

Abstract
The genesis of the ischaemia intolerant phenotype in aged myocardium is poorly understood. We tested the hypothesis that impaired adenosine-mediated protection contributes to ischaemic intolerance, and examined whether this is countered by A1 adenosine receptor (A1AR) overexpression. Responses to 20 min ischaemia and 45 min reperfusion were assessed in perfused hearts from young (2–4 months) and moderately aged (16–18 months) mice. Post-ischaemic contractility was impaired by ageing with elevated ventricular diastolic (32 ± 2 vs. 18 ± 2 mmHg in young) and reduced developed (37 ± 3 vs. 83 ± 6 mmHg in young) pressures. Lactate dehydrogenase (LDH) loss was exaggerated (27 ± 2 vs. 16 ± 2 IU g−1in young) whereas the incidence of tachyarrhythmias was similar in young (15 ± 1 %) and aged hearts (16 ± 1 %). Functional analysis confirmed equipotent effects of 50 μm adenosine at A1 and A2 receptors in young and aged hearts. Nonetheless, while 50 μm adenosine improved diastolic (5 ± 1 mmHg) and developed pressures (134 ± 7 mmHg) and LDH loss (6 ± 2 IU g−1) in young hearts, it did not alter these variables in the aged group. Adenosine did attenuate arrhythmogenesis for both ages (to ∼10 %). In contrast to adenosine, 50 μm diazoxide reduced ischaemic damage and arrhythmogenesis for both ages. Contractile and anti-necrotic effects of adenosine were limited by 100 μm 5-hydroxydecanoate (5-HD) and 3 μm chelerythrine. Anti-arrhythmic effects were limited by 5-HD but not chelerythrine. Non-selective (100 μm 8-sulfophenyltheophylline) and A1-selective (150 nm 8-cyclopentyl-1,3-dipropylxanthine) adenosine receptor antagonism impaired ischaemic tolerance in young but not aged hearts. Quantitative real-time PCR and radioligand analysis indicated that impaired protection is unrelated to changes in A1AR mRNA transcription, or receptor density (∼8 fmol mg−1 protein in both age groups). However, A1AR overexpression improved tolerance for both ages, restoring adenosine-mediated protection. These data reveal impaired protection via exogenous and endogenous adenosine contributes to ischaemic intolerance with ageing. This is independent of A1AR expression, and involves ineffective activation of a 5-HD-/diazoxide-sensitive process. The effects of A1AR overexpression indicate that the age-related failure in signalling can be overcome.
There is increasing evidence of a decline in myocardial tolerance to injury with ageing. A reduction in the ‘intrinsic’ tolerance to ischaemic insult is supported by data from animal models (Pahor et al. 1985; Frolkis et al. 1991; Misare et al. 1992; Lesnefsky et al. 1994; Tani et al. 1997; Headrick, 1998; Abete et al. 1999; Rosenfeldt et al. 2002) and humans (Mariani et al. 2000; Rosenfeldt et al. 2002). The molecular basis of this intolerant phenotype is unclear, but it may involve multiple alterations including mitochondrial abnormalities (Lesnefsky et al. 2001), impaired anti-oxidant responses (Boucher et al. 1998; Coombes et al. 2000; Lesnefsky et al. 2001), loss of proteasome function (Bulteau et al. 2002) and modified Ca2+ handling (Cain et al. 1998). One possibility receiving increased attention is impairment of intrinsic cardioprotective responses (Abete et al. 1996; Gao et al. 2000; Schulman et al. 2001; Lee et al. 2002). This may be a particularly important factor since it may impact directly on the therapeutic approach to ischaemic injury in aged hearts. It is increasingly evident that conventional therapeutic strategies developed through findings in young tissues and subjects may not be relevant in aged subjects (Rosenfeldt et al. 2002).
Adenosine is an important determinant of ischaemic (Zhao et al. 1993, 1994; Peart & Headrick, 2000) or hypoxic tolerance (Matherne et al. 1996). We previously acquired evidence that altered adenosine handling might contribute to impaired ischaemic tolerance with age (Headrick, 1998), and more recent evidence supports an age-related decline in adenosine-mediated protection (Gao et al. 2000). However, this latter study failed to establish whether functionally equipotent levels of adenosine were effective in different ages. The aims of the current study were to characterise age-related changes in ischaemic tolerance in mouse heart, to examine the efficacy of exogenous and endogenous adenosine in protecting against ischaemic injury, and to test the hypothesis that enhanced expression of protective A1 adenosine receptors (A1ARs) will reverse the detrimental effects of ageing on ischaemic tolerance.
METHODS
Perfused mouse heart model
Investigations conformed with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85–23, revised 1996). Hearts were acquired from young (2–4 months, 23.3 ± 2.4 g body weight) and aged (16–18 months, 38.1 ± 2.0 g body weight) male and female wild-type C57/Bl6 mice, and mice overexpressing cardiac A1ARs. Details of the generation and characterisation of transgenic mice have previously been provided (Matherne et al. 1997; Gauthier et al. 1998). Mice were anaesthetised with 50 mg kg−1 sodium pentobarbitone, a thoracotomy was performed and hearts were rapidly excised into ice-cold perfusion fluid. The aorta was immediately cannulated and perfused at a pressure of 80 mmHg with modified Krebs bicarbonate buffer containing (mm): NaCl, 118; NaHCO3, 25; KCl, 4.7; KH2PO4, 1.2; CaCl2, 2.5; MgSO4, 1.2; glucose, 11; and EDTA, 0.6. Perfusate was equilibrated with 95 % O2, 5 % CO2 at 37 °C to give a pH of 7.4 and a PO2 of > 550 mmHg at the aortic cannula. The perfusate was passed through an in-line 0.45 μm Sterivex-HV filter cartridge (Millipore, Bedford, MA, USA). The left ventricle was vented with an apical drain and hearts were instrumented for functional assessment. Hearts were placed in a water-jacketed chamber continuously superfused with warmed buffer maintained at 37 °C.
Contractile function was assessed via an intra-ventricular balloon, as described previously (Peart & Headrick, 2000; Headrick et al. 2001). Coronary flow was monitored using an ultrasonic flow-probe in the aortic perfusion line connected to a T106 flowmeter (Transonic Systems Inc, Ithaca, NY, USA). Functional data were recorded at 1 kHz on a 4-channel MacLab (ADInstruments, Castle Hill, Australia). The ventricular pressure signal was digitally processed to give systolic and diastolic pressures, +dP/dt (reflecting inotropic state), –dP/dt (reflecting lusitropic state) and heart rate (Peart & Headrick, 2000; Headrick et al. 2001).
