

Abstract
The human NBC1 (SLC4A4) gene encodes the electrogenic sodium bicarbonate cotransporters kNBC1 and pNBC1, which are highly expressed in the kidney and pancreas, respectively. The HCO3−:Na+ stoichiometry of these cotransporters is an important determinant of the direction of ion flux. Recently we showed in a mouse proximal tubule (mPCT) cell line expressing kNBC1, that 8-Br-cAMP shifts the stoichiometry of the cotransporter from 3:1 to 2:1 via protein kinase A (PKA)-dependent phosphorylation of Ser982. pNBC1 has the identical carboxy-terminal consensus phosphorylation PKA site (KKGS1026), and an additional site in its amino-terminus (KRKT49). In this study we determined the potential role of these sites in regulating the function of pNBC1. The results demonstrated that in mPCT cells expressing pNBC1, PKA-dependent phosphorylation of Ser1026 following 8-Br-cAMP treatment shifted the stoichiometry from 3:1 to 2:1. The effect was electrostatic in nature as replacing Ser1026 with Asp resulted in a similar stoichiometry shift. In addition to shifting the stoichiometry, 8-Br-cAMP caused a significant increase in the 4,4′-dinitrostilbene-2,2′-disulfonic acid (DNDS)-sensitive basolateral membrane conductance (GDS) of cells expressing pNBC1, but not kNBC1. Although, the effect did not involve phosphorylation of Thr49, which was endogenously phosphorylated, replacing this residue with Asp or Ala abolished the 8-Br-cAMP-induced increase in GDS. In the mPEC pancreatic duct cell line, where endogenous pNBC1 functions with a HCO3−:Na+ stoichiometry of 2:1, 8-Br-cAMP increased GDS by ∼90 % without altering the stoichiometry or inducing phosphorylation of the cotransporter. The results demonstrate that phosphorylation of Ser1026 mediates the cAMP-dependent shift in the stoichiometry of pNBC1, whereas Thr49 plays an essential role in the cAMP-induced increase in GDS.
Of the currently characterized sodium bicarbonate cotransporters, NBC1 and NBC4 are known to be electrogenic (Romero et al. 1997; Gross et al. 2001a; Sassani et al. 2002). An important thermodynamic property of electrogenic sodium bicarbonate cotransporters is their HCO3−:Na+ stoichiometry, which determines the reversal potential of these proteins (Gross & Kurtz, 2002). The magnitude of the reversal potential relative to the membrane potential determines whether the cotransporter functions in an efflux or influx mode.
In humans, the NBC1 (SLC4A4) gene encodes two known electrogenic sodium bicarbonate cotransport proteins, kNBC1 and pNBC1, which differ in their amino-terminus (Abuladze et al. 2000). Both transporters possess 10 transmembrane segments with the N- and C-termini localised in the cytosol (Tatishchev et al. 2003). kNBC1 mediates basolateral sodium bicarbonate efflux in the proximal tubule (Burnham et al. 1997; Romero et al. 1997; Abuladze et al. 1998). In this nephron segment, kNBC1 functions with a 3:1 stoichiometry which places the reversal potential of the cotransporter positive relative to the basolateral membrane potential, driving the efflux of sodium and bicarbonate. pNBC1 mediates basolateral sodium bicarbonate influx in the pancreatic duct, and is also widely expressed in various other cell types including the duodenum, colon, heart, salivary gland, eye, brain, prostate, testis and thyroid (Abuladze et al. 1998; Marino et al. 1999; Bok et al. 2001; Gross et al. 2001a). We have recently shown that in the mPEC cell line derived from mouse pancreatic duct, the HCO3−:Na+ stoichiometry of the cotransporter is 2:1 (Gross et al. 2001a). This stoichiometry places the reversal potential of the cotransporter negative relative to the basolateral membrane potential, allowing pNBC1 to transport sodium and bicarbonate in the influx direction.
We have previously shown that the stoichiometry of pNBC1 is either 3:1 or 2:1 depending on the cell type in which it is expressed. In the mPEC and mCD (mouse collecting duct) cell lines, pNBC1 functions with a 2:1 stoichiometry whereas in mPCT cells, the cotransporter functions in the 3:1 mode (Gross et al. 2001a, b). These finding raise the possibility, that pNBC1 can potentially mediate bicarbonate influx or efflux depending on the cell type in which it is expressed in vivo. This variable stoichiometry is not unique to pNBC1 since the stoichiometry of kNBC1 has also been shown to be cell-dependent (Gross et al. 2001b). Moreover, we recently have shown that cAMP-mediated PKA-dependent phosphorylation of Ser982 shifts the stoichiometry of kNBC1 from 3:1 to 2:1 (Gross et al. 2001c).
Several hormones, which increase intracellular cAMP, decrease renal tubular bicarbonate absorption and increase pancreatic duct bicarbonate secretion (McKinney & Myers, 1980; Liu & Cogan, 1989; Ishiguro et al. 1996b). The shift in stoichiometry from 3:1 to 2:1 by protein kinase A- (PKA)-dependent phosphorylation of kNBC1 provides a potential mechanism by which parathyroid hormone (PTH) and dopamine decrease bicarbonate absorption in the proximal tubule (McKinney & Myers 1980; Kunimi et al. 2000). In pancreatic ducts, secretin stimulates transepithelial bicarbonate secretion (Ishiguro et al. 1996b) and basolateral sodium bicarbonate influx (Ishiguro et al. 1996a) via an increase in intracellular cAMP. The exact mechanism by which cAMP increases bicarbonate secretion in the pancreas is not known. It has been suggsted that cAMP may stimulate bicarbonate secretion (Ishiguro et al. 1996b) and Na+ and bicarbonate influx (Ishiguro et al. 1996a) in pancreatic ducts by activating apical cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channels, which results in depolarization of the basolateral membrane potential, thereby indirectly stimulating basolateral electrogenic sodium bicarbonate cotransport(Shumaker et al. 1999).
In the present study we demonstrate an alternative mechanism for stimulation of bicarbonate secretion in the pancreas that involves a direct, membrane potential-independent, stimulation of the cotransporter by cAMP. Here, we provide evidence that cAMP increases the conductance and Vmax of pNBC1. Mutagenesis experiments indicate that Thr49, at the amino-terminal consensus PKA phosphorylation site KRKT49, plays an essential role in the cAMP-induced increase in the cotransporter's conductance. In addition, we found that phosphorylation of Ser1026 in pNBC1 by cAMP-dependent PKA shifts the stoichiometry of the cotransporter from 3:1 to 2:1. Our data provide the first evidence for the distinct roles of the amino- and carboxy-termini in regulating the function of an electrogenic sodium bicarbonate cotransporter. A preliminary account of this work has been published in abstract form (Gross et al. 2002b).
