

Abstract
Calcium waves in layer 2/3 and layer 5 neocortical somatosensory pyramidal neurons were examined in slices from 2- to 8-week-old rats. Repetitive synaptic stimulation evoked a delayed, all-or-none [Ca2+]i increase primarily on the main dendritic shaft. This component was blocked by 1 mm (R,S)-α-methyl-4-carboxyphenylglycine (MCPG), 10 μm ryanodine, 1 mg ml−1 internal heparin, and was not blocked by 400 μm internal Ruthenium Red, indicating that it was due to Ca2+ release from internal stores by inositol 1,4,5-trisphosphate (IP3) mobilized via activation of metabotropic glutamate receptors. Calcium waves were initiated on the apical shaft at sites between the soma to around the main branch point, mostly at insertion points of oblique dendrites, and spread in both directions along the shaft. In the proximal dendrites the peak amplitude of the resulting [Ca2+]i change was much larger than that evoked by a train of Na+ spikes. In distal dendrites the peak amplitude was comparable to the [Ca2+]i change due to a Ca2+ spike. IP3-mediated Ca2+ release also was observed in the presence of the metabotropic agonists t-ACPD and carbachol when backpropagating spikes were generated. Ca2+ entry through NMDA receptors was observed primarily on the oblique dendrites. The main differences between waves in neocortical neurons and in previously described hippocampal pyramidal neurons were, (a) Ca2+ waves in L5 neurons could be evoked further out along the main shaft, (b) Ca2+ waves extended slightly further out into the oblique dendrites and (c) higher concentrations of bath-applied t-ACPD and carbachol were required to generate Ca2+ release events by backpropagating action potentials.
Two of the most extensively studied principal neurons in the central nervous system are the pyramidal neurons from the hippocampus and neocortex. Many of their anatomical and physiological properties are similar although there are some clear differences. For example, layer 5 (L5) neocortical pyramidal neurons have a much longer main dendritic shaft than pyramidal neurons from the CA1 region of the hippocampus. This middle dendritic region separates the proximal apical region near the soma from the distal apical tufted arborization, forming zones of Na+ and Ca2+ electrogenesis that are more distinct in the neocortex than in the hippocampus (Schiller et al. 1997; Golding et al. 1999; Larkum et al. 2001). Since backpropagating Na+ spikes and dendritic Ca2+ spikes each produce transient increases in intracellular calcium concentration ([Ca2+]i) due to Ca2+ entry through voltage-sensitive Ca2+ channels, the characteristic spatial distributions of these events can lead to different kinds of Ca2+ signalling in discrete dendritic regions. In addition, regenerative electrical events can propagate in both directions along the dendrite leading to synergistic interactions among inputs arriving at different parts of the dendritic arborization. These widespread voltage- dependent sources of Ca2+ complement the more localized [Ca2+]i increases due to Ca2+ entry through ligand-gated receptors when these neurons are activated synaptically.
Recently, we found that repetitive synaptic stimulation could evoke large amplitude (several μm), regenerative Ca2+ waves in the proximal apical dendritic region of hippocampal CA1 pyramidal neurons (Nakamura et al. 1999, 2002; Zhou & Ross, 2002). The rise in [Ca2+]i following this kind of stimulation is much larger than the [Ca2+]i increase due to influx through voltage dependent Ca2+ channels. These waves are produced by the activation of group I metabotropic glutamate receptors (mGluRs) activating phopholipase C, mobilizing IP3 which releases Ca2+ from the endoplasmic reticulum (ER). The waves are found on the main dendritic shaft and not on the oblique dendrites and usually initiate at dendritic branch points (Nakamura et al. 2002) before propagating in both directions along the apical dendrite.
Due to the importance of postsynaptic Ca2+ signalling in a variety of neuronal processes, including synaptic plasticity and gene expression, it was of interest to determine if Ca2+ waves could be evoked in neocortical pyramidal neurons. Previous experiments suggested that Ca2+ could be released from internal stores in L5 neurons (Markram, 1997) and L2/3 pyramidal neurons (Yamamoto et al. 2000, 2002). If similar waves could be detected, we were interested to know if the spatial characteristics of these events or other properties differed from those found in hippocampal neurons. In particular, we wanted to compare the Ca2+ signals from these waves with the Ca2+ signals resulting from Ca2+ and Na+ spikes in the dendrites. Such a comparison could give insights into the importance of these different signals in regulating synaptic plasticity and other processes in the cortex.
Using similar techniques to those employed in the hippocampal experiments (Nakamura et al. 1999, 2002) we found that indeed large-amplitude Ca2+ waves could be generated in both L5 and L2/3 neocortical pyramidal neuron dendrites. The [Ca2+]i changes generated by these waves were much larger and more localized than the [Ca2+]i changes generated by dendritic Ca2+ and Na+ spikes. Most of the properties of the waves were similar to those found in CA1 pyramidal neurons. Among the differences were: (a) Ca2+ waves in L5 pyramidal neurons could be evoked much further out along the main shaft, which may be related to its greater length; (b) Ca2+ waves extended slightly further out into the oblique dendrites and (c) higher concentrations of bath-applied metabotropic agonists t-ACPD (trans-1-amino-cyclopentyl-1, 3-dicarboxylate) and CCh (carbachol) were required to generate release events by backpropagating action potentials.
METHODS
Slice preparation, whole-cell recording and synaptic stimulation
Experiments were performed on pyramidal neurons from somatosensory neocortical slices prepared from 2- to 4- and 6- to 8-week-old Sprague-Dawley rats (Nakamura et al. 1999; Larkum et al. 2001). Animals were anaesthetized with isoflurane and decapitated using procedures approved by the Institutional Animal Care and Use Committee of New York Medical College. Recordings were made from slices submerged in a chamber. The chamber was perfused with warmed and oxygenated artificial cerebrospinal fluid (ACSF) at 32–34 °C. The composition of the ACSF was (mm): 124 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose; pH was 7.4 when bubbled with 95 % O2–5%CO2.
Submerged and superfused slices were mounted on a stage rigidly bolted to an air table and were viewed with a ×20, 40 or 60 water-immersion lens (Olympus, Melville, NY, USA) in an Olympus BX50WI microscope mounted on an X-Y translation stage. Somatic and dendritic whole-cell recordings were made using patch pipettes pulled from 1.5 mm outer diameter thick-walled borosilicate glass tubing (1511-M, Friderick and Dimmock, Millville, NJ). Tight seals on pyramidal cell somata and dendrites were made with the ‘blow and seal’ technique using video-enhanced DIC optics to visualize the cells (Sakmann & Stuart, 1995; Larkum et al. 2001). For most experiments the pipette solution contained (mm): 140 potassium gluconate, 4 NaCl, 4 Mg-ATP, 0.3 Na-GTP, and 10 Hepes, pH adjusted to 7.2–7.4 with KOH. This solution was supplemented with 150–200 μm bis-fura-2, 300 μm fura-6F or 500 μm furaptra (Molecular Probes, Eugene, OR, USA). For some experiments (detailed in Results) low molecular weight heparin or Ruthenium Red were added to this solution. Synaptic stimulation was evoked with 100 μs pulses using a bipolar tungsten electrode that had one sharpened tip (WPI, Sarasota, FL, USA, model TM33B01KT) about 1 mm in front of the other. The tip of this electrode was placed carefully on the slice usually about 20–50 μm to the side of the main apical dendritic shaft at various distances from the soma. Placement of the electrode close to the dendrite made it easier to evoke Ca2+ release (Zhou & Ross, 2002).
APV ((±)-2-amino-5-phosphonopentanoic acid), CNQX (6-cyano-7-nitroquinoxaline-2,4-dione), MCPG ((R, S)-α-methyl-4-carboxyphenylglycine), t-ACPD, and TTX (tetrodotoxin) were obtained from Sigma-RBI (St Louis, MO, USA). All other chemicals were obtained from Fisher Scientific (Piscataway, NJ, USA).
Dynamic [Ca2+]i measurements
Time-dependent [Ca2+]i measurements from different regions of the pyramidal neuron were made as previously described (Lasser-Ross et al. 1991). Briefly, changes in Ca2+ indicator fluorescence were detected with a Photometrics (Tucson, AZ, USA) AT200 cooled CCD camera, operated in the frame transfer mode. A few experiments used a higher resolution Quantix camera (Photometrics). Custom software controlled readout parameters and synchronization with electrical recordings. A second custom program was used to analyse the data. For all experiments in this paper the readout of the AT200 camera was binned in groups of 5 × 5 pixels, creating square superpixels 2.4 μm × 2.4 μm using the × 60 lens that defined the spatial resolution of most of our measurements. The superpixels were 3.6 μm × 3.6 μm and 7.2 μm × 7.2 μm when we used the × 40 and × 20 lenses. Static images in the figures showing the cell structure were taken without binning to achieve higher spatial resolution. Typical readout rates were 30 frames s−1. Fluorescence changes of bis-fura-2, fura-6F, and furaptra were measured with single wavelength excitation (382 ± 10 nm) and emission > 455 nm. [Ca2+]i changes are expressed as -ΔF/F where F is the fluorescence intensity when the cell is at rest and ΔF is the change in fluorescence during activity. Corrections were made for indicator bleaching during trials by subtracting the signal measured under the same conditions when the cell was not stimulated. To measure the resting fluorescence (F) accurately we subtracted the background fluorescence of the slice from the cell image. This background level was estimated by measuring the fluorescence of an equivalent position in the slice that contained no indicator-injected neurons.