Experimental protocol
After 20 min stabilisation hearts were switched to pacing at 400 beats min−1 (silver electrodes, pacing 20 % above threshold, 1 ms square pulses; Peart & Headrick, 2000; Headrick et al. 2001) and permitted to stabilise for a further 10 min. Hearts were excluded if they met one of the following criteria: (i) left ventricular systolic pressure below 100 mmHg; (ii) coronary flow equal to or exceeding 5 ml min−1 (maximal dilatation or aortic tear); (iii) unstable (fluctuating) contractile function; or (iv) significant arrhythmias. This amounted to less than 3 % of hearts perfused. Baseline measurements were made and hearts were subjected to 20 min global normothermic ischaemia and 45 min aerobic reperfusion. Pacing was stopped on induction of ischaemia and resumed after 2 min reperfusion (Peart & Headrick, 2000; Headrick et al. 2001). Functional parameters were assessed throughout, and coronary venous effluent collected on ice for analysis of LDH efflux using a commercially available kit (Sigma Chemical Co., St Louis, MO, USA; Headrick et al. 2001). Efflux is expressed as units per gram wet weight. The degree of ectopy (tachyarrhythmias) during the initial 10 min reperfusion was calculated as the sum of premature and tachycardic beats divided by the total number of beats during this period, as described previously (Headrick, 1998):
The protocol was performed in young (n = 11) and aged wild-type hearts (n = 10) and young (n = 9) and aged transgenic hearts (n = 10) overexpressing A1ARs. Adenosine-mediated cardioprotection was studied after first verifying that 50 μm adenosine was functionally equipotent in young and aged hearts. Adenosine was infused incrementally at concentrations of 1, 50 and 100 μm in young (n = 8) and aged (n = 6) hearts, and A1AR-mediated bradycardia and A2 adenosine receptor-mediated dilatation measured at each concentration. For adenosine-mediated cardioprotection, 50 μm adenosine was infused 10 min prior to ischaemia and re-instated upon reperfusion. The effects of adenosine were measured in young (n = 9) and aged wild-type hearts (n = 10) and young (n = 9) and aged transgenic hearts (n = 9).
To identify the role of endogenously released adenosine at A1ARs and other receptors, we studied the effects of treatment with 150 nm of the selective and potent A1AR antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) or 100 μm of the non-selective antagonist 8-ρ-sulfophenyltheophylline (8-SPT) in ischaemic- reperfused hearts from young wild-type (n = 8 for DPCPX, n = 9 for 8-SPT) and aged wild-type mice (n = 9 for DPCPX, n = 8 for 8-SPT). Infusion of antagonist was initiated 10 min prior to ischaemia and was re-instated upon reperfusion since we have shown protective effects of adenosine pre- and post-ischaemia (Peart & Headrick, 2000).
The protective effects of the putative mito KATP channel activator diazoxide (50 μm) were assessed in young (n = 9) and aged wild-type (n = 6) hearts. Diazoxide was infused as for adenosine. To assess the contributions of mito KATP channels and protein kinase C (PKC) in adenosine and diazoxide-mediated protection, young wild-type hearts were treated with 100 μm 5-hydroxydecanoic acid (5-HD), a mito KATP channel inhibitor, or 3 μm chelerythrine, a non-isoform-specific PKC inhibitor. Inhibitors were infused for 15 min prior to ischaemia, and re-instated during reperfusion. This was performed in untreated hearts (n = 7 for 5-HD, n = 8 for chelerythrine), 50 μm adenosine-treated hearts (n = 8 for 5-HD, n = 8 for chelerythrine) and 50 μm diazoxide-treated hearts (n = 8 for 5-HD, n = 7 for chelerythrine). These experiments were undertaken in young hearts since adenosine failed to modify ischaemic tolerance in the aged group.
Analysis of A1AR transcription and expression
The effects of age on A1AR gene transcription and protein expression were assessed in normoxic myocardium. Wild-type and transgenic hearts were perfused for 30 min under normoxic conditions and snap-frozen in aluminium tongs cooled in liquid N2. For analysis of A1AR transcription, samples of ventricular myocardium were homogenised and total RNA was extracted using standard TRIzol and DNase treatment with subsequent spin column purification (Qiagen, Hilden, Germany). RNA integrity was verified by formaldehyde agarose gel electrophoresis and total RNA stored at −80 °C until analysed. Expression of mRNA for the A1AR was assessed via quantitative real-time PCR using the iCycler iQ real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA, USA). 18S ribosomal RNA was used as an endogenous control to correct for minor experimental variations. PCR primers used were:
18S rRNA: forward primer, 5′-CTCAACACGGGAAACCTCAC-3′; reverse primer, 5′-AAATCGCTCCACCAACTAAGAA-3′.
A1AR: forward primer, 5′-CATTGGGCCACAGACCTACT-3′; reverse primer, 5′-ACCGGAGAGGGATCTTGACT-3′.
Quantitative real-time PCR is based on the measurement of the cycle threshold (Ct) for transcripts of interest. This Ct value is the minimum PCR cycle number at which accumulation of amplicon can be accurately quantified. Hence, higher levels of a transcript in a sample will reach the Ct value earlier. To normalise transcription levels between samples and groups, the Ct for the A1AR transcript was calculated relative to the Ct for the endogenous house-keeping gene 18S rRNA in each sample – the value ΔCt represents the difference in Ct for A1AR transcription vs. 18S rRNA transcription. A lower ΔCt value reflects higher levels of transcription, and vice versa.
For analysis of A1AR expression, ventricular myocardium was homogenised in 10 volumes of ice-cold buffer (10 mm EDTA, 10 mm Hepes, 0.1 mm benzamidine, pH 7.4), the homogenate was centrifuged at 48 000 g for 10 min, and the pellet was resuspended in 3 ml of buffer with EDTA reduced to 1 mm. The pellet was washed an additional two times by resuspension and centrifugation, with the final pellet resuspended in 1 ml of ligand binding buffer (50 mm Tris-HCl, 5 mm MgCl2, pH 7.4). For ligand binding, 50 μl aliquots of membrane suspension, containing 100 μg of protein for wild-types and 10 μg of protein for transgenics, were incubated with 2 U ml−1 adenosine deaminase and the A1AR-selective ligand [3H]DPCPX (Dupont NEN, Boston, MA, USA). After 2 h incubation at 21 °C, 3 ml of ice-cold rinse buffer (10 mm Tris-HCl, 5 mm MgCl2, pH 7.4) was added to each sample and membranes were collected onto Whatman GF/C glass-fibre filters. Filters were washed with ice-cold buffer and trapped radioactivity was counted. Non-specific binding was determined by addition of 500 nmN6-cyclohexyladenosine to incubations. Specific binding was fitted to a single site binding model using non-linear least-squares curve fitting of un-transformed data to calculate receptor density (Bmax) and the dissociation constant (KD).
Statistical analysis
Functional responses to ischaemia–reperfusion were assessed via multi-way ANOVA with repeated measures. LDH efflux and receptor expression data were analysed by one-way ANOVA. In all cases, a Tukey post hoc test was applied for specific comparisons when significance was detected. Significance was accepted for P < 0.05; data shown are means ± s.e.m.
RESULTS
Functional response to ischaemia–reperfusion
Heart mass increased from 110 ± 9 mg in the young group to 159 ± 14 mg in the aged group. Contractile function was similar in the two age groups. Intrinsic heart rate was moderately reduced with age, as was coronary flow rate (Table 1). Global normothermic ischaemia rapidly reduced contractile function with no detectable systolic pressure development after the first 2–3 min. End-diastolic pressure rose gradually throughout the ischaemic episode. The time to ischaemic contracture (rise of 20 mmHg) was similar in young and aged hearts, whereas peak ischaemic contracture achieved was lower in aged vs. young hearts (Fig. 1).
Table 1.
