

Abstract
To investigate the role of nitric oxide (NO) in fetal cerebral circulatory responses to acute hypoxia, near-term fetal sheep were instrumented with laser Doppler probes placed in the parasagittal parietal cortices and vascular catheters in the sagittal sinus and brachiocephalic artery. After a 3 day recovery period, responses of cortical blood flow (CBF) to hypoxia were compared with and without inhibition of nitric oxide synthase (NOS). After an initial 30 min baseline period, fetuses were given a bolus followed by a continuous infusion of Nω-nitro-l-arginine methyl ester (l-NAME), or saline vehicle as control. After administration of l-NAME, CBF decreased by 14 ± 6 % (P < 0.01) despite increases in arterial blood pressure of 15 mmHg, resulting in an ∼60 % increase in cerebrovascular resistance. Thirty minutes following initiation of l-NAME or vehicle infusion, fetal systemic hypoxia was induced by allowing the ewes to breathe 10–11 % oxygen. In control fetuses CBF increased progressively to 145 ± 9 % of baseline (P < 0.01) after 30 min, while cortical release of cyclic guanylate cyclase (cGMP), an index of NOS activity, increased 26 ± 8 % (P < 0.05). In contrast, CBF in l-NAME-treated fetuses increased to only 115 % of the reduced CBF baseline, whereas cortical release of cGMP did not change significantly. In summary, basal levels of NO lower resting cortical vascular resistance by ∼15 % in the fetal sheep. Inhibition of NO synthesis attenuates hypoxic cerebral relaxation but does not completely prevent the characteristic increases in CBF. Hypoxic increases in NO directly increase cortical production of cGMP and inhibition of NO synthesis ablates these changes in cGMP.
The mammalian fetus has several mechanisms that serve to protect the organs most vulnerable to hypoxia. During hypoxia, arterial blood pressure increases and the resistance to blood flow increases in the periphery. At the same time the resistance to blood flow decreases in the cerebral, myocardial and adrenal beds, and cardiac output is redistributed to these organs (Jensen et al. 1999). These responses appear adequate to maintain oxygen delivery to the brain, heart and adrenals at the expense of peripheral organs during moderate hypoxia (Jones et al. 1977).
In the adult, there is strong evidence suggesting that nitric oxide (NO) produced by both endothelial and neuronal nitric oxide synthase (NOS) is involved in the regulation of the cerebral circulation (Faraci, 1991; Watkins, 1995; Hudetz et al. 1998). It clearly is an important mediator of cerebral vasodilatation in response to hypercapnia (Iadecola, 1992; Pelligrino et al. 1993) and may influence but not be a necessary component of the mechanisms involved in hypoxic responses (Pelligrino et al. 1993; McPherson et al. 1994). NO has also been shown to have an important role in the regulation of blood pressure, peripheral resistance, placental perfusion and maintenance of cardiac function in the fetus (Green et al. 1996; Smolich, 1998; Gardner et al. 2002). NO has a short half-life and is produced widely throughout the cerebral vascular bed. In addition, functional NOS is present in fetal cerebral tissue as early as 0.4 gestation (Northington et al. 1997).
A number of investigators have sought to determine the role of NO in the control of cerebral blood flow in the ovine fetus (van Bel et al. 1995; Green et al. 1996; McCrabb & Harding, 1996; Northington et al. 1997; Harris et al. 2001). These studies have typically used the microsphere method to measure cerebral blood flow, a technique that is limited by the small number of spot measurements that are possible. The majority of these studies have provided evidence for a significant contribution of NO synthesis in the maintenance of basal cerebrovascular tone. However, the role of NO in hypoxic responses has been less clear. A recent study by Harris et al. for example, did not find an appreciable role for NO synthesis in cerebrovascular modulation during hypoxia (Harris et al. 2001), which was in contrast to work by others showing NO as a major mediator of cerebrovascular relaxation during hypoxia (Green et al. 1996).
The present study was undertaken to test the hypothesis that NO modulates baseline cortical blood flow (CBF) and contributes to the increase in CBF that occurs during hypoxia in the late-term ovine fetus. To that end, the experimental design was structured to directly and continuously record CBF responses using laser Doppler flowmetry (LDF), to measure cortical production of cyclic guanylate cyclase (cGMP) as an index of responses activated by NO, and to estimate what fraction of total responses to hypoxia can be attributed to NO. Production of cGMP by the fetal brain in response to hypoxia is also reported for the first time as a direct indicator of NO release.
METHODS
Surgical and postoperative procedures
Animal protocols were approved by the Institutional Animal Research Committee of Loma Linda University. Ten Western ewes and their fetuses were obtained from Nebeker Ranch (Lancaster, CA, USA), and instrumented at 131–136 days of gestation (term ∼147 days). Following induction of anaesthesia with thiopental sodium (10 mg kg−1i.v.), the ewe was placed in the supine position and intubated. Anaesthesia was maintained throughout the surgical procedure with inhalation of 1.5–2.5 % halothane in oxygen. All surgical procedures were carried out under aseptic conditions.