In a typical experiment changes in [Ca2+]i were measured in a window of about 3 s around the time of synaptic or intrasomatic stimulation. Measurements were repeated every 1–2 min, changing the stimulation conditions from trial to trial. In the interval between trials we often evoked action potentials at 5 Hz with brief intrasomatic pulses. This stimulation appeared to make it easier to evoke Ca2+ release in the subsequent trial, probably by priming the endoplasmic reticulum with Ca2+ that entered the cell following the spikes (Jaffe & Brown, 1994; Finch & Augustine, 1998).
All measurements are quoted as means ± standard deviation (s.d.).
RESULTS
Synaptic activation of Ca2+ waves
Repetitive synaptic stimulation (50 pulses, 100 μs each, at 100 Hz) consistently evoked Ca2+ waves in the dendrites of both layer 2/3 (n = 4/4; Fig. 1 top) and layer 5 pyramidal neurons (n = 84/92; Fig. 1 bottom). We found no major differences between the waves evoked in the proximal apical dendrite of either type of neocortical neuron. Therefore, we focused the major part of our study on L5 neurons. Most experiments were performed on 2 to 4-week-old rats but similar waves were also evoked in older (6–8 week) animals (n = 6/9; data not shown). At all ages and in both cell types the waves appeared after a delay from the start of synaptic stimulation (516 ± 32 ms, range: 73 to 1409 ms; measured at the time of half-peak amplitude; n = 61). Once initiated, the waves generally propagated an equal distance in opposite directions along the apical dendrite although there was a slight preference for propagation towards the cell body. (For 50 measured waves, 31 propagated equally within 10 μm, 15 propagated more towards the soma, and 4 propagated more away from the soma.) The duration of the Ca2+ release transient was not the same at all locations (see Fig. 1). For 29 of these waves, the maximum duration occurred at the site of initiation, whereas for the rest, the maximum duration was at a point closer to the soma. The maximum duration of individual propagating waves averaged 1054 ± 79 ms (also determined at half-peak amplitude; n = 61) when measured with bis-fura-2. These properties distinguish these events from the [Ca2+]i changes due to ligand-gated or voltage-gated Ca2+ entry. Those changes began without a significant delay from the start of the corresponding electrical events and have a much shorter duration (Helmchen et al. 1996; Sabatini et al. 2002).
Figure 1. Repetitive synaptic stimulation evokes Ca2+ waves in layer 2/3 and layer 5 neocortical pyramidal neurons.
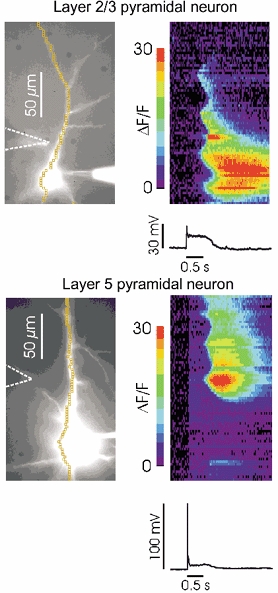
Left, the images show bis-fura-2 filled neurons. Patch electrodes are visible on the somata; the stimulating electrode is indicated in both images by dashed lines. Right, fluorescence changes at different locations in the cells in response to 100 Hz stimulation for 0.5 s. The abscissa of the pseudocolour images correspond to the time axis of the electrical traces below, recorded at the soma. The ordinate corresponds to the chain of ‘superpixels’ through the dendrites and somata. The ‘line scan’ images show that in each cell a large [Ca2+]i increase occurred in the dendrites and (in the L2/3 neuron) in the soma. These [Ca2+]i changes initiated at several locations after a delay of several hundred milliseconds from the start of synaptic stimulation. They propagated over a restricted region of the cell and had a duration of ∼0.5–1.5 s at different locations. In the layer 5 neuron the response generated an action potential at the beginning of the train. This spike evoked a smaller [Ca2+]i change that occurred almost simultaneously at all locations.
A few action potentials were sometimes evoked at the beginning of the synaptic response and provided a source of comparison between the rise in [Ca2+]i due to voltage-dependent Ca2+ channels and Ca2+ release. However, we generally found that we needed to use stimulation intensities that were lower than threshold for evoking action potentials but slightly higher than threshold for evoking synaptic potentials (e.g. Fig. 9) although we did not explore all the stimulation parameters (but see Zhou & Ross, 2002). In one L5 neuron we successfully tested that theta-burst stimulation (10 repetitions of 4 pulses at 100 Hz separated by 200 ms) generated a Ca2+ wave in the dendrites (data not shown).
Figure 9. APV and MCPG differently affect the spatial distribution of dendritic [Ca2+]i increases.
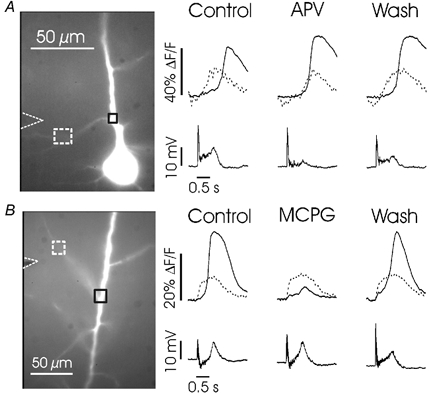
A, left (Control), responses in branch and shaft locations to 100 Hz (0.5 s) synaptic stimulation close to the branch. The [Ca2+]i increase on the branch peaked earlier than the increase on the shaft. Middle (APV), 100 μm APV added to the bath blocked the early part of the branch signal without any significant effect on the shaft signal. Right (Wash), recovery on return to normal ACSF. B, similar experiment on another cell in response to 1 mm MCPG. This mGluR antagonist reversibly eliminated the large shaft [Ca2+]i increase without any significant effect on the branch signal near the stimulating electrode. The electrical traces show the somatically recorded membrane potential changes.
Pharmacology
We found that the pharmacological profile of the Ca2+ waves was similar to that observed with Ca2+ waves in hippocampal neurons. MCPG (1 mm), an antagonist of group I mGluRs (Conn & Pin, 1997), reversibly blocked synaptically evoked waves (n = 8/9; Figs. 2 and 9B). Heparin (1–2 mg ml−1), which blocks IP3 receptors (Ghosh et al. 1988; Kobayashi et al. 1988), prevented release when it was included in the internal pipette solution (n = 12/13). These experiments were performed by first recording in whole-cell mode without heparin and then repeating the procedure after replacing the electrode with a heparin-filled pipette. In five experiments we replaced the first pipette with another pipette containing exactly the same solution (without heparin) to show that repeated whole-cell recordings from a neuron did not prevent the release process. Figure 3 shows an experiment in which we recorded from the same dendritic region with four different pipettes. The first three pipettes contained normal internal solution. In each case a large Ca2+ release transient was evoked by synaptic stimulation. For the fourth recording heparin was included in the pipette solution. Immediately after obtaining a whole-cell recording, Ca2+ release was still observed. But 10 min later, after heparin diffused into the dendrite, release was blocked. This experiment shows that repeated patching did not prevent Ca2+ release and that blockage was due to the included heparin.
Figure 2. MCPG reversibly blocks the generation of Ca2+ waves in the dendrites.
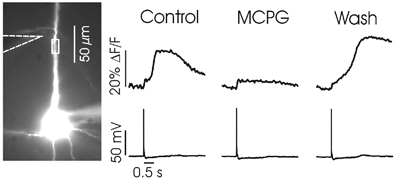
The image on the left shows the bis-fura-2 filled L5 pyramidal neuron with the position of the stimulating electrode indicated with dashed lines. The region of interest used for the measurements of [Ca 2+]i (shown to the right of the image) is indicated by a small white box near the stimulating electrode. Simultaneous electrical recordings are shown under the fluorescence measurements. In Control, synaptic stimulation evoked a sharp [Ca2+]i increase at the time of the spike (seen as a small step change of ∼5 % ΔF/F) followed by a larger [Ca2+]i increase due to the Ca2+ release wave. In MCPG (after 1 mm was added to ACSF) the wave was blocked but the spike signal remained. After wash and return to normal ACSF, synaptic stimulation again evoked a Ca2+ wave, but with different temporal characteristics.
Figure 3. Intradendritic heparin blocks synaptically evoked Ca2+ release.