METHODS
Cell culture
Most experiments were carried out with the mouse proximal tubule mPCT cell line, which lacks endogenous electrogenic sodium bicarbonate cotransport (Gross et al. 2001b). In some experiments, the mPEC cell line derived from mouse pancreatic ducts (Gross et al. 2001a), which endogenously expresses pNBC1, was used to determine the effect of 8-Br-cAMP treatment on GDS. Cell isolation, culture and general characterization were described previously (Gross et al. 2001a, b). Cells were studied between passages 15 and 25.
Transfection
Cells were transiently transfected with pNBC1, kNBC1 or with the indicated pNBC1 constructs as previously described (Gross et al. 2001b). For transfection, cells were grown on permeable support inserts, as previously described (Gross et al. 2001b). Cells were transfected with the corresponding plasmid using Lipofectamine (Gibco, Grand Island, NY, USA) according to the manufacturer's protocol. Mock-transfected cells were transfected with the vector only. All plasmids were purified with Endofree plasmid purification kit (Qiagen, Valencia, CA, USA) prior to their use.
Mutagenesis
The coding region of human wt-pNBC1 was cloned into the EcoRI and EcoRV sites in the pcDNA3.1 vector (Clontech, Palo Alto, CA, USA). An amino-terminal enhanced green fluorescent protein (EGFP) fusion protein was produced by inserting pNBC1 into the EcoRI and ApaI sites in the EGFP-C2 vector (Clontech). The QuikChange site-directed mutagenesis kit (Invitrogen, Carlsbad, CA, USA) was used to generate the mutant pNBC1 and EGFP-pNBC1 constructs. The mutations were verified by DNA sequencing.
Phosphorylation assays
EGFP-wt-pNBC1, EGFP-S1026A-pNBC1, and EGFP-pNBC1-T49A-S1026A mutants were transiently expressed in mPCT cells. Cells treated with 0.1 mm 8-Br-cAMP for 15 min or untreated cells were washed with PBS, collected by centrifugation at 2000 g for 10 min, and homogenized in 1 ml IPT buffer (60 mm Hepes, pH 7.4, 150 mm NaCl, 3 mm KCl, 25 mm sodium pyrophosphate, 5 mm EDTA, 5 mm EGTA, 1 mm Na3VO4, 50 mm NaF and 1 % Triton X-100) containing complete protease inhibitor cocktail and pepstatin A (Roche, Indianapolis, IN, USA). The cells were homogenized in a glass homogenizer and incubated for 1 h at 0 °C on a rotating rocker. After cetrifugation for 15 min at 14 000 g, the supernatants were precleared with 50 μl protein A–Sepharose and 50 μl protein G–Sepharose (both Amersham Biosciences, Piscataway, NJ, USA) by rocking for 2 h at 4 °C. The Sepharose beads then were spun down at 14 000 g for 15 s. To each supernatant, 1.5 μl rabbit polyclonal anti-GFP antibody (Abcam, Cambridge, UK) was added, and after incubation for 2 h at 4 °C, 15 μl protein A–Sepharose was added. The samples were incubated for 2 h at 4 °C on a rotating rocker, spun down at 14 000 g for 15 s and the beads were washed once with IPT buffer. Then the beads were washed twice with each of the following solutions: (1) 50 mm Tris-HCl, pH 7.5, 1 m NaCl, 1 % Triton X-100, and 1 % deoxycholate; (2) 50 mm Tris-HCl, pH 7.5, 1 m NaCl, and 0.1 % Triton X-100; (3) 50 mm Tris-HCl, pH 7.5, 100 mm NaCl, 1 % Triton X-100, 0.1 % SDS, and 0.5 % deoxycholate; (4) 50 mm Tris-HCl, pH 7.5, 100 mm NaCl, and 0.5 % SDS; and (5) 10 mm Tris-HCl, pH 7.5, and 150 mm NaCl. The immunoprecipitated proteins were eluted from the beads with 50 μl 30 mm glycine, pH 2.8, and immediately neutralized with 6 μl 200 mm Tris, pH 11. Samples were phosphorylated at pH 7.5 for 30 min at 30 °C in the presence of 100 mm Tris, pH 7.5, 10 mm glycine, 50 μm magnesium acetate, 180–200 U of the catalytic subunit of PKA (Promega, Madison, WI, USA) and 20 μCi [γ-32P]ATP (Amersham Biosciences). The reaction was terminated by adding 50 μl 2 × Laemmli buffer and incubated for 8 min at 65 °C. The samples were separated on 7.5 % polyacrylamide gel (Bio-Rad, Hercules, CA, USA) in the presence of sodium dodecyl sulfate. The proteins from the gel were electrotransferred onto nitrocellulose membranes (Amersham Biosciences). The membranes were blocked with 5 % dried milk solution in 20 mm Tris-HCl, pH 7.5, containing 140 mm NaCl (TBS) for 1 h. Affinity-purified rabbit polyclonal pNBC1-specific antibody was diluted in TBS at 1:1000. Mouse anti-rabbit HR-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) was used at 1:10 000 dilution. The bands were visualized with ECL kit and Hyperfilm-ECL from Amersham Biosciences. To visualize radioactivity, the nitrocellulose film was exposed to Kodak Biomax MS film (Kodak, Rochester, NY, USA).
Dephosphorylation assay
The EGFP-S1026A-pNBC1 and EGFP-T49-S1026A-pNBC1 mutant proteins were expressed in mPCT cells. The proteins were immunoprecipitated (see phosphorylation assay) using an anti-GFP antibody (Abcam) and protein A–Sepharose 4B (Amersham Biosciences). Immunoprecipitated EGFP-pNBC1 mutants attached to protein A–Sepharose beads were mixed with 1 ml dephosphorylation buffer (Roche) and were dephosphorylated with 5 U calf intestine alkaline phosphatase (Roche) at 20 °C for 18 h. The beads were washed 10 times with 50 mm Tris-HCl, pH 7.5, and the immunoprecipitated constructs were eluted from the beads and phosphorylated in vitro using the PKA catalytic subunit and [γ-32P] ATP as described in the ‘Phosphorylation assays’ section. The amount of EGFP-tagged pNBC1 protein was quantified using Western blotting with an anti-pNBC1 antibody (Bok et al. 2001).
Electrophysiology
HCO3−:Na+ stoichiometry
The stoichiometry of the cotransporter was determined from Erev and eqn (1), as described previously (Gross et al. 2001b):
| (1) |
where n is the number of bicarbonate anions cotransported with each sodium cation, and the subscripts i and o represent intra- and extracellular concentrations of the indicated ion. R, T and F have their usual meaning. Briefly, Erev of the cotransporter was determined from measurements of the current–voltage (I–V) relationships at a 5-fold Na+ sodium concentration gradient, as previously described (Gross et al. 2001b). Current through the cotransporter was defined as the 4,4′-dinitrostilbene-2,2′-disulfonic acid (DNDS)-sensitive current. Only high-resistance monolayers with transepithelial resistance of ≥ 1 kΩ cm2 were used in this study. For these monolayers, the DNDS-sensitive current was at least 20 % of the total transepithelial current. For experiments with transfected cells, only monolayers in which the DNDS-sensitive current was at least 10-fold larger than that of the corresponding mock-transfected cells were included in this study. About 30 % of all transfected cell monolayers met the inclusion criteria.
