means ±s.e.m. Proton fluxes were calculated by multiplying the initial steep pHi slope determined by regression analysis after addition of Na+ or removal of NH4Cl, with the total buffering capacity (βt) at the initial pHi (Bachmann et al. 1998). Statistical significance was determined using Student's t test in its paired or unpaired version as appropriate.
RESULTS
NKCC1 expression levels in colonic crypts of CFTR (+/+) and (−/−) mice
The expression levels of NKCC1 were measured in colonic crypts from CFTR (+/+) and CFTR (−/−) mice using a semiquantitative PCR technique. Primers were chosen that displayed a similar amplification rate as the primers for 18 s rRNA, which was used as an internal control (Fig. 2A). As anticipated from the in situ expression data (Grubb et al. 2000), NKCC1 mRNA abundance was 12- and 18-fold higher in crypts than in surface cells in CFTR (+/+) and CFTR (−/−) mice, respectively (Fig. 2B). We found a slight but not statistically significant reduction in NKCC1 expression levels in CFTR (−/−) crypts (Fig. 2B). Thus, the absence of functional CFTR does not cause a severe reduction in the NKCC1 expression level in mouse colon.
Figure 2. Method of the semiquantitative RT-PCR (A) and expression analysis of NKCC1 in colonic crypts and surface cells from CFTR (+/+) and CFTR (−/−) mice (B).
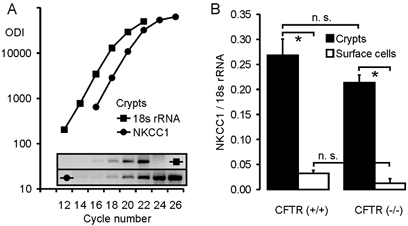
A, determination of NKCC1 expression in colonic crypts from CFTR (+/+) mice, using 18 s rRNA as an internal control. Amplification curves were created for both the NKCC1 and 18 s rRNA primer pairs, and the virtual relationship optical density integrated (ODI) of the studied gene vs. the ODI of 18 s rRNA was used as a measure for the expression level of the gene of interest after correction of the values for the different PCR products according to their length. B, relative expression levels of NKCC1 in colonic crypts (▪) and surface cells (□) from CFTR (+/+) (left panel) and CFTR (−/−) mice (right panel). NKCC1 was significantly more abundant in crypts than in surface cells (P < 0.01, Student's unpaired t test); the expression levels in CFTR (−/−) tissues were slightly, but not significantly reduced (n = 4–6 mice).
Specificity of azosemide for Na+-K+-2Cl− cotransport
Azosemide was used to inhibit NKCC1 (Heitzmann et al. 2000) because unlike bumetanide it does not exhibit autofluorescence. In previous experiments in isolated rat colonic mucosa, its inhibitory potency on ISC stimulated by cAMP activation was similar to that of bumetanide (Schultheiss et al. 1998). The inhibitory potency and specificity of azosemide for NKCC1 inhibition in murine proximal colon was tested in isolated colonic mucosa of normal mice (C57/B6) as well as in NKCC1-knockout mice and their normal littermates, in Ussing-chamber experiments. The addition of 100 μm azosemide to the serosal side of normal colonic tissue prior to stimulation of electrogenic anion secretion by dibutyryl-cAMP (db-cAMP) or forskolin resulted in the same reduction in ISC response compared to control than the application of 100 or 500 μm bumetanide (Fig. 3). When either 100 μm azosemide was added after 500 μm bumetanide, followed by forskolin, or when 500 μm bumetanide was added after 100 μm azosemide, followed by forskolin, no decrease in ISC was seen, indicating that both substances fully inhibited NKCC1 (Fig. 3B and C). Neither azosemide nor bumetanide had any effect on NKCC1 in the (−/−) colon (Fig. 3D and E), as reported by others for bumetanide (Flagella et al. 1999). The lack of effect of azosemide on ISC and the HCO3− secretory response in bumetanide-pretreated normal colonic mucosa, or in the NKCC1 (−/−) mice suggests that azosemide does not interfere with the alternative Cl− uptake pathways or with the cAMP-activated K+ channels in the colon, and can be used to assess NKCC1 activity with the same specificity as bumetanide.
Figure 3. ISC measurements in Ussing-chamber experiments with isolated colonic mucosa.
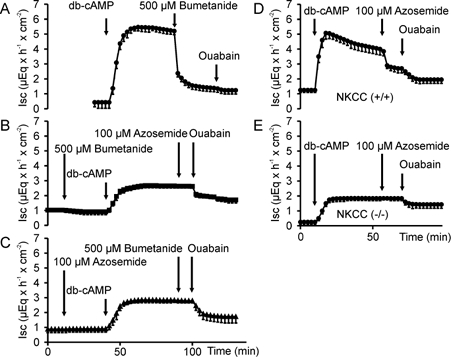
ISC measurements in Ussing-chamber experiments with isolated colonic mucosa from normal mice (Fig. 3A–C), from NKCC1 (−/−) mice and from their NKCC1 (+/+) littermates (Fig. 3D and E). cAMP-dependent stimulation of ISC (A) was inhibited to the same extent by 100 μm (not shown) or 500 μm bumetanide (B) as by 100 μm azosemide (C), and neither compound resulted in further ISC inhibition after the other one was added (B and C). Furthermore, 100 μm azosemide had no effect on ISC (D and E) or HCO3− flux (not shown) in NKCC1 (−/−) colon (n = 3–5).
cAMP-dependent stimulation of NKCC1 activity
Application of 40 mm NH3/NH4+ caused rapid alkalization to pH 7.61 ± 0.2 and a subsequent slower recovery, which was azosemide sensitive, and therefore in part attributable to NH4+ uptake by the Na+-K+-2Cl− cotransporter (Fig. 4A and B). In order to better standardize the results and compare the forskolin-stimulated NKCC1 transport rates with unstimulated NKCC1 rates, obtained in different experiments, we calculated the base efflux rates after application of forskolin and azosemide in relation to the total base efflux after NH3/NH4+ application. The procedure is explained in Fig. 4. The azosemide-sensitive part of the base efflux expressed as a percentage of the control value reflects NKCC1 transport during stimulated and unstimulated conditions.