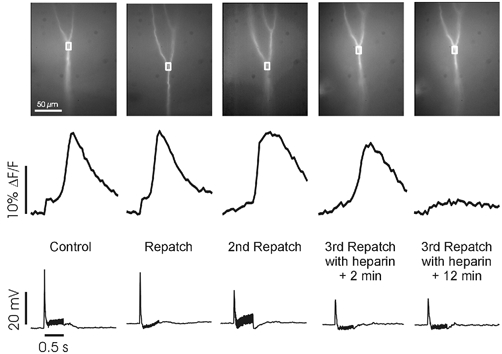
The images show a region of a L5 neuron near the primary branch point (350 μm from the soma). The cell was patched four times about 50 μm below the branch; the electrode was slowly removed after each trial. In Control and first two repatches the electrodes contained the normal internal solution. Repetitive synaptic stimulation (100 Hz for 0.5 s) evoked a large, delayed [Ca2+]i increase, which did not diminish after repatching over the course of ∼1.5 h. In the 3rd repatch the internal solution contained 2 mg ml−1 low MW heparin. Immediately after establishing a whole-cell recording from the dendrite, but before much heparin diffused into the dendrite, Ca2+ release was observed again. 10 min later, the same stimulation did not evoke Ca2+ release.
Ryanodine (5–20 μm), which opens ryanodine receptors on the ER (Rousseau et al. 1987), prevented synaptically evoked release (Fig. 4A; n = 4). Activation of either ryanodine receptors or IP3 receptors can open channels in the ER. The ryanodine experiments and the heparin experiments suggest that both intracellular channel types are found in the ER in these cells and that the IP3 receptors participate in the release of Ca2+. This result is consistent with the observation that group I mGluRs are coupled to phospholipase C and their activation mobilizes IP3 (Conn & Pin, 1997). Although the experiments with heparin showed that IP3 receptor activation is necessary for release it was still possible that ryanodine receptors also participate in the observed Ca2+ release. To test this possibility, we included Ruthenium Red (100–400 μm), a blocker of ryanodine receptors (Smith et al. 1988) in the internal pipette solution. Synaptically activated Ca2+ release was still observed in this condition (n = 7). To test whether the Ruthenium Red was effectively blocking the ryanodine receptors we added ryanodine (5–10 μm) to the bath (Fig. 4B). In this case we could still evoke Ca2+ release. Therefore, even though ryanodine receptors are found in these cells, probably on the same membranes as the IP3 receptors, they do not participate significantly in this release process.
Figure 4. Ca2+ release comes from ryanodine-sensitive stores but ryanodine receptors do not mediate release.
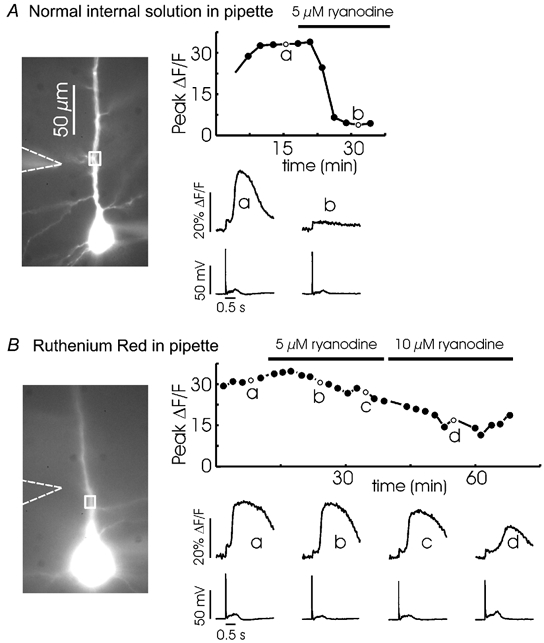
A, extracellular synaptic stimulation (100 Hz for 0.5 s) evoked regenerative Ca2+ release in the apical dendrites. This experiment was repeated several times at 2 min intervals. The peak fluorescence change in each trial is plotted in the graph. One trial, t = 15 min (a), is shown below. After 17 min, 5 μm ryanodine was added to the bath. In subsequent trials the same stimulation did not evoke release. One example, at 31 min, is shown in b. B, a similar experiment except 400 μm Ruthenium Red was included in the pipette. When 5 μm ryanodine and later 10 μm ryanodine was added to the bath Ca2+ release was still observed. The peak amplitude of release gradually declined during the experiment (a common observation) possibly due to depletion of stores.
Amplitude of the [Ca2+]i increase due to Ca2+ release
The amplitude of the Ca2+ release transient was usually bigger than the transient evoked by backpropagating action potentials. However, the fluorescence change often had a flat top when bis-fura-2 was used as the Ca2+ indicator in the pipette (e.g. Fig. 5, left), suggesting that the response saturated the indicator and therefore did not accurately indicate the peak [Ca2+]i change. To estimate the true amplitude of the [Ca2+]i change and to reduce the buffering effect of the high-affinity indicator bis-fura-2 (KD= 0.4 μm), we repeated the experiments using two lower affinity indicators, fura-6F (KD= 6 μm) and furaptra (KD= 40 μm). Figure 5 shows typical experiments comparing the fluorescence signal due to a train of 10 spikes to the fluorescence signal due to synaptically evoked Ca2+ release. It is clear that the relative fluorescence change from Ca2+ release using furaptra was much greater than with the higher affinity indicators. Since the percentage change in fluorescence using furaptra was small, the response of the indicator was in the linear range and it was easy to calibrate the magnitude of the [Ca2+]i increase (Nakamura et al. 1999). In four experiments we measured a peak fluorescence change of 9.5 ± 1.2 % for the Ca2+ release transient and 2.0 ± 0.1 % for a train of 10 action potentials, both measured in the proximal apical dendrites. These changes correspond to increases of about 4.8 μm (release) and 1.0 μm (spikes), values similar to those observed in corresponding locations in hippocampal neurons (Nakamura et al. 1999). From these experiments we conclude that using furaptra gives quantitatively more accurate estimates of the Ca2+ transients in these cells but using bis-fura-2 produces larger percentage changes in fluorescence making it easier to detect the transients.
Figure 5. The relative fluorescence change due to Ca2+ release is large compared with the change from voltage-gated Ca2+ entry when measured with low-affinity indicators.
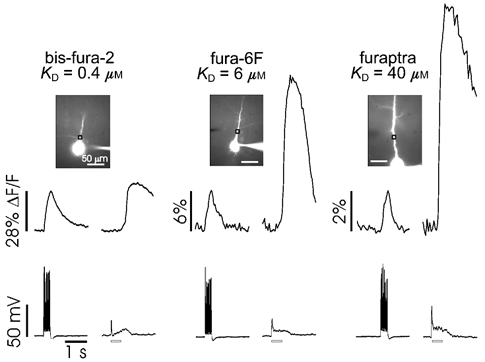
Layer 5 pyramidal neurons were filled with bis-fura-2, fura-6F, or furaptra (KDs indicated below the names). Each cell was first stimulated with a train of 10 backpropagating action potentials (evoked by 2 ms intrasomatic pulses every 30 ms), followed by synaptic stimulation (100 Hz for 0.5 s, indicated by the hollow horizontal bars under the electrical traces). The relative amplitudes of the fluorescence changes, measured at the indicated regions of interest, are shown in the top panels. To better compare the release transients using the different indicators the spike signals were normalized to the same amplitude. The electrical responses from recordings made at the soma are shown below. Note that the scales for the fluorescence changes are different for each indicator.
Synergistic interaction of synaptic input with backpropagating APs
Several other aspects of the release signals were measured and compared with those found in hippocampal pyramidal neurons. In CA1 neurons we found that backpropagating action potentials synergistically enhanced the likelihood of synaptically evoking Ca2+ release if the action potentials occurred in a time window of about 0.5 s following the end of the synaptic train (Nakamura et al. 1999). We found a similar synergy following synaptic activation of L5 neurons (Fig. 6; n = 9), but we did not determine the time window for this effect. The most probable explanation for this synergy is that the Ca2+ entering the cytoplasm from the spikes upregulates the sensitivity of IP3 receptors to the IP3 mobilized by tetanic stimulation of mGluRs (Iino, 1990; Bezprozvanny et al. 1991; Finch et al. 1991; Yao & Parker, 1992) causing regenerative Ca2+ release. We note, however, that exogenous Ca2+ was not required to generate Ca2+ waves since in other cells they could be evoked in ACSF containing 10 μm CNQX and 50 μm APV (n = 7; data not shown). In this solution Ca2+ entry was blocked both by the presence of APV and because the membrane potential hyperpolarized since only inhibitory conductances were synaptically activated. The hyperpolarization enhances the Mg2+ block of the NMDA receptors and prevents the opening of voltage-dependent Ca2+ channels. It is probable that the IP3 receptors can still be opened without an additional source of Ca2+ if mGluR activation mobilizes enough IP3 (Moraru et al. 1999).
Figure 6. Backpropagating spikes enhance synaptically activated Ca2+ release.
