

Abstract
CNS glycogen, contained predominantly in astrocytes, can be converted to a monocarboxylate and transported to axons as an energy source during aglycaemia. We analysed glycogen regulation and the role of glycogen in supporting neural activity in adult mouse optic nerve, a favourable white matter preparation. Axon function was quantified by measuring the compound action potential (CAP) area. During aglycaemia, axon function persisted for 20 min, then declined in conjunction with glycogen content. Lactate fully supported CAPs in the absence of glucose, but was unable to sustain glycogen content; thus, axon failure occurred rapidly when lactate was withdrawn. Glycogen content in the steady state was directly proportional to bath glucose concentration. Increasing [K+]o to 10 mm caused a rapid decrease in glycogen content. Latency to onset of CAP failure during aglycaemia was directly proportional to glycogen content and varied from about 2 to 30 min. Intense neural activity reduced glycogen content in the presence of 10 mm bath glucose and CAP area gradually declined. CAP area declined more rapidly during high frequency stimulation if monocarboxylate transport was inhibited. This suggested that astrocytic glycogen was broken down to a monocarboxylate(s) that was used by rapidly discharging axons. Likewise, depleting glycogen by brief periods of high frequency axon stimulation accelerated onset of CAP decline during aglycaemia. In summary, these experiments indicated that glycogen content was under dynamic control and that glycogen was used to support the energy needs of CNS axons during both physiological as well as pathological processes.
Glycogen is located almost exclusively in astrocytes in the central nervous system (CNS) (Peters & Palay, 1976; Cataldo & Broadwell, 1986; Magistretti et al. 1993) but its function is not well understood. Recent evidence suggests that glycogen may act as an energy source to sustain neural elements during periods of energy deprivation. Specifically, it is hypothesised that glycogen breaks down to a monocarboxylate, probably lactate, that is shuttled from astrocytes to neural elements where it is metabolised oxidatively (Dringen et al. 1993; Izumi et al. 1997a; Magistretti et al. 1999; Wender et al. 2000). This is an attractive hypothesis for several reasons. First, brain glycogen would have a similar role to its role in muscle and liver, namely as an energy source (Stryer, 1995). Second, it explains the heterogeneous cellular distribution of the various enzymes and transporters involved in the metabolism and transport of glycogen and its metabolites (Izumi et al. 1997a; Magistretti et al. 1999; Wender et al. 2000). Third, it explains the ability of in vitro brain tissue preparations to survive for extended periods of time in the absence of an exogenous energy substrate (Swanson & Choi, 1993; Izumi et al. 1997a; Wender et al. 2000; Cater et al. 2001).
In the adult rat optic nerve, a central white matter tract, the glycogen content of the nerve, is intimately associated with nerve function. During periods of aglycaemia, CAP function is initially maintained while glycogen content steadily declines. When glycogen reaches its nadir, CAP function fails (Wender et al. 2000). Higher levels of glycogen were associated with longer periods of function (i.e. CAP persistence) during aglycaemia. These data suggest that glycogen is used to maintain function during periods of energy deprivation (Wender et al. 2000). Lactate probably is the glycogen breakdown product shuttled to axons as oxidative fuel (Wender et al. 2000; Brown et al. 2001b). Transporters and enzymes for monocarboxylates (lactate and pyruvate) are selectively localised on astrocytes and neural elements in a configuration that favours the transport of lactate from astrocytes to axons, where it is converted to pyruvate and oxidatively metabolised (Jackson & Halestrap, 1996; Broer et al. 1999; Pierre et al. 2002, but see Chih et al. 2001).
The role of glycogen in brain function under non-pathological conditions remains unclear. It is known that glycogen has a high turnover rate in the CNS, and that the rate of ‘turnover’ is enhanced by increased adjacent neural activity (Pentreath & Kai-Kai, 1982; Swanson et al. 1992). In this study we analysed certain aspects of glycogen regulation and the behaviour of glycogen during aglycaemia, hypoglycaemia and intense stimulation in the adult mouse optic nerve (MON). This work extends our prior studies on the rat optic nerve and introduces the MON preparation. We have moved to the mouse for important technical reasons (e.g. its smaller size reduces diffusion distances), to verify our previous results in another species and to prepare for future genetic studies that favour mouse models. Our results indicated that glycogen content was modulated by glucose concentration, extracellular K+ concentration ([K+]o) and neural activity. As in the rat, glycogen was used in the MON to support function during aglycaemia. Manipulations that lowered glycogen content accelerated CAP failure during intense activity or aglycaemia. Intense axonal discharge in normoglycaemic conditions caused glycogen breakdown and axonal discharge failed if monocarboxylate transport was blocked, implying axonal utilisation of the glycogen breakdown product lactate, or possibly pyruvate. These experimental findings indicated that astrocytic glycogen was dynamically regulated and played an important role in helping to supply energy to CNS axons under both pathological and physiological conditions. A preliminary account of this work has been published (Brown & Ransom, 2001).
METHODS
Preparation
All experiments were carried out in accordance with the guidelines for Animal Care of the University of Washington. Adult male Swiss Webster mice (20–25 g) were deeply anaesthetised with CO2 then decapitated. The optic nerves were exposed, gently freed from their dural sheaths and placed in an interface perfusion chamber (Medical Systems Corp., Greenvale, NY, USA). Nerves were maintained at 37 °C and perfused with control artificial cerebrospinal fluid (aCSF) that contained (mm): 153 Na+, 3 K+, 2 Mg2+, 2 Ca2+, 143 Cl−, 26 HCO3−, 1.25 HPO42- and 10 glucose. In experiments with increased extracellular [K+], osmolarity was maintained by equimolar reduction of NaCl. The aCSF was bubbled with a 5 % CO2-containing gas mixture (95 % O2, 5 % CO2) to maintain pH at 7.45. The tissue was continuously exposed to a moisturised gas mixture containing 95 % O2, 5 % CO2. Suction electrodes filled with aCSF (of the same composition as the test perfusate for each experiment) were attached to the nerve for stimulation and recording of the CAP after the nerves were equilibrated for 60 min in control aCSF. CAPs were evoked every 30 s and stimulus strength was adjusted to evoke the maximum amplitude CAP and then increased another 25 % to ensure that stimulus strength was always supramaximal.
Data were acquired online (Digidata 1200A, Axon Instruments, CA, USA) using proprietary software (Axoscope, Axon Instruments). CAP area was calculated using Clampfit (Axon Instruments). Curve fitting procedures to determine latency to CAP fall and percentage CAP decline were carried out as previously described (Wender et al. 2000; Brown, 2001).