Under aseptic conditions, the uterus was opened and the fetal head was delivered. Two burr holes were drilled bilaterally in the fetal skull 5 mm lateral to the midline and 5 mm posterior to the coronal suture. Laser Doppler probes were placed as described previously 5 mm below the dura in the parasagittal parietal cortex (Lan et al. 2000). A polyvinyl catheter was next placed in the sagittal sinus approximately midway between the points where the coronal and lamboid sutures intersect the sagittal suture.
Catheters were placed in the fetal brachiocephalic artery and vein for blood sampling, pressure measurement and administration of drugs. A catheter was also placed in the amniotic fluid for administration of antibiotics in the postoperative period and measurement of amniotic pressure during the experiments. Leads of the instrumentation and catheter ports were passed through a maternal flank incision for storage in a nylon pouch.
Postoperatively, the ewe was given 900 000 u penicillin intramuscularly for 3 days, and 500 mg ampicillin and 40 mg gentamicin were instilled daily in the amniotic fluid until the experiments were completed. During the postoperative period (0–24 h) the ewes were monitored for signs of discomfort such as restlessness, teeth grinding, abnormal posture or respiratory rate and changes in mental alertness. Any animal that displayed these signs was treated with buprenorphine hydrochloride (Buprenex, Reckitt & Colman Products Ltd, Hull, UK, 0.005 mg kg−1i.m. every 4–6 h). Typically ewes resumed normal feeding 4–10 h following surgery. Fetal arterial blood gases were monitored daily during postoperative recovery.
Experimental procedures
Experiments were carried out 3–7 days after surgery while ewes stood in a metabolic cart with a room temperature of 20–22 °C. Access to water and alfalfa pellets was provided throughout the study.
Control protocol
During an initial 60 min baseline period, fetal arterial and sagittal sinus blood samples were taken at 0, 15, 30, 40, 50 and 60 min (Fig. 1). Hypoxia was induced by having the ewe breathe 10–12 % O2 in nitrogen. This was administered by passing a 50: 50 air: nitrogen gas mixture at 30 l min−1 through a transparent bag placed over the ewe's head during which fetal blood samples were taken after 5, 10, 20 and 30 min. The ewes were returned to breathing air and fetal blood samples were taken after 10, 20, 30, 45 and 60 min of recovery. An infusion of saline was given beginning 30 min before hypoxia and ending 30 min after as a vehicle control.
Figure 1. Protocol.

Time sequence of vehicle or l-NAME infusion, hypoxia and recovery stimulated by l-arginine is shown.
Nitric oxide inhibition protocol
The protocol was similar in this group of fetuses except that a bolus (45 mg kg−1 estimated fetal weight) and continuing infusion (1.1 mg kg−1 min−1) of l-NAME was given to inhibit NO synthesis. The infusion was begun 30 min before hypoxia and ended 30 min after termination of hypoxia (Fig. 1). To conclude the experiment in this group a bolus (30 mg kg−1) and continuing infusion of l-arginine (3.3 mg kg−1 min−1) were given, beginning after a 30 min recovery period.
Laser Doppler flowmetry
Measurement of local CBF was performed with a Biopac LDF100A laser Doppler flowmeter (Biopac Systems, Inc., Santa Barbara, CA, USA), as described previously (Lan et al. 2000). The LDF probes were calibrated with an aseptically prepared, colloidal solution of suspended latex spheres undergoing Brownian motion. The laser Doppler signal provides an index of relative blood perfusion on a continuous basis that correlates satisfactorily with microsphere measurements of flow in that region of the cortex in both the ovine fetus and adult animals (Skarphedinsson et al. 1988; Haberl et al. 1989; Kirkeby et al. 1995; Muller et al. 2002; Bishai et al. 2002).
After completion of the experiment, the fetus, and subsequently the ewe, were humanely killed with a proprietary killing solution (T-61, Hoechst-Roussel). The fetal body was towel-dried and weighed to the nearest 10 g. The location of the LDF probes and all catheters was verified.
Blood sampling
Fetal arterial blood samples (0.3 ml) were analysed for blood gases using microelectrodes (ABL3, Radiometer, Copenhagen) at 39 °C. The observed values were corrected to the body temperature of the fetus. Oxyhaemoglobin saturation and haemoglobin concentration were measured using an OSM2 Hemoximeter (Radiometer).
Determination of plasma cGMP
Cortical cGMP release was calculated as the difference between sagittal sinus and arterial cGMP concentrations multiplied by LDF flow at the time of blood sample collection. Blood samples (1–2 ml) were collected in chilled vacutainer tubes containing EDTA (7.5 mm). Tubes were centrifuged at 4000 g for 15 min. The plasma was removed and stored at −70 °C. At the time of assay, samples were defrosted at 4 °C. A 200 μl volume of plasma was diluted 1: 5 with 6 % trichloroacetic acid, vortexed and centrifuged at 4000 g for 15 min. The resulting supernatant was washed with water-saturated diethyl ether for a minimum of three times to remove trichloroacetic acid. Any remaining ether was allowed to evaporate, and aliquots of the aqueous phase were then lyophilized. Samples were reconstituted with 200 μl of 50 mm acetate buffer and were acetylated. The cGMP content of each sample was determined using a commercially available radioimmunoassay kit (RPA 525 Amersham Corp., IL, USA) as previously described (Pearce et al. 1994).