Figure 4. NKCC1 activity in colonic crypts, measured as the azosemide-sensitive fraction of pHi recovery from a NH3/NH4+ load.
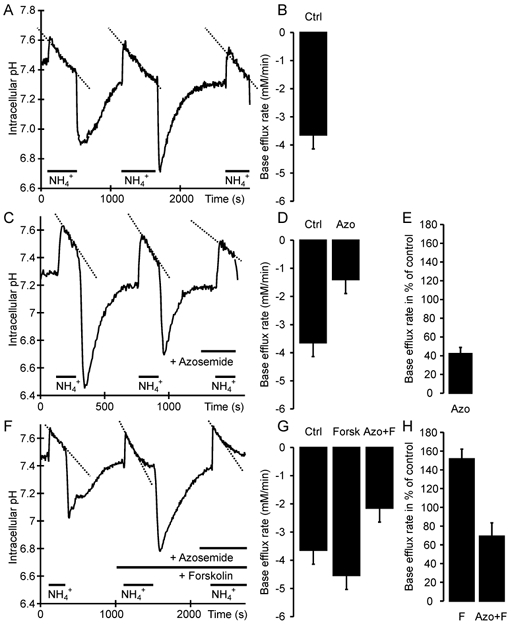
A, C and F, time course of changes in pHi; B, D and G, corresponding base efflux rates; E and H, relative NKCC1 activity. After demonstrating equal base efflux rates during multiple NH4+ pulses (A, B; -3.65 ± 0.5 mm min−1), NKCC1 activity was measured using azosemide, which decreased the ΔpHi/Δt (C) and the flux rate (D; -1.4 ± 0.3 mm min−1). Cotransport activity is expressed as a percentage of the initial flux(es) in each experiment as an internal control, to better standardize the results (E; 42 ± 7 %). When stimulated, the total and azosemide-sensitive ΔpHi/Δt (F) as well as the base efflux rates (G; -4.54 ± 0.67 and -2.15 ± 0.45 mm min−1) and the relative NKCC1 activity (H; 70 ± 14 %) was higher (Ctrl = control, Azo = azosemide, F/Forsk = forskolin; n = 8–10 crypts from 5–7 mice).
DIDS also inhibited a fraction of NH4+-induced base efflux (Table 1), suggesting that in addition to NKCC1, a DIDS-sensitive base-extruding process is also present, most likely a Cl−-OH− exchanger, and both contribute to pHi recovery after NH4+ prepulse. The combination of both virtually abolished base efflux, demonstrating that in the presence of Ba2+, NH4+ entry via other routes plays a minor role in this model.
Table 1.
Azosemide- and DIDS-sensitive base efflux rates in colonic crypts in the presence of Ba2+ upon the addition of NH4CI
| Control flux rate for each group | Flux rate after inhibition | % Inhibition by the substance† | |
|---|---|---|---|
| (mm min−1) | (mm min−1) | ||
| Azosemide | −3.35 ± 0.50 | −1.34 ± 0.17* | 58 ± 7 |
| DIDS | −2.56 ± 0.44 | −1.63 ± 0.22* | 35 ± 5 |
| Azosemide+DIDS | −2.67 ± 0.35 | −0.36 ± 0.08* | 84 ± 5 |
n = 8–10 crypts from 5–7 mice,
P < 0.01 compared with the respective control group (Student's paired t test).
Calculated individually for each experiment in a group.
Na+-K+-2Cl− cotransporter activity was stimulated by a rise in intracellular cAMP
Administration of 10 μm forskolin 5 min prior to alkalization increased the relative azosemide-sensitive NH4+ influx rates by 73 % in the absence of 1 mm Ba2+, and by 63 % in its presence (Fig. 5C). The rationale for the use of 1 mm Ba2+ was taken from the literature (Heitzmann et al. 2000), and was done to minimize NH4+ influx via K+ channels. However, 1 mm Ba2+ will not inhibit all K+ channels, and K+ channel inhibition may induce cell swelling, which in turn could inhibit NKCC1 activity. To assess the magnitude of these potentially conflicting parameters, we also performed experiments in the absence of Ba2+ (Fig. 5). A higher total base efflux rate as well as the azosemide-sensitive fraction of NH4+ influx was observed in the absence of 1 mm Ba2+, indicating that Ba2+ inhibits NH4+ influx both by blocking K+ channels and by slightly inhibiting the NKCC1 cotransporter, probably by increasing [K+]i and/or cell volume. Nevertheless, the azosemide-sensitive stimulatory effect of forskolin on the relative azosemide-sensitive NH4+ influx rate was virtually identical in both conditions.
Figure 5. Base efflux rates in murine colonic crypts in the absence and presence of Ba2+.