Glycogen and protein assays
Nerves were immediately placed in 2 ml ice-cold 85 % ethanol/15 % 30 mm HCl, which instantly stops glycogen metabolism. The tissue (in solution) was stored at -20 °C until assays were performed. Each determination required 10 optic nerves (five pairs of nerves, ∼8 mg wet tissue total). The nerves were transferred to 400 μl ice-cold 0.1 m NaOH/0.01 % sodium lauryl sulphate plus 54 μl 1 m HCl (final acid concentration 0.03 m) and sonicated to suspension using a Fisher 60 Sonic Dismembrator (Fisher Scientific, CA, USA). A 40 μl sample of the suspension was removed and used for protein assay using the Pierce BCA Protein kit 23225 (Pierce, IL, USA) with BSA standards to generate a curve against which the samples were compared. Fifteen microlitres of BSA standard and samples were added to 300 μl of the working solution and incubated for 105 min at 37 °C. A Packard Spectracount (Packard Biosciences, CA, USA) was used to read protein values at 546 nm.
The remainder of the nerve homogenate was divided into two 200 μl fractions. One fraction was added to a solution containing 0.08 units of the enzyme amylo-α-1,4-α-1,6-glucosidase (AG: EC 3.2.1.3) while the other sample was added to an AG-free solution. Samples were agitated for 1 h at 37 °C. AG completely hydrolyses glycogen to glucose, thus the AG-treated sample contains background glucose plus the glucose produced from glycogen breakdown, whereas the other sample only contains the background glucose. Subtraction of the latter sample from the AG-treated sample yields the glucose due solely to glycogen breakdown; in fact, the background glucose values were negligible (i.e. the amount of free glucose in the tissue was negligible) due to glucose dissolving in the ethanol/HCl storage solution. The contribution from glucose-6-phosphate is considered negligible due to its low intracellular concentration (Garrett & Grisham, 1999). Each sample was combined with 260 μm ATP, 50 μm NADP and 2.27 units and 6.25 units, respectively, of the enzymes glucose-6-phosphate dehydrogenase (EC 1.1.1.49) and hexokinase (EC 2.7.1.1). Hexokinase converts glucose to glucose-6-phosphate, which is subsequently converted to 6-phosphogluconolactone by glucose-6-phosphate dehydrogenase. This reaction catalyses the formation of NADPH from NADP. Measurement of NADPH fluorescence with a Bio-Rad VersaFluor fluorometer (excited at 360 nm, emission at 415 nm) was used to calculate the glucose concentration in each sample when compared with a standard curve generated using known concentrations of glucose. It is assumed that all glycogen is converted to glucose, so that glucose determination yields the glycogen concentration in equivalent glucosyl units (Passonneau et al. 1967).
Data analysis
Data are presented as means and standard error of the mean (s.e.m.). Significance was determined by analysis of variance (ANOVA) using Tukey's post hoc test or Student's t test, where P < 0.05 was taken to indicate statistical significance.
RESULTS
Glycogen content of MON: effects of aglycaemia
MONs in control aCSF containing 10 mm glucose had stable robust evoked CAPs for several hours. The glycogen content of the nerve at fixed time points in the presence of 10 mm glucose was also stable at about 7 pmol glycogen (μg protein)−1. There was no significant difference in the glycogen content at 0, 60, 120 or 180 min intervals (n = 3; data not shown). These data were slightly different from those found in rat optic nerve; glycogen content in the acutely isolated rat optic nerve (RON) fell during the first hour and then stabilised (Wender et al. 2000). For consistency, in all subsequent experiments MONs were pre-incubated in 10 mm glucose for 60 min prior to test conditions.
The effect of glucose deprivation on CAP area and glycogen content was investigated. During a 60 min period of glucose deprivation CAP was maintained for about 20 min before it began to fail; with continued glucose deprivation the CAP fell rapidly to zero. Following aglycaemia, the CAP recovered to 44.1 ± 8.2 % (n = 14; data not shown) of its control level indicating that irreversible injury had occurred, and that about half of the MON axons were affected. Prior to aglycaemia, the glycogen content in nerves perfused with 10 mm glucose was stable between 6 and 7 pmol glycogen (μg protein)−1. On exposure to aglycaemia the glycogen content fell until it reached its nadir after about 20 min. In the presence of continued aglycaemia, glycogen remained stable at this level (Fig. 1A). The glycogen content of nerves was allowed to recover for 60 min in control aCSF after the 60 min period of aglycaemia remained at this low level (n = 3, data not shown). These data suggested that during glucose withdrawal glycogen was used to maintain axon function, and that axon failure coincided with exhaustion of usable glycogen stores.
Figure 1. MON function and glycogen content.
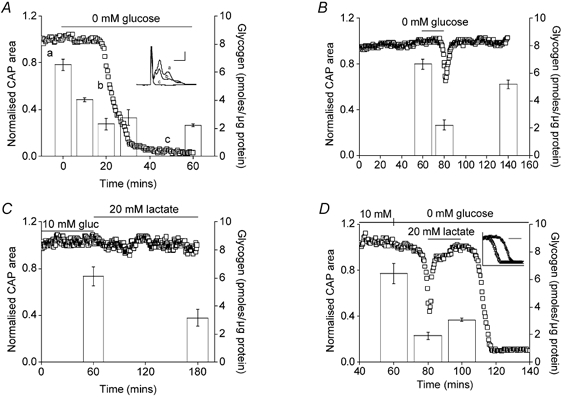
A, aglycaemia resulted in onset of failure of the CAP after 17.8 ± 1.6 min (n = 4). The inset shows representative CAPs recorded from one of the nerves averaged in A at the indicated time points (a-c; calibration 0.5 mV, 1 ms). Prior to aglycaemia, the glycogen content was 6.52 ± 0.40 pmol glycogen (μg protein)−1 (n = 3). Glycogen fell to 4.40 ± 0.13 pmol glycogen (μg protein)−1 after 10 min of glucose deprivation (n = 3; P < 0.01vs. 0 min group) and to 2.30 ± 0.41 pmol glycogen (μg protein)−1 (n = 3; P < 0.01vs. 10 min group; P < 0.01vs. 0 min group) after 20 min of glucose deprivation and remained roughly at this latter value in spite of continuing aglycaemia (30 and 60 min, 2.77 ± 0.53 and 2.22 ± 0.09 pmol glycogen (μg protein)−1, respectively; n = 3, P < 0.001vs. 0 min group). Subsequent recovery in control aCSF had no significant effect on glycogen content (2.87 ± 0.12 pmol glycogen (μg protein)−1; n = 3; n.s. vs. 60 min group; data not shown); ANOVA with Tukey's post hoc test. B, glucose-free aCSF for 20 min led to fully reversible CAP reduction and loss of glycogen. Function was fully restored by 10 mm glucose (n = 3). The glycogen content of nerves after 1 h of incubation in 10 mm glucose was 6.66 ± 0.36 pmol glycogen (μg protein)−1 (n = 3), which fell to 2.22 ± 0.38 pmol glycogen (μg protein)−1 (n = 3; P < 0.01 compared with 60 min) after 20 min of aglycaemia. Glycogen content was significantly restored after 1 h recovery in 10 mm glucose aCSF (5.18 ± 0.32 pmol glycogen (μg protein)−1, n = 3; P < 0.05 compared with 80 min). The difference between the glycogen content at 60 and 140 min was not significant (P > 0.05; ANOVA with Tukey's post hoc test). C, bath lactate (120 min) sustained CAP amplitude (n = 3), but not glycogen content, which fell significantly when incubated in 20 mm lactate for 2 h (6.13 ± 0.66 to 3.14 ± 0.60 pmol glycogen (μg protein)−1, n = 3; P < 0.05; Student's t test). D, glycogen was depleted by exposure to 20 min of aglycaemia (6.44 ± 0.73 to 1.92 ± 0.27 pmol glycogen (μg protein)−1, n = 3; P < 0.01). CAP function, but not glycogen (3.05 ± 0.20 compared with 1.92 ± 0.27 pmol glycogen (μg protein)−1, n = 3, n.s.), was restored by 20 mm lactate aCSF; ANOVA with Tukey's post hoc test. Lactate withdrawal now resulted in rapid CAP failure (n = 3). The inset illustrates the accelerated CAP failure after glycogen depletion (open triangles) compared with control where MONs were bathed in 10 mm glucose for 100 min (open squares; latency to CAP failure = 19.5 ± 2.2 vs. 8.5 ± 1.7 min, P < 0.01, n = 3; Student's t test). The scale bar is 10 min and the x axis starts at onset of aglycaemia. For A-D the left axis indicates average CAP area (open squares) and the right axis indicates the glycogen content of the nerve (open columns). Error bars indicate the s.e.m.