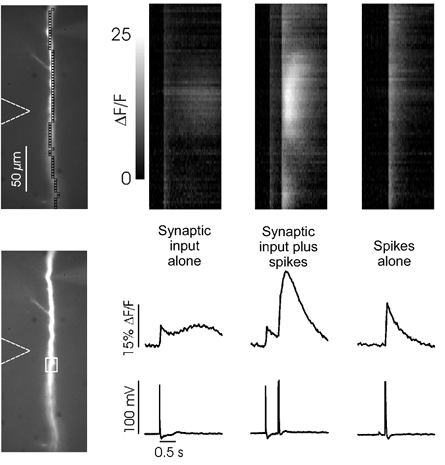
The image shows a region of the apical dendrite of a layer 5 neuron that was relatively devoid of oblique branches. The soma is just below the bottom of the image. Line scans of ΔF/F are shown in the top row and the fluorescence change in the ROI shown in the bottom image are shown in the middle row with the corresponding electrical recordings underneath them. Synaptic input alone (100 Hz for 0.5 s) evoked a spike at the beginning of the postsynaptic response. The [Ca2+]i signal from the spike was comparable with the weak release signal that followed. Synaptic input plus spikes (stimulation at same intensity plus two action potentials separated by 30 ms evoked by 2 ms pulses in the soma) generated a large Ca2+ release transient in the region opposite the stimulating electrode. Spikes alone (two action potentials separated by 30 ms) produced a sharper and smaller [Ca2+]i increase. The scale for the greyscale line scans (top row) is the same for all three panels.
Spatial distribution of Ca2+ waves
In CA1 neurons most synaptically activated Ca2+ waves initiate at a location on the main dendritic shaft close to a branch point and there is little Ca2+ release on small, oblique dendritic branches (Nakamura et al. 2002). We measured the initiation site on the main shaft for 65 Ca2+ waves in a selection of L5 neurons by determining the site of the earliest Ca2+ signal. Almost all the waves initiated close to branch points (Fig. 7A and B). Figure 7C shows that there was no correlation of the initiation site with the position of the stimulating electrode on a scale of about 40 μm. On a larger scale the waves generally initiated in the region of the dendrites near the stimulation site and the position of the wave changed as the stimulation electrode was moved along the dendrite. A comparable large-scale correlation between stimulation site and wave location was observed in hippocampal neurons (Nakamura et al. 1999; Fig. 3).
Figure 7. Synaptically activated Ca2+ waves initiate near branch points on the main apical shaft and not directly opposite the position of the stimulating electrode.

A, Ca2+ wave evoked by repetitive synaptic stimulation (100 Hz for 0.5 s). The dashed line connects the site of wave initiation with the corresponding position on the apical shaft. The initiation site is close to a branch point. The traces below the images show the electrical recording and the fluorescence change from the ROI. B, histogram showing the distribution of Ca2+ release events as a function of the separation between the initiation site and the closest branch point. Most waves start close to branch points. C, histogram showing the distribution of events as a function of the separation between the initiation site and the position of the tip of the stimulating electrode. The correlation is not as close as with branch points. Note that the scales for the abscissas in B and C are different.
We observed many examples of waves (n > 15) initiating at more than one site in the dendrites even though synaptic stimulation was via a focal stimulating electrode (Fig. 1). In most cases, all the separate waves initiated at branch points. We made similar observations of multiple site initiation of Ca2+ waves in CA1 pyramidal neurons (Nakamura et al. 2002).
One difference between L5 neocortical pyramidal neurons and hippocampal pyramidal neurons is the long, relatively unbranched apical shaft in the middle of the dendrites of neocortical cells. In several experiments we placed the stimulating electrode near this region. Although a higher stimulation intensity was required than near branch points, it was still possible to generate Ca2+ release waves in this unbranched region (e.g. see Fig. 6; n = 7). Therefore, it appears that initiation at a branch point is not an absolute requirement. Rather, waves will initiate preferentially at a branch point if there is one near the stimulation site.
Several examples of regenerative Ca2+ waves were observed restricted to the initial part of an oblique branch (Fig. 8, middle panel; n = 5). These events had a clear threshold for regenerative activation and were delayed by 0.2–0.5 s compared with the electrical response in the soma. In all these cases, increasing the intensity of synaptic stimulation produced waves that invaded the main dendritic shaft (Fig. 8, right panel, n = 10). In most (but not all) these examples, Ca2+ release on the branch preceded the larger release on the shaft. The waves propagated along the oblique branches in a manner comparable to waves travelling along the apical shaft, suggesting that they were not the result of Ca2+ entry through ligand-gated receptors along the branch. This result suggests that regenerative Ca2+ release is not always restricted to the main dendritic shaft in neocortical neurons. However, synaptically activated Ca2+ release was only observed in the first 10–30 μm of the oblique branches and generally began with a delay relative to the start of the synaptic train. Ca2+ signals further out on the branches usually began simultaneously with the synaptic response (e.g. black trace in Fig. 8).
Figure 8. Threshold synaptic stimulation sometimes evokes Ca2+ release in an oblique dendrite close to the main shaft.
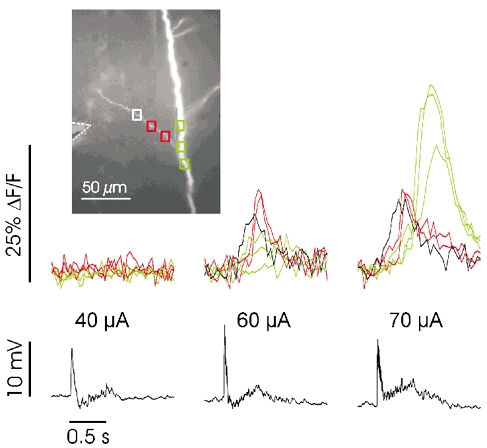
Stimulation at three intensities (100 Hz for 0.5 s). At 40 μA an electrical response was recorded in the soma but there was no [Ca2+]i increase on the branch or shaft near the stimulating electrode. At 60 μA there was a delayed regenerative response at the two locations close to the shaft (red boxes), but little or no response on the shaft (green boxes). The immediate response at the third position (white box, black trace) was probably due to ligand-gated Ca2+ entry since it was blocked by 50 μm APV (data not shown; see also Fig. 9A). At 70 μA a strong regenerative and more delayed response was observed on the shaft. Note that the electrical response was still below action potential threshold.
Spatial separation of different sources of dendritic [Ca2+]i increases
In hippocampal pyramidal neurons the main source of synaptically activated [Ca2+]i increases on the oblique branches is Ca2+ entry through NMDA receptors while the primary component on the main shaft is Ca2+ release from stores (Nakamura et al. 2002). The propagation of waves in the proximal parts of oblique dendrites of neocortical neurons (e.g. Fig. 8) suggests that the segregation of Ca2+ sources is less definitive in these cells. Pharmacological experiments support this conclusion. Figure 9A (typical of seven tested neurons) shows that 50–100 μm APV strongly reduced the early [Ca2+]i increase (during the time of stimulation) on the branch (50 μm from the shaft) but did not eliminate the late signal or the regenerative response on the shaft. There was little early signal on the shaft and this component did not change in the presence of APV. This experiment implies that most of the early [Ca2+]i increase is probably due to Ca2+ entry through NMDA receptors, although we cannot rule out some contribution from entry through voltage-dependent Ca2+ channels (Miyakawa et al. 1992) since the synaptic potential was smaller in the presence of APV.
A blocker of group I mGluRs, MCPG, had a variable effect on branch [Ca2+]i increases. In some cells the amplitude of the increase, especially the late component (due to Ca2+ release), was reduced (e.g. the branch signal in Fig. 9A; data not shown). However, in other neurons (e.g. Fig. 9B) there was little change in the branch signal even when the regenerative Ca2+ release component on the shaft was almost completely blocked. Thus, both the experiments with APV and the experiments with MCPG suggest that in some cases, but not all, there is a component of Ca2+ released in the oblique branches near the main shaft.
Ca2+ waves in distal dendrites
We searched for Ca2+ waves in the distal regions of neocortical neurons, using dendritic whole-cell recording and distal-synaptic stimulation. In CA1 pyramidal neurons we found it difficult to evoke Ca2+ waves in the distal dendrites more than 150 μm from the soma, where the apical shaft breaks into many sub branches (Nakamura et al. 1999). Figure 10 shows that we could evoke a Ca2+ wave in the distal dendrites of a L5 neuron about 400 μm from the soma. The electrical response to the standard stimulation protocol (100 Hz for 0.5 s) shows an initial voltage jump that evoked a small [Ca2+]i change, which was synchronous over the dendritic region shown in the high resolution image at the top (dashed box in bottom image). This initial response due to voltage dependent Ca2+ entry was followed by a larger asynchronous [Ca2+]i increase that had characteristics similar to Ca2+ waves recorded in the proximal dendrites. The region of interest (ROI) traces above the electrical recording show that the wave propagated from the left branch to the branch point but not into or from the right branch. This pattern was typical of many waves (n = 10) recorded in the distal dendrites. It was harder to evoke waves past the branch point, but many examples of post-branch waves (as in this Figure) were noted. The distal waves appeared to result from the same IP3-dependent mechanism as the proximal waves since they were blocked by MCPG (n = 3) and intradendritic heparin (n = 3; Fig. 3).