Short-circuit current (Isc) recording
To study the time course of the Isc response to apical adition of 8-Br-cAMP, mPEC and mPCT cells were grown on filters, mounted in an Ussing chamber and permeabilized apically with amphotericin B, as described previously (Gross et al. 2001b). A 5-fold Na+ concentration gradient (basolateral/apical = 50/10) was applied as described previously (Gross et al. 2001b). In Na+-free solutions, Na+ was replaced by N-methyl-d-glucamine (NMDG+). At the indicated times, 8-Br-cAMP (100 μm) was added acutely to the apical solution, while DNDS (1 mm) was added to the basolateral solution (Fig. 3A). In some experiments, mPCT cells expressing wt-pNBC1 were pretreated with the PKA inhibitor H-89 (Calbiochem, San Diego, CA, USA) or the inactive analogue H-85 (Seikagaku, Fulmouth, MA, USA).
Figure 3. Effect of 8-Br-cAMP on sodium and bicarbonate transport in mPEC cells.
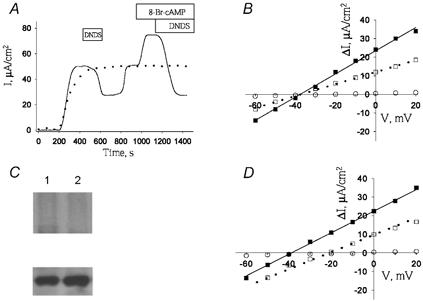
A, time course of Isc response to apical addition of 8-Br-cAMP (100 μm) in mPEC cells, in a bicarbonate-containing solution (continuous trace) or a bicarbonate-free solution (dotted trace). B, I–V relationship of sodium bicarbonate cotransport in mPEC cells in the absence (□) or presence (▪) of 8-Br-cAMP (100 μm), or in the nominal absence of bicarbonate (○). C upper panel, phosphorylation of wt-EGFP-pNBC1 (lanes 1 and 2) isolated from untreated mPEC cells (lane 1) or mPEC1 cells treated with 100 μm 8-Br-cAMP (lane 2). Lower panel, Western blots showing the amount of EGFP-tagged pNBC1 proteins used in the in vitro phosphorylation experiments using an affinity-purified rabbit polyclonal pNBC1 antibody. D, I–V relationship of wt-kNBC1 in mPCT cells in the absence (□) or presence (▪) of 8-Br-cAMP (100 μm), or of mock-transected mPCT cells (○). All Isc and I–V relationships were measured at 5-fold Na+ concentration gradient (basolateral/apical = 50/10) in apically permeabilized cells, as described in the Methods section.
DNDS-sensitive conductance (GDS) of the basolateral membrane
GDS was calculated from the slope of the ΔI–V plots in apically-permeabilized cell monolayers, where ΔI is the DNDS-sensitive current. For kinetic studies, Na+ concentrations were varied symmetrically in both the apical and basolateral solutions by replacing Na+ with NMDG+.
Materials
Amphotericin B, N-methyl-d-glucamine (NMDG), and all other salts were purchased from Sigma, (St Louis, MO, USA). DNDS was obtained from Pfaltz and Bauer (Waterbury, CT, USA). Filters were purchased from Millipore (Bedford, MA, USA).
Statistics
Experiments were performed at least four times. The results for the reversal potential and stoichiometry are presented as means ± s.e.m. Student's unpaired t tests were used for statistical analysis, with P < 0.05 considered significant.
RESULTS
When expressed in mPCT cells, wt-pNBC1 exhibited a reversal potential (Erev) of −21 mV which corresponded to a HCO3−:Na+ stoichiometry of 3:1 (Fig. 1A; Table 1). 8-Br-cAMP treatment shifted the cotransporter's Erev from −21 mV to −40 mV, P < 0.05 (Fig. 1A, Table 1). The change in Erev represents a corresponding shift in the HCO3−:Na+ stoichiometry from 3:1 to 2:1 (Table 1). In mPCT cells expressing EGFP-wt-pNBC1, the transport stoichiometry was also 3:1 and shifted to 2:1 following treatment with 8-Br-cAMP (Table 1). Using this construct, experiments were perfomed to determine whether 8-Br-cAMP treatment phosphorylates the cotransporter. As shown in Fig. 2A, 8-Br-cAMP induced the phosphorylation of EGFP-wt-pNBC1 in transformed mPCT cells expressing the cotransporter. To determine whether PKA mediated the effect of 8-Br-cAMP on the stoichiometry of wt-pNBC1, we pretreated mPCT cells expressing wt-pNBC1 with the PKA inhibitor H-89 for 15 min. In cells pre-incubated with H-89, 8-Br-cAMP failed to shift Erev of wt-pNBC1 significantly (−24 mV in the presence of H-89 vs.−21 mV in untreated cells, P = n.s.) (Table 1). In contrast, in cells pretreated for 15 min with the inactive analogue H-85, the effect of 8-Br-cAMP was not blocked (Table 1). These results suggested that the phosphorylation-induced shift in stoichiometry was mediated by PKA.
Figure 1. Current–voltage relationships of pNBC1 constructs.
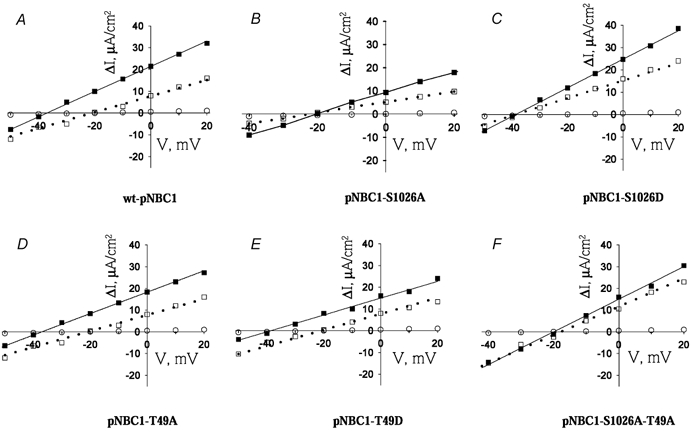
Current–voltage relationships of wt-pNBC1 (A), pNBC1-S1026A (B), pNBC1-S1026D (C), pNBC1-T49A (D), pNBC1-T49D (E) and pNBC1-S1026A-T49A (F), in mPCT cells in the absence (□) or presence (▪) of 8-Br-cAMP (100 μm) or in mock-transfected cells (○). Cells were pretreated with 8-Br-cAMP for 15 min prior to collection of I–V data. All I–V relationships were measured at 5-fold Na+ concentration gradient (basolateral/apical = 50/10) in apically permeabilized cells, as described in the Methods section.