The data described above indicated that after 60 min of aglycaemia there was no significant recovery in glycogen content, suggesting that once glycogen content is depleted it cannot be replenished. However 60 min of aglycaemia produced significant tissue damage (Brown et al. 2001a) and therefore the ability of the tissue to generate glycogen may be compromised. We tested for capacity to replenish glycogen content under conditions where there was no obvious tissue damage. Figure 1B illustrates that 20 min of aglycaemia resulted in fully reversible failure of the CAP and reversible depletion of glycogen (n = 3). The glycogen content of nerves after 1 h of incubation in 10 mm glucose fell significantly after 20 min of aglycaemia but was subsequently significantly restored after 1 h recovery in 10 mm glucose aCSF.
Lactate and glycogen
Although glucose is viewed as the energy substrate of choice of the brain, other metabolic substrates have been shown to sustain neural function in the absence of glucose (McIlwain, 1953; Izumi et al. 1997a,1997b; Brown et al. 2001b). Thus, the finding that 20 mm lactate (carbon equivalent to 10 mm glucose) sustained the CAP for 2 h was no surprise. However, glycogen content fell significantly when tissue was incubated in 20 mm lactate for 2 h (Fig. 1C).
We tested the expectation that depleting glycogen content would accelerate CAP failure during aglycaemia. After the standard 1 h incubation period, a 20 min period of aglycaemia resulted in CAP failure (n = 4) and glycogen depletion. Introduction of aCSF containing 20 mm lactate for 20 min restored CAP area to baseline levels, but did not replenish glycogen (Fig. 1D). Withdrawing lactate resulted in rapid CAP failure. The inset in Fig. 1D compares the rates of CAP failure under control conditions where nerves were perfused with 10 mm glucose for 100 min and when glycogen was depleted as described above. The glycogen levels measured under control conditions were stable at ∼6.5 pmol glycogen (μg protein)−1 prior to substrate withdrawal.
Ambient glucose concentrations and [K+]o determine glycogen content
The data illustrated in Fig. 1 suggest that latency to CAP failure during aglycaemia is dependent upon the concentration of glycogen in the nerve at the onset of aglycaemia. To test this hypothesis we attempted to systematically manipulate glycogen content. After the standard 1 h in 10 mm glucose, nerves were incubated in aCSF containing concentrations of glucose ranging between 2 and 30 mm for 2 h. Then 1 h of aglycaemia was imposed and the latency to CAP failure was measured. Nerves bathed for 2 h in 2, 5, 10, 20 or 30 mm glucose, respectively, had increasing latencies to CAP failure (Fig. 2A and B). To confirm that the data observed with increased extracellular glucose concentration were due to increased glycogen, and not due to loading of the extracellular space with glucose, we performed experiments with 25 mm fructose. Fructose sustains the CAP (Brown et al. 2001b) but does not induce glycogenesis (Wiesinger et al. 1997). There was no significant difference in latency to CAP failure in nerves bathed in 25 mm fructose or 10 mm glucose for 1 h (23.7 ± 5.5 vs. 25.6 ± 2.1 min; n = 3; n.s.) or glycogen content (6.87 ± 1.34 vs. 7.04 ± 0.99 pmol glycogen (μg protein)−1; n = 3; n.s.; data not shown). These results suggest the effects of hyperglycaemic extracellular glucose concentration were attributable to glycogen, and not to excess glucose in the extracellular space.
Figure 2. Effects of pre-incubation glucose concentration on glycogen content and latency to CAP failure during aglycaemia.
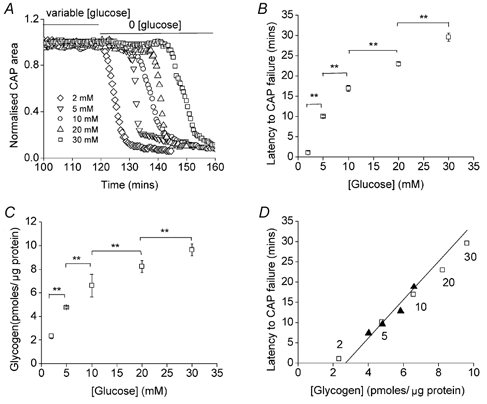
A and B, nerves bathed for 2 h in 2, 5, 10, 20 or 30 mm glucose had latencies to CAP failure of 1.0 ± 0.5 min (n = 3), 10.1 ± 0.3 min (n = 3; P < 0.001vs. 2 mm), 16.9 ± 0.8 min (n = 3; P < 0.001vs. 5 mm), 23.0 ± 0.5 min (n = 3; P < 0.001vs. 10 mm) and 29.6 ± 1.1 min (n = 3; P < 0.001vs. 20 mm), respectively (** indicates P < 0.001); ANOVA with Tukey's post hoc test. C, nerves bathed for 2 h in 2, 5, 10, 20 or 30 mm glucose had glycogen content of 2.34 ± 0.25 pmol glycogen (μg protein)−1 (n = 3), 4.77 ± 0.10 pmol glycogen (μg protein)−1 (n = 3; P < 0.001vs. 2 mm), 6.57 ± 0.23 pmol glycogen (μg protein)−1 (n = 3; P < 0.001vs. 5 mm), 8.23 ± 0.49 pmol glycogen (μg protein)−1 (n = 3; P < 0.001vs. 10 mm) and 9.63 ± 0.50 pmol glycogen (μg protein)−1 (n = 3; P < 0.001vs. 20 mm), respectively (** indicates P < 0.001); ANOVA with Tukey's post hoc test. D, straight line shows calculated relationship between the latency to onset of CAP failure during aglycaemia and glycogen content (see text for details). Open squares indicate actual data points from B and C, and numbers by the squares indicate pre-incubation glucose concentrations. Filled triangles indicate the data from Fig. 7B and C (see text). Error bars in B and C indicate the s.e.m.