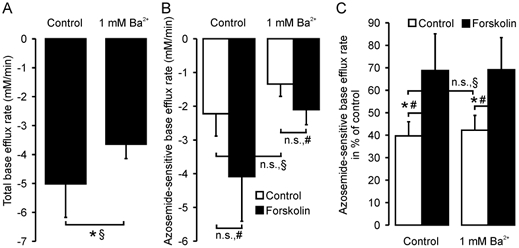
Total (A) and azosemide-sensitive (B and C) base efflux rates in murine colonic crypts. In C, the azosemide-sensitive flux rates are expressed in relation to the first NH4 pulse as an internal control (see Fig. 4A). Ba2+ slightly decreased total flux rates (A, P < 0.05) as well as azosemide-sensitive base efflux (n.s.; B and C), indicating an inhibitory effect of 1 mm Ba2+ on both non-NKCC1-related base efflux and on basal NKCC1 activity. However, forskolin stimulation increased the azosemide-sensitive base efflux rates to a similar extent both in the presence and absence of Ba2+ (C; 42 ± 7 % vs. 69 ± 14 % and 40 ± 6 % vs. 69 ± 16 %, n = 6–8 crypts from 4–5 mice, *P < 0.05: § Student's unpaired t test, # Student's paired t test). When values are given as the flux rate as a percentage of the control measured within the same experiment (C), the variability is somewhat smaller, and therefore forskolin stimulation was significant at a relatively low value of n.
NKCC activity in CFTR (+/+) and CFTR (−/−) mice
Na+-K+-2Cl− cotransporter activity was slightly reduced in CFTR (−/−) crypts compared to (+/+) crypts, although this difference was not statistically significant (42 ± 7 % in (+/+) vs. 24 ± 3 % in (−/−) crypts, Fig. 6). Five minutes after the addition of 10 μm forskolin, NKCC1 activity was stimulated by 63.6 % in (+/+) and 87.3 % in (−/−) crypts. Interestingly, a 20 min period of forskolin stimulation, the effect of which was no different to that of a 5 min period in (+/+) crypts, was able to increase the absolute NKCC1 activity in (−/−) crypts to the level found in forskolin-stimulated (+/+) crypts. This indicates that the reduced basal activity of Na+-K+-2Cl− cotransporter in (−/−) crypts is not due to an absolute lack of NKCC1 protein, but to some downregulation of function. This is in agreement with our finding of almost normal expression levels for NKCC1 in CFTR (−/−) crypts.
Figure 6. NKCC activity in colonic crypts from CFTR (+/+) and (−/−) mice, in the presence and absence of forskolin stimulation.
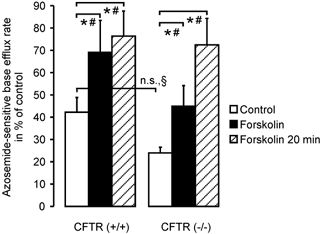
cAMP-dependent stimulation with forskolin markedly enhanced the relative azosemide-sensitive flux rate in CFTR (+/+) crypts (42 ± 4 % to 69 ± 14 %, P < 0.05: Student's paired t test; n = 8 crypts from 5 mice) as well as in CFTR (−/−) crypts (24 ± 2 % to 45 ± 9 %, P < 0.05: Student's paired t test, n = 8 crypts from 6 mice). Interestingly, a longer duration of stimulation with forskolin yielded a much more pronounced stimulation on top of the brief forskolin stimulation in CF mice (72 ± 12 %, n = 10 crypts from 8 mice) than in normal mice (76 ± 11 %, n = 9 crypts from 6 mice; *P < 0.05: § Student's unpaired t test, # Student's paired t test).
Changes in intracellular volume and [Cl−]i in colonic crypts of CFTR (+/+) and CFTR (−/−) mice upon cAMP-dependent stimulation
It has been suggested that Na+-K+-2Cl− cotransporter activity, including stimulation by cAMP, is mediated by changes in intracellular volume and [Cl−]i caused by apical Cl− secretion (Matthews et al. 1994; Lytle & Forbush, 1996). Although several investigators have demonstrated that cAMP does not stimulate Cl− secretion in the murine CFTR (−/−) intestine (Clarke et al. 1992; Cuthbert et al. 1994; Seidler et al. 1997), changes in cellular volume or [Cl−]i that are mediated by intracellular cAMP may still occur in CFTR (−/−) crypts. We therefore measured volume changes in colonic crypts of CFTR (+/+) and (−/−) mice, using the protocol of Heitzmann et al.(2000). Figure 7A shows the time course of changes in the cross-sectional area of colonic crypts from CFTR (+/+) and CFTR (−/−) mice. Azosemide, added to prevent rapid volume recovery, did not change the cross-sectional area, but forskolin caused a decrease by 9.4 ± 1.2 % in (+/+) crypts and no change in (−/−) crypts (Fig. 7A and B). This shows that a rise in intracellular cAMP decreases crypt cell volume only when CFTR expression is intact. This finding is in agreement with previous data from Valverde et al. (1993) for murine small intestinal crypts.
Figure 7. Changes in crypt volume and [Cl−]i in response to forskolin stimulation in CFTR (+/+) and (−/−) crypts.