Figure 10. Distal synaptic stimulation evokes Ca2+ release in the distal dendrites.
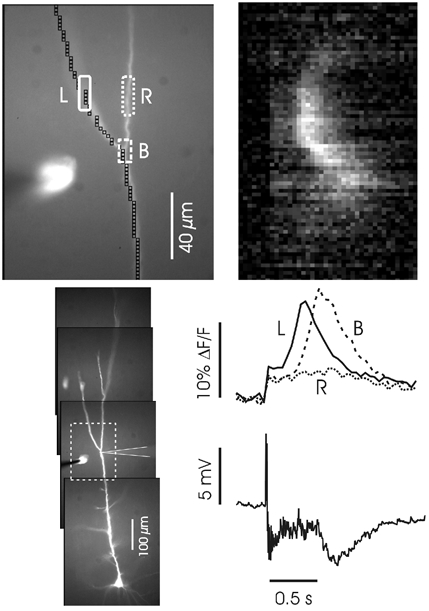
The cell was patched on the dendrites just below a branch about 400 μm from the soma; the stimulating electrode was placed in the same region. The image top left is from the section in dashed lines below. Synaptic stimulation (100 Hz for 0.5 s) evoked a wave in this region. The ‘line scan’ and ROI traces below show that the wave was initiated above the branch point and propagated downwards in the left branch into the shaft but not into the right branch.
Ca2+ waves evoked by backpropagating spikes and metabotropic agonists
We tested the effect of bath-applied t-ACPD (an mGluR agonist) and CCh (carbachol, a cholinergic agonist) on neocortical pyramidal neurons. The effects of these agonists may relate to their neuromodulatory role in the cortex (e.g. McCormick, 1993; Wang & McCormick, 1993; Gil et al. 1997). When applied slowly through the superfusion system (∼1 ml min−1) neither compound caused large amplitude changes in [Ca2+]i. However, if short trains of backpropagating action potentials were evoked in the presence of these agonists, Ca2+ release transients were generated (Fig. 11). The same effect occurred in both L2/3 pyramidal neurons (t-ACPD: n = 5/6, CCh: n = 3/3; see also Yamamoto et al. 2000) and L5 pyramidal neurons (t-ACPD: n = 14/18, CCh: n = 6/10). Ca2+ release mediated by either agonist was blocked by the inclusion of 1–2 mg ml−1 heparin in the patch pipette (n = 5/7). This result is similar to that found in hippocampal pyramidal neurons (Nakamura et al. 2000). We found that t-ACPD and CCh induced Ca2+ release less consistently in neocortical pyramidal neurons than in CA1 pyramidal neurons when we used the same agonist concentrations that were effective in the hippocampus. We had more consistent results by doubling the concentration of both agonists (t-ACPD from 25 to 50 μm and CCh from 10 to 20 μm). Even with these concentrations we often had to use a protocol of double spike trains (Fig. 11A) to see Ca2+ release. In addition, the amplitudes of the [Ca2+]i increases evoked by this protocol were not as large as the ones observed in hippocampal neurons. In CA1 neurons, the Ca2+ release transients were much larger than the [Ca2+]i increases caused by the spikes that evoked them (e.g. Fig. 1 in Nakamura et al. 2000) and often saturated the response of the indicator when bis-fura-2 was used. In the neocortical cells the change in fluorescence caused by Ca2+ release was rarely more than twice the change caused by a train of spikes in normal ACSF and did not saturate the indicator. Interestingly, the amplitude of the synaptically activated regenerative Ca2+ transients was similar in both hippocampal and L5 pyramidal neurons (compare the furaptra signal in Fig. 5 in this paper with the signal in Fig. 6 in Nakamura et al. 1999).
Figure 11. Backpropagating action potentials evoke Ca2+ release in the presence of the metabotropic agonists t-ACPD and CCh.
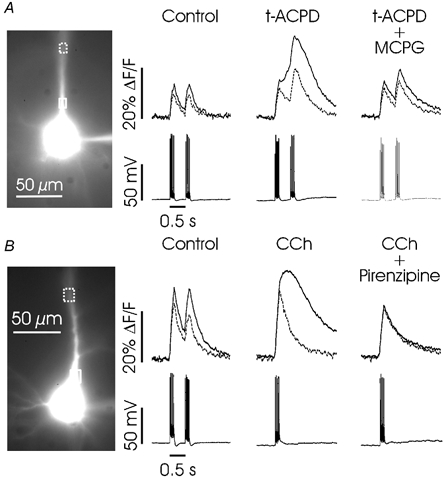
A, in control conditions (normal ACSF) trains of backpropagating spikes evoked sharp increases in [Ca2+]i in a L5 pyramidal neuron both adjacent to the soma and in the proximal apical dendrite (ROIs shown over image of cell). The dashed traces correspond to the dashed ROIs. In t-ACPD (50 μm) the same action potentials induced a larger, rounded [Ca2+]i increase in the proximal dendrite, but the increase in the distal location was still sharp. Addition of MCPG (1 mm) to this solution blocked the rounded increase. B, a similar experiment using 20 μm CCh in another L5 pyramidal neuron. The Ca2+ release transient was larger in the proximal location and was blocked by 0.5 μm pirenzipine.
The pharmacology of spike-induced Ca2+ release was tested in L5 pyramidal neurons and was similar to the profile found in hippocampal neurons. The t-ACPD evoked Ca2+ release was blocked by 1 mm MCPG (n = 3) and the CCh evoked release was blocked by 0.5–1.0 μm pirenzipine (n = 3).
One interesting finding about Ca2+ release evoked by spikes and metabotropic agonists was that it appeared to be primarily in the proximal 100–150 μm of the apical dendrite (Fig. 11), even though the agonists were bath applied over the entire slice and the backpropagating spikes evoked [Ca2+]i increases at much more distal locations (Fig. 12). We did not check this result systematically. Although a similar spatial distribution was observed in hippocampal neurons (Nakamura et al. 1999, 2000) this result was not expected since synaptically evoked Ca2+ release waves could be observed more than 400 μm from the soma in neocortical neurons (Fig. 10).
Figure 12. Backpropagating action potentials evoke [Ca2+]i increases at all dendritic locations.
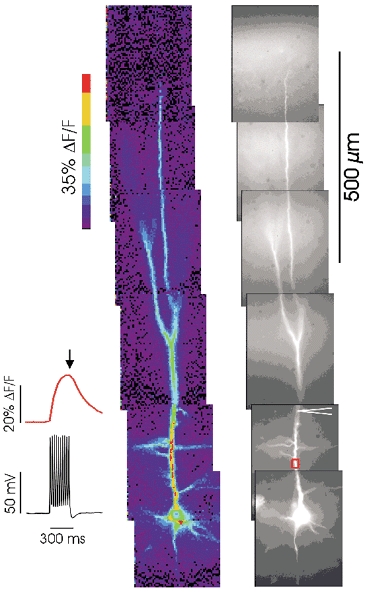
The fluorescence image on the right shows a montage of pictures, taken with the × 20 lens, that together span the entire apical dendritic arbor. The dendrites were patched with an electrode containing 150 μm bis-fura-2. The traces on the left show that a train of action potentials (10 spikes at 30 ms intervals) evoked a [Ca2+]i increase at the indicated ROI in the proximal dendrites that began at the start of the train and declined immediately after the train (average of 10 trials). The pseudocolour image shows the fluorescence change at the end of the train (arrow). To make the montage the experiment was repeated as the cell was moved to different positions. The largest changes are in the proximal apical dendrites, but fluorescence increases were detected at all locations where fluorescence was detected, including the apical tuft and oblique branches. Data from a 19-day-old rat.
Comparison between Ca2+ release and spike-evoked [Ca2+]i changes in distal dendrites
A distinctive feature of L5 pyramidal neurons is that under some conditions, such as synaptic activation, high frequency backpropagating action potentials, or direct intradendritic stimulation, Ca2+ action potentials can be evoked in the distal dendrites of these cells. Ca2+ spikes have been proposed as a potential signalling mechanism in the distal arborization (Reuveni et al. 1993; Schiller et al. 1997). Therefore, we measured the [Ca2+]i changes associated with these spikes and compared them with the changes resulting from Ca2+ release in the same distal dendritic region. Figure 13 shows an example of this kind of comparison, using the high-affinity indicator bis-fura-2. The amplitude of the Ca2+ release transient was slightly higher than the peak [Ca2+]i change generated by the Ca2+ spike. In general, the peak [Ca2+]i change during the calcium wave when measured near the branch point was similar to the peak [Ca2+]i following a Ca2+spike in the same region. For 16 cells, the average ratio between [Ca2+]i for release to [Ca2+]i during a Ca2+ spike was 1.04 ± 0.40. The changes in [Ca2+]i were usually smaller in both cases near the branch point than the peak [Ca2+]i change measured during release in the proximal apical dendrite, suggesting that we were not saturating the indicator. The release transient near the branch point was more localized than the spike-evoked [Ca2+]i change. Ca2+ release typically extended over a region of less than 100 μm and occasionally was restricted to a region of about 10 μm. In contrast, the Ca2+ spike signal often extended over the full extent of the camera field (140 μm using the ×60 objective).