The glycogen content of the nerves was directly proportional to glucose concentration (Fig. 2C). These data strongly suggested that the glycogen concentration in the nerves at the onset of aglycaemia determined the latency to onset of CAP failure. Based on the data presented in Fig. 1A we developed a simple predictive model of onset to CAP failure based on glycogen content (straight line in Fig. 2D). The rate of decrease in glycogen content during aglycaemia was 0.211 pmol glycogen (μg protein)−1 min−1 (i.e. 6.52–2.30 pmol glycogen (μg protein)−1 divided by 20 min). The ‘threshold’ glycogen concentration at which CAP began to decline was reached at 18 min and was calculated to be (6.52 - 0.211 pmol glycogen (μg protein)−1× 18 min) 2.72 pmol glycogen (μg protein)−1. This value became the x intercept for our model. Then applying a rate of decrease in glycogen content during aglycaemia of 0.211 pmol glycogen (μg protein)−1 min−1 gives us a prediction to latency of CAP failure (straight line). The open squares and filled triangles, respectively, in Fig. 2D are the actual values derived from Fig. 2B and C, and Fig. 7B and C, respectively, and they closely fit the predicted relationship.
Figure 7. High frequency axonal discharge depleted glycogen content and accelerated CAP failure during aglycaemia.
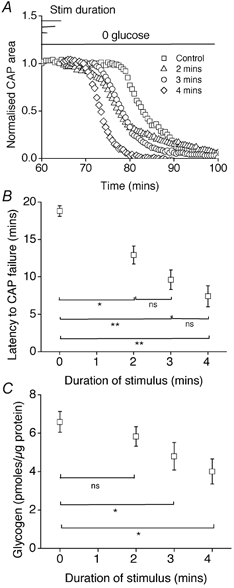
A and B, the latency to CAP failure in the control group was 18.8 ± 0.7 min (n = 4). With 100 Hz stimulation for 2, 3 or 4 min, latency to onset of CAP failure was 12.9 ± 1.2 min (n = 4; P < 0.05vs. control), 9.6 ± 1.3 min (n = 4; P < 0.001vs. control; vs. 2 min n.s.) and 7.4 ± 1.4 min (n = 4; P < 0.001vs. control; vs. 3 min n.s.), respectively (* indicates P < 0.05; ** indicates P < 0.001); ANOVA with Tukey's post hoc test. C, under control conditions, at the onset of aglycaemia, the glycogen content was 6.59 ± 0.54 (n = 3). Glycogen content immediately following 2, 3 or 4 min of 100 Hz stimulation was 5.83 ± 0.51 (n = 3), 4.80 ± 0.72 (n = 3) or 4.01 ± 0.65 pmol glycogen (μg protein)−1 (n = 3), respectively. The 3 and 4 min periods of stimulation produced statistically significant reductions in glycogen (P < 0.05) compared with control, (n = 3 for each condition; * indicates P < 0.05); ANOVA with Tukey's post hoc test. Error bars in B and C indicate the s.e.m.
Previous studies have suggested that ambient [K+]o can affect brain glycogen concentration (Hof et al. 1988). Because [K+]o can fluctuate with activity in RONs (Connors et al. 1982), it was important to know if this would affect glycogen content. Switching [K+]o from 3 to 10 mm for 20 min caused a significant drop in glycogen content from 6.32 ± 0.44 to 3.74 ± 0.23 pmol glycogen (μg protein)−1 (n = 3, P < 0.05; Student's t test; data not shown).
Glucose concentration and MON function
We tested the ability of glucose concentrations considered hypoglycaemic (Frier & Fisher, 1999) to sustain MON function (NB in the absence of vascular perfusion in our model the diffusion barrier would render glucose < 4 mm hypoglycaemic in the tissue). Perfusion of MONs with aCSF containing 2 mm glucose or greater resulted in robust CAPs for 2 h. Reducing glucose to 1 mm caused onset of CAP failure after 26.7 ± 1.1 min and reduced CAP area to 22.3 ± 2.4 % of baseline after 2 h (Fig. 3A). Lower levels of glucose caused greater loss of CAP area after 2 h. Because glycogen levels fall when shifting from 10 to 5 or 2 mm glucose, we wondered if glycogen was supplying substrate for nerves during incubation in 2 mm glucose. Indeed if glycogen were ‘pre-depleted’ prior to switching from 10 to 0 mm glucose, 2 mm cannot maintain CAP area (Fig. 3B). Glycogen was depleted here by 20 min of aglycaemia (cf. Fig. 1B). Given that glycogen supports axons by being broken down to lactate (or possibly pyruvate) we tested for axonal use of lactate by applying the monocarboxylate transport (MCT) blocker cinnamic acid (CIN). CIN preferentially inhibits the MCT2 transporter found predominantly on neural elements and which is best suited for lactate uptake (Pierre et al. 2002). CIN (150 μm) resulted in reversible decline of the CAP during perfusion with 2 mm glucose (Fig. 3C), but not 10 mm glucose (Izumi et al. 1997a; Wender et al. 2000), indicating that lactate was used as an energy source by axons being perfused with 2 mm glucose (but see McKenna et al. 2001). Additional experiments carried out with d-lactate, which is transported by the monocarboxylate transporter, but is not metabolised by l-lactate dehydrogenase, support our results with CIN. In the presence of 10 mm glucose, inclusion of 20 mm d-lactate had no effect but when perfused in the presence of 2 mm glucose led to functional failure (n = 3, data not shown).
Figure 3. Ambient glucose concentration and MON function.