Table 1.
Stoichiometry (n) of pNBC1 constructs expressed in mPCT cells
| wt | S1026A | S1026D | T49A | T49D | S1026A–T49A | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | Erev | n | Erev | n | Erev | n | Erev | n | Erev | n | Erev | n |
| None | −21.2±2 | 3.0±0.3 | −23.1 ±2 | 2.8 ±0.3 | −38.9 ±4 | 2.1 ±0.2* | −22.1 ±2 | 2.9 ±0.3 | −21.9 ±2 | 2.9 ±0.3 | −22.6 ±2 | 2.9 ±0.3 |
| (4) | (5) | (8) | (6) | (4) | (6) | |||||||
| (EGFP-tagged constructs) | −20.6 ±2 | 3.0 ±0.3 | −21.5 ±2 | 2.9 ±0.3 | −20.2 ±2 | 3.1 ±0.3 | ||||||
| (3) | (5) | (6) | ||||||||||
| 8-Br-cAMP | −39.7 ±4 | 2.1 ±0.2* | −22.2±2 | 2.9 ±0.3 | −43.8 ±4 | 1.9 ±0.2* | −37.8 ±4 | 2.1 ±0.3* | −39.1 ±4 | 2.0 ±0.2* | −21.3 ±2 | 3.0 ±0.3 |
| (5) | (4) | (8) | (5) | (4) | (4) | |||||||
| (EGFP-tagged constructs) | −36.7 ±4 | 2.1 ±0.2 | −18.1 ±2 | 3.3 ±0.3 | −40.1 ±4 | 2.0 ±0.3* | ||||||
| (4) | (4) | (4) | ||||||||||
| H-89→ 8-Br-cAMP | −23.8 ±2 (4) | 2.8 ±0.3 | ||||||||||
| (4) | ||||||||||||
| H-85→ 8-Br-cAMP | −41.7 ±4 | 2.0 ±0.2* | ||||||||||
| (6) | ||||||||||||
Erev values were calculated from current–voltage relationships measured at 5-fold Na+ concentration gradient (basolateral/apical = 50/10). Number. of experiments is shown in parentheses.
P <0.05 compared with the corresponding wild-type (wt)-pNBC1 construct in cells not treated with 8-Br-cAMP.
Figure 2. Phosphorylation of pNBC1 by PKA.
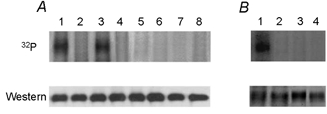
A upper panel, in vitro phosphorylation of EGFP-wt-pNBC1 (lanes 1 and 2), EGFP-pNBC1-T49A (lanes 3 and 4), EGFP-pNBC1-S1026A (lanes 5 and 6), EGFP-pNBC1-T49A-S1026A (lanes 7 and 8) isolated from untreated mPCT cells (lanes 1, 3, 5 and 7) or mPCT cells treated with 100 μm 8-Br-cAMP (lanes 2, 4, 6 and 8) for 15 min. Immunoprecipitated EGFP-pNBC1 constructs were eluted from protein A–Sepharose beads and then phosphorylated in vitro using the PKA catalytic subunit and [γ-32P]ATP. Representative results are shown. Lower panel, Western blots showing the amount of EGFP-tagged pNBC1 proteins used in the in vitro phosphorylation experiments with an affinity-purified rabbit polyclonal pNBC1 antibody. Lane assignments are as in upper panel. Treatment of the cells with 8-Br-cAMP did not effect the expression level of EGFP-tagged pNBC1 proteins. B upper panel, in vitro phosporylation of EGFP-pNBC1-S1026A (lanes 1 and 2) and EGFP-pNBC1-T49A-S1026A (lanes 3and 4) isolated from mPCT cells. Immunoprecipitated EGFP-pNBC1-S1026A and EGFP-pNBC1-T49A-S1026A bound to protein A–Sepharose beads were dephosphorylated in vitro with calf intestine alkaline phosphatase (lanes 1and 3) or were not treated with the alkaline phosphatase (lanes 2 and 4). The immunoprecipitated constructs were eluted from the beads and phosphorylated in vitro using the PKA catalytic subunit and [γ-32P]ATP. Representative results are shown. Lower panel, Western blots showing the amount of EGFP-tagged pNBC1 proteins used in the in vitro phosphorylation experiments using an affinity purified rabbit polyclonal pNBC1 antibody. Lane assignments are as in upper panel.
pNBC1 has two PKA consensus phosphorylation sites: KKGS1026, in its carboxy-terminus and KRKT49 in its amino-terminus. Mutagenesis experiments were done to determine the importance of these sites in mediating the shift in stoichiometry of the cotransporter. Ser1026 was substituted with alanine and the S1026A mutant was expressed the in mPCT cells. The S1026A mutant had an Erev of −23 mV (Fig. 1B) in these cells, which under the conditions of the experiment corresponded to a 3:1 stoichiometry (Table 1). Treatment of these cells with 8-Br-cAMP did not alter the cotransporter's Erev (−22 mV) nor the stoichiometry (Fig. 1B and Table 1). To determine whether 8-Br-cAMP induces phosphorylation of Ser1026, we compared the phosphorylation status of the T49A and T49A-S1026A mutant constructs to that of wt-pNBC1 using back phosphorylation experiments. As shown in Fig. 2A, treatment with 8-Br-cAMP induced the phosphorylation of only the wt-pNBC1 and T49A constructs, indicating that Ser1026 was phosphorylated. These findings suggest that phosphorylation of Ser1026 plays an important role in mediating the shift in stoichiometry from 3:1 to 2:1 in response to addition of 8-Br-cAMP.
To further examine whether the added negative charge of phosphorylated Ser1026 is responsible for the shift in stoichiometry of pNBC1 from 3:1 to 2:1, we replaced Ser1026 with aspartate. Aspartate is commonly used to mimic the negative charge of a phosphorylated serine residue (Hoeffler et al. 1994; Kwak et al. 1999; Lin et al. 2000; Gross et al. 2002a). In mPCT cells the S1026D mutant had an Erev of −39 mV (Fig. 1C) which corresponded to a HCO3−:Na+ stoichiometry of 2:1. Treatment of these cells with 8-Br-cAMP did not have a significant effect on Erev (−44 mV, P = n.s., Fig. 1C) or on the stoichiometry (2:1, P = n.s., Table 1).