Data handling
LDF signals, fetal arterial blood pressure, sagittal sinus pressure, heart rate and fetal core temperature were recorded continuously using the data acquisition software Acqknowledge (Biopac Systems, Inc., Santa Barbara, CA, USA). Averages were calculated over 3 min time spans. Because LDF provides only a relative measure of blood flow, data are presented as percentage change from initial control values. Cortical vascular resistance (CVR) was calculated by subtracting sagittal sinus from arterial blood pressure and dividing the result by CBF. Vascular resistance was also expressed as a percentage change from baseline.
Statistical analysis
Results are expressed as means ±s.e.m. Differences in response to l-NAME inhibition were assessed using two-way ANOVA with time as a repeated measure. When an effect of time was found its significance was assessed using Fisher's least significant difference for multiple comparisons test. Significant differences were accepted at the 0.05 level. The statistical software used was DATAMSTR (courtesy of R. A. Brace, UC San Diego, USA).
RESULTS
A total of 16 experiments were carried out on 10 sheep. Six sheep were studied under the control protocol and then, after a 3 day recovery period, they were studied in the experimental protocol. Two sheep were studied only as controls and the remaining two sheep were studied under the experimental protocol alone.
CBF and vascular resistance
Figure 2 (top panel) shows the changes in CBF throughout the course of the experiment and permits a comparison of responses in those fetuses receiving l-NAME as opposed to the saline vehicle. Infusion of saline did not alter CBF, but in response to a 30 min period of hypoxia CBF increased progressively to 145 ± 10 % of baseline (P < 0.05). In those fetuses receiving l-NAME, CBF decreased to 86 ± 4 % of baseline (P < 0.01) in the initial period of observation after drug administration. Upon initiation of hypoxia CBF then increased from this reduced level to reach 0103 ± 5 % of the initial baseline, or 115 % as calculated from the new baseline established by l-NAME infusion.
Figure 2. Comparison of CBF (top panel) and cortical vascular resistance (bottom panel) responses in untreated and NOS-inhibited late-gestation fetal sheep.
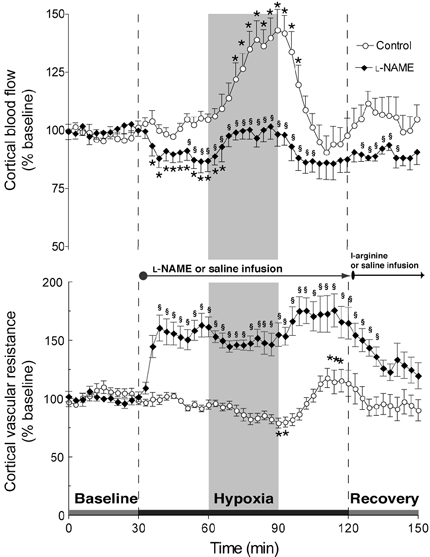
Results (means ±s.e.m.) are shown for eight control and eight l-NAME-treated fetuses. * Significantly different from initial baseline; § significantly different between groups. The rise in CBF in NOS-inhibited fetuses from the lowered baseline after l-NAME infusion was not significantly different.
Changes in CVR are shown in the bottom panel of Fig. 2. CVR of untreated fetuses decreased to 80 ± 4 % of baseline during hypoxia (P < 0.01). CVR in the l-NAME group increased after the infusion of the drug to reach 163 ± 9 % of baseline values (P < 0.01) and then subsequently declined to 144 ± 9 % of baseline after initiation of hypoxia. Thus hypoxia led to ∼20 % reduction in CVR in untreated fetuses in contrast to ∼12 % reductions in those in which NO synthesis was blocked.
Mean arterial blood pressure and heart rate
Figure 3 shows mean arterial blood pressure (MABP) and heart rate responses in untreated and l-NAME-treated fetuses. In untreated fetuses, MABP gradually rose ∼5 mmHg following induction of hypoxia (P < 0.05) and then returned to baseline values during recovery from hypoxia. In the treatment group, infusion of l-NAME caused a rapid and significant increase in MABP from 42 ± 4 mmHg during the baseline period to 58 ± 5 mmHg (P < 0.01). Then during hypoxia, MABP rose further and progressively to reach 64 ± 6 mmHg (not statistically significant). This elevated blood pressure was maintained until the termination of the l-NAME infusion and infusion of l-arginine 30 min following hypoxia.
Figure 3. Changes in mean arterial blood pressure (top panel) and heart rate (bottom panel).

Results (means ±s.e.m.) are shown for eight control and eight l-NAME-treated fetuses. * Significantly different from initial baseline; § significantly different between groups.