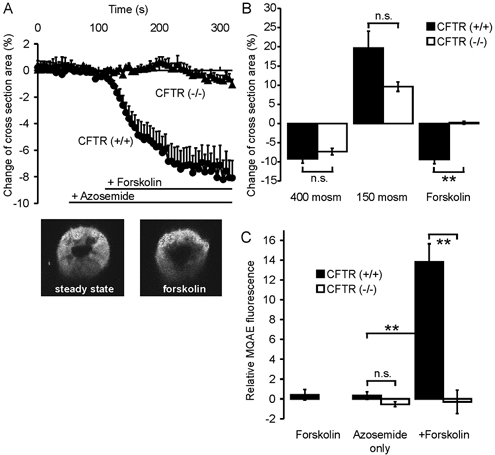
A, time course of changes in the crypt cross-sectional area. B, maximal change upon forskolin stimulation or incubation with non-isotonic solutions. The volume change after a hyperosmotic challenge was almost equal in CF and normal mice. Hyposmotic challenge produced a stronger change in crypt cross-section area in CFTR (+/+) mice. Upon forskolin stimulation, crypt cross-section area decreased by 9.4 ± 1.2 % in CFTR (+/+) crypts, while there was no change in CFTR (−/−) crypts (n = 10 crypts from 6–7 mice in each group, P < 0.01). C shows MQAE fluorescence at t = 10 min, which was used to follow changes in [Cl−]i in CFTR (+/+) and CFTR (−/−) crypts. Azosemide alone caused no change in MQAE fluorescence in CF or normal mice. Subsequent forskolin stimulation caused a substantial increase in MQAE fluorescence (13.8 ± 0.34 %) in CFTR (+/+) crypts, and no change in CFTR (−/−) crypts (n = 5–7 crypts from 3–5 mice in each group; **P < 0.01: Student's unpaired t test). While a portion of the increase in MQAE fluorescence in the normal crypts could be due to the fact that they shrink, the complete lack of change in signal intensity rules out a forskolin-induced change in [Cl−]i in the CF crypts.
Measurements with the fluorescent dye MQAE were performed to exclude a possible decrease of [Cl−]i by mechanisms other than CFTR-mediated Cl− secretion. Forskolin caused no significant change in MQAE quenching in colonic crypts in the absence of azosemide, but in its presence, subsequent forskolin stimulation increased the emitted fluorescence intensity by 14.8 ± 0.35 % in CFTR (+/+) crypts, compared to unstimulated crypts. There was no fluorescence change in forskolin-treated vs. untreated crypts from CFTR (−/−) crypts (Fig. 7C). Although the change in MQAE fluorescence may be due, at least in part, to cellular volume changes and does not give a quantitative measurement for the magnitude of the change in [Cl−]i, the lack of change in CFTR (−/−) crypts rules out that a change in [Cl−]i is the reason for the forskolin-induced increase in NKCC1 activity in CFTR (−/−) crypts.
Role of basolateral K+ channels
K+ channel activation can potentially activate Na+-K+- 2Cl− cotransporter via cellular shrinkage and/or a decrease in [K+]i. This could be the mechanism by which cAMP activates Na+-K+-2Cl− cotransporter. Since the Ba2+ sensitivity of the colonic K+ channels activated by cAMP has not been established, we used the chromanol Hoe 293b, which has been shown to inhibit the basolateral cAMP-dependent K+ channels involved in intestinal Cl− secretion (Barrett & Keely, 2000; Greger, 2000; Kunzelmann et al. 2001b). In the presence of 100 μm Hoe 293b, which is fully inhibitory for the KCNQ1 K+ channel (Kunzelmann et al. 2001a), Na+-K+-2Cl− cotransport rates were significantly reduced (Fig. 8A). This could either be due to a higher [K+]i and/or [Cl−]i in the crypt, or to cell swelling. We therefore measured crypt volume in response to addition of Hoe 293b. The compound caused a minor increase in cross-sectional area of 2.01 ± 0.31 % (Fig. 8B), suggesting that these K+ channels are only minimally active in the basal state. Forskolin still increased Na+-K+-2Cl− cotransporter activity 1.6-fold in the presence of Hoe 293b (Fig. 8A), demonstrating that the stimulatory effect of cAMP on Na+-K+-2Cl− cotransporter activity is fully preserved after Hoe 293b-sensitive K+ channel blockade.
Figure 8. Effect of Hoe 293b, an inhibitor of cAMP-activated K+ channels, on cotransporter activity.

A, the volume effect of Hoe 293b alone, which is a slight swelling of the crypt (2.03 ± 0.3 %, n = 5 crypts from 3 mice, n.s.: Student's t test), measured confocally as the change of cross-section area. The basal NKCC activity is decreased in the presence of Hoe 293b (B), but NKCC1 (B) is activated by a rise in intracellular cAMP upon forskolin stimulation in both cases (n = 6–8 crypts from 3–5 mice in each group; *P < 0.05: §Student's unpaired t test, # Student's paired t test).
Discussion
Na+-K+-2Cl− cotransport, mediated by the NKCC1 isoform in enterocytes, is considered to be the predominant Cl− uptake mechanism underlying intestinal anion secretion (Haas, 1994). Recent studies have found reduced expression levels and/or defective activation of NKCC1 in a CFTR-deficient pancreatic cell line (Shumaker & Soleimani, 1999). This finding was of great interest since several attempts to stimulate anion secretion in CF epithelia failed, despite functional, molecular or immunohistochemical evidence for the presence of non-CFTR apical anion conductances (Hagos et al. 1999; Joo et al. 1999; Gaspar et al. 2000). Failure to activate apical anion secretion may be caused by the absence of the rate-limiting basolateral anion uptake proteins. It was therefore important to clarify whether downregulation of NKCC1 also occurs in native intestinal epithelia in the absence of functional CFTR. As early as 1993, Haas et al. showed using [3H]bumetanide binding that the number of active Na+-K+-2Cl− cotransporters increased in CF nasal epithelial cells upon stimulation with a cAMP analogue, even though 36Cl− efflux did not alter (Haas et al. 1993). The different results of these two groups are possibly due to differences in the expression of the Na+-K+-2Cl− cotransporter in these cells, since Slotki et al. (1993) reported extremely low Na+-K+-2Cl− cotransporter activity in CF-P1-derived artificial chromosome (PAC) cells, irrespective of CFTR expression. Another possibility is differences in the expression of non-CFTR Cl− channels in different cell types, which are also dependent on Na+-K+-2Cl− cotransport for Cl− supply and may regulate its activity and/or expression. A third possibility is that epithelial cells from CF patients have residual CFTR expression levels that may be sufficient to prevent the downregulation of NKCC1 expression.