To determine the role of the amino-terminus consensus phosphorylation site at Thr49 in modulating the function of pNBC1, we replaced Thr49 with alanine. In mPCT cells expressing the pNBC1-T49A mutant Erev was −22 mV (Fig. 1D), which corresponded to stoichiometry of 3:1 (Table 1). Treatment of the cells with 8-Br-cAMP shifted Erev to −38 mV (Fig. 1D) and the stoichiometry of this mutant from 3:1 to 2:1 (Table 1). Furthermore, unlike EGFP-wt-pNBC1, the EGFP-pNBC1-S1026A mutant isolated from mPCT cells failed to incorporate 32P in vitro (Fig. 2A). This result suggests that Thr49 may be either constitutively phosphorylated in vivo or inaccesible for phosphorylation in vitro. To distinguish between the two possibilities, we dephosphorylated in vitro with alkaline phosphatase the S1026A and S1016A-T49A constructs expressed in mPCT cells, followed by in vitro phosphorylation using [γ-32P]-ATP and the catalytic subunit of PKA (see Methods section). The data indicate that while the S1026A mutant incorporated 32P, the S1016A-T49A mutant did not, suggesting that Thr49 is endogenously phosphorylated in untreated mPCT cells (Fig. 2B).
The functional data obtained with the EGFP-pNBC1-T49A mutant suggested that the amino-terminal PKA consensus phosphorylation site is not involved in the regulation of the stoichiometry of the cotransporter. To determine whether replacing Thr49 with a negative residue to mimic the negative charge of Thr49-phosphate would affect the stoichiometry of the cotransporter, we replaced Thr49 with an aspartate residue. When expressed in mPCT cells, the pNBC1-T49D mutant exhibited an Erev of −22 mV (Fig. 1E) which corresponded to a stoichiometry of 3:1 (Table 1). Treatment of the cells with 8-Br-cAMP shifted Erev of this mutant to −39 mV (Fig. 1E) and the stoichiometry to 2:1 (Table 1).
To confirm the results obtained with the amino- and carboxy-terminal mutants of pNBC1, we replaced both Thr49 and Ser1026 in pNBC1 with alanine residues and expressed the pNBC1-S1026A-T49A mutant in mPCT cells. This mutant exhibited an Erev of −23 mV (Fig. 1F) which corresponded to a stoichiometry of 3:1 (Table 1). Treatment of the cells with 8-Br-cAMP did not affect the Erev significantly (−21 mV, Fig. 1F) nor the stoichiometry (3:1, Table 1). Furthermore, 8-Br-cAMP did not induce phosphorylation of this mutant (Fig. 2A).
It is known that secretin stimulates pancreatic duct bicarbonate secretion via a cAMP-dependent mechanism (Ishiguro et al. 1996b). Since we have previously shown that the stoichiometry of wt-pNBC1 expressed endogenously in the pancreatic duct mPEC cell line is 2:1 (Gross et al. 2001a), experiments were performed using mPEC cells to determine whether 8-Br-cAMP treatment has an additional effect on pNBC1 function operating in the 2:1 mode, which differs from its ability to shift the cotransporter's stoichiometry. Figure 3A shows the effect on Isc of apical addition of 8-Br-cAMP to mPEC cells. As can be seen, the agonist increased the DNDS-sensitive component of Isc∼2-fold (upper trace). The increase in Isc was acute (continuous line) and was bicarbonate dependent (dotted line in Fig. 3A). The HCO3−:Na+ stoichiometry of pNBC1 in mPEC cells was 2:1 (Erev 38 mV, Fig. 3B). 8-Br-cAMP treatment did not have a significant effect on Erev (−36 mV, P = n.s., Fig. 3A) or on the stoichiometry (2:1, P = n.s., Fig. 3B). Unlike in mPCT cells, 8-Br-cAMP did not induce phosphorylation of the EGFP-wt-pNBC1 construct expressed in mPEC cells, suggesting that in these cells Ser1026 in pNBC1 is endogenously phosphorylated (Fig. 3C).
While 8-Br-cAMP did not change the HCO3−:Na+ cotransport stoichiometry in mPEC cells, or the phosphorylation state of EGFP-wt-pNBC1 in these cells, it increased the DNDS-sensitive conductance GDS by 88 % (Fig. 3A and B, Table 2). The two known variants of the human NBC1 gene, pNBC1 and kNBC1, differ only in their extreme amino-terminus. In contrast to the profound effect of 8-Br-cAMP on GDS in mPEC cells expressing wt-pNBC1, the agonist did not affect GDS in mPCT cells expressing wt-kNBC1 (Fig. 3D, Table 2).
Table 2.
Effect of 8-Br-cAMP on basolateral membrane DNDS-sensitive conductance (GDS)
| Cell line | pNBC1 mutant | GDS before cAMP (mS cm−2) | GDS after cAMP (mS cm−2) | Change (%) |
|---|---|---|---|---|
| mPCT | wt | 0.31 ± 0.03 (10) | 0.41 ± 0.04 | +32* |
| S1026A | 0.27 ± 0.03 (8) | 0.38 ± 0.03 | +41* | |
| S1026D | 0.28 ± 0.03 (9) | 0.39 ± 0.04 | +39* | |
| T49A | 0.36 ± 0.04 (12) | 0.40 ± 0.04 | +11 | |
| T49D | 0.34 ± 0.03 (8) | 0.35 ± 0.04 | +3.0 | |
| S1026A–T49D | 0.40 ± 0.04 (10) | 0.43 ± 0.04 | +7.5 | |
| wt–kNBC1† | 0.32 ± 0.03 (12) | 0.36 ± 0.04 | +12 | |
| mPEC | 0.32 ± 0.03 (4) | 0.60 ± 0.05 | +88* |
Number of experiments is shown in parentheses. Change in GDS reported for same cells before and after treatment for 15 min with apical 8-Br-cAMP (100 μM). Data for EGFP-tagged and untagged mutants were pooled together.
P < 0.05.
GDS for mPCT cells expressing wt-kNBC1 was calculated from I–V data in Gross et al. (2001c).
The 8-Br-cAMP-evoked increase in GDS was not unique to mPEC cells, but was also observed in mPCT cells expressing wt-pNBC1 (Fig. 1, Table 2). In order to determine the molecular mechanism of the 8-Br-cAMP-induced increase in GDS, further experiments were performed using various pNBC1 mutant constructs expressed in mPCT cells, which lack endogenous electrogenic sodium bicarbonate cotransport. The data are summarized in Table 2. 8-Br-cAMP induced a significant increase in GDS in cells expressing wt-pNBC1, pNBC1-S1026A, pNBC1-S1026D and their corresponding EGFP-tagged counterparts, but no significant change in GDS of cells expressing pNBC1-T49A, pNBC1-S1026A-T49A or their corresponding EGFP-tagged counterparts. To determine whether a negative charge at position Thr49 would mimic the Thr49-phosphate, we replaced Thr49 with aspartate. When the pNBC1-T49D mutant was expressed in mPCT cells, there was no significant change in the conductance in cells treated with 8-Br-cAMP. Therefore unlike the carboxy-terminus PKA consensensus phosphorylation site, the added negative charge in the pNBC1-T49D mutant does not mimic Thr49-phosphate.