Heart rate decreased in control fetuses after initiation of hypoxia, and returned to baseline values by the end of the hypoxic period (Fig. 3). In the treatment group, infusion of l-NAME caused a rapid and significant decrease in heart rate from 164 ± 4 beats min−1 during the baseline period to 125 ± 8 beats min−1 (P < 0.01). After initiation of hypoxia, heart rate decreased further to ∼115 beats min−1 and then gradually returned to ∼130 beats min−1 by the end of the hypoxic period. After termination of hypoxia, heart rate returned to the level prior to infusion of l-NAME.
cGMP responses
Arterial cGMP concentration was 92.1 ± 6.2 fmol ml−1 in control animals and 103.0 ± 8.2 fmol ml−1 during the baseline period. Changes in cGMP release from the brain in response to hypoxia in control and NOS-inhibited fetuses are shown in Fig. 4. In control fetuses, cortical cGMP release was unaffected by saline infusion, rose 26 ± 8 % above baseline (P < 0.05) during hypoxia and then returned to baseline values during the recovery period. In the experimental group, cortical cGMP release did not change following NOS inhibition. In contrast to controls, cGMP release did not increase during hypoxia and was significantly less than controls during hypoxia. Cortical cGMP release during recovery in the l-NAME group was not significantly different from baseline or the control group.
Figure 4. Changes in cortical production of cGMP.
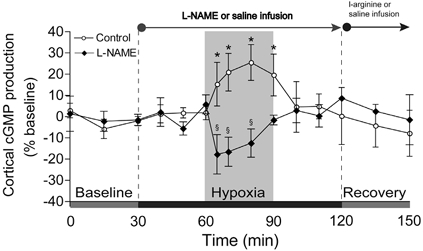
Results (means ±s.e.m.) are shown for five control and five l-NAME-treated fetuses. * Significantly different from initial baseline; § significantly different between groups.
Fetal blood gases, pH, oxyhaemoglobin saturation, glucose and lactate values
Figure 5 shows changes in PO2, PCO2 and pH in the vehicle and l-NAME groups measured during the course of the experiment. Infusion of saline in control experiments and of l-NAME in the experimental group did not change blood gas values appreciably in the initial baseline period. After initiation of hypoxia fetal arterial PO2 fell from ∼21 to ∼11 mmHg in controls and from ∼20 to ∼11 mmHg in l-NAME-treated fetuses; the responses in both groups were similar. Arterial PCO2 decreased to a somewhat greater extent in control fetuses than in l-NAME-treated fetuses. Arterial pH declined progressively during hypoxia and the acidosis was more pronounced with NOS blockade. Blood gases values returned to baseline levels during recovery from hypoxia irrespective of treatment with l-NAME.
Figure 5. Changes in blood gases and pH.
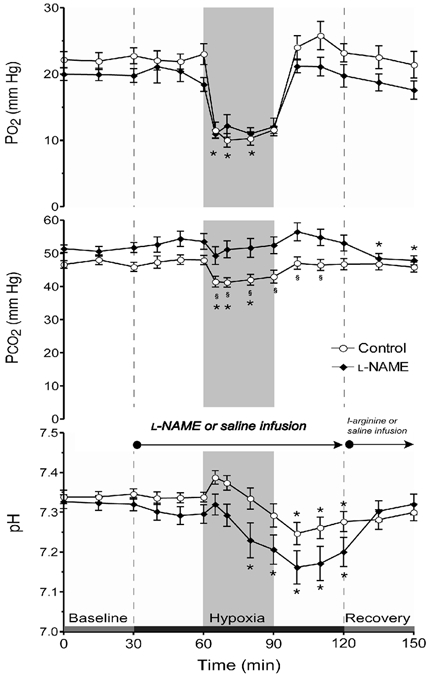
Results (means ±s.e.m.) are shown for eight control and eight l-NAME-treated fetuses. * Significantly different from initial baseline; § significantly different between groups.
Figure 6 shows responses of plasma glucose and lactate to l-NAME or saline infusion and hypoxia. Blood glucose became elevated to a comparable extent in both groups during hypoxia and then returned towards baseline during recovery. Hyperlactaemia developed in both groups and the increase of plasma lactate was measurably greater and more persisting in those receiving NOS blockade.
Figure 6. Changes in glucose and lactate.
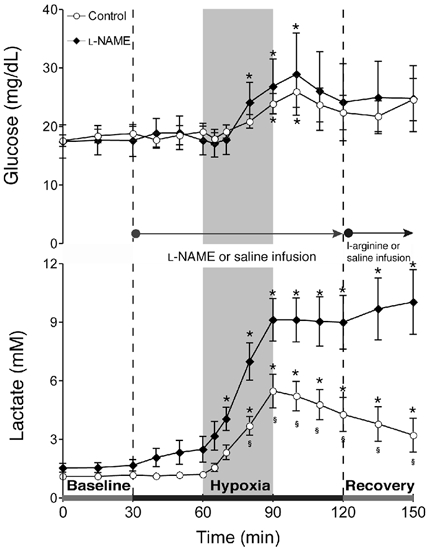
Results (means ±s.e.m.) are shown for eight control and eight l-NAME-treated fetuses. * Significantly different from initial baseline; § significantly different between groups.
Discussion
In agreement with a number of earlier studies this investigation has shown that NO helps maintain a state of vasodilatation in cortical resistance vessels under normoxic conditions. Thus, when the synthesis of NO was inhibited by l-NAME, the resistance of the cerebrovasculature increased indicating a direct action of NO on cortical resistance vessels under baseline conditions. The second principal finding is that the action of NO contributes to the increase of CBF evoked by hypoxia. A causal and local role for NO is strongly supported by the finding that the release of cGMP from the brain increased in response to hypoxia.