Baseline functional parameters in untreated and adenosine-treated hearts from young and aged wild-type and transgenic mice
| Group | Treatment | LVDP (mmHg) | +dP/dt (mmHg s−1) | −dP/dt (mmHg s−1) | Heart rate (beats min−1) | Flow (ml min g−1) |
|---|---|---|---|---|---|---|
| Young wild-type (n = 11) | Untreated | 159 ± 6 | 6869 ± 360 | 4511 ± 242 | 381 ± 9 | 28 ± 2 |
| Young wild-type (n = 9) | Adenosine (50 μm) | 169 ± 7 | 7084 ± 291 | 4100 ± 228 | — | 35 ± 2† |
| Aged wild-type (n = 10) | Untreated | 160 ± 8 | 6762 ± 240 | 4882 ± 185 | 350 ± 10* | 23 ± 2* |
| Aged wild-type (n = 10) | Adenosine (50 μm) | 165 ± 9 | 7147 ± 427 | 4568 ± 246 | — | 33 ± 2† |
| Young transgenic (n = 9) | Untreated | 167 ± 6 | 6575 ± 210 | 4390 ± 163 | 343 ± 11*‡ | 27 ± 1 |
| Young transgenic (n = 9) | Adenosine (50 μm) | 179 ± 9 | 6989 ± 384 | 4658 ± 243 | — | 33 ± 1† |
| Aged transgenic (n = 10) | Untreated | 162 ± 9 | 6958 ± 419 | 4492 ± 215 | 321 ± 8*‡ | 23 ± 1* |
| Aged transgenic (n = 9) | Adenosine (50 μm) | 177 ± 8 | 7358 ± 296 | 4893 ± 195 | — | 35 ± 1*† |
All values represent means ± s.e.m. All parameters except intrinsic hearts rate were measured after 30 min aerobic perfusion in wild-type hearts and hearts overexpressing A1ARs. Intrinsic heart rate was assessed immediately prior to ventricular pacing (20 min aerobic perfusion). LVDP, left ventricular developed pressure.
P < 0.05vs. young wild-type hearts
P < 0.05vs. untreated
P < 0.05vs. wild-type hearts.
Figure 1. Contracture development during global ischaemia in young and aged hearts.
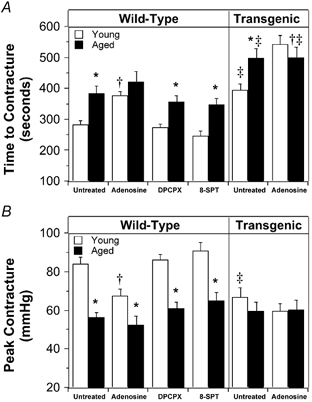
A, time to ischaemic contracture. B, peak ischaemic contracture. Wild-type and transgenic hearts overexpressing A1ARs were untreated or treated with 50 μm adenosine.Wild-type hearts were also treated with 150 nm of the A1AR antagonist DPCPX or 100 μm of the non-specific antagonist 8-SPT. All values represent means ± s.e.m. * P < 0.05vs. young heart; †P < 0.05vs. untreated heart; ‡P < 0.05vs. wild-type heart.
During reperfusion there was an initial rapid recovery in contractile function followed by a gradual decline then slow recovery over the remaining 30 min of reperfusion (Fig. 2). At the end of reperfusion there remained a sustained elevation in diastolic pressure and decline in contractility in both age groups (Fig. 2A). Post-ischaemic contractile dysfunction was significantly worsened in aged vs. young hearts (Fig. 2 and Fig. 3). Coronary flow initially recovered to pre-ischaemic levels during early reperfusion, then gradually declined to ∼75 % of pre-ischaemia (Fig. 2D and Fig. 3D). In aged hearts, coronary flow recovery during early reperfusion was similar, although flow tended to be slightly lower in aged vs. young hearts. The incidence of reperfusion-induced arrhythmias was similar in the two groups (Fig. 4A). Impaired functional recovery with ageing was not associated with differences in the incidence of ectopy during reperfusion (Fig. 4A), but was matched by greater loss of myocardial LDH (Fig. 4B).
Figure 2. Time course of contractile recoveries following 20 min ischaemia in young and aged wild-type hearts.
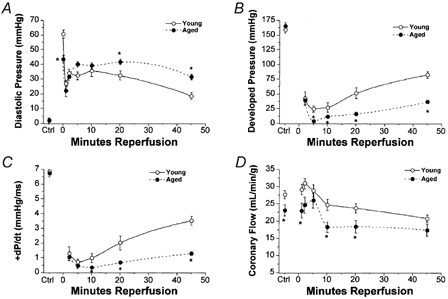
A, left ventricular diastolic pressure. B, left ventricular developed pressure. C, +dP/dt. D, coronary flow. All values represent means ± s.e.m. * P < 0.05vs. young heart.
Figure 3. Effects of adenosine, adenosine receptor antagonism and A1AR overexpression on post-ischaemic functional recoveries.
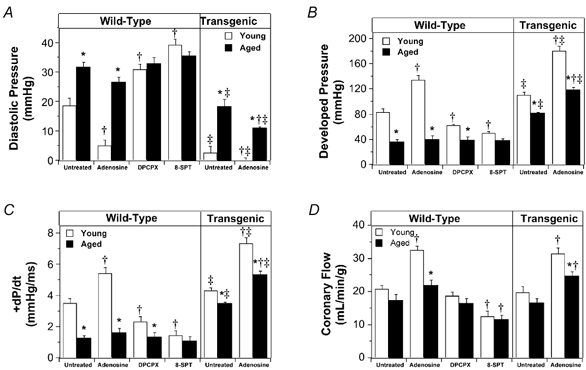
Functional recoveries were assessed after 45 min reperfusion following 20 min ischaemia in young and aged hearts. A, left ventricular diastolic pressure. B, left ventricular developed pressure. C, +dP/dt. D, coronary flow. Wild-type and transgenic hearts overexpressing A1ARs were untreated or treated with 50 μm adenosine. Wild-type hearts were also treated with 150 nm of the A1AR antagonist DPCPX or 100 μm of the non-specific antagonist 8-SPT. All values represent means ± s.e.m. * P < 0.05vs. young heart; †P < 0.05vs. untreated heart; ‡P < 0.05vs. wild-type heart.
Figure 4. Arrhythmogenesis and LDH efflux in post-ischaemic hearts.
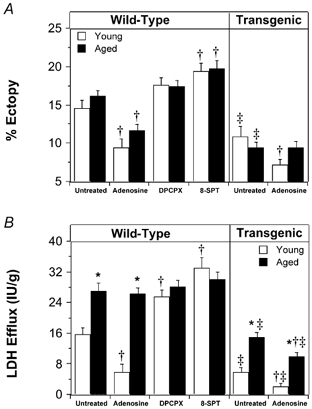
A, incidence of tachyarrhythmias was assessed in the initial 10 min of reperfusion following 20 min ischaemia. B, myocardial LDH efflux was assessed over the entire 45 min post-ischaemic period. Wild-type and transgenic hearts overexpressing A1ARs were untreated or treated with 50 μm adenosine. Wild-type hearts were also treated with 150 nm of the A1AR antagonist DPCPX or 100 μm of the non-specific antagonist 8-SPT. All values represent means ± s.e.m. * P < 0.05vs. young heart; †P < 0.05vs. untreated heart; ‡P < 0.05vs. wild-type heart.