The increase in GDS of cells expressing pNBC1 in response to treatment with 8-Br-cAMP could result from an increase in Vmax and/or an increase in substrate affinity of the cotransporter. To address these two possibilities, we characterized the Km and Vmax of wt-pNBC1 in mPCT and mPEC cells for Na+. Our data indicate that in both cell lines, the DNDS-sensitive conductance to Na+ is saturable for both treated and untreated cells (Fig. 4A and C). Double-reciprocal analysis of the data revealed that 8-Br-cAMP increased theVmax in mPEC cells for Na+ by 85 %, and in mPCT cells by 37 % (Fig. 4B and D, Table 3). Although a small increase in the Michaelis constant (Km) for Na+ in response to 8-Br-cAMP was observed, the effect was not significant (Table 3). In separate biotinylation experiments, there was no significant change in the membrane expression of pNBC1 after treatment with 8-Br-cAMP (data not shown).
Figure 4. Effect of 8-Br-cAMP on the kinetics of wt-pNCB1.
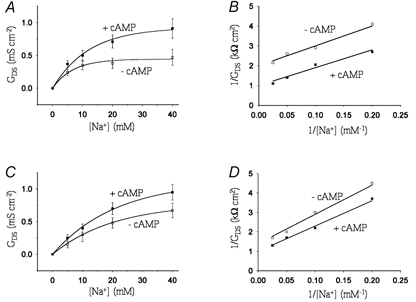
A and C, DNDS-sensitive conductance (GDS) as a function of Na+ concentration in the absence (□) and presence (▪) of 8-Br-cAMP (100 μm) in mPEC (A) and mPCT (C) cells. B and D, double reciprocal plot analyses of the data for mPEC (B) and mPCT (D) cells.
Table 3.
Effect of 8-Br-cAMP on Vmax and Km of wt-pNBC1 for Na+
| Cell line | Parameter | Control | + 8-Br-cAMP | Change (%) |
|---|---|---|---|---|
| mPEC | Vmax (mS cm−2) | 0.54 ± 0.1 (4) | 1.00 ± 0.2 (4) | +85* |
| Km (mM) | 6.23 ± 1.3 (4) | 7.21 ± 1.3 (4) | +16† | |
| mPCT | Vmax (mScm−2) | 0.73 ± 0.2 (6) | 1.00 ± 0.2 (6) | +37* |
| Km(mM) | 9.2 ± 2.1 (6) | 10.4 ± 2.3 (6) | +13† |
Number of experiments is shown in parentheses. Vmax of cells treated with 8-Br-cAMP were normalized to a value of 1.00.
P < 0.05.
n.s.
DISCUSSION
In the present study we determined the effects of the cAMP agonist 8-Br-cAMP on the HCO3−:Na+ cotransport stoichiometry and the basolateral conductance of cells expressing pNBC1. The data indicate that both the amino and carboxy-terminal consensus PKA sites can be phosphorylated, and that their PKA-dependent phosphorylation has different effects on the transport characteristics of pNBC1. PKA-dependent phosphorylation of pNBC1-Ser1026 shifts the HCO3−:Na+ stoichiometry of the cotransporter from 3:1 to 2:1 whereas the amino-terminal PKA consensus phosphorylation site does not play a role in mediating the cAMP-induced shift in the cotransporter stoichiometry. The unique amino-terminus of pNBC1 was found to play an important role in modulating the cAMP-induced increase in the current through the cotransporter. The dual effect of cAMP in modulating both the stoichiometry and the current through pNBC1 is unique to this variant of the NBC1 (SLC4A4) gene, as cAMP induces only a stoichiometric shift in the kNBC1 cotransporter.
kNBC1 possesses a PKA phosphorylation site in its carboxy-terminus which is identical to the carboxy-terminal site in pNBC1. In the proximal tubule, PKA-dependent phosphorylation of kNBC1-Ser982 modulates the function of the cotransporter by shifting the HCO3−:Na+ stoichiometry from 3:1 to 2:1 thereby providing a potential mechanism for decreasing net transepithelial bicarbonate absorption (Gross et al. 2001c). Hormones which raise intracellular cAMP levels, such as dopamine (Kunimi et al. 2000; Wiederkehr et al. 2001) or parathyroid hormone (McKinney & Meyers, 1980; Collazo et al. 2000) can decrease net basolateral bicarbonate efflux by shifting the stoichiometry of kNBC1 from 3:1 to 2:1. Unlike kNBC1 in the proximal tubule, which normally mediates basolateral bicarbonate efflux, pNBC1 functioning with a 2HCO3−:1Na+ stoichiometry in pancreatic duct cells would mediate Na+ and HCO3− influx (Gross et al. 2001a), as long as the basolateral membrane potential is positive relative to the reversal potential for the cotransporter. Our results are compatible with the hypothesis that in pancreatic ducts in vivo, pNBC1 normally functions with a 2:1 stoichiometry because Ser1026 is endogenously phosphorylated, whereas in the proximal tubule, kNBC1-Ser982 is unphosphorylated under basal conditions and therefore the cotransporter functions in the 3:1 mode. Whether these stoichiometric differences are due to cellular differences in endogenous cAMP concentration or compartmentalization requires further study. Furthermore, whether there are tissue-specific differences in the complexation of the carboxy-terminus of these cotransporters with adenylyl cyclase/PKA complex and/or specific phosphatases by anchoring/scaffolding proteins such as A-kinase anchoring proteins (Tumlin, 1997) remains to be determined.
Both in vivo and in vitro studies have shown that kNBC1 can function in either a 2:1 or 3:1 mode in mammalian cells (Muller-Berger et al. 1997; Gross et al. 2001b). Moreover, we have previously shown in in vitro studies that pNBC1 can also function in both modes in the same cell (Gross et al. 2001b). There are no in vivo studies in which the stoichiometry of pNBC1 has been directly measured in any tissue. Additionally, it is unknown whether in vivo the stoichiometry of pNBC1 can be physiologically regulated. In the in vivo pancreatic duct under basal conditions, the stoichiometry of pNBC1 is inferred to be 2:1 because pNBC1 mediates cellular sodium bicarbonate influx. Our results raise the possibility that the stoichiometry of pNBC1 may shift in vivo providing the pancreatic duct with an additional mechanism for modulating the magnitude of transepithelial bicarbonate transport. Additional studies are needed to address whether pNBC1 in the native pancreatic duct can ever function in the 3:1 mode. In this regard, it would be important in future studies to determine the in vivo stoichiometry of pNBC1 in the various tissues in which it is expressed.
Secretin stimulates transepithelial bicarbonate secretion and basolateral sodium bicarbonate influx in pancreatic ducts (Ishiguro et al. 1996a, b) via an increase in intracellular cAMP. Studying pancreatic ducts from the rat perfused in vitro, Novak & Pahl (1993) reported that secretin depolarized the basolateral membrane potential by 35 mV. Although the magnitude of this depolarization is sufficient in principle to stimulate basolateral bicarbonate influx through an electrogenic sodium bicarbonate cotransport process, it is not clear whether a depolarization of that magnitude also occurs in vivo since these experiments were done with low Cl−-containing solutions. Our finding that 8-Br-cAMP increases GDS in mPEC cells by ∼90 % provides an alternative mechanism for secretin-evoked stimulation of bicarbonate secretion in this tissue.