In contrast to biliary, bronchial and pancreatic epithelia, the intestines of CFTR null mutant mice do not display functional evidence of major non-CFTR anion conductances, and there is no functional CFTR protein expression in these mice (Ratcliff et al. 1993). We therefore chose this tissue from CFTR (−/−) mice to investigate NKCC1 expression and Na+-K+-2Cl− cotransporter regulation in the absence or presence of CFTR expression and function. To measure the expression and activity of NKCC1 in the same cellular system, we isolated colonic crypts from the proximal colon of a CFTR null mutant mouse strain and their normal wild-type littermates. CFTR (−/−) and (+/+) crypts were morphologically indistinguishable. NKCC1 expression levels were only slightly reduced in crypts from CFTR (−/−) mice. NKCC1 activity, however, was reduced in CFTR (−/−) crypts compared to crypts of normal littermates. The decrease in transport rates was more than the decrease in expression levels, but not nearly as much as described for CF-PAC cells (Shumaker & Soleimani, 1999). One possible explanation for the increase in NKCC1 expression level in the CFTR-retransfected cell line compared to the CF-PAC cell line could be a difference in growth rate and/or differentiation status between the two, leading to differences in expression levels for the ion transport proteins involved in vectorial ion transport. Of note is that the same group has also found an upregulation of expression in the CFTR-retransfected cell line compared to the CF-PAC cell line for the Na+-HCO3− cotransporter NBC1 and the apical anion exchanger DRA (Shumaker et al. 1999; Wheat et al. 2000). In native murine epithelia with a complete lack of CFTR expression, we did not find major differences in the expression levels of any of these transport proteins (Bachmann et al. 2003; also B. Riederer & U. Seidler, unpublished results). Thus, in the native intestine, factors other than CFTR are able to maintain a high expression of these anion transporters.
Even in the presence of normal NKCC1 expression and transport rates, cAMP-dependent activation could be defective in the absence of CFTR expression. In CFTR-deficient epithelia, several other ion-transport processes appear to be deregulated (Schwiebert et al. 1999), and it appears not so much the basal activity, but the cAMP-dependent regulation that is defective. Thus, in CFTR-deficient tissues or cells, the epithelial Na+ channel ENaC is not inhibited by a rise in cAMP (Stutts et al. 1995), the outwardly rectifying depolarization-induced Cl− channel (ORDIC) is not activated by cAMP (Jovov et al. 1995), electroneutral Na+ absorption in the jejunum is not inhibited by cAMP (Clarke & Harline, 1996) and VIP-dependent K+ channel opening is not observed (Loussouarn et al. 1996).
We therefore investigated the effect of raising intracellular cAMP with forskolin on the Na+-K+-2Cl− cotransporter in the cells that form the colonic crypts of CFTR (−/−) and normal mice. As already shown for rat colonic crypts (Heitzmann et al. 2000), an increase in NKCC1 transport rate occurred upon stimulation of normal crypts with forskolin. Significantly, the same degree of activation was observed in CFTR (−/−) crypts, although the time to achieve maximal stimulation was slower than in normal crypts, suggesting that the decrease in basal activity observed in the CFTR (−/−) crypts is largely due to post-transcriptional events. These results demonstrate clearly that the absence of CFTR expression does not result in a general cellular defect in the cAMP-dependent regulation of the Na+-K+-2Cl− cotransporter.
For NKCC activation, evidence from a number of epithelial and non-epithelial cell types argues for complex and possibly multiple regulatory mechanisms, among them a fall in cytosolic Cl− activity, transient cell shrinkage, phosphorylation of the cotransporter or other phosphorylation events (Haas et al. 1993, 1995; Matthews et al. 1994; Lytle & Forbush, 1996; Lytle, 1997,1998; Heitzmann et al. 2000). Recently, volume-sensitive protein kinases have been discovered, and it has been suggested that they are the link between cAMP-mediated secretion and phosphorylation of basolateral anion transporters (Lytle, 1997; Roig et al. 2000). Since we have observed no forskolin-induced anion secretion in the isolated colon of these mice in the Ussing chamber, we would not have expected a secretion-related volume or [Cl−]i decrease in CFTR (−/−) crypts. It was feasible, however, that the activation of basolateral K+ channels, or unknown transport events, could result in cell shrinkage and/or a decrease in [Cl−]i even in CFTR-deficient crypts. To address this, the crypt cross-sectional area was measured by confocal microscopy (Heitzmann et al. 2000). Addition of forskolin in the presence of azosemide in CFTR (+/+) crypts resulted in a marked decrease in this parameter, but no change was observed in CFTR (−/−) crypts. Although this already makes a cellular Cl− loss unlikely, we nevertheless assessed changes in [Cl−]i using the fluorescent dye MQAE. Again, in CFTR (+/+) crypts in the presence of azosemide there was a marked increase in MQAE fluorescence, indicative of cellular Cl− loss, whereas this increase did not occur in CFTR (−/−) crypts. Finally, we studied whether NKCC1 activation still occurred after blockade of the cAMP-dependent, chromanol-sensitive K+ channels in the colon. Although Hoe 293b decreased basal Na+-K+-2Cl− cotransporter activity, activation by forskolin still occurred.