After administration of l-NAME, blood pressure increased and heart rate declined. Induction of hypoxia during NOS inhibition further increased blood pressure indicating that the fetal vascular system remained responsive to pressor stimuli such as catecholamines known to increase in response to hypoxia (Jensen et al. 1999). In control fetuses cortical perfusion increased gradually but progressively during the first 15–20 min of exposure to hypoxia to reach an apparent plateau about 45 % above baseline after 30 min. Earlier work in our laboratory has shown little further change after this time (Bishai et al. 2002).
Comparison with other fetal animal studies
The majority of previous studies of the role of NO in regulation of the fetal cortical vasculature have supported a role for NO in control of resting tone in fetal cortical resistance vessels (van Bel et al. 1995; Green et al. 1996; Harris et al. 2001). There is disagreement, however, about the importance of NO in mediating increases of CBF during hypoxia. van Bel and coworkers studied the role of NO regulation of CBF in the 130-day-old ovine fetus during periods of normoxia and hypoxaemia using the radioactive microsphere method (van Bel et al. 1995). They found that (1) NO was of importance in maintaining basal CBF and (2) that NO was required but was probably not alone in being responsible for hypoxia-induced cortical vasodilatation. In a subsequent study Green and coworkers studied sheep fetuses of between 123 and 129 days gestation using carotid blood flow as an indicator of cerebral blood flow (Green et al. 1996). Their results largely corroborated the results of van Bel et al. (1995) and they concluded that the rise in carotid blood flow and fall in carotid vascular resistance seen during hypoxia were absent following inhibition of NO synthesis. McCrabb and coworkers found that inhibition of NO synthesis with the drug N-nitro-l-arginine had little effect on basal cerebral blood flow in late-term fetal lambs, and that inhibition of NO synthesis at the beginning of moderate hypoxia prevented CBF from increasing as much as in control animals (McCrabb & Harding, 1996). Ioroi et al. investigated the role of NO in the regulation of CBF in the striatum of 7- and 14-day-old rats and measured CBF using LDF and striatal NO concentration using a NO electrode (Ioroi et al. 1998). The normal increase in CBF during hypoxia was associated with an increase in striatal NO production in both age groups, and both CBF and NO production were attenuated by NOS inhibition. These investigators speculated that NO plays a major role in cerebral vasodilatation during hypoxia.
Recent work by Harris et al. provided conflicting results to the above studies (Harris et al. 2001). Using fetal sheep at both 0.6 and 0.9 gestation these authors used radioactive microspheres to measure blood flow distributions to the brain and other organs before and during hypoxia. A significant role for NO synthesis in regulation of basal CBF at both 0.6 and 0.9 gestational ages was observed. Inhibition of NO synthesis attenuated hypoxic increases in absolute CBF but did not reduce the percentage increase in CBF during hypoxia. These authors concluded that NOS inhibition decreases CBF during normoxia and hypoxia but does not mediate hypoxic cerebral vasodilatation in the fetal brain.
In contrast the results of the present study reinforce the view that NO plays an important role in the maintenance of basal CBF and contributes to hypoxic cerebral vasodilatation. In the present study we found that although hypoxia increased CBF from the lowered baseline produced by l-NAME administration, this response was much smaller than that found in control animals (Fig. 2). In addition the hypoxic increases in cortical cGMP production were completely eliminated by l-NAME, strongly suggesting a direct role for NO in hypoxic cortical relaxation. These data also suggest that basal cGMP levels are not highly sensitive to basal release of NO. This in vivo finding is in agreement with our previous in vitro work using isolated ovine arteries, wherein endothelial denudation had no significant effect on cGMP content (White & Pearce, 1996). Although the present results are different than the findings and interpretation of Harris et al. (2001) there are multiple reasons for this inconsistency, including major differences in the methods used to measure CBF, the timing of CBF measurements (one microsphere injection during control, l-NAME infusion and hypoxia period in the study of Harris et al.), and most importantly in dose and route of administration of l-name (single bolus in the study of Harris et al. vs. bolus followed by injection in the present study). Also, laser Doppler measurements of flow show greater reproducibility and less variability than the spot measurements possible with labelled spheres (Bishai et al. 2002). However, a limitation of this study is that while LDF has the important advantage of providing continuous measurement, it provides only relative changes in tissue perfusion within a relatively small volume of tissue. In this study flow is measured from a local region of grey matter in the parietal cortices; however, flow in white matter and other regions of the brain can vary appreciably during basal and hypoxic conditions. This limitation means that calculations of resistance and arteriovenous (a-v) production based on sagittal sinus sampling represent global aspects and may not be directly comparable to local cortical responses. However, we have previously shown that local flow responses in the sampled region of the parietal cortex are reasonably comparable to surrounding cortical areas, including contralateral regions, when examined with microspheres in tissue samples of several cubic millimetres volume (Bishai et al. 2002). This earlier study thus provides strong evidence that the responses measured here are a valid indicator of parietal flow in general.