Effects of adenosine and A1AR overexpression on responses to ischaemia–reperfusion
The data in Fig. 5 demonstrate that adenosine reduces heart rate and coronary resistance in young and aged hearts, and that the 50 μm concentration of adenosine employed in ischaemic studies produced near-maximal and equipotent effects in the two age groups (Fig. 5). Treatment of ischaemic-reperfused hearts with 50 μm adenosine did not significantly alter pre-ischaemic contractile function in any experimental groups (Table 1), although coronary flow was elevated. In young hearts adenosine reduced contracture development (Fig. 1), and markedly enhanced post-ischaemic contractile recovery (Fig. 3). Coronary reflow during reperfusion was significantly enhanced by adenosine in young hearts, and was enhanced to a lesser extent in the aged group (Fig. 3D). Adenosine also reduced reperfusion arrhythmias and LDH efflux in young hearts (Fig. 4). In aged hearts adenosine failed to alter contractile dysfunction or necrosis, but it did reduce arrhythmogenesis to an extent similar to that for young hearts (Fig. 4A). Blockade of A1ARs with DPCPX did not alter baseline function in young or aged hearts (data not shown), but it impaired ischaemic tolerance in young but not aged hearts (Fig. 3 and Fig. 4). Similarly, non-selective blockade of adenosine receptors with 8-SPT impaired ischaemic tolerance (Fig. 3 and Fig. 4). The effects of DPCPX and 8-SPT were similar although DPCPX failed to alter reflow whereas 8-SPT reduced reflow (Fig. 3).
Figure 5. Concentration–response data for A1 (bradycardia) and A2 (vasodilatation) receptor-mediated effects of adenosine.
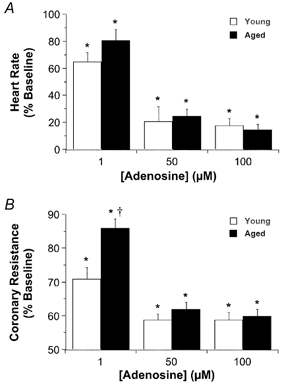
Adenosine was infused cumulatively at concentrations of 1, 50 and 100 μm in young and moderately aged hearts. Maximal functional changes were measured at each concentration. All values represent means ± s.e.m. * P < 0.05vs. baseline; †P < 0.05vs. young hearts.
Normoxic contractile function in both young and aged hearts was unaltered by overexpression of A1ARs, although intrinsic heart rate was reduced in both age groups (Table 1). A1AR overexpression improved ischaemic tolerance in young and aged hearts, reducing contracture development (Fig. 1), contractile dysfunction (Fig. 3), arrhythmogenesis and LDH efflux (Fig. 4). In contrast to aged wild-type hearts, aged transgenic hearts displayed a significant protective response to 50 μm adenosine. Adenosine treatment in aged transgenic hearts further enhanced contractile recovery, and reduced LDH efflux and arrhythmogenesis (Figs 3 and 4). Nonetheless, post-ischaemic recovery remained much higher in young transgenic hearts treated with adenosine. A1AR overexpression failed to alter the extent of post-ischaemic coronary reflow in both young and aged hearts untreated or treated with adenosine (Fig. 3D).
Effects of diazoxide, 5-HD and chelerythrine on ischaemic tolerance
Treatment with diazoxide enhanced recovery from ischaemia in both young and aged hearts, reducing contractile dysfunction (Fig. 6A), arrhythmogenesis (Fig. 6B) and LDH efflux (Fig. 6C). The effects of diazoxide on contractile recovery and necrosis were inhibited by the PKC inhibitor chelerythrine and the putative mito KATP channel inhibitor 5-HD (Fig. 7). However, while the anti-arrhythmic effects of diazoxide were also blocked by 5-HD, they were resistant to chelerythrine (Fig. 7B). The cardioprotective effects of adenosine were all abolished by 5-HD (Fig. 7). As with diazoxide, chelerythrine abrogated the effects of adenosine on contractile function and LDH efflux, but failed to modify the effects on reperfusion-induced tachyarrhythmias (Fig. 7B).
Figure 6. Cardioprotective responses to diazoxide in young and aged hearts.
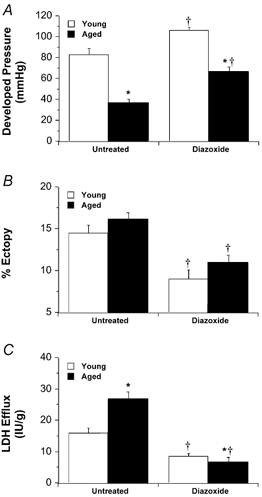
Protective effects of 50 μm diazoxide were assessed in young and aged hearts subjected to 20 min ischaemia and 45 min reperfusion. A, final recovery of left ventricular developed pressure. B, incidence of tachyarrhythmias in the initial 10 min of reperfusion. C, post-ischaemic LDH efflux. All values represent means ± s.e.m. * P < 0.05vs. young heart; †P < 0.05vs. untreated heart.
Figure 7. Effects of chelerythrine and 5-HD on cardioprotective effects of adenosine and diazoxide.
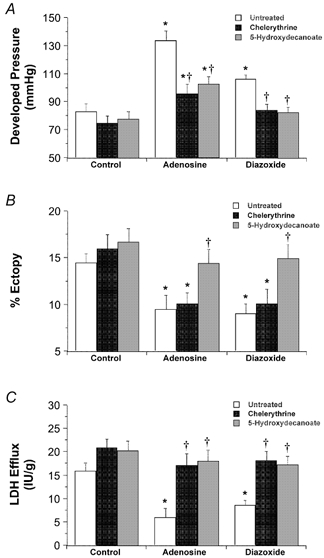
Ability of 100 μm 5-HD and 3 μm chelerythrine to modify cardioprotection with 50 μm adenosine or 50 μm diazoxide was assessed in young wild-type hearts. A, ventricular pressure development. B, reperfusion-induced tachyarrhythmias. C, LDH efflux. All values represent means ± s.e.m. * P < 0.05vs. Control; †P < 0.05vs. hearts untreated with chelerythrine or 5-HD.
Effects of age on myocardial A1AR transcription and expression
Analysis of ventricular myocardium revealed no change in transcription of the A1AR gene with age in wild-type hearts (Fig. 8A). Curiously, we detected a modest age-related increase in A1AR transcription in transgenic hearts (reflected by reduced ΔCt relative to 18S rRNA). Radioligand binding analysis failed to detect a difference in A1AR density in young vs. aged wild-type hearts (Fig. 8B). However, there was a modest decline in A1AR density with age in transgenic hearts despite the apparent increase in A1AR gene transcription. No changes in the dissociation constant for DPCPX were observed. The KD was 1.2 ± 0.2 and 1.4 ± 0.2 nm in young and aged wild-type hearts, respectively, and was 1.3 ± 0.2 and 1.2 ± 0.2 nm in young and aged transgenic hearts, respectively.
Figure 8. Effects of age on A1AR gene transcription and protein expression.
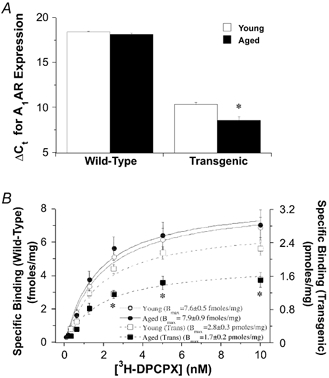
Relative A1AR gene transcription and myocardial A1AR density were assessed in young and aged wild-type hearts. A, A1AR gene transcription determined via quantitative real-time PCR, and presented as the ΔCt value relative to 18S rRNA transcription (lower ΔCt value reflects higher transcript levels – see Methods). B, [3H]DPCPX binding to myocardial A1ARs. Density of A1ARs (Bmax) is also provided in B for all four groups. All values represent means ± s.e.m. * P < 0.05vs. young. No differences were detected between Bmax values in young vs. aged hearts in B.