The stimulation of Na+-K+-2Cl− cotransport activity in CFTR (−/−) cells in the absence of changes in cell volume and [Cl−]i show that cell shrinkage is not required for NKCC1 activation. In contrast, an increase in intracellular cAMP clearly activates the Na+-K+-2Cl− cotransporter in a direct fashion, supporting the results of Haas et al. (1993). One such activation pathway could be a protein kinase A (PKA)-mediated phosphorylation of the protein, since NKCC1 contains highly conserved PKA consensus sites in their cytoplasmic domains. However, it is not yet known whether these consensus sites are indeed phosphorylated by PKA, whether NKCC1 phosphorylation results in changes in transport activity, or what additional cAMP-dependent events are necessary to fully activate the transporter.
Thus, it appears that cAMP-dependent NKCC1 activation does not require either altered chemical gradients or secretion-associated events such as shrinkage or ionic disequilibrium. The data imply that receptor-mediated signal transduction events activate both the apical and basolateral ion transport pathways required for the initiation and maintenance of the secretory process. This is consistent with the observation of Heitzmann et al. (2000) in rat and our work with murine colonic crypts, that forskolin-associated shrinkage is small in the absence of Na+-K+-2Cl− cotransport inhibition. The situation is similar to that observed by us in rabbit parietal cells, where Ca2+-dependent and cAMP-dependent secretagogues, although both resulting in acid production and cellular shrinkage with rapid volume recovery, resulted in a differential activation of the Na+-H+ exchanger isoforms NHE1 and NHE4 (Bachmann et al. 1998). This suggests strongly that secretagogue-induced Na+-H+ exchange activation in these cells is not simply secondary to secretion-associated shrinkage, but is induced directly by the secretagogue-associated cellular signalling pathways.
In summary, our results demonstrate normal NKCC1 expression, slightly reduced basal Na+-K+-2Cl− cotransporter rates, and fully preserved cAMP-dependent activation in native colonic crypts from CFTR-deficient mice. Since our functional data from the CFTR-knockout mouse colon are in agreement with those from nasal epithelial cells of CF patients, a severe dysfunction of this anion importer is unlikely to contribute to the secretory defect in CF epithelia. Furthermore, the cAMP-dependent activation of NKCC in CFTR (−/−) crypts occurred in the absence of cellular volume and [Cl−]i loss, ruling out the latter as the final common pathway for NKCC1 activation.
Acknowledgments
The authors thank Christina Neff for expert technical assistance, Professor G. Shull, University of Cincinnati Medical School, for providing NKCC1 (+/-) breeder pairs, and Professor Dr Michael Sessler for constructive criticism. This work was supported by DFG grants Se 460/13-1 and Se 460/13-2, provided by the Mukoviszidose Foundation e.V., and by a grant from the IZKF Tübingen (Project IIIC1). This article includes experimental work performed by Katrin Wüchner in fulfilment of the requirements for her doctoral thesis.
References
- Bachmann O, Rossmann H, Berger UV, Colledge WH, Ratcliff R, Evans MJ, Gregor M, Seidler U. cAMP-mediated regulation of murine intestinal/pancreatic Na+/HCO3− cotransporter subtype pNBC1. Am J Physiol. 2003;284:G37–45. doi: 10.1152/ajpgi.00209.2002. [DOI] [PubMed] [Google Scholar]
- Bachmann O, Sonnentag T, Siegel WK, Lamprecht G, Weichert A, Gregor M, Seidler U. Different acid secretagogues activate different Na+/H+ exchanger isoforms in rabbit parietal cells. Am J Physiol. 1998;275:G1085–1093. doi: 10.1152/ajpgi.1998.275.5.G1085. [DOI] [PubMed] [Google Scholar]
- Barrett KE, Keely SJ. Chloride secretion by the intestinal epithelium: molecular basis and regulatory aspects. Annu Rev Physiol. 2000;62:535–572. doi: 10.1146/annurev.physiol.62.1.535. [DOI] [PubMed] [Google Scholar]
- Boyarsky G, Ganz MB, Sterzel RB, Boron WF. pH regulation in single glomerular mesangial cells. I. Acid extrusion in absence and presence of HCO3−. Am J Physiol. 1988;255:C844–856. doi: 10.1152/ajpcell.1988.255.6.C844. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Clarke LL, Grubb BR, Gabriel SE, Smithies O, Koller BH, Boucher RC. Defective epithelial chloride transport in a gene-targeted mouse model of cystic fibrosis. Science. 1992;257:1125–1128. doi: 10.1126/science.257.5073.1125. [DOI] [PubMed] [Google Scholar]