Another important complication of most previous studies of the cerebral circulation in the fetus including the present one are the large changes in MABP and heart rate that occur after systemic blockade of NOS. These systemic changes alter cerebral perfusion pressure of the brain, cardiac output and placental perfusion, and thereby may indirectly influence cerebrovascular myogenic tone and blood flow without acting directly on cerebrovascular smooth muscle (Smolich, 1998; Gardner et al. 2002). While it is clear that that NO is not the only mediator of hypoxic fetal cerebral vasodilatation the data reinforce the view that NO is a key player.
Comparison with adult animal studies
Most earlier studies of the effects of NOS inhibition on hypoxic cerebral vasodilatation in adult animals have not reported any significant effects. In the adult rat, for example, Buchanan and coworkers found that NO does not play a major role in hypoxia-evoked cerebral vasodilatation (Buchanan & Phillis, 1993). Pelligrino et al. showed in adult rats that NO synthesis is important in the CBF responses to hypercapnia but not hypoxia (Pelligrino et al. 1993). However, recent studies have challenged this view. Reid and co-workers have suggested a role for NO in hypoxic cerebral vasodilatation using the adult rat (Reid et al. 1995), and Armstead showed that cerebrospinal fluid concentrations of cGMP increase during hypoxia (Armstead, 1997). In a later study Pelligrino et al. showed that NO contributes to cerebral vasodilatation during severe hypoxia and that this NO-dependent portion of the vascular response is dependant on activation of neuronal NMDA receptors (Pelligrino et al. 1995). Work by Hudetz et al. took these findings further by indicating that neuronal NOS is an important modulator of increases in cerebral capillary flow during hypoxia (Hudetz et al. 1998). In addition studies in adult humans indicate that baseline CBF is not affected by blockade of NO but the normal increase in CBF in response to hypoxia is completely abolished (Van Mil et al. 2002). The trend of recent studies indicates that NO plays a role in hypoxic cerebral relaxation but the importance of this role is dependant on the duration and intensity of hypoxia, the brain area studied, and the density of NMDA receptors in that region.
Mechanisms of hypoxic cerebrovascular relaxation
NO produces vascular relaxation by interacting with the haeme group of soluble guanylate cyclase (sGC), the enzyme that synthesizes the second messenger cGMP, which in turn promotes vasodilatation in many vascular beds (see Moncada & Higgs, 1993 for review). The tonic release of NO is governed by continuous activation of endothelial cells by stimuli such as pulsatile flow and sheer stress (Moncada & Higgs, 1993). These sources of NO directly stimulate cerebrovascular sGC which is highly abundant in cerebral arteries particularly in the fetus (Pearce et al. 1991; White et al. 2000). The resulting cGMP is an extremely potent vasodilator in cerebral arteries. Pearce et al. have shown that fetal cerebral arteries are more sensitive to the vasodilator effects of cGMP than adult arteries, whereas the sensitivity of cGMP production to nitric oxide is the same for both fetal and adult common carotid and basilar arteries (Pearce et al. 1994). Thus, the vasorelaxant capacity of the cGMP pathway appears to be attenuated by maturation (Pearce, 1995; Nauli et al. 2001).
Taken alone these data suggest a more important role for the NO-cGMP pathway in maintenance of cerebrovascular tone during the fetal period. The results of the present and previous studies (Blood et al. 2002) clearly indicate that multiple mediators such as adenosine also contribute to cerebral vasorelaxation during hypoxia and are likely to interact with NO.
Conclusions and perspective
In summary, basal levels of NO lower resting cortical vascular resistance ∼15 % in the fetal sheep. Inhibition of NO synthesis attenuates hypoxic cortical relaxation but does not completely prevent the characteristic increases in CBF. Hypoxic increases in NO directly increase cortical production of cGMP and inhibition of NO synthesis ablates these changes in cGMP. Thus NO contributes to hypoxic cortical vasodilatation through activation of the cGMP relaxation pathway. In comparison of the effects of hypoxia on the adult cerebral circulation, this role for NO appears to be particularly prominent in the fetus.
Hypoxia is a ubiquitous environmental stress throughout all biology. Accordingly, numerous mechanisms have evolved to maintain homeostasis during hypoxic challenges, given that fetal oxygen tensions are dramatically lower than those found in the adult it follows that the pathways activated by hypoxia would be quite different in the fetus and adult. The present studies demonstrate that hypoxic activation of NO synthesis is a mechanism that distinguishes fetal from adult responses to hypoxia. How low oxygen tension is coupled to activation of NO synthesis remains a fascinating but unresolved question. Similarly, the manner in which hypoxic increases in NO interact and integrate with other hypoxic mediators remains a virtually unstudied but promising area of investigation. The present study illustrates the value of continuous measurements of CBF in the chronically instrumented, unanaesthetized fetal preparations for future studies of this important topic.
Acknowledgments
The authors thank Shannon Bragg for expert technical assistance, John M. Bishai for experimental and analytical assistance, and Alistair Gunn for helpful critical suggestions and discussion. This work was supported in part by USPHS NIH (HL 654941 (G.G.P.) and HL 64867 (W.J.P)).