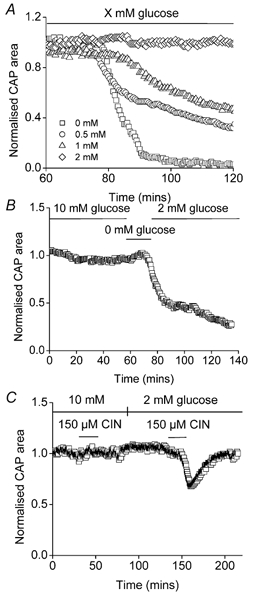
A, glucose concentrations between 0 and 2 mm were tested for their ability to sustain MON function measured as the CAP. A glucose concentration of 2 mm supported axon function for at least 120 min (n = 3). Lower glucose concentrations were unable to fully support axon function (1 mm, n = 3; 0.5 mm, n = 6; 0 mm, n = 4). B, prior incubation in 0 mm glucose for 20 min rendered 2 mm glucose ineffective to maintain axon function (n = 3). C, the monocarboxylate transport inhibitor CIN (150 μm) had no effect on CAP area in 10 mm glucose, but reversibly depressed CAP area in 2 mm glucose (n = 3).
High frequency stimulation of MON and CAP area: role of glycogen
In MONs bathed in 10 mm glucose, CAP area initially increased and then declined to a level below baseline during extended 100 Hz stimulation (Fig. 4A). In MONs bathed in 30 mm glucose and exposed to 100 Hz stimulation, the CAP area did not fall below baseline (Fig. 4B). The decline of CAP area during 100 Hz stimulation in 10 mm glucose was corrected by switching to 30 mm glucose (Fig. 4C). These data indicated that during high frequency stimulation delivery of sufficient energy substrate to the nerve was necessary to fully support axon excitability.
Figure 4. CAP area is affected by substrate availability during high frequency stimulation.
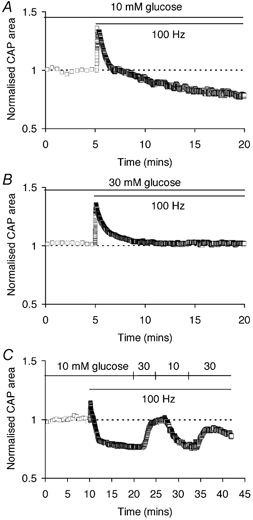
A, CAP area gradually declined during extended 100 Hz stimulation in 10 mm glucose. B, in 30 mm glucose CAP area showed an initial increase but quickly stabilised during 100 Hz stimulation. C, CAP area varied reversibly during 100 Hz stimulation when glucose concentration was switched repeatedly between 10 and 30 mm.
We wondered if astrocytic glycogen provided energy support to CNS axons during times of increased activity. In 10 mm glucose aCSF, nerves were subjected to 100 Hz stimulation for 4 min (a total of 24 000 action potentials). Representative CAPs during the period of stimulation are illustrated (Fig. 5B). While the first peak appeared to be maintained, the second and third peaks decreased in amplitude but increased in duration, resulting in only small net changes in CAP area (Fig. 5A). Glycogen content significantly decreased over the 4 min period of stimulation, suggesting that glycogen is used to sustain function during intense stimulation (Fig. 5A). Consistent with this notion, nerves with depleted glycogen due to incubation in 2 mm glucose showed a marked decline in CAP area during stimulation (Fig. 5C and D). Because 2 mm glucose, independent of glycogen content, contributed to the rapid CAP decline, an additional set of experiments was carried out where glycogen was reduced by a 15 min period (from 35 to 50 min) in aglycaemia prior to 4 min of 100 Hz stimulation; the stimulation, itself was in the presence of 10 mm glucose. Again, CAP area fell during high frequency stimulation (Fig. 5E and F), although during the 15 min pre-incubation in 0 mm glucose there was no change in CAP area.
Figure 5. High frequency axon discharge and glycogen in the MON.
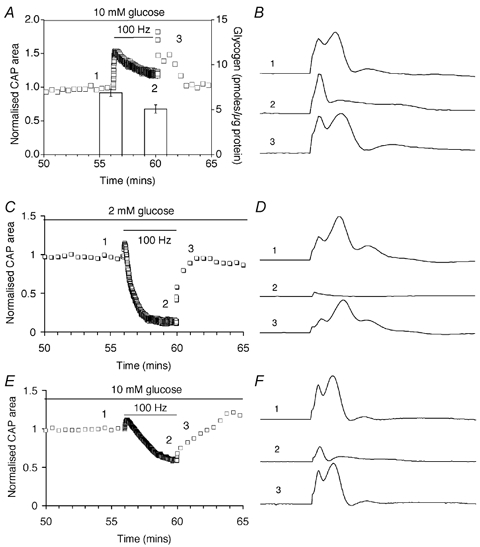
A, CAP area (open squares, left axis) during a 4 min period of 100 Hz stimulation in 10 mm glucose. Note that CAP area does not decline. Glycogen content (columns, right axis) decreased from 6.86 ± 0.39 to 5.08 ± 0.45 pmol glycogen (μg protein)−1 (n = 3; P < 0.05; Student's t test) B, representative CAPs recorded from one of the nerves averaged in A at the indicated time points (1–3). CAP shape changes considerably during high frequency stimulation and recovery. C, CAP area fell rapidly during 100 Hz stimulation in nerves perfused in 2 mm glucose for 1 h. D, representative CAPs recorded from one of the nerves averaged in C at the indicated time points (1–3). E, CAP area fell during high frequency stimulation in nerves perfused with 10 mm glucose when glycogen content was depleted by prior aglycaemia (for 15 min; i.e. minute ‘35 to 50′). F, representative CAPs recorded from one of the nerves averaged in E at the indicated time points (1–3). Error bars in A indicate the s.e.m.
To further investigate the role of glycogen in supporting function during high frequency firing under normoglycaemic conditions we blocked lactate transport (Wender et al. 2000). In the presence of 150 μm CIN, which presumably blocks lactate uptake into axons, CAP area declined rapidly (Fig. 6A and B). Similarly quercitin (50 μm), a compound that inhibits export of astrocytic lactate (Belt et al. 1979; Volk et al. 1997) caused more rapid CAP decline during 100 Hz stimulus (Fig. 6C and D), compared with control (i.e. Fig. 5A). These data suggested that 10 mm glucose must be supplemented with glycogen-derived lactate in order for function to be fully maintained during periods of intense activity.
Figure 6. Monocarboxylate transport blockers and CAP area.