- Clarke LL, Harline MC. CFTR is required for cAMP inhibition of intestinal Na+/absorption in a cystic fibrosis mouse model. Am J Physiol. 1996;270:G259–267. doi: 10.1152/ajpgi.1996.270.2.G259. [DOI] [PubMed] [Google Scholar]
- Cuthbert AW, MacVinish LJ, Hickman ME, Ratcliff R, Colledge WH, Evans MJ. Ion-transporting activity in the murine colonic epithelium of normal animals and animals with cystic fibrosis. Pflugers Arch. 1994;428:508–515. doi: 10.1007/BF00374572. [DOI] [PubMed] [Google Scholar]
- Flagella M, Clarke LL, Miller ML, Erway LC, Giannella RA, Andringa A, Gawenis LR, Kramer J, Duffy JJ, Doetschman T, Lorenz JN, Yamoah EN, Cardell EL, Shull GE. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J Biol Chem. 1999;274:26946–26955. doi: 10.1074/jbc.274.38.26946. [DOI] [PubMed] [Google Scholar]
- Gaspar KJ, Racette KJ, Gordon JR, Loewen ME, Forsyth GW. Cloning a chloride conductance mediator from the apical membrane of porcine ileal enterocytes. Physiol Genomics. 2000;3:101–111. doi: 10.1152/physiolgenomics.2000.3.2.101. [DOI] [PubMed] [Google Scholar]
- Greger R. Role of CFTR in the colon. Annu Rev Physiol. 2000;62:467–491. doi: 10.1146/annurev.physiol.62.1.467. [DOI] [PubMed] [Google Scholar]
- Grubb BR, Boucher RC. Pathophysiology of gene-targeted mouse models for cystic fibrosis. Physiol Rev. 1999;79:S193–214. doi: 10.1152/physrev.1999.79.1.S193. [DOI] [PubMed] [Google Scholar]
- Grubb BR, Lee E, Pace AJ, Koller BH, Boucher RC. Intestinal ion transport in NKCC1-deficient mice. Am J Physiol. 2000;279:G707–718. doi: 10.1152/ajpgi.2000.279.4.G707. [DOI] [PubMed] [Google Scholar]
- Haas M. The Na-K-Cl cotransporters. Am J Physiol. 1994;267:C869–885. doi: 10.1152/ajpcell.1994.267.4.C869. [DOI] [PubMed] [Google Scholar]
- Haas M, McBrayer D, Lytle C. [Cl−]i-dependent phosphorylation of the Na-K-Cl cotransport protein of dog tracheal epithelial cells. J Biol Chem. 1995;270:28955–28961. doi: 10.1074/jbc.270.48.28955. [DOI] [PubMed] [Google Scholar]
- Haas M, McBrayer DG, Yankaskas JR. Dual mechanisms for Na-K-Cl cotransport regulation in airway epithelial cells. Am J Physiol. 1993;264:C189–200. doi: 10.1152/ajpcell.1993.264.1.C189. [DOI] [PubMed] [Google Scholar]
- Hagos Y, Krick W, Burckhardt G. Chloride conductance in HT29 cells: investigations with apical membrane vesicles and RT-PCR. Pflugers Arch. 1999;437:724–730. doi: 10.1007/s004240050838. [DOI] [PubMed] [Google Scholar]
- Heitzmann D, Warth R, Bleich M, Henger A, Nitschke R, Greger R. Regulation of the Na+/2Cl− K+/cotransporter in isolated rat colon crypts. Pflugers Arch. 2000;439:378–384. doi: 10.1007/s004249900156. [DOI] [PubMed] [Google Scholar]
- Jacob P, Rossmann H, Neff C, Weichert A, Gregor M, Seidler U. Multiple transport pathways are involved in cellular Cl− uptake during colonic anion secretion. Gastroenterology. 2001;120:A-699. [Google Scholar]
- Joo NS, Clarke LL, Han BH, Forte LR, Kim HD. Cloning of ClC-2 chloride channel from murine duodenum and its presence in CFTR knockout mice. Biochim Biophys Acta. 1999;1446:431–437. doi: 10.1016/s0167-4781(99)00110-4. [DOI] [PubMed] [Google Scholar]
- Joo NS, London RM, Kim HD, Forte LR, Clarke LL. Regulation of intestinal Cl− and HCO3− secretion by uroguanylin. Am J Physiol. 1998;274:G633–644. doi: 10.1152/ajpgi.1998.274.4.G633. [DOI] [PubMed] [Google Scholar]
- Jovov B, Ismailov II, Berdiev BK, Fuller CM, Sorscher EJ, Dedman JR, Kaetzel MA, Benos DJ. Interaction between cystic fibrosis transmembrane conductance regulator and outwardly rectified chloride channels. J Biol Chem. 1995;270:29194–29200. doi: 10.1074/jbc.270.49.29194. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Bleich M, Warth R, Levy-Holzman R, Garty H, Schreiber R. Expression and function of colonic epithelial KvLQT1 K+ channels. Clin Exp Pharmacol Physiol. 2001a;28:79–83. doi: 10.1046/j.1440-1681.2001.03407.x. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Hubner M, Schreiber R, Levy-Holzman R, Garty H, Bleich M, Warth R, Slavik M, Von Hahn T, Greger R. Cloning and function of the rat colonic epithelial K+ channel KVLQT1. J Membr Biol. 2001b;179:155–164. doi: 10.1007/s002320010045. [DOI] [PubMed] [Google Scholar]
- Loussouarn G, Demolombe S, Mohammad-Panah R, Escande D, Baro I. Expression of CFTR controls cAMP-dependent activation of epithelial K+/currents. Am J Physiol. 1996;271:C1565–1573. doi: 10.1152/ajpcell.1996.271.5.C1565. [DOI] [PubMed] [Google Scholar]