Our results demonstrate that 8-Br-cAMP significantly increases the DNDS-sensitive basolateral conductance (GDS) of cells expressing pNBC1. Kinetic analysis of the effect of the agonist revealed that the increase in conductance was due to an increase in Vmax, without a significant effect on the Km of the cotransporter to Na+ (Table 3). The increase in Vmax is probably due to a kinetic change of individual transporters since biotinylation experiments failed to detect a change in transporter number following 8-Br-cAMP treatment. Similarly, Weinman et al. (2001) have recently shown that cAMP does not alter the surface expression of NBC1 in BSC1 cells. To determine which of the two PKA consensus phosphorylation sites might be responsible for the increase in conductance, we analysed the changes in current through pNBC1 mutants expressed in mPCT cells. These cells do not express functional electrogenic sodium bicarbonate cotransport (Gross et al. 2001b). As can be seen from Table 2, the largest increase in conductance following 8-Br-cAMP treatment was obtained with the two carboxy-terminal mutants (S1026A and S1026D) followed by wt-pNBC1, with no significant change in conductance detected for the three amino-terminal mutants. These data suggest that the increase in conductance requires phosphorylation of the amino-terminal, but not the carboxy-terminal, PKA consensus phosphorylation site. Of interest, the change in conductance with both carboxy-terminal mutants was ∼35 % larger than the change obtained with wt-pNBC1 suggesting that the phosphorylation of the carboxy-terminal PKA consensus site attenuates the increase in flux mediated by phosphorylation of the amino-terminal site. The mechanism of the apparent crosstalk between the amino and carboxy termini remains to be determined. The intracellular localization of both the amino- and carboxy-termini of pNBC1 (Tatishchev et al. 2003) is compatable with this finding.
Unlike the effect on pNBC1, 8-Br-cAMP shifted the stoichiometry of kNBC1 without altering its conductance significantly. These results have potentially important implications regarding the in vivo effect of hormones such as dopamine and PTH which decrease proximal tubule basolateral sodium bicarbonate efflux. Specifically, it would be predicted that by shifting the stoichiometry of kNBC1 from 3:1 to 2:1 without significantly altering the flux, the inhibition of basolateral bicarbonate efflux by dopamine and PTH is less than it would be were the conductance also to have increased as we found for pNBC1. Therefore, the differences between various cells types regarding their specific need to modulate the stoichiometry or flux through NBC1 cotransporters in response to cAMP might account for their tissue-specific expression which has thus far remained unexplained.
Our data indicate that the amino-terminal PKA concensus phosphorylation site of pNBC1 is endogenously phosphorylated in mPCT cells prior to treatment with 8-Br-cAMP. The analysis of several pNBC1 mutants also demonstrated that the Thr49 residue is required for 8-Br-cAMP-induced increase in the current through the cotransporter. These data are compatible with a model wherein cAMP treatment phosphorylates one or more additional proteins, which interact with the phosphorylated pNBC1 amino terminus thereby altering the flux through the cotransporter via changes in its structure and/or membrane insertion. The finding that 8-Br-cAMP treatment did not increase the current through the pNBC1-T49D mutant suggests that, in addition to the negative charge on Thr49-phosphate, specific size constraints are involved. Further studies are required to determine which putative protein(s) can potentially interact with the amino-terminus of pNBC1. Generalizing this model to the in vivo pancreatic duct, we would predict that (1) endogenous ductal cell cAMP levels might be sufficient to induce the phosphorylation of both the amino and carboxy terminal pNBC1 consensus phosphorylation sites, and that (2) secretin, by further increasing cAMP levels, results in the phosphorylation of one or more proteins which interact with the pNBC1 amino-terminus thereby increasing the flux through the cotransporter. Additional experiments are required to explore the specific details of this model in isolated pancreatic ducts.
Acknowledgments
This work was supported by grants from the Cystic Fibrosis Foundation, and the American Heart Association to EG. IK is supported by NIH grants, DK-58563, DK-63125, DK-07789, the Max Factor Family Foundation, the Richard and Hinda Rosenthal Foundation, and the NKF of Southern California J891002.
References
- Abuladze N, Lee I, Newman D, Hwang J, Boorer K, Pushkin A, Kurtz I. Molecular cloning, chromosomal localization, tissue distribution, and functional expression of the human pancreatic sodium bicarbonate cotransporter. J Biol Chem. 1998;273:17689–17695. doi: 10.1074/jbc.273.28.17689. [DOI] [PubMed] [Google Scholar]
- Abuladze N, Song M, Pushkin A, Newman D, Lee I, Nicholas S, Kurtz I. Structural organization of the human NBC1 gene: kNBC1 is transcribed from an alternative promoter in intron 3. Gene. 2000;251:109–122. doi: 10.1016/s0378-1119(00)00204-3. [DOI] [PubMed] [Google Scholar]
- Bok D, Schibler MJ, Pushkin A, Sassani P, Abuladze N, Naser Z, Kurtz I. Immunolocalization of electrogenic sodium-bicarbonate cotransporters pNBC1 and kNBC1 in the rat eye. Am J Physiol Renal Physiol. 2001;281:F920–935. doi: 10.1152/ajprenal.2001.281.5.F920. [DOI] [PubMed] [Google Scholar]
- Burnham CE, Amlal H, Wang Z, Shull GE, Soleimani M. Cloning and functional expression of a human kidney Na+:HCO3− cotransporter. J Biol Chem. 1997;272:19111–19114. doi: 10.1074/jbc.272.31.19111. [DOI] [PubMed] [Google Scholar]
- Collazo R, Fan L, Hu MC, Zhao H, Wiederkehr MR, Moe OW. Acute regulation of Na+/H+ exchanger NHE3 by parathyroid hormone via NHE3 phosphorylation and dynamin-dependent endocytosis. J Biol Chem. 2000;275:31601–31608. doi: 10.1074/jbc.M000600200. [DOI] [PubMed] [Google Scholar]