References
- Armstead WM. Role of nitric oxide, cyclic nucleotides, and the activation of ATP-sensitive K+ channels in the contribution of adenosine to hypoxia-induced pial artery dilation. J Cereb Blood Flow Metab. 1997;17:100–108. doi: 10.1097/00004647-199701000-00013. [DOI] [PubMed] [Google Scholar]
- Bishai JM, Blood AB, Hunter CJ, Longo LD, Power GG. Fetal lamb cerebral blood flow (CBF) and oxygen tensions during hypoxia: a comparison of laser Doppler and microsphere measurements of CBF. J Physiol. 2002;546:869–878. doi: 10.1113/jphysiol.2002.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blood AB, Hunter CJ, Power GG. The role of adenosine in regulation of cerebral blood flow during hypoxia in the near-term fetal sheep. J Physiol. 2002;543:1015–1023. doi: 10.1113/jphysiol.2002.023077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan JE, Phillis JW. The role of nitric oxide in the regulation of cerebral blood flow. Brain Res. 1993;610:248–255. doi: 10.1016/0006-8993(93)91408-k. [DOI] [PubMed] [Google Scholar]
- Faraci FM. Role of endothelium-derived relaxing factor in cerebral circulation: large arteries vs. microcirculation. Am J Physiol. 1991;261:H1038–1042. doi: 10.1152/ajpheart.1991.261.4.H1038. [DOI] [PubMed] [Google Scholar]
- Gardner DS, Fowden AL, Giussani DA. Adverse intrauterine conditions diminish the fetal defense against acute hypoxia by increasing nitric oxide activity. Circulation. 2002;106:2278–2283. doi: 10.1161/01.cir.0000033827.48974.c8. [DOI] [PubMed] [Google Scholar]
- Green LR, Bennet L, Hanson MA. The role of nitric oxide synthesis in cardiovascular responses to acute hypoxia in the late gestation sheep fetus. J Physiol. 1996;497:271–277. doi: 10.1113/jphysiol.1996.sp021766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberl RL, Heizer ML, Ellis EF. Laser-Doppler assessment of brain microcirculation: effect of local alterations. Am J Physiol. 1989;256:H1255–1260. doi: 10.1152/ajpheart.1989.256.4.H1255. [DOI] [PubMed] [Google Scholar]
- Harris AP, Helou S, Gleason CA, Traystman RJ, Koehler RC. Fetal cerebral and peripheral circulatory responses to hypoxia after nitric oxide synthase inhibition. Am J Physiol Regul Integr Comp Physiol. 2001;281:R381–390. doi: 10.1152/ajpregu.2001.281.2.R381. [DOI] [PubMed] [Google Scholar]
- Hudetz AG, Shen H, Kampine JP. Nitric oxide from neuronal NOS plays critical role in cerebral capillary flow response to hypoxia. Am J Physiol. 1998;274:H982–989. doi: 10.1152/ajpheart.1998.274.3.H982. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Does nitric oxide mediate the increases in cerebral blood flow elicited by hypercapnia? Proc Natl Acad Sci U S A. 1992;89:3913–3916. doi: 10.1073/pnas.89.9.3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioroi T, Yonetani M, Nakamura H. Effects of hypoxia and reoxygenation on nitric oxide production and cerebral blood flow in developing rat striatum. Pediatr Res. (1998);43:733–737. doi: 10.1203/00006450-199806000-00004. [DOI] [PubMed] [Google Scholar]
- Jensen A, Garnier Y, Berger R. Dynamics of fetal circulatory responses to hypoxia and asphyxia. Eur J Obstet Gynecol Reprod Biol. 1999;84:155–172. doi: 10.1016/s0301-2115(98)00325-x. [DOI] [PubMed] [Google Scholar]
- Jones M, Sheldon RE, Peeters LL, Meschia G, Battaglia FC, Makowski EL. Fetal cerebral oxygen consumption at different levels of oxygenation. J Appl Physiol. 1977;43:1080–1084. doi: 10.1152/jappl.1977.43.6.1080. [DOI] [PubMed] [Google Scholar]
- Kirkeby OJ, Rise IR, Nordsletten L, Skjeldal S, Hall C, Risoe C. Cerebral blood flow measured with intracerebral laser-Dopplerflow probes and radioactive microspheres. J Appl Physiol. 1995;79:1479–1486. doi: 10.1152/jappl.1995.79.5.1479. [DOI] [PubMed] [Google Scholar]
- Lan J, Hunter CJ, Murata T, Power GG. Adaptation of laser-Doppler flowmetry to measure cerebral blood flow in the fetal sheep. J Appl Physiol. 2000;89:1065–1071. doi: 10.1152/jappl.2000.89.3.1065. [DOI] [PubMed] [Google Scholar]
- McCrabb GJ, Harding R. Role of nitric oxide in the regulation of cerebral blood flow in the ovine foetus. Clin Exp Pharmacol Physiol. 1996;23:855–860. doi: 10.1111/j.1440-1681.1996.tb01133.x. [DOI] [PubMed] [Google Scholar]