Figure 13. Synaptically activated Ca2+ release generates similarly large but broader and more localized [Ca2+]i increases than dendritic Ca2+ spikes generate.
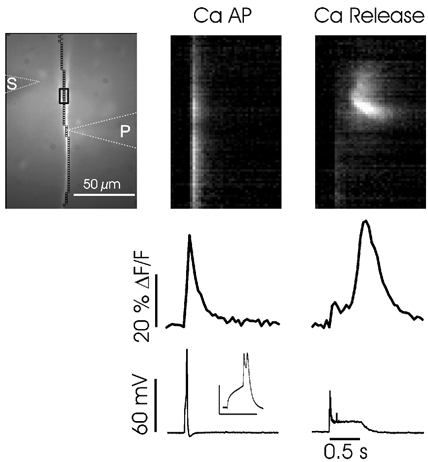
Intradendritic depolarization evoked a Ca2+ spike (middle panels). Synaptic stimulation (100 Hz for 0.5 s) evoked a fluorescence change of similar amplitude at the same location (right panels). The patch electrode contained bis-fura-2 (150 μm). The positions of the stimulating electrode (S) and patch electrode (P) are shown with dotted lines. The Ca2+ spike evoked a [Ca2+]i increase at all locations in the camera field while the Ca2+ wave occurred in a more restricted region of the dendrites. Dendritic region 300–400 μm from the soma. Inset over electrical trace shows the expanded Ca2+ spike. Scale for inset: 50 mV, 100 ms.
We have two reservations in interpreting these results. First, there is a region usually around the principal bifurcation in L5 neurons where the threshold for generating Ca2+ APs is lowest (Larkum & Zhu, 2002), which may indicate a high density of Ca2+ channels. However, backpropagating APs also elicit increases in dendritic [Ca2+]i and are usually evoked by dendritic Ca2+ APs. Therefore, the spatial distribution of the [Ca2+]i change seen during Ca2+ APs may reflect not just the Ca2+ AP near the region of high density of Ca2+ channels, but may also include [Ca2+]i increases from the backpropagating spikes. In the experiments presented here, it was hard to detect the presence and number of back-propagating APs within a Ca2+ AP since we did not simultaneously record the potential in the soma. Second, the peak amplitude of dendritic Ca2+ APs and their associated [Ca2+]i increases are sensitive functions of distance from the soma (Schiller et al. 1997). The amplitude of the Ca2+ release transient also appears to be a function of the position in the dendrites. Therefore, the approximate equality of amplitude of these two signals applies just to the sample of events we studied. A more precise comparison might show that the ratio is different at different locations. Similarly, the ratio may depend on the age of the animal since the ability to generate Ca2+ APs matures during the first six weeks after birth (Zhu, 2000).
In another series of experiments (n = 4) we compared the Ca2+ release signal in the dendrites to the [Ca2+]i change evoked by a train of backpropagating action potentials. Figure 5 showed that the release signal was much bigger than the spike signal when measured in the proximal dendrites. Figure 12 showed that a 30 Hz train of Na+ spikes evoked [Ca2+]i changes all over the pyramidal neuron from a young rat, including the apical tuft and the oblique dendrites (Stuart & Sakmann, 1994; Schiller et al. 1995). In older rats (> P 28), higher-frequency trains of Na+ spikes were needed to evoke [Ca2+]i changes all over the neuron (Larkum et al. 1999). Close to the main apical branch point the magnitude of the Ca2+ release signal was about five times larger than the spike train signal using the linearly responding indicator furaptra (n = 3; data not shown).
DISCUSSION
These experiments show that repetitive synaptic activation evokes Ca2+ waves in the apical dendrites of both layer 2/3 and layer 5 neocortical pyramidal neurons. The waves were observed in both 2- to 4- and 6- to 8-week-old animals. Thus the ability to generate Ca2+ waves is a stable feature of these cells even though during this period significant changes occur in the expression of Ca2+ channels (Lorenzon & Foehring, 1995) and the ability to generate Ca2+ action potentials (Zhu, 2000). The Ca2+ waves have many properties similar to those described in pyramidal neurons from the CA1 region (Nakamura et al. 1999, 2002; Power & Sah, 2002) and from the CA3 region (Kapur et al. 2001) of the hippocampus. Synaptically activated Ca2+ release occurs in cerebellar Purkinje neurons (Finch & Augustine, 1998; Takechi et al. 1998). However, the release transients in Purkinje cells are confined to spines and do not spread as a wave in the dendrites. IP3-mediated Ca2+ release has been demonstrated in midbrain dopamine neurons (Morikawa et al. 2000) but waves in these cells have not been described. Preliminary experiments (n = 9; S. Zhou and W.N. Ross, unpublished observations) have not been able to demonstrate Ca2+ waves in hippocampal interneurons even though there is some evidence for Ca2+ release from internal stores in these cells (Woodhall et al. 1999). The reasons for this variation among neuron types are not known. Possible explanations include different kinds of IP3 receptor isoforms (Thrower et al. 2001), differing distribution of IP3 receptors within the dendrites (Sharp et al. 1993), different Ca2+ buffering molecules in the cytoplasm (Baimbridge et al. 1982; Lee et al. 2000), and different organisation of Ca2+ signalling microdomains (Delmas et al. 2002). We conclude that most pyramidal cells of the CNS have the ability to generate Ca2+ waves in the soma and apical dendrite, but their appearance in other cells types is yet to be demonstrated.
Neocortex vs. hippocampus
There are no major differences between the properties of Ca2+ waves in hippocampal CA1 pyramidal neurons and L5 neocortical pyramidal neurons, the two best studied cell types. In both kinds of neurons the waves result from activation of Group I mGluRs that mobilize IP3 and act on IP3 receptors to release Ca2+. Activation of NMDA or AMPA receptors is not required to induce Ca2+ release although the rise in [Ca2+]i caused by Ca2+ entry through ligand-gated receptors or through voltage-sensitive Ca2+ channels may synergistically enhance the probability of release by shifting the sensitivity of IP3Rs to IP3 (Fig. 6; Nakamura et al. 1999, 2002). The large amplitude waves propagate along the main apical dendrite and appear to initiate near branch points on these thick processes. Dendritic Ca2+ waves may actually initiate on the oblique branches before exploding into larger waves when they reach the main shaft since we observed earlier small waves on the initial part of many oblique branches. Waves in oblique dendrites were observed less frequently in hippocampal pyramidal neurons. These branch waves were rarely observed more than 30 μm into the oblique dendrites, although we did not examine this point in detail in these experiments on L5 neurons and not at all in L2/3 neurons. Another difference between hippocampal and L5 neocortical cells is that Ca2+ waves could be generated several hundred microns from the soma in L5 neurons; in CA1 pyramidal neurons these waves were only observed up to 100–150 μm from the soma (Nakamura et al. 1999). The reasons for these differences are not clear. In fact, the reasons why Ca2+ waves are observed in only a restricted part of the dendrites in either kind of pyramidal neuron are not known. Some possible explanations are discussed in Nakamura et al. (2002). One consistent parameter in both neuron types is that the waves propagate within the thick apical dendrite and are not easily observed in the smaller oblique dendrites, basal dendrites (beyond 30 μm from the soma), and apical tuft. In the layer 5 neocortical cells the thick apical shaft extends much further from the soma. A third small difference between CA1 pyramidal neurons and layer 5 neocortical neurons is that higher concentrations of metabotropic agonists were required to generate Ca2+ release when a train of backpropagating action potentials was generated in the soma. In addition, when stimulated this way, the peak amplitude of the [Ca2+]i change was smaller in neocortical cells. The reason is not that neocortical neurons are not capable of large Ca2+ release transients. Indeed, Fig. 5 shows that the synaptically activated Ca2+ transients are as large as the transients evoked in hippocampal pyramidal neurons. The causes of this difference in sensitivity to metabotropic agonists are not known. One possibility is that the lifetime of IP3 is shorter in neocortical pyramidal neurons, requiring a higher rate of IP3 production to achieve the same equilibrium concentration.