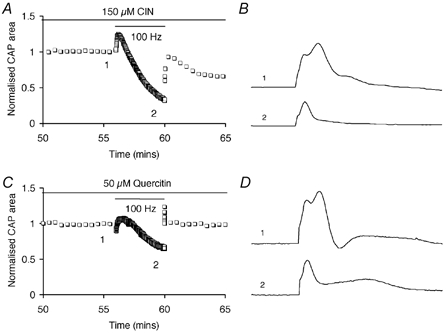
A, the MCT blocker cinnamic acid (CIN) caused CAP area to decline during stimulation in 10 mm glucose. Control CAP area (0.94 ± 0.04, n = 4) vs. area after stimulation (0.41 ± 0.03, n = 4) was significantly different (P < 0.0001; Student's t test). B, representative CAPs recorded from one of the nerves averaged in A at the two indicated time points. C, the MCT blocker quercitin caused CAP area to decline during stimulation in 10 mm glucose. Control CAP area (1.01 ± 0.03, n = 3) vs. area after stimulation (0.65 ± 0.05, n = 3) was significantly different (P < 0.005; Student's t test). D, representative CAPs recorded from one of the nerves averaged in C at the two indicated time points. These results indicate that lactate transport into axons was required to fully support function during intense activity, even in the presence of glucose.
High frequency electrical stimulation and CAP failure during aglycaemia
Given that neural activity depleted glycogen (see Fig. 5A), we hypothesised that aglycaemia would more rapidly impair axonal function if it occurred in conjunction with high frequency stimulation. Indeed, aglycaemia coupled with 2, 3 or 4 min of 100 Hz supramaximal stimulation produced, in a graded fashion, shorter latency to CAP failure (Fig. 7A and B). The latency to CAP failure decreased with increasing duration of stimulus (Fig. 7A and B). As anticipated, the above results were accompanied by changes in glycogen content (Fig. 7C). Under control conditions, at the onset of aglycaemia, the glycogen content was 6.59 ± 0.54 pmol glycogen (μg protein)−1 (n = 3). Glycogen content immediately following 2, 3 or 4 min of 100 Hz stimulation decreased, the degree of decrease being proportional to the duration of stimulus (Fig. 7C). The 3 and 4 min periods of stimulation produced statistically significant reductions in glycogen (P < 0.05) compared with control.
The glycogen content values and latencies to onset of CAP decline shown here were plotted as filled triangles in Fig. 2D. These data closely fit the theoretical relationship between glycogen content and onset of CAP failure, adding additional credibility to this relationship.
Discussion
The results presented in this paper show that glycogen content of mammalian brain is under dynamic control and that CNS glycogen supports the energy needs of white matter axons during periods of heightened activity, as well as during hypoglycaemia or aglycaemia. To our knowledge, this is the first study to show direct experimental evidence of glycogen breakdown and breakdown product (probably lactate) utilisation during physiological activity (i.e. high frequency optic nerve axon discharge). Our conclusions in this regard are based on the following observations: (1) during intense activity glycogen content decreased significantly; (2) inhibition of lactate transport out of astrocytes, or into axons, reduced function; (3) down-regulation of glycogen impaired high frequency firing during normoglycaemia.
Glycogen content in vitro
The glycogen content of MONs perfused with 10 mm glucose, the ‘control’ glucose concentration, was stable and about the same as when nerves were just harvested. This agrees with our findings in rats with the exception that in rats there was a decrease in glycogen content within the first hour after removal (Wender et al. 2000). The difference between rat and mouse may relate to the fact that the RON is about twice the diameter of the MON and therefore glucose from the bathing solution has further to diffuse. Experiments indicate that the energy demands of the RON, and presumably substrate need for glycogen synthesis, cannot be entirely met by 10 mm bath glucose (Baltan Tekkök et al. 2002).
Glycogen was quickly broken down during 1 h of aglycaemia. There was no restoration of glycogen during the subsequent recovery period in 10 mm glucose. This was most likely to be a consequence of aglycaemia-induced tissue injury. When glycogen was depleted by a shorter period of aglycaemia (i.e. 15 min) not associated with permanent functional deficit, glycogen content recovered (Fig. 1B). This makes biological sense, and is in agreement with tissue culture studies where glycogen content recovers (in fact, rebounds to higher levels) following depletion by neurotransmitters (Sorg & Magistretti, 1992). While not seen under the conditions studied here, future experiments should determine if this recovery overshoot is detectable in white matter treated with adrenergic neurotransmitters.
Lactate could substitute for glucose as an effective fuel to maintain the CAP (Schurr et al. 1988; Izumi et al. 1997a; Brown et al. 2001b). However, it could not substitute for glucose as a substrate for glycogen synthesis. Thus, even when there was adequate metabolic substrate to support axon function, glycogen content was still depleted. As both axons and astrocytes can take up and metabolise lactate, our data suggested that glycogen was constantly metabolised, even under conditions where it appears it was not required to support function. Indeed, it has been reported that the turnover rate of glycogen under non-stimulated conditions is very rapid (Watanabe & Passonneau, 1973).
Glycogen content and ambient glucose
The maintenance of the CAP for about 20 min during aglycaemia agrees qualitatively with prior data from the RON where function was maintained for about 30 min during aglycaemia (Wender et al. 2000). The increased latency to CAP failure in rat compared with mouse could be due to a variety of factors including, but not limited to, increased glycogen content in rat, increased metabolic rate in mouse, or variations in distribution of monocarboxylate transporters, glycogen phosphorylase etc. The glycogen content that was consistently found under our control conditions in mouse was about 6 pmol glycogen (μg protein)−1. The values in rat were more variable but roughly in the same concentration range. It would seem, therefore, that glycogen content per se was not the explanation.
Glycogen content varied with the concentration of glucose perfusing the nerve. Given that glycogen is formed from glucose, it is not surprising that perfusing nerves with low concentrations of glucose resulted in decreased glycogen, and this is in agreement with data from the rat (Wender et al. 2000) and cultured astrocytes (Magistretti et al. 1993). The relationship between glucose and glycogen content was not linear, consistent with glycogen synthesis being a saturable process (Champe & Harvey, 1994).
The relationship between latency to functional failure and glycogen content prior to aglycaemia was linear and consistent under different test paradigms. These data indicated that astrocytic glycogen provided axons with energy substrate in an orderly and predictable manner. So long as the demand side, that is axon activity, was constant, then the glycogen content immediately prior to aglycaemia determined the latency to functional failure. Indeed, the predictability of this relationship was highlighted by our model of glycogen usage based on the directly measured rate of glycogen loss during aglycaemia (Fig. 2D). Thus, a possible clinical strategy to stave off the CNS injury that can be associated with severe hypoglycaemia, a common side-effect of insulin therapy to treat Type-1 diabetes, may be to increase CNS glycogen content.