- Lytle C. Activation of the avian erythrocyte Na-K-Cl cotransport protein by cell shrinkage, cAMP, fluoride, and calyculin-A involves phosphorylation at common sites. J Biol Chem. 1997;272:15069–15077. doi: 10.1074/jbc.272.24.15069. [DOI] [PubMed] [Google Scholar]
- Lytle C. A volume-sensitive protein kinase regulates the Na-K-2Cl cotransporter in duck red blood cells. Am J Physiol. 1998;274:C1002–1010. doi: 10.1152/ajpcell.1998.274.4.C1002. [DOI] [PubMed] [Google Scholar]
- Lytle C, Forbush B. Regulatory phosphorylation of the secretory Na-K-Cl cotransporter: modulation by cytoplasmic Cl. Am J Physiol. 1996;270:C437–448. doi: 10.1152/ajpcell.1996.270.2.C437. [DOI] [PubMed] [Google Scholar]
- Matthews JB, Smith JA, Tally KJ, Awtrey CS, Nguyen H, Rich J, Madara JL. Na-K-2Cl cotransport in intestinal epithelial cells. Influence of chloride efflux and F-actin on regulation of cotransporter activity and bumetanide binding. J Biol Chem. 1994;269:15703–15709. [PubMed] [Google Scholar]
- Ratcliff R, Evans MJ, Cuthbert AW, MacVinish LJ, Foster D, Anderson JR, Colledge WH. Production of a severe cystic fibrosis mutation in mice by gene targeting. Nat Genet. 1993;4:35–41. doi: 10.1038/ng0593-35. [DOI] [PubMed] [Google Scholar]
- Roig J, Huang Z, Lytle C, Traugh JA. p21-activated protein kinase gamma-PAK is translocated and activated in response to hyperosmolarity. Implication of Cdc42 and phosphoinositide 3-kinase in a two-step mechanism for gamma-PAK activation. J Biol Chem. 2000;275:16933–16940. doi: 10.1074/jbc.M001627200. [DOI] [PubMed] [Google Scholar]
- Rossmann H, Alper SL, Nader M, Wang Z, Gregor M, Seidler U. Three 5′-variant mRNAs of anion exchanger AE2 in stomach and intestine of mouse, rabbit, and rat. Ann N Y Acad Sci. 2000;915:81–91. doi: 10.1111/j.1749-6632.2000.tb05226.x. [DOI] [PubMed] [Google Scholar]
- Rossmann H, Bachmann O, Vieillard-Baron D, Gregor M, Seidler U. Na+/HCO3− cotransport and expression of NBC1 and NBC2 in rabbit gastric parietal and mucous cells. Gastroenterology. 1999;116:1389–1398. doi: 10.1016/s0016-5085(99)70503-2. [DOI] [PubMed] [Google Scholar]
- Schultheiss G, Horger S, Diener M. The bumetanide-resistant part of forskolin-induced anion secretion in rat colon. Acta Physiol Scand. 1998;164:219–228. [PubMed] [Google Scholar]
- Schwiebert EM, Benos DJ, Egan ME, Stutts MJ, Guggino WB. CFTR is a conductance regulator as well as a chloride channel. Physiol Rev. 1999;79:S145–166. doi: 10.1152/physrev.1999.79.1.S145. [DOI] [PubMed] [Google Scholar]
- Seidler U, Blumenstein I, Kretz A, Viellard-Baron D, Rossmann H, Colledge WH, Evans M, Ratcliff R, Gregor M. A functional CFTR protein is required for mouse intestinal cAMP-, cGMP- and Ca2+-dependent HCO3− secretion. J Physiol. 1997;505:411–423. doi: 10.1111/j.1469-7793.1997.411bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumaker H, Amlal H, Frizzell R, Ulrich CD, Soleimani M. CFTR drives Na+-nHCO3− cotransport in pancreatic duct cells: a basis for defective HCO3− secretion in CF. Am J Physiol. 1999;276:C16–25. doi: 10.1152/ajpcell.1999.276.1.C16. [DOI] [PubMed] [Google Scholar]
- Shumaker H, Soleimani M. CFTR upregulates the expression of the basolateral Na+-K+-2Cl− cotransporter in cultured pancreatic duct cells. Am J Physiol. 1999;277:C1100–1110. doi: 10.1152/ajpcell.1999.277.6.C1100. [DOI] [PubMed] [Google Scholar]
- Slotki IN, Breuer WV, Greger R, Cabantchik ZI. Long-term cAMP activation of Na+-K+-2Cl− cotransporter activity in HT-29 human adenocarcinoma cells. Am J Physiol. 1993;264:C857–865. doi: 10.1152/ajpcell.1993.264.4.C857. [DOI] [PubMed] [Google Scholar]
- Soleimani M, Ulrich CD. How cystic fibrosis affects pancreatic ductal bicarbonate secretion. Med Clin North Am. 2000;84:641–655. doi: 10.1016/s0025-7125(05)70247-9. [DOI] [PubMed] [Google Scholar]
- Stutts MJ, Canessa CM, Olsen JC, Hamrick M, Cohn JA, Rossier BC, Boucher RC. CFTR as a cAMP-dependent regulator of sodium channels. Science. 1995;269:847–850. doi: 10.1126/science.7543698. [DOI] [PubMed] [Google Scholar]
- Valverde MA, O'Brien JA, Sepulveda FV, Ratcliff R, Evans MJ, Colledge WH. Inactivation of the murine cftr gene abolishes cAMP-mediated but not Ca2+-mediated secretagogue-induced volume decrease in small-intestinal crypts. Pflugers Arch. 1993;425:434–438. doi: 10.1007/BF00374869. [DOI] [PubMed] [Google Scholar]
- Wheat VJ, Shumaker H, Burnham C, Shull GE, Yankaskas JR, Soleimani M. CFTR induces the expression of DRA along with Cl−/HCO3− exchange activity in tracheal epithelial cells. Am J Physiol. 2000;279:C62–71. doi: 10.1152/ajpcell.2000.279.1.C62. [DOI] [PubMed] [Google Scholar]