DISCUSSION
The results of this study indicate that ageing substantially limits the tolerance to ischaemic insult, and suggest an abnormality in adenosine-mediated cardioprotection plays an important role. The abnormal adenosine response appears to result from impaired activation of mito KATP channels, and is unrelated to altered transcription or expression of A1ARs. Enhanced expression of A1ARs substantially improves tolerance in aged hearts, and restores the protective response to adenosine.
Effects of ageing on functional responses to myocardial ischaemia–reperfusion
An increasing number of studies support reduced ischaemic tolerance with ageing (Pahor et al. 1985; Frolkis et al. 1991; Misare et al. 1992; Lesnefsky et al. 1994; Tani et al. 1997; Headrick, 1998; Abete et al. 1999; Mariani et al. 2000; Rosenfeldt et al. 2002) and implicate a variety of mechanisms (Boucher et al. 1998; Cain et al. 1998; Headrick, 1998; Coombes et al. 2000; Lesnefsky et al. 2001; Bulteau et al. 2002). Our data demonstrate substantially worsened post-ischaemic functional recovery and enhanced enzyme leakage in aged vs. young hearts (Fig. 2 and Fig. 4). As opposed to studies in rat (Boucher et al. 1998, 2000; Headrick 1998), we observe reduced ischaemic contracture in aged mouse heart (Fig. 1). Nonetheless, post-ischaemic diastolic pressure was elevated in aged hearts (Fig. 2A). These observations have parallels in other investigations. In an elegant study, Cross et al. (1996) showed that glycogen content and extent of ATP depletion are key determinants of contracture and post-ischaemic diastolic pressure. High pre-ischaemic glycogen limits ischaemic contracture yet worsens recovery due to acidosis and Na+–H+ exchange. Elevations in myocardial glycogen with age (Tani et al. 1999) may therefore contribute to reduced contracture development (Fig. 1). In addition, since contracture continues to develop until ATP available to myosin-ATPase is consumed, higher pre-ischaemic ATP may enhance contracture (Cross et al. 1996). Though the data are equivocal, myocardial ATP may decline with ageing (Ramani et al. 1986), limiting ischaemic contracture development.
Whether impaired functional recovery reflects greater stunning vs. necrosis is difficult to ascertain. Abete and colleagues observed changes in both end-points (Abete et al. 1999). Since we observed elevated LDH loss, enhanced necrosis must contribute to poor functional outcome with age. In terms of reflow, we found no evidence for a substantial role in the decline in tolerance: while reflow was modestly reduced in aged hearts, the difference was small (10–20 %) relative to differences in contractile recovery (50–60 %). Moreover, contractile function and LDH efflux appear independent of reflow. Adenosine increased reflow in aged hearts yet contractile recovery was unaltered (Fig. 3), and A1AR overexpression enhanced ischaemic tolerance in all groups without altering reflow (Fig. 3D). These data agree with earlier findings regarding protection with adenosine and A1AR activation (Matherne et al. 1997; Headrick et al. 2000), and the data of Kolocassides et al. (1996) regarding the lack of an important role for reflow in the effects of cardioplegia and preconditioning.
Effects of ageing on adenosine-mediated cardioprotection
We and others have acquired evidence implicating adenosine as an endogenous determinant of ischaemic tolerance in immature and mature myocardium (Zhao et al. 1993, 1994; Matherne et al. 1996; Peart & Headrick, 2000), and a recent study suggests adenosine-mediated protection may decline with age (Gao et al. 2000). Unfortunately, the latter study examined an adenosine stimulus selectively modifying heart rate and flow in young but not aged hearts. The lack of protection may therefore have reflected an age-related reduction in activation of effector mechanisms (Cai et al. 1997; Gao et al. 1997). In the current study we showed that an equipotent and near-maximally effective concentration of adenosine enhanced ischaemic tolerance in young hearts yet failed to modify contractile recovery and enzyme efflux in aged myocardium (Fig. 3 and Fig. 4). Furthermore, blockade of endogenous adenosine with A1AR-selective or non-selective antagonists revealed endogenous adenosine also exerts protection in young but not aged hearts (Fig. 3 and Fig. 4). Thus, the effects of both exogenous and endogenous adenosine are abrogated with ageing. Curiously, the impact of age is selective as adenosine does exert comparable anti-arrhythmic effects in both groups (Fig. 4). This may reflect unique signalling in different forms of protection (see below).
Roles of mito KATP channels and PKC in adenosine-mediated protection
We have acquired evidence that protection with A1AR activation depends upon mito KATP channel activation (Headrick et al. 2000), in agreement with other studies (Van Winkle et al. 1994; Miura et al. 1999). We show here that the mito KATP channel inhibitor 5-HD and PKC inhibitor chelerythrine both limit protection with adenosine (Fig. 7). Additionally, both agents eliminate protection with the mito KATP channel opener diazoxide (Fig. 7), which mimicked the effects of adenosine in young hearts (Fig. 6). However, a recent study suggests diazoxide and 5-HD may exert effects on β-oxidation and respiratory chain function (Hanley et al. 2002). These authors hypothesise that protection with diazoxide may occur via partial inhibition of respiratory chain complexes. In the light of their observations it might be appropriate to conclude that adenosine-mediated protection occurs via a 5-HD-sensitive process which may involve mito KATP channels (Garlid et al. 1996; Liu et al. 1998) and/or a change in respiratory chain function, as proposed by Hanley and colleagues (2002). Furthermore, since the effects of diazoxide and adenosine are sensitive to 5-HD and chelerythrine, and diazoxide protects aged hearts whereas adenosine does not, we conclude that adenosine activates the same signalling path activated by diazoxide, PKC acts downstream of the site(s) modified by adenosine and diazoxide, and the failure of adenosine-mediated protection with ageing involves ineffective upstream activation of the diazoxide-/5-HD-sensitive process.
That the failure in adenosine- and A1AR-mediated protection is proximal to the site targeted by diazoxide agrees with recent studies demonstrating impaired protection with preconditioning and PKC activation and preserved protection with diazoxide (Tani et al. 2001; Lee et al. 2002). Moreover, studies of Korzick et al. (2001) and Takayama et al. (2001) support an ageing-related disruption of PKC signalling and translocation, potentially explaining the impaired responses to protective stimuli. However, these observations contrast with the data of Schulman et al. (2001), who documented a loss of protection with transient A1 agonism, PKC activation and diazoxide. The reason for this discrepancy is unclear, but may involve the different protective stimulus (i.e. transient activation) assessed by Schulman and colleagues (2001). Our data, and the findings of Tani et al. (2001) and Lee et al. (2002), support a failure in signalling proximal to molecular processes targeted by diazoxide. It is tempting to speculate that this may involve the PI3 kinase pathway, since myocardial PI3 kinase activity falls with ageing (Martineau et al. 1999), the enzyme may activate cardioprotection (Tong et al. 2000), and it is located upstream of PKC and mito KATP channels (Tong et al. 2000). However, while adenosine receptors may activate PI3-kinase (Krieg et al. 2002), data indicate that the enzyme does not play an essential role in the anti-ischaemic effects of adenosine (Qin et al. 2003). Future work might address the precise role of this path in adenosine-mediated cardioprotection.