The data indicating that function can be supported for 2 h in the presence of 2 mm glucose, a concentration considered extremely hypoglycaemic, was at first surprising. Cognitive impairment is seen in patients when blood glucose is 70 mg ml−1, equivalent to ∼4 mm glucose (Frier & Fisher, 1999); the concentration of glucose in brain extracellular space would be lower, of course (Silver & Erecinska, 1994). Given that glycogen is used to maintain function during aglycaemia it is consistent that glycogen is also used to support function during hypoglycaemia. Consistent with this hypothesis are the following observations: (1) depletion of glycogen prevented full maintenance of function in 2 mm glucose; (2) lactate transport blockers prevented normal function during hypoglycaemia.
A physiological role for glycogen
A physiological role for glycogen has been suggested by several groups based on the following: (1) it seems unlikely that glycogen would be available solely as a safeguard against hypoglycaemia, a rare physiological event prior to the advent of insulin therapy for diabetes; (2) glycogen content increases during anaesthesia thus correlating increased glycogen content with decreased brain activity; (3) the content of glycogen is under dynamic, regional control mediated, in part, by neurotransmitters and their receptors on astrocytes (Swanson, 1992; Swanson et al. 1992; Magistretti et al. 1999; Shulman et al. 2001). It has been hypothesised that glycogen may fuel astrocytes in grey matter during periods of intense synaptic activity that require enhanced, energy-depleting, glutamate uptake by these cells (Shulman et al. 2001). The main evidence for this hypothesis is the decrease in the ratio of oxygen consumption to glucose consumption and increased lactate output during periods of high energy demand (Fox et al. 1988; Prichard et al. 1991), implying that an increased percentage of glucose is metabolised anaerobically. However, no studies have directly investigated the role of glycogen during such activity.
Our data provide proof of principle that glycogen has a role during intense physiological activity. Astrocytic glycogen declined during high frequency optic nerve stimulation in the presence of normal glucose concentration. The evidence supported transfer of a monocarboxylate, probably lactate, from astrocytes to axons during the period of increased energy demand associated with rapid axon discharge. Specifically, depletion of glycogen or blockade of lactate transport impaired optic nerve axon excitability during the stimulation period. The vulnerability of axon excitability during periods of rapid discharge was exaggerated in the presence of hypoglycaemic concentrations of glucose (i.e. 2 mm). While the CAP was stable during low frequency firing (0.033 Hz) for at least 2 h, it declined precipitously at 100 Hz stimulation. There is a need to expand these observations to other regions of the CNS and to determine what ‘signals’ the astrocyte to produce lactate for consumption by nearby neural elements. The signal could be as simple as a decrease in ambient glucose concentration. Another viable candidate would be activity-dependent increase in [K+]o as increases in [K+]o quantitatively indicate the magnitude of nearby neural activity (Connors et al. 1982) and cause glycogen breakdown (Hof et al. 1988). Alternatively active neurons may release a neurotransmitter which may act as a signal for astrocytic glycogenolysis. A suitable candidate is noradrenaline, which has been shown to promote glycogenolysis not only in cell culture (Magistretti et al. 1993), but also in the intact rat optic nerve (Wender et al. 2000).
Acknowledgments
The authors thank Dr Ray Swanson and Kevin Farrell for technical assistance with glycogen assays, Dr Jeff Chamberlain and Dr Dennis Hartigan-O'Connor for the use of a fluorometer, Dr Thomas Möller for assistance with protein assays, and Paul Newman for technical assistance. This research was supported by the National Institutes of Health NS15589 (B.R.R.) and the Eastern Paralyzed Veterans Association (A.M.B. and B.R.R.).
References
- Baltan Tekkök S, Brown AM, Ransom BR. Persistence of axonal function during anoxic insult in mouse optic nerve. Glia. 2002;38(suppl. 1):245. [Google Scholar]
- Belt JA, Thomas JA, Buchsbaum RN, Racker E. Inhibition of lactate transport and glycolysis in Ehrlich ascites tumor cells by bioflavonoids. Biochemistry. 1979;18:3506–3511. doi: 10.1021/bi00583a011. [DOI] [PubMed] [Google Scholar]
- Broer S, Broer A, Schneider HP, Stegen C, Halestrap AP, Deitmer JW. Characterization of the high-affinity monocarboxylate transporter MCT2 in Xenopus laevis oocytes. Biochem J. 1999;341:529–535. doi: 10.1042/0264-6021:3410529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM. A step-by-step guide to non-linear regression analysis of experimental data using a Microsoft Excel spreadsheet. Comp Meth Prog Biomed. 2001;65:191–200. doi: 10.1016/s0169-2607(00)00124-3. [DOI] [PubMed] [Google Scholar]
- Brown AM, Ransom BR. Astrocytic glycogen maintains axon function during periods of hypoglycaemia in central white matter. JPhysiol. 2001;536.P:118P. [Google Scholar]
- Brown AM, Wender R, Ransom BR. Ionic mechanisms of aglycemic axon injury in mammalian central white matter. J Cereb Blood Flow Met. 2001a;21:385–395. doi: 10.1097/00004647-200104000-00007. [DOI] [PubMed] [Google Scholar]
- Brown AM, Wender R, Ransom BR. Metabolic substrates other than glucose support axon function in central white matter. J Neurosci Res. 2001b;66:839–843. doi: 10.1002/jnr.10081. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Broadwell RD. Cytochemical identification of cerebral glycogen and glucose-6-phosphatase activity under normal and experimental conditions. I. Neurons and glia. J Elec Micro Tech. 1986;3:413–437. doi: 10.1007/BF01611733. [DOI] [PubMed] [Google Scholar]
- Cater HL, Benham CD, Sundstrom LE. Neuroprotective role of monocarboxylate transport during glucose deprivation in slice cultures of rat hippocampus. J Physiol. 2001;531:459–466. doi: 10.1111/j.1469-7793.2001.0459i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champe PC, Harvey RA. Lippincott's Illustrated Reviews: Biochemistry. Philadelphia: Williams and Wilkins; 1994. [Google Scholar]
- Chih C, Lipton P, Roberts EL. Do active cerebral neurons really use lactate rather than glucose? Trends Neurosci. 2001;24:573–578. doi: 10.1016/s0166-2236(00)01920-2. [DOI] [PubMed] [Google Scholar]