Functional implications
The peak amplitude of the propagating Ca2+ waves reached a value of several μm in the proximal and distal dendrites. In the somatic region and in the proximal dendritic region these [Ca2+]i increases are much larger than the [Ca2+]i increases generated by trains of backpropagating Na+ action potentials. In the distal dendritic region, near the branch point, the increases in [Ca2+]i are similar for Ca2+ release and Ca2+ spikes although values in this region are generally smaller than the maximum values found closer to the soma during release events. Ca2+ waves are more localized and longer lasting than the electrically generated Ca2+ transients. Unlike voltage-gated signals the [Ca2+]i changes from release are not highly localized just under the plasma membrane. For all these reasons the waves may activate downstream mechanisms that are untouched by the spike-induced [Ca2+]i changes. For example, calmodulin has a KD for binding Ca2+ in the micromolar range and slow rate constants for this reaction (e.g. Klee, 1988). Spike-induced [Ca2+]i increases do not reach these values and recover rapidly (Helmchen et al. 1996; Sabatini et al. 2002). Synaptic activation of NMDA receptors also generates [Ca2+]i increases in the μm range (Petrozzino et al. 1995; Sabatini et al. 2002). These increases are localized to spines and generally do not overlap spatially with the regions where waves are generated (Nakamura et al. 2002; this paper). Therefore, they may activate different signalling mechanisms than the Ca2+ waves.
It is probable that the large and long-lasting [Ca2+]i increases at the peak of Ca2+ waves activate many cellular functions. One simple consequence is that the Ca2+ modulates dendritic conductances (e.g. g(K)Ca) and affects synaptic integration and electrogenesis in the dendrites in the immediate time frame following wave generation. This idea remains to be tested. In other neuron types a variety of more complex functions have been shown to depend on intracellular Ca2+ release. For example, in retinal ganglion cells intradendritic Ca2+ release controls the stabilization of new dendritic processes during development (Lohmann et al. 2002). In spinal motoneurons Ca2+ waves are important in axon outgrowth (e.g. Lautermilch & Spitzer, 2000). In pyramidal neurons Ca2+ release from stores may be important in controlling synaptic plasticity (e.g. Reyes & Stanton, 1996; Nishiyama et al. 2000; Daw et al. 2002).
Many of these mechanisms require activation of gene expression and protein synthesis. In this regard, it has been suggested (Berridge, 1998; Nakamura et al. 1999) that the dendritic Ca2+ waves propagate towards the soma and activate these processes in the nucleus. In hippocampal neurons we found that stimulation near the soma evoked waves that invaded the cell body (Nakamura et al. 1999). Power & Sah (2002), using confocal imaging, found that synaptic activation of muscarinic receptors activated Ca2+ waves that entered the nucleus. However, most of the Ca2+ waves activated by our experiments in both hippocampal and neocortical pyramidal neurons do not propagate to the soma or nucleus. One possible explanation is that the focal stimulation protocol we used mobilized IP3 in a restricted region of the dendrites and the unavailability of IP3 prevented the propagation of the regenerative Ca2+ wave beyond that region. Furthermore, IP3 is rapidly diluted when it diffuses into a large volume soma. The concentration in the soma may then be too low to evoke Ca2+ release. Stimulation near the soma (Nakamura et al. 1999) or widespread activation of presynaptic fibres with a bipolar stimulating electrode (Power & Sah, 2002) directly generates IP3 in the somatic region, allowing the wave to enter the soma (Oertner & Svoboda, 2002).
Synaptically activated Ca2+ waves also could fail to enter the nuclear region in our experiments because the patch electrode on the soma dialysed the somatic cytoplasm, washing out critical components necessary for wave propagation to this region. The fact that waves entered the soma in some circumstances (Nakamura et al. 1999, 2000; Power & Sah, 2002; our observations on neocortical neurons (not shown)), even with a patch electrode on the soma, makes this explanation unlikely. However, to test this idea, we did two experiments where we made whole-cell recordings on the dendrites 200–300 μm from the soma and generated Ca2+ waves in the region between the soma and the patch electrode. Even in these experiments, where somatic washout was unlikely, the Ca2+ waves did not invade the soma if the electrode, stimulating fibres in a restricted region near the tip, was positioned opposite the dendrites. Consequently, it is unclear if and when dendritically generated Ca2+ waves enter the nucleus in physiological conditions.
Acknowledgments
This study was supported in part by NIH grant NS16295. We thank Jack Waters and Fritjof Helmchen for their comments on the manuscript and Bert Sakmann for his encouragement.
REFERENCES
- Baimbridge KG, Miller JJ, Parkes CO. Calcium-binding protein distribution in the rat brain. Brain Res. 1982;239:519–525. doi: 10.1016/0006-8993(82)90526-1. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium response curves of Ins(1, 4, 5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and function of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Daw MI, Bortolotto ZA, Saulle E, Zaman S, Collingridge GL, Isaac JT. Phosphatidylinositol 3 kinase regulates synapse specificity of hippocampal long-term depression. Nature Neurosci. 2002;5:835–836. doi: 10.1038/nn903. [DOI] [PubMed] [Google Scholar]
- Delmas P, Wanaverbecq N, Abogadie FC, Mistry M, Brown DA. Signaling microdomains define the specificity of receptor-mediated InsP3 pathways in neurons. Neuron. 2002;34:209–20. doi: 10.1016/s0896-6273(02)00641-4. [DOI] [PubMed] [Google Scholar]
- Finch EA, Augustine GJ. Local calcium signaling by inositol-1, 4, 5-trisphosphate in Purkinje cell dendrites. Nature. 1998;396:753–756. doi: 10.1038/25541. [DOI] [PubMed] [Google Scholar]
- Finch EA, Turner TJ, Goldin SM. Calcium as za coagonist of inositol trisphosphate-induced calcium release. Science. 1991;252:443–446. doi: 10.1126/science.2017683. [DOI] [PubMed] [Google Scholar]
- Ghosh TK, Eis PS, Mullaney JM, Ebert CL, Gill DL. Competitive, reversible, and potent antagonism of inositol 1, 4, 5-trisphosphate-activated calcium release by heparin. J Biol Chem. 1988;263:11075–11079. [PubMed] [Google Scholar]
- Gil Z, Connors BW, Amitai Y. Differential regulation of neocortical synapses by neuromodulators and activity. Neuron. 1997;19:679–686. doi: 10.1016/s0896-6273(00)80380-3. [DOI] [PubMed] [Google Scholar]
- Golding NL, Jung HY, Mickus T, Spruston N. Dendritic calcium spike initiation and repolarization are controlled by distinct potassium channel subtypes in CA1 pyramidal neurons. J Neurosci. 1999;19:8789–8798. doi: 10.1523/JNEUROSCI.19-20-08789.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F, Imoto K, Sakmann B. Ca2+ buffering and action potential-evoked Ca2+ signaling in dendrites of pyramidal neurons. Biophys J. 1996;70:1069–1081. doi: 10.1016/S0006-3495(96)79653-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino M. Biphasic Ca2+ dependence of inositol 1, 4, 5-trisphosphate-induced Ca2+ release in smooth muscle cells of the guinea pig Taenia caeci. J Gen Physiol. 1990;95:1103–1122. doi: 10.1085/jgp.95.6.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe DB, Brown TH. Metabotropic glutamate receptor activation induces calcium waves within hippocampal dendrites. J Neurophysiol. 1994;72:471–474. doi: 10.1152/jn.1994.72.1.471. [DOI] [PubMed] [Google Scholar]
- Kapur A, Yeckel M, Johnston D. Hippocampal mossy fiber activity evokes Ca2+ release in CA3 pyramidal neurons via a metabotropic glutamate receptor pathway. Neuroscience. 2001;107:59–69. doi: 10.1016/s0306-4522(01)00293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klee CB. Interaction of calmodulin with Ca2+ and target proteins. In: Cohen P, Klee CB, editors. Calmodulin. Amsterdam: Elsevier; 1988. pp. 35–56. [Google Scholar]
- Kobayashi S, Somlyo AV, Somlyo AP. Heparin inhibits the inositol 1, 4, 5-trisphosphate-dependent, but not the independent, calcium release induced by guanine nucleotide in vascular smooth muscle. Biochem Biophys Res Commun. 1988;153:625–631. doi: 10.1016/s0006-291x(88)81141-0. [DOI] [PubMed] [Google Scholar]