Despite our evidence for a role for PKC in protective signalling, recent reports question an obligatory function for the enzyme in cardioprotection. These studies support roles for other kinase cascades (Qin et al. 2003), lending support to the notion that adenosine and other protective stimuli activate multiple parallel kinase-dependent pathways (Tanno et al. 2000; Cohen et al. 2001; Li & Sato, 2001; Takeishi et al. 2001; Qin et al. 2003). As discussed by Cohen et al. (2001), this would provide some level of redundancy and explain why blockade of individual paths is not always effective in eliminating protective responses. In this respect, since we show adenosine fails to protect aged hearts, any redundancy provided by multiple signalling paths is clearly ineffective in countering the effects of ageing. This, in turn, implicates an age-related failure at an upstream site in the signalling cascade.
As noted above, the effects of age on arrhythmogenesis vs. other end-points differ, suggesting unique signalling. This is verified by the observation that chelerythrine and 5-HD reduce the functional and anti-necrotic effects of adenosine and diazoxide whereas only 5-HD altered anti-arrhythmic responses (Fig. 7). Dissociation of arrhythmogenesis from contractile recovery and necrosis in these groups indicates necrosis and contractile dysfunction are unlikely contributors to arrhythmogenesis. Our findings are consistent with prior studies that showed no role for PKC in post-ischaemic arrhythmogenesis (Du et al. 1995) and in anti-arrhythmic effects of preconditioning (Kita et al. 1998). Other studies support distinct signalling in the protection against necrosis vs. arrhythmogenesis (Wang et al. 2001).
Effects of modulating A1AR expression
We hypothesised that enhanced A1AR overexpression might restore ischaemic tolerance in aged hearts, based on observations that adenosine receptors are important in determining intrinsic tolerance in younger hearts (Zhao et al. 1994; Matherne et al. 1996; Peart & Headrick, 2000). Overexpression of A1ARs does improve ischaemic tolerance (Figs 1, 3 and 4), and recoveries in aged transgenic hearts were equivalent to those for young wild-type hearts. Furthermore, the protective response to adenosine was restored. These findings indicate that mediators and end-effectors of adenosine- and A1AR-dependent protection are functional in aged myocardium and can be harnessed if the amplitude of the initial stimulus (A1AR activation or coupling) is sufficiently enhanced. This is consistent with the observations of McCully and colleagues (1998), who found preconditioning and adenosine are individually ineffective in aged hearts while the two together are protective, possibly via additive activation of protective signalling. Our data also indicate that aged myocardium is not necessarily the ‘victim’ of unavoidable accumulation of cellular damage, and it can possess an apparently normal phenotypic response to ischaemia. Nonetheless, while A1AR overexpression in aged hearts produces an ischaemia-tolerant phenotype, A1AR overexpression additionally enhances tolerance in young hearts (Fig. 3 and Fig. 4). Since the detrimental effects of ageing are not eliminated by A1AR overexpression, factors other than impaired adenosine-mediated cardioprotection may also play a role in reduced ischaemic tolerance.
The role of A1AR transcription and expression
As noted above, the data for A1AR overexpression demonstrate signalling downstream from A1ARs is intact in aged heart. Thus, a site of ‘failure’ in aged myocardium may be the A1AR itself. While prior studies suggest there is no change in cardiac A1AR expression with ageing (Cai et al. 1997), studies in non-cardiac tissue support reduced A1AR density (Cunha et al. 2001). We observed no age-related change in either A1AR transcription or expression in wild-type mouse hearts (Fig. 8). Thus, the age-related failure in A1AR-mediated cardioprotection does not occur at the level of A1AR gene transcription or expression, further implicating impaired activation of downstream effector mechanisms. Interestingly, we did observe a modest decline in A1AR expression in aged transgenic hearts, despite an apparent increase in A1AR transcription. The cause of this apparent dissociation between transcription and expression in transgenic hearts is unclear, but may reflect age-related changes in expression amplification via the α-MHC promoter.
Conclusions
This study documents a substantial decline in myocardial ischaemic tolerance with moderate ageing, which involves a failure in cardioprotection mediated by endogenous and exogenous adenosine. The data indicate that the effects of ageing are selective for protection against contractile dysfunction and necrosis vs. arrhythmogenesis and support differing signalling in these responses. The inability of adenosine to protect against contractile dysfunction and necrosis is unrelated to changes in A1AR transcription or translation, and appears to involve a failure in one or more signalling elements upstream of mito KATP channels. Overexpression of A1ARs improves ischaemic tolerance in aged hearts, and restores protection with exogenous adenosine. However, since an age-related difference in ischaemic tolerance persists in transgenic hearts, it is probable that factors additional to impaired adenosine-mediated cardioprotection play some role in the ischaemia-intolerant aged phenotype.
Acknowledgments
This work was supported by grants from the National Heart Foundation of Australia (G 98B 0080 and G 99B 0246) and the National Health and Medical Research Council of Australia (145310). John Headrick was the recipient of a career research fellowship from the National Heart Foundation of Australia
References
- Abete P, Cioppa A, Calabrese C, Pascucci I, Cacciatore F, Napoli C, Carnovale V, Ferrara N, Rengo F. Ischemic threshold and myocardial stunning in the aging heart. Exp Gerontol. 1999;34:875–884. doi: 10.1016/s0531-5565(99)00060-1. [DOI] [PubMed] [Google Scholar]
- Abete P, Ferrara N, Cioppa A, Ferrara P, Bianco S, Calabrese C, Cacciatore F, Longobardi G, Rengo F. Preconditioning does not prevent postischemic dysfunction in aging heart. J Am Coll Cardiol. 1996;27:1777–1786. doi: 10.1016/0735-1097(96)00070-8. [DOI] [PubMed] [Google Scholar]
- Boucher F, Tanguy S, Toufektsian MC, Besse S, De Leiris J. Effects of fasting and exogenous glucose delivery on cardiac tolerance to low-flow ischemia in adult and senescent rats. Mech Ageing Dev. 2000;116:15–32. doi: 10.1016/s0047-6374(00)00125-1. [DOI] [PubMed] [Google Scholar]