- Connors BW, Ransom BR, Kunis DM, Gutnick MJ. Activity-dependent K+ accumulation in the developing rat optic nerve. Science. 1982;216:1341–1343. doi: 10.1126/science.7079771. [DOI] [PubMed] [Google Scholar]
- Dringen R, Gebhardt R, Hamprecht B. Glycogen in astrocytes: possible function as lactate supply for neighboring cells. Brain Res. 1993;623:208–214. doi: 10.1016/0006-8993(93)91429-v. [DOI] [PubMed] [Google Scholar]
- Fox PT, Raichle ME, Mintun MA, Dence C. Nonoxidative glucose consumption during focal physiologic neural activity. Science. 1988;241:462–464. doi: 10.1126/science.3260686. [DOI] [PubMed] [Google Scholar]
- Frier BM, Fisher BM. Hypoglycaemia in Clinical Diabetes. New York: John Wiley and Sons; 1999. [Google Scholar]
- Garrett RH, Grisham CM. Biochemistry. Fort Worth: Saunders College Publishing; 1999. [Google Scholar]
- Hof PR, Pascale E, Magistretti PJ. K+ at concentrations reached in the extracellular space during neuronal activity promotes a Ca2+-dependent glycogen hydrolysis in mouse cerebral cortex. J Neurosci. 1988;8:1922–1928. doi: 10.1523/JNEUROSCI.08-06-01922.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Benz AM, Katsuki H, Zorumski CF. Endogenous monocarboxylates sustain hippocampal synaptic function and morphological integrity during energy deprivation. J Neurosci. 1997a;17:9448–9457. doi: 10.1523/JNEUROSCI.17-24-09448.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Katsuki H, Zorumski CF. Monocarboxylates (pyruvate and lactate) as alternative energy substrates for the induction of long-term potentiation in rat hippocampal slices. Neurosci Lett. 1997b;232:17–20. doi: 10.1016/s0304-3940(97)00567-3. [DOI] [PubMed] [Google Scholar]
- Jackson VN, Halestrap AP. The kinetics, substrate, and inhibitor specificity of the monocarboxylate (lactate) transporter of rat liver cells determined using the fluorescent intracellular pH indicator, 2′,7′-bis(carboxyethyl)-5(6)-carboxyfluorescein. J Biol Chem. 1996;271:861–868. doi: 10.1074/jbc.271.2.861. [DOI] [PubMed] [Google Scholar]
- McIlwain H. Substances which support respiration and metabolic response to electrical impulses in human cerebral tissues. J Neurol Neurosurg Psychiatry. 1953;16:257–266. doi: 10.1136/jnnp.16.4.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna MC, Hopkins IB, Carey A. α-Cyano-4-hydroxycinnamate decreases both glucose and lactate metabolism in neurons and astrocytes: implications for lactate as an energy substrate for neurons. J Neurosci Res. 2001;66:747–754. doi: 10.1002/jnr.10084. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Pellerin L, Rothman DL, Shulman RG. Energy on demand. Science. 1999;283:496–497. doi: 10.1126/science.283.5401.496. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Sorg O, Martin J-L. Regulation of glycogen metabolism in astrocytes: physiological, pharmacological, and pathological aspects. In: Murphy S, editor. Astrocytes: Pharmacology and Function. San Diego CA: Academic Press Inc.; 1993. pp. 243–265. [Google Scholar]
- Passonneau JV, Gatfield PD, Schulz DW, Lowry OH. An enzymic method for measurement of glycogen. Anal Biochem. 1967;19:315–326. doi: 10.1016/0003-2697(67)90167-4. [DOI] [PubMed] [Google Scholar]
- Pentreath VW, Kai-Kai MA. Significance of the potassium signal from neurones to glial cells. Nature. 1982;295:59–61. doi: 10.1038/295059a0. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL. Fine Structure of the Nervous System: The Neurons and Supporting Cells. Philadelphia: W. B. Saunders Company; 1976. [Google Scholar]
- Pierre K, Magistretti PJ, Pellerin L. MCT2 is a major neuronal monocarboxylate transporter in the adult mouse brain. J Cereb Blood Flow Metab. 2002;22:586–595. doi: 10.1097/00004647-200205000-00010. [DOI] [PubMed] [Google Scholar]
- Prichard J, Rothman D, Novotny E, Petroff O, Kuwabara T, Avison M, Howseman A, Hanstock C, Shulman R. Lactate rise detected by 1HNMR in human visual cortex during physiologic stimulation. Proc Natl Acad Sci U S A. 1991;88:5829–5831. doi: 10.1073/pnas.88.13.5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurr A, West CA, Rigor BM. Lactate-supported synaptic function in the rat hippocampal slice preparation. Science. 1988;240:1326–1328. doi: 10.1126/science.3375817. [DOI] [PubMed] [Google Scholar]
- Shulman RG, Hyder F, Rothman DL. Cerebral energetics and the glycogen shunt: neurochemical basis of functional imaging. Proc Natl Acad Sci U S A. 2001;98:6417–6422. doi: 10.1073/pnas.101129298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver IA, Erecinska M. Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J Neurosci. 1994;14:5068–5076. doi: 10.1523/JNEUROSCI.14-08-05068.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg O, Magistretti PJ. Vasoactive intestinal peptide and noradrenaline exert long-term control on glycogen levels in astrocytes: blockade by protein synthesis inhibition. J Neurosci. 1992;12:4923–31. doi: 10.1523/JNEUROSCI.12-12-04923.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryer L. Biochemistry. New York: W.H. Freeman & Co; 1995. [Google Scholar]
- Swanson RA. Physiologic coupling of glial glycogen metabolism to neuronal activity in brain. Can JPhysiol Pharmacol. 1992;70:S138–44. doi: 10.1139/y92-255. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Choi DW. Glial glycogen stores affect neuronal survival during glucose deprivation in vitro. J Cereb Blood Flow Metab. 1993;13:162–169. doi: 10.1038/jcbfm.1993.19. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Morton MM, Sagar SM, Sharp FR. Sensory stimulation induces local cerebral glycogenolysis: demonstration by autoradiography. Neuroscience. 1992;51:451–461. doi: 10.1016/0306-4522(92)90329-z. [DOI] [PubMed] [Google Scholar]
- Volk C, Kempski B, Kempski OS. Inhibition of lactate export by quercetin acidifies rat glial cells in vitro. Neurosci Lett. 1997;223:121–124. doi: 10.1016/s0304-3940(97)13420-6. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Passonneau JV. Factors affecting the turnover of cerebral glycogen and limit dextrin in vivo. J Neurochem. 1973;20:1543–1554. doi: 10.1111/j.1471-4159.1973.tb00272.x. [DOI] [PubMed] [Google Scholar]
- Wender R, Brown AM, Fern R, Swanson RA, Farrell K, Ransom BR. Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J Neurosci. 2000;20:6804–6810. doi: 10.1523/JNEUROSCI.20-18-06804.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesinger H, Hamprecht B, Dringen R. Metabolic pathways for glucose in astrocytes. Glia. 1997;21:22–34. doi: 10.1002/(sici)1098-1136(199709)21:1<22::aid-glia3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]