- Gross E, Abuladze N, Pushkin A, Kurtz I, Cotton CU. The stoichiometry of the electrogenic sodium bicarbonate cotransporter pNBC1 in mouse pancreatic duct cells is 2HCO3−:1Na+ J Physiol. 2001a;531:375–382. doi: 10.1111/j.1469-7793.2001.0375i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross E, Hawkins K, Abuladze N, Pushkin A, Cotton CU, Hopfer U, Kurtz I. The stoichiometry of the electrogenic sodium bicarbonate cotransporter NBC1 is cell-type dependent. J Physiol. 2001b;531:597–603. doi: 10.1111/j.1469-7793.2001.0597h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross E, Hawkins K, Pushkin A, Sassani P, Dukkipati R, Abuladze N, Hopfer U, Kurtz I. Phosphorylation of Ser982 in the sodium bicarbonate cotransporter kNBC1 shifts the HCO3−:Na+ stoichiometry from 3:1 to 21 in murine proximal tubule cells. J Physiol. 2001c;537:659–665. doi: 10.1111/j.1469-7793.2001.00659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross E, Kurtz I. Structural determinants and significance of regulation of electrogenic Na+-HCO3− cotransporter stoichiometry. Am J Physiol Renal Physiol. 2002;283:F876–887. doi: 10.1152/ajprenal.00148.2002. [DOI] [PubMed] [Google Scholar]
- Gross E, Pushkin A, Abuladze N, Fedotoff O, Kurtz I. Regulation of the sodium bicarbonate cotransporter kNBC1 function: Role of Asp986, Asp988 and kNBC1-carbonic anhydrase II binding. J Physiol. 2002a;544:679–685. doi: 10.1113/jphysiol.2002.029777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross E, Pushkin A, Abuladze N, Hawkins K, Dukkipati R, Kurtz I. Phosphorylation of Ser1026 in pNBC1 shifts the HCO3−:Na+ stoichiometry from 3:1 to 2:1. J Am Soc Nephrol. 2002b;13:61A. [Google Scholar]
- Hoeffler WK, Levinson AD, Bauer EA. Activation of c-Jun transcription factor by substitution of a charged residue in its N-terminal domain. Nucleic Acids Res. 1994;22:1305–1312. doi: 10.1093/nar/22.7.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro H, Steward MC, Lindsay AR, Case RM. Accumulation of intracellular HCO3− by Na+-HCO3− cotransport in interlobular ducts from guinea-pig pancreas. J Physiol. 1996a;495:169–178. doi: 10.1113/jphysiol.1996.sp021582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro H, Steward MC, Wilson RW, Case RM. Bicarbonate secretion in interlobular ducts from guinea-pig pancreas. J Physiol. 1996b;495:179–191. doi: 10.1113/jphysiol.1996.sp021583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunimi M, Seki G, Hara C, Taniguchi S, Uwatoko S, Goto A, Kimura S, Fujita T. Dopamine inhibits renal Na+:HCO3− cotransporter in rabbits and normotensive rats but not in spontaneously hypertensive rats. Kidney Int. 2000;57:534–543. doi: 10.1046/j.1523-1755.2000.00873.x. [DOI] [PubMed] [Google Scholar]
- Kwak YG, Hu N, Wei J, George AL, Jr, Grobaski TD, Tamkun MM, Murray KT. Protein kinase A phosphorylation alters Kvbeta1. 3 subunit-mediated inactivation of the Kv1.5 potassium channel. J Biol Chem. 1999;274:13928–13932. doi: 10.1074/jbc.274.20.13928. [DOI] [PubMed] [Google Scholar]
- Lin YF, Jan YN, Jan LY. Regulation of ATP-sensitive potassium channel function by protein kinase A-mediated phosphorylation in transfected HEK293 cells. EMBO J. 2000;19:942–955. doi: 10.1093/emboj/19.5.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu FY, Cogan MG. Angiotensin II stimulates early proximal bicarbonate absorption in the rat by decreasing cyclic adenosine monophosphate. J Clin Invest. 1989;84:83–91. doi: 10.1172/JCI114174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney TD, Myers P. Bicarbonate transport by proximal tubules: effect of parathyroid hormone and dibutyryl cyclic AMP. Am J Physiol. 1980;238:F166–174. doi: 10.1152/ajprenal.1980.238.3.F166. [DOI] [PubMed] [Google Scholar]
- Marino CR, Jeanes V, Boron WF, Schmitt BM. Expression and distribution of the Na+-HCO3− cotransporter in human pancreas. Am J Physiol. 1999;277:G487–494. doi: 10.1152/ajpgi.1999.277.2.G487. [DOI] [PubMed] [Google Scholar]
- Muller-Berger S, Nesterov VV, Fromter E. Partial recovery of in vivo function by improved incubation conditions of isolated renal proximal tubule. II. Change of Na-HCO3 cotransport stoichiometry and of response to acetazolamide. Pflugers Arch. 1997;434:383–391. doi: 10.1007/s004240050411. [DOI] [PubMed] [Google Scholar]
- Novak I, Pahl C. Effect of secretin and inhibitors of HCO3−/H+ transport on the membrane voltage of rat pancreatic duct cells. Pflugers Arch. 1993;425:272–279. doi: 10.1007/BF00374178. [DOI] [PubMed] [Google Scholar]
- Romero MF, Hediger MA, Boulpaep EL, Boron WF. Expression cloning and characterization of a renal electrogenic Na/HCO3 cotransporter. Nature. 1997;387:409–413. doi: 10.1038/387409a0. [DOI] [PubMed] [Google Scholar]
- Sassani P, Pushkin A, Gross E, Gomer A, Abuladze N, Dukkipati R, Carpenito G, Kurtz I. Functional characterization of NBC4: a new electrogenic sodium-bicarbonate cotransporter. Am J Physiol Cell Physiol. 2002;282:C408–416. doi: 10.1152/ajpcell.00409.2001. [DOI] [PubMed] [Google Scholar]
- Shumaker H, Amlal H, Frizzell R, Ulrich CD, 2nd, Soleimani M. CFTR drives Na+-nHCO3− cotransport in pancreatic duct cells: a basis for defective HCO3− secretion in CF. Am J Physiol. 1999;276:C16–25. doi: 10.1152/ajpcell.1999.276.1.C16. [DOI] [PubMed] [Google Scholar]
- Tatishchev S, Abuladze N, Pushkin A, Newman D, Liu W, Weeks D, Sachs G, Kurtz I. Identification of membrane topography of the electrogenic sodium bicarbonate cotransporter pNBC1 by in vitro transcription/translation. Biochemistry. 2003;42:755–765. doi: 10.1021/bi026826q. [DOI] [PubMed] [Google Scholar]
- Tumlin JA. Expression and function of calcineurin in the mammalian nephron: physiological roles, receptor signaling, and ion transport. Am J Kidney Dis. 1997;30:884–895. doi: 10.1016/s0272-6386(97)90100-1. [DOI] [PubMed] [Google Scholar]
- Weinman EJ, Evangelista CM, Steplock D, Liu MZ, Shenolikar S, Bernardo A. Essential role for NHERF in cAMP-mediated inhibition of the Na+-HCO3− co-transporter in BSC-1 cells. J Biol Chem. 2001;276:42339–42346. doi: 10.1074/jbc.M106153200. [DOI] [PubMed] [Google Scholar]
- Wiederkehr MR, Di SF, Collazo R, Quinones H, Fan L, Murer H, Helmle-Kolb C, Moe OW. Characterization of acute inhibition of Na/H exchanger NHE-3 by dopamine in opossum kidney cells. Kidney Int. 2001;59:197–209. doi: 10.1046/j.1523-1755.2001.00480.x. [DOI] [PubMed] [Google Scholar]