- Boucher F, Tanguy S, Toufektsian MC, Besse S, Tressallet N, Favier A, De Leiris J. Age-dependent changes in myocardial susceptibility to zero flow ischemia and reperfusion in isolated perfused rat hearts: relation to antioxidant status. Mech Ageing Dev. 1998;103:301–316. doi: 10.1016/s0047-6374(98)00050-5. [DOI] [PubMed] [Google Scholar]
- Bulteau AL, Szweda LI, Friguet B. Age-dependent declines in proteasome activity in the heart. Arch Biochem Biophys. 2002;397:298–304. doi: 10.1006/abbi.2001.2663. [DOI] [PubMed] [Google Scholar]
- Cai GP, Wang HY, Gao EH, Horwitz J, Snyder DL, Pelleg A, Roberts J, Friedman E. Reduced adenosine A1 receptor and Gα protein coupling in rat ventricular myocardium during aging. Circ Res. 1997;81:1065–1071. [PubMed] [Google Scholar]
- Cain BS, Meldrum DR, Joo KS, Wang J-F, Meng X, Cleveland JC, Jr, Banerjee A, Harken AH. Human SERCA2a levels correlate inversely with age in senescent human myocardium. J Am Coll Cardiol. 1998;32:458–467. doi: 10.1016/s0735-1097(98)00233-2. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Liu GS, Heusch G, Downey JM. Acetylcholine, bradykinin, opioids, and phenylephrine, but not adenosine, trigger preconditioning by generating free radicals and opening mitochondrial KATP channels. Circ Res. 2001;89:273–278. doi: 10.1161/hh1501.094266. [DOI] [PubMed] [Google Scholar]
- Coombes JS, Powers SK, Hamilton KL, Demirel HA, Shanely RA, Zergeroglu MA, Sen CK, Packer L, Ji LL. Improved cardiac performance after ischemia in aged rats supplemented with vitamin E and alpha-lipoic acid. Am J Physiol Reg Int Comp Physiol. 2000;279:R2149–2155. doi: 10.1152/ajpregu.2000.279.6.R2149. [DOI] [PubMed] [Google Scholar]
- Cross HR, Opie LH, Radda GK, Clarke K. Is a high glycogen content beneficial or detrimental to the ischemic rat heart? A controversy resolved. Circ Res. 1996;78:482–491. doi: 10.1161/01.res.78.3.482. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Constantino MD, Fonseca E, Ribeiro JA. Age-dependent decrease in adenosine A1 receptor binding sites in the rat brain - Effect of cis unsaturated free fatty acids. Eur J Biochem. 2001;268:2939–2947. doi: 10.1046/j.1432-1327.2001.02183.x. [DOI] [PubMed] [Google Scholar]
- Du XJ, Anderson K, Jacobsen A, Woodcock E, Dart A. Suppression of ventricular arrhythmias during ischaemia-reperfusion by agents inhibiting Ins(1,4,5)P3 release. Circulation. 1995;91:2712–2716. doi: 10.1161/01.cir.91.11.2712. [DOI] [PubMed] [Google Scholar]
- Frolkis VV, Frolkis RA, Mkhitarian LS, Fraifeld VE. Age-dependent effects of ischemia and reperfusion on cardiac function and Ca2+ transport in myocardium. Gerontology. 1991;37:233–239. doi: 10.1159/000213266. [DOI] [PubMed] [Google Scholar]
- Gao E, Snyder DL, Johnson MD, Friedman E, Roberts J, Horwitz J. The effect of age on adenosine A1 receptor function in the rat heart. J Mol Cell Cardiol. 1997;29:593–602. doi: 10.1006/jmcc.1996.0302. [DOI] [PubMed] [Google Scholar]
- Gao F, Christopher TA, Lopez BL, Friedman E, Cai G, Ma XL. Mechanism of decreased adenosine protection in reperfusion injury of aging rats. Am J Physiol Heart Circ Physiol. 2000;279:H329–338. doi: 10.1152/ajpheart.2000.279.1.H329. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Paucek P, Yarov-Yarovoy V, Sun X, Schindler PA. The mitochondrial KATP channel as a receptor for potassium channel openers. J Biol Chem. 1996;271:8796–8799. doi: 10.1074/jbc.271.15.8796. [DOI] [PubMed] [Google Scholar]
- Gauthier NS, Morrison RR, Byford AM, Jones R, Headrick JP, Matherne GP. Functional genomics of transgenic overexpression of A1 adenosine receptors in the heart. Drug Dev Res. 1998;45:402–409. [Google Scholar]
- Hanley PJ, Mickel M, Löffler M, Brandt U, Daut J. KATP channel-independent targets of diazoxide and 5-hydroxydecanoate in the heart. J Physiol. 2002;542:735–741. doi: 10.1113/jphysiol.2002.023960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Headrick JP. Aging impairs functional, metabolic and ionic recovery from ischemia-reperfusion and hypoxia-reoxygenation. J Mol Cell Cardiol. 1998;30:1415–1430. doi: 10.1006/jmcc.1998.0710. [DOI] [PubMed] [Google Scholar]
- Headrick JP, Gauthier NS, Morrison RR, Matherne GP. Cardioprotection by KATP channels in wild-type hearts and hearts overexpressing A1 adenosine receptors. Am J Physiol Heart Circ Physiol. 2000;279:H1690–1697. doi: 10.1152/ajpheart.2000.279.4.H1690. [DOI] [PubMed] [Google Scholar]
- Headrick JP, Peart J, Hack B, Flood A, Matherne GP. Functional properties and responses to ischemia reperfusion in Langendorff perfused mouse hearts. Exp Physiol. 2001;86:703–716. doi: 10.1111/j.1469-445x.2001.tb00035.x. [DOI] [PubMed] [Google Scholar]
- Kita H, Miura T, Tsuchida A, Hasegawa T, Shimamoto K. Suppression of reperfusion arrhythmias by preconditioning is inhibited by an ATP-sensitive potassium channel blocker, 5-hydroxydecanoate, but not by protein kinase C blockers in the rat. J Cardiovasc Pharmacol. 1998;32:791–797. doi: 10.1097/00005344-199811000-00016. [DOI] [PubMed] [Google Scholar]
- Kolocassides KG, Galinanes M, Hearse DJ. Ischemic preconditioning, cardioplegia or both? Differing approaches to myocardial and vascular protection. J Mol Cell Cardiol. 1996;28:623–634. doi: 10.1006/jmcc.1996.0058. [DOI] [PubMed] [Google Scholar]
- Korzick DH, Holiman DA, Boluyt MO, Laughlin MH, Lakatta EG. Diminished alpha1-adrenergic-mediated contraction and translocation of PKC in senescent rat heart. Am J Physiol Heart Circ Physiol. 2001;281:H581–589. doi: 10.1152/ajpheart.2001.281.2.H581. [DOI] [PubMed] [Google Scholar]
- Krieg T, Qin QN, McIntosh EC, Cohen MV, Downey JM. ACh and adenosine activate PI3-kinase in rabbit hearts through transactivation of receptor tyrosine kinases. Am J Physiol Heart Circ Physiol. 2002;283:H2322–2330. doi: 10.1152/ajpheart.00474.2002. [DOI] [PubMed] [Google Scholar]
- Lee TM, Su SF, Chou TF, Lee YT, Tsai CH. Loss of preconditioning by attenuated activation of myocardial ATP-sensitive potassium channels in elderly patients undergoing coronary angioplasty. Circulation. 2002;105:334–340. doi: 10.1161/hc0302.102572. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Gallo DS, Ye J, Whittingham TS, Lust WD. Aging increases ischemia-reperfusion injury in the isolated, buffer-perfused heart. J Lab Clin Med. 1994;124:843–851. [PubMed] [Google Scholar]
- Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: Ischemia-reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001;33:1065–1089. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- Li Y, Sato T. Dual signaling via protein kinase C and phosphatidylinositol 3′-kinase/AKT contributes to bradykinin B2 receptor-induced cardioprotection in guinea pig hearts. J Mol Cell Cardiol. 2001;33:2047–2053. doi: 10.1006/jmcc.2001.1455. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sato T, O'Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels:novel effectors of cardioprotection? Circulation. 1998;97:2463–2469. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- Mariani J, Ou RC, Bailey M, Rowland M, Nagley P, Rosenfeldt F, Pepe S. Tolerance to ischemia and hypoxia is reduced in aged human myocardium. J Thorac Cardiovasc Surg. 2000;120:660–667. doi: 10.1067/mtc.2000.106528. [DOI] [PubMed] [Google Scholar]