- Larkum ME, Kaiser KM, Sakmann B. Calcium electrogenesis in distal apical dendrites of layer 5 pyramidal cells at a critical frequency of back-propagating action potentials. Proc Nat Acad Sci U S A. 1999;96:14600–14604. doi: 10.1073/pnas.96.25.14600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ. Signaling of layer 1 and whisker-evoked Ca 2+ and Na+ action potentials in distal and terminal dendrites of rat neocortical pyramidal neurons in vitro and in vivo. J Neurosci. 2002;22:6991–7005. doi: 10.1523/JNEUROSCI.22-16-06991.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ, Sakmann B. Dendritic mechanisms underlying the coupling of the dendritic with the axonal action potential initiation zone of adult rat layer 5 pyramidal neurons. J Physiol. 2001;533:447–466. doi: 10.1111/j.1469-7793.2001.0447a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasser-Ross N, Miyakawa H, Lev-Ram V, Young SR, Ross WN. High time resolution fluorescence imaging with a CCD camera. J Neurosci Methods. 1991;36:253–261. doi: 10.1016/0165-0270(91)90051-z. [DOI] [PubMed] [Google Scholar]
- Lautermilch NJ, Spitzer NC. Regulation of calcineurin by growth cone calcium waves controls neurite extension. J Neurosci. 2000;20:315–325. doi: 10.1523/JNEUROSCI.20-01-00315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Rosenmund C, Schwaller B, Neher E. Differences in Ca2+ buffering properties between excitatory and inhibitory hippocampal neurons from the rat. J Physiol. 2000;525:405–418. doi: 10.1111/j.1469-7793.2000.t01-3-00405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann C, Myhr KL, Wong ROL. Transmitter-evoked local calcium release stabilizes developing dendrites. Nature. 2002;418:177–181. doi: 10.1038/nature00850. [DOI] [PubMed] [Google Scholar]
- Lorenzon NM, Foehring RC. Characterization of pharmacologically identified voltage-gated calcium channel currents in acutely isolated rat neocortical neurons. II. Postnatal development. J Neurophysiol. 1995;73:1443–1451. doi: 10.1152/jn.1995.73.4.1443. [DOI] [PubMed] [Google Scholar]
- McCormick DA. Actions of acetylcholine in the cerebral cortex and thalamus and implications for function. Prog Brain Res. 1993;98:303–308. doi: 10.1016/s0079-6123(08)62412-7. [DOI] [PubMed] [Google Scholar]
- Markram H. A network of tufted layer 5 pyramidal neurons. Cereb Cortex. 1997;7:523–533. doi: 10.1093/cercor/7.6.523. [DOI] [PubMed] [Google Scholar]
- Miyakawa H, Ross WN, Jaffe D, Callaway JC, Lasser-Ross N, Lisman JE, Johnston D. Synaptically activated increases in Ca2+ concentration in hippocampal CA1 pyramidal cells are primarily due to voltage-gated Ca2+ channels. Neuron. 1992;9:1163–1173. doi: 10.1016/0896-6273(92)90074-n. [DOI] [PubMed] [Google Scholar]
- Moraru II, Kaftan EJ, Ehrlich BE, Watras J. Regulation of type 1 inositol 1, 4, 5-trisphosphate-gated calcium channels by InsP3 and calcium: Simulation of single channel kinetics based on ligand binding and electrophysiological analysis. J Gen Physiol. 1999;113:837–849. doi: 10.1085/jgp.113.6.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa H, Imani F, Khodakhah K, Williams JT. Inositol 1, 4, 5-trisphosphate-evoked responses in midbrain dopamine neurons. J Neurosci. 2000;20:RC103. doi: 10.1523/JNEUROSCI.20-20-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Barbara J-G, Nakamura K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727, 737. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Lasser-Ross N, Nakamura K, Ross WN. Spatial segregation and interaction of calcium signalling mechanisms in rat hippocampal CA1 pyramidal neurons. J Physiol. 2002;543:465–480. doi: 10.1113/jphysiol.2002.020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Nakamura K, Lasser-Ross N, Barbara J-G, Sandler VM, Ross WN. Inositol 1, 4, 5-trisphosphate (IP3)-mediated Ca2+ release evoked by metabotropic agonists and backpropagating action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 2000;20:8365–8376. doi: 10.1523/JNEUROSCI.20-22-08365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature. 2000;408:584–588. doi: 10.1038/35046067. [DOI] [PubMed] [Google Scholar]
- Oertner TG, Svoboda K. Subliminal messages in hippocampal pyramidal cells. J Physiol. 2002;543:397. doi: 10.1113/jphysiol.2002.023606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrozzino JJ, Pozzo Miller LD, Connor JA. Micromolar Ca2+ transients in dendritic spines of hippocampal pyramidal neurons in brain slice. Neuron. 1995;14:1223–1231. doi: 10.1016/0896-6273(95)90269-4. [DOI] [PubMed] [Google Scholar]
- Power JM, Sah P. Nuclear calcium signaling evoked by cholinergic stimulation in hippocampal CA1 pyramidal neurons. J Neurosci. 2002;22:3454–3462. doi: 10.1523/JNEUROSCI.22-09-03454.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuveni I, Friedman A, Amitai Y, Gutnick MJ. Stepwise repolarization from Ca2+ plateaus in neocortical pyramidal cells: evidence for nonhomogeneous distribution of HVA Ca2+ channels in dendrites. J Neurosci. 1993;13:4609–4621. doi: 10.1523/JNEUROSCI.13-11-04609.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes M, Stanton PK. Induction of hippocampal long-term depression requires release of Ca2+ from separate presynaptic and postsynaptic intracellular stores. J Neurosci. 1996;16:5951–5960. doi: 10.1523/JNEUROSCI.16-19-05951.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau E, Smith SJ, Meissner G. Ryanodine modifies conductance and gating behavior of single Ca2+ release channel. Am J Physiol. 1987;253:C364–368. doi: 10.1152/ajpcell.1987.253.3.C364. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca2+ ions in dendritic spines. Neuron. 2002;33:439–452. doi: 10.1016/s0896-6273(02)00573-1. [DOI] [PubMed] [Google Scholar]
- Sakmann B, Stuart G. Patch-pipette recordings from the soma, dendrites, and axon of neurons in brain slices. In: Sakmann B, Neher E, editors. Single Channel Recording. New York: Plenum Press; 1995. pp. 199–211. [Google Scholar]
- Schiller J, Helmchen F, Sakmann B. Spatial profile of dendritic calcium transients evoked by action potentials in rat neocortical pyramidal neurones. J Physiol. 1995;487:583–600. doi: 10.1113/jphysiol.1995.sp020902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Stuart G, Sakmann B. Calcium action potentials restricted to the distal apical dendrites of rat neocortical pyramidal neurons. J Physiol. 1997;505:605–616. doi: 10.1111/j.1469-7793.1997.605ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AH, McPherson PS, Dawson TM, Aoki C, Campbell KP, Snyder SH. Differential immunochemical localization of inositol 1, 4, 5-trisphosphate- and ryanodine-sensitive Ca2+ release channels in rat brain. J Neurosci. 1993;13:3051–3063. doi: 10.1523/JNEUROSCI.13-07-03051.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SJ, Imagawa T, Ma J, Fill M, Campbell KP, Coronado R. Purified ryanodine receptor from rabbit skeletal muscle is the calcium-release channel from sarcoplasmic reticulum. J Gen Physiol. 1988;92:1–26. doi: 10.1085/jgp.92.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart GJ, Sakmann B. Active propagation of somatic action potentials into neocortical pyramidal cell dendrites. Nature. 1994;367:69–72. doi: 10.1038/367069a0. [DOI] [PubMed] [Google Scholar]
- Takechi H, Eilers J, Konnerth A. A new class of synaptic response involving calcium release in dendritic spines. Nature. 1998;396:757–760. doi: 10.1038/25547. [DOI] [PubMed] [Google Scholar]
- Thrower EC, Hagar RE, Ehrlich BE. Regulation of Ins(1, 4, 5)P3 receptor isoforms by endogenous modulators. Trends Pharmacol Sci. 2001;22:580–586. doi: 10.1016/s0165-6147(00)01809-5. [DOI] [PubMed] [Google Scholar]
- Wang Z, McCormick DA. Control of firing mode of corticotectal and corticopontine layer V burst-generating neurons by norepinephrine, acetylcholine, and 1S, 3R-ACPD. J Neurosci. 1993;13:2199–2216. doi: 10.1523/JNEUROSCI.13-05-02199.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhall G, Gee CE, Robitaille R, Lacaille JC. Membrane potential and intracellular Ca2+ oscillations activated by mGluRs in hippocampal stratum oriens/alveus interneurons. J Neurophysiol. 1999;81:371–382. doi: 10.1152/jn.1999.81.1.371. [DOI] [PubMed] [Google Scholar]
- Yao Y, Parker I. Potentiation of inositol trisphosphate-induced Ca2+ mobilization in Xenopus oocytes by cytosolic Ca2+ J Physiol. 1992;458:319–338. doi: 10.1113/jphysiol.1992.sp019420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Hashimoto K, Isomura Y, Shimohana S, Kato N. An IP3-assisted form of Ca2+-induced Ca2+ release in neocortical neurons. Neuroreport. 2000;11:535–539. doi: 10.1097/00001756-200002280-00022. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Hashimoto K, Isomura Y, Shimohana S, Kato N. A distinct form of calcium release down-regulates membrane excitability in neocortical pyramidal cells. Neuroscience. 2002;109:665–676. doi: 10.1016/s0306-4522(01)00486-9. [DOI] [PubMed] [Google Scholar]
- Zhou S, Ross WN. Threshold conditions for synaptically evoking Ca2+ waves in hippocampal pyramidal neurons. J Neurophysiol. 2002;87:1799–1804. doi: 10.1152/jn.00601.2001. [DOI] [PubMed] [Google Scholar]
- Zhu JJ. Maturation of layer 5 neocortical pyramidal neurons: amplifying salient layer 1 and layer 4 inputs by Ca2+ action potentials in adult rat tuft dendrites. J Physiol. 2000;526:571–587. doi: 10.1111/j.1469-7793.2000.00571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]