- McPherson RW, Koehler RC, Traystman RJ. Hypoxia, alpha 2-adrenergic, and nitric oxide-dependent interactions on canine cerebral blood flow. Am J Physiol. 1994;266:H476–482. doi: 10.1152/ajpheart.1994.266.2.H476. [DOI] [PubMed] [Google Scholar]
- Moncada S, Higgs A. The l-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- Muller T, Lohle M, Schubert H, Bauer R, Wicher C, Antonow-Schlorke I, Sliwka U, Nathanielsz PW, Schwab M. Developmental changes in cerebral autoregulatory capacity in the fetal sheep parietal cortex. J Physiol. 2002;539:957–967. doi: 10.1113/jphysiol.2001.012590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauli SM, Zhang L, Pearce WJ. Maturation depresses cGMP-mediated decreases in [Ca2+]i and Ca2+ sensitivity in ovine cranial arteries. Am J Physiol Heart Circ Physiol. 2001;280:H1019–1028. doi: 10.1152/ajpheart.2001.280.3.H1019. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Tobin JR, Harris AP, Traystman RJ, Koehler RC. Developmental and regional differences in nitric oxide synthase activity and blood flow in the sheep brain. J Cereb Blood Flow Metab. 1997;17:109–115. doi: 10.1097/00004647-199701000-00014. [DOI] [PubMed] [Google Scholar]
- Pearce WJ. Mechanisms of hypoxic cerebral vasodilatation. Pharmacol Ther. 1995;65:75–91. doi: 10.1016/0163-7258(94)00058-b. [DOI] [PubMed] [Google Scholar]
- Pearce WJ, Hull AD, Long DM, Longo LD. Developmental changes in ovine cerebral artery composition and reactivity. Am J Physiol. 1991;261:R458–465. doi: 10.1152/ajpregu.1991.261.2.R458. [DOI] [PubMed] [Google Scholar]
- Pearce WJ, Hull AD, Long DM, White CR. Effects of maturation on cyclic GMP-dependent vasodilation in ovine basilar and carotid arteries. Pediatr Res. 1994;36:25–33. doi: 10.1203/00006450-199407001-00005. [DOI] [PubMed] [Google Scholar]
- Pelligrino DA, Koenig HM, Albrecht RF. Nitric oxide synthesis and regional cerebral blood flow responses to hypercapnia and hypoxia in the rat. J Cereb Blood Flow Metab. 1993;13:80–87. doi: 10.1038/jcbfm.1993.10. [DOI] [PubMed] [Google Scholar]
- Pelligrino DA, Wang Q, Koenig HM, Albrecht RF. Role of nitric oxide, adenosine, N-methyl-d-aspartate receptors, and neuronal activation in hypoxia-induced pial arteriolar dilation in rats. Brain Res. 1995;704:61–70. doi: 10.1016/0006-8993(95)01105-6. [DOI] [PubMed] [Google Scholar]
- Reid JM, Davies AG, Ashcroft FM, Paterson DJ. Effect of l-NMMA, cromakalim, and glibenclamide on cerebral blood flow in hypercapnia and hypoxia. Am J Physiol. 1995;269:H916–922. doi: 10.1152/ajpheart.1995.269.3.H916. [DOI] [PubMed] [Google Scholar]
- Skarphedinsson JO, Harding H, Thoren P. Repeated measurements of cerebral blood flow in rats. Comparisons between the hydrogen clearance method and laser Doppler flowmetry. Acta Physiol Scand. 1988;134:133–142. doi: 10.1111/j.1748-1716.1988.tb08469.x. [DOI] [PubMed] [Google Scholar]
- Smolich JJ. NO modulates fetoplacental blood flow distribution and whole body oxygen extraction in fetal sheep. Am J Physiol. 1998;274:R1331–1337. doi: 10.1152/ajpregu.1998.274.5.R1331. [DOI] [PubMed] [Google Scholar]
- van Bel F, Sola A, Roman C, Rudolph AM. Role of nitric oxide in the regulation of the cerebral circulation in the lamb fetus during normoxemia and hypoxemia. Biol Neonate. 1995;68:200–210. doi: 10.1159/000244238. [DOI] [PubMed] [Google Scholar]
- Van Mil AH, Spilt A, Van Buchem MA, Bollen EL, Teppema L, Westendorp RG, Blauw GJ. Nitric oxide mediates hypoxia-induced cerebral vasodilation in humans. J Appl Physiol. 2002;92:962–966. doi: 10.1152/japplphysiol.00616.2001. [DOI] [PubMed] [Google Scholar]
- Watkins LD. Nitric oxide and cerebral blood flow: an update. Cerebrovasc Brain Metab Rev. 1995;7:324–337. [PubMed] [Google Scholar]
- White CR, Hao X, Pearce WJ. Maturational differences in soluble guanylate cyclase activity in ovine carotid and cerebral arteries. Pediatr Res. 2000;47:369–375. doi: 10.1203/00006450-200003000-00014. [DOI] [PubMed] [Google Scholar]
- White CR, Pearce WJ. Effects of maturation on cyclic GMP metabolism in ovine carotid arteries. Pediatr Res. 1996;39:25–31. doi: 10.1203/00006450-199601000-00004. [DOI] [PubMed] [Google Scholar]