- Martineau LC, Chadan SG, Parkhouse WS. Age-associated alterations in cardiac and skeletal muscle glucose transporters, insulin and IGF-1 receptors, and PI3-kinase protein contents in the C57BL/6 mouse. Mech Ageing Dev. 1999;106:217–232. doi: 10.1016/s0047-6374(98)00106-7. [DOI] [PubMed] [Google Scholar]
- Matherne GP, Berr SS, Headrick JP. Integration of vascular, contractile and metabolic responses to hypoxia: Effects of maturation and the role of adenosine. Am J Physiol. 1996;270:R895–905. doi: 10.1152/ajpregu.1996.270.4.R895. [DOI] [PubMed] [Google Scholar]
- Matherne GP, Linden J, Byford AM, Gauthier NS, Headrick JP. Transgenic A1 adenosine receptor over-expression increases myocardial resistance to ischemia. Proc Nat Acad Sci U S A. 1997;94:6541–6546. doi: 10.1073/pnas.94.12.6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misare BD, Krukenkamp IB, Levitsky S. Age-dependent sensitivity to unprotected cardiac ischemia: the senescent myocardium. J Thorac Cardiovasc Surg. 1992;103:60–64. [PubMed] [Google Scholar]
- Miura T, Liu Y, Kita H, Ogawa T, Shimamoto K. Roles of mitochondrial ATP-sensitive K+ channels and PKC in anti-infarct tolerance afforded by adenosine A1 receptor activation. J Am Coll Cardiol. 1999;35:238–245. doi: 10.1016/s0735-1097(99)00493-3. [DOI] [PubMed] [Google Scholar]
- Pahor M, Di Gennaro M, Cocchi A, Bernabei R, Carosella L, Carbonin P. Age-related incidence of reperfusion- and reoxygenation-induced ventricular tachyarrhythmias in the isolated rat heart. Gerontology. 1985;31:15–26. doi: 10.1159/000212677. [DOI] [PubMed] [Google Scholar]
- Peart J, Flood A, Linden J, Matherne GP, Headrick JP. Adenosine-mediated cardioprotection in ischemic-reperfused mouse heart. J Cardiovasc Pharmacol. 2002;39:117–129. doi: 10.1097/00005344-200201000-00013. [DOI] [PubMed] [Google Scholar]
- Peart J, Headrick JP. Intrinsic activation of A1 adenosine receptors during ischemia and reperfusion improves ischemic tolerance. Am J Physiol Heart Circ Physiol. 2000;279:H2166–2175. doi: 10.1152/ajpheart.2000.279.5.H2166. [DOI] [PubMed] [Google Scholar]
- Qin QN, Downey JM, Cohen MV. Acetylcholine but not adenosine triggers preconditioning through PI3-kinase and a tyrosine kinase. Am J Physiol Heart Circ Physiol. 2003;284:H727–734. doi: 10.1152/ajpheart.00476.2002. [DOI] [PubMed] [Google Scholar]
- Ramani K, Lust WD, Whittingham TS, Lesnefsky EJ. ATP catabolism and adenosine generation during ischemia in the aging heart. Mech Ageing Dev. 1986;89:113–124. doi: 10.1016/0047-6374(96)01732-0. [DOI] [PubMed] [Google Scholar]
- Rosenfeldt FL, Pepe S, Linnane A, Nagley P, Rowland M, Ou RC, Marasco S, Lyon W. The effects of ageing on the response to cardiac surgery: protective strategies for the ageing myocardium. Biogerontology. 2002;3:37–40. doi: 10.1023/a:1015299127969. [DOI] [PubMed] [Google Scholar]
- Schulman D, Latchman DS, Yellon DM. Effect of aging on the ability of preconditioning to protect rat hearts from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2001;281:H1630–1636. doi: 10.1152/ajpheart.2001.281.4.H1630. [DOI] [PubMed] [Google Scholar]
- Takayama M, Ebihara Y, Tani M. Differences in the expression of protein kinase-C isoforms and its translocation after stimulation with phorbol ester between young-adult and middle-aged ventricular cardiomyocytes isolated from Fischer 344 rats. Jpn Circ J. 2001;65:1071–1076. doi: 10.1253/jcj.65.1071. [DOI] [PubMed] [Google Scholar]
- Takeishi Y, Huang Q, Wang T, Glassman M, Yoshizumi M, Baines CB, Lee JD, Kawakatsu H, Che W, Lerner-Marmarosh N, Zhang C, Yan C, Ohta S, Walsh RA, Berk BC, Abe J. Src family kinase and adenosine differentially regulate multiple MAP kinases in ischemic myocardium: modulation of MAP kinases activation by ischemic preconditioning. J Mol Cell Cardiol. 2001;33:1989–2005. doi: 10.1006/jmcc.2001.1463. [DOI] [PubMed] [Google Scholar]
- Tani M, Honma Y, Hasegawa H, Tamaki K. Direct activation of mitochondrial KATP channels mimics preconditioning but protein kinase C activation is less effective in middle-aged rat hearts. Cardiovasc Res. 2001;49:56–68. doi: 10.1016/s0008-6363(00)00240-6. [DOI] [PubMed] [Google Scholar]
- Tani M, Honma Y, Takayama M, Hasegawa H, Shinmura K, Ebihara Y, Tamaki K. Loss of protection by hypoxic preconditioning in aging Fischer 344 rat hearts related to myocardial glycogen content and Na+ imbalance. Cardiovasc Res. 1999;41:594–602. doi: 10.1016/s0008-6363(98)00256-9. [DOI] [PubMed] [Google Scholar]
- Tani M, Suganuma Y, Hasegawa H, Shinmura K, Hayashi Y, Guo X-D, Nakamura Y. Changes in ischemic tolerance and effects of ischemic preconditioning in middle-aged rat hearts. Circulation. 1997;95:2559–2566. doi: 10.1161/01.cir.95.11.2559. [DOI] [PubMed] [Google Scholar]
- Tanno M, Tsuchida A, Nozawa Y, Matsumoto T, Hasegawa T, Miura T, Shimamoto K. Roles of tyrosine kinase and protein kinase C in infarct size limitation by repetitive ischemic preconditioning in the rat. J Cardiovasc Pharmacol. 2000;35:345–352. doi: 10.1097/00005344-200003000-00001. [DOI] [PubMed] [Google Scholar]
- Tong HY, Chen WN, Steenbergen C, Murphy E. Ischemic preconditioning activates phosphatidylinositol-3-kinase upstream of protein kinase C. Circ Res. 2000;87:309–315. doi: 10.1161/01.res.87.4.309. [DOI] [PubMed] [Google Scholar]
- Van Winkle DM, Chien GL, Wolff RA, Soifer BE, Kuzume K, Davis RF. Cardioprotection provided by adenosine receptor activation is abolished by blockade of the KATP channel. Am J Physiol Heart Circ Physiol. 1994;266:H829–839. doi: 10.1152/ajpheart.1994.266.2.H829. [DOI] [PubMed] [Google Scholar]
- Wang GY, Wu S, Pei JM, Yu XC, Wong TM. Kappa- but not delta-opioid receptors mediate effects of ischemic preconditioning on both infarct and arrhythmia in rats. Am J Physiol Heart Circ Physiol. 2001;280:H384–391. doi: 10.1152/ajpheart.2001.280.1.H384. [DOI] [PubMed] [Google Scholar]
- Zhao ZQ, McGee S, Nakanishi K, Toombs WE, Johnston CF, Ashar MS, Vinten-Johansen J. Receptor-mediated cardioprotective effects of endogenous adenosine are exerted primarily during reperfusion after coronary occlusion in the rabbit. Circulation. 1993;88:709–719. doi: 10.1161/01.cir.88.2.709. [DOI] [PubMed] [Google Scholar]
- Zhao ZQ, Nakanishi K, McGee DS, Tan P, Vinten-Johansen J. A1 receptor mediated myocardial infarct size reduction by endogenous adenosine is exerted primarily during ischaemia. Cardiovasc Res. 1994;28:270–279. doi: 10.1093/cvr/28.2.270. [DOI] [PubMed] [Google Scholar]