

Abstract
Pancreatic polypeptides (PPs) such as neuropeptide Y (NPY) and peptide YY (PYY) exert profound, vagally mediated effects on gastrointestinal (GI) motility and secretion. Whole-cell patch clamp recordings were made from brainstem slices containing identified GI-projecting rat dorsal motor nucleus of the vagus (DMV) neurons to determine the mechanism of action of PPs. Electrical stimulation of nucleus tractus solitarii (NTS) induced excitatory postsynaptic currents (EPSCs) that were reduced in a concentration-dependent manner by NPY and PYY (both at 0.1–300 nm) in 65 % of the neurons. An increase in the paired-pulse ratio without changes in the postsynaptic membrane input resistance or EPSC rise and decay time suggested that the effects of PPs on EPSCs were due to actions at presynaptic receptors. The Y1 and Y2 receptor selective agonists [Leu31,Pro34]NPY and NPY(3–36) (both at 100 nm) mimicked the inhibition of NPY and PYY on the EPSC amplitude. The effects of 100 nm NPY, but not PYY, were antagonized partially by the Y1 receptor selective antagonist BIBP3226 (0.1 μm). In addition, the inhibition of the EPSC amplitude induced by NPY, but not PYY, was attenuated partially by pretreatment with the α2 adrenoceptor antagonist yohimbine (10 μm), and occluded partially by the α2 adrenoceptor agonist UK14,304 (10 μm) as well as by pretreatment with reserpine. Pretreatment with a combination of BIBP3226 and yohimbine almost completely antagonized the NPY-mediated effects on EPSCs. Contrary to the inhibition of EPSCs, perfusion with PPs had no effect on the amplitude of inhibitory postsynaptic currents (IPSCs) and a minimal effect on a minority of DMV neurons. Differences in the receptor subtypes utilized and in the mechanism of action of NPY and PYY may indicate functional differences in their roles within the circuitry of the dorsal vagal complex (DVC).
Nucleus tractus solitarii (NTS) is the recipient of sensory information from the gastrointestinal (GI) tract relayed centrally via vagal afferent nerves (Rogers et al. 1999; reviewed by Gillis et al. 1989; Travagli & Rogers, 2001; Travagli et al. 2003). The NTS then integrates and transfers this information to the dorsal motor nucleus of the vagus (DMV) using, in the main, GABA and glutamate as neurotransmitters (Travagli et al. 1991; Hornby, 2001). Subsequently, the DMV provides the final modulated vagal parasympathetic motor output to the subdiaphragmatic viscera (reviewed by Travagli & Rogers, 2001; Travagli et al. 2003).
Neuropeptide Y (NPY) and peptide YY (PYY), both members of the pancreatic polypeptide family of peptides, have been shown to act centrally to exert profound, vagally mediated actions on GI function (Geoghegan et al. 1993; Chen & Rogers, 1995, 1997; Chen et al. 1996, 1997; Yoneda et al. 1997; Fujimiya et al. 2000; Fujimiya & Inui, 2000; Kawakubo et al. 2002; Yang, 2002).
Binding sites with similar affinities for NPY and PYY, specifically Y1 and Y2 receptors, have been identified within the dorsal vagal complex (DVC, i.e. the NTS and DMV) (Leslie et al. 1988; Lynch et al. 1989; Dumont et al. 1990). Furthermore, a putative Y3 receptor that recognizes NPY but not PYY has been proposed to be present in the NTS (Grundemar et al. 1991; Glaum et al. 1997; Lee & Miller, 1998).
The lack of readily available pharmacological tools has prevented investigation of the effects of Y3 receptor activation on GI function; however, Y1 and Y2 receptor-mediated differential effects of NPY and PYY on GI function have been demonstrated in previous studies. For example, application of PYY either by intravenous injection or by microinjection directly into the DVC, has been shown to inhibit gastric motility, gastric acid secretion and pancreatic secretion, in addition to increasing intestinal transit time (Adrian et al. 1985; Buell & Harding, 1989; Masuda et al. 1994; Chen & Rogers, 1995; Naruse et al. 2002; Yang, 2002). Such actions were abolished by vagotomy and were mimicked by selective Y2 receptor agonists (Chen & Rogers, 1995; Chen et al. 1997). Extracellular recordings in vivo and in vitro suggested that the principal actions of PYY were to inhibit cholinergic vagal efferent outflow to the GI tract via direct action at Y2 receptors located on DMV neuronal cell bodies, even though the activation of an inhibitory non-adrenergic, non-cholinergic pathway has also been postulated (Chen & Rogers, 1997). When administered in high concentrations, however, PYY has been shown to activate gastric function (Chen & Rogers, 1995; Yang et al. 1998).
Similar differences in the GI responses to pancreatic polypeptides (PPs) have also been noted with NPY. For example, an increase in GI motility and secretion was seen following NPY administration (Geoghegan et al. 1993; Chen et al. 1997; Yoneda et al. 1997). Other studies, however, have noted a decrease in GI motility and secretion (Matsuda et al. 1993; Chen et al. 1997; Yang et al. 2000; Ishiguchi et al. 2001). Further investigation revealed that the effects of NPY on gastric motility depend upon the activity of the GI tract at the time of application. In fact, under basal conditions, application of NPY to the DVC causes an increase in gastric motility; however, if gastric motility is stimulated, NPY has no additional stimulatory effects, indeed NPY reduces gastric motility (Chen et al. 1997).
It was suggested that such brainstem-mediated diverse effects on gastric motility were related to different actions at Y1 versus Y2 receptors, with activation of Y1 receptors by NPY causing GI stimulation while activation of Y2 receptors by PYY caused GI inhibition (Chen et al. 1997). To date, however, the cellular determinants of the actions of NPY and PYY in the dorsal vagal complex (DVC; i.e. NTS and DMV) have not been examined.
The aims of this study were: (1) to investigate the effects of NPY and PYY on the membrane of identified GI-projecting DMV neurons; (2) to investigate their effects on excitatory and inhibitory synaptic transmission within the DVC; and (3) to investigate the receptor subtype(s) responsible for their effect. Preliminary accounts have been presented at the 2001 Digestive Disease Week meeting (Atlanta, GA, USA).
METHODS
Retrograde tracing
Retrograde tracers were applied to discrete GI regions as described previously (Browning et al. 1999). Briefly, 12-day-old Sprague-Dawley rat pups of either sex were anaesthetized deeply with a 6 % solution of halothane with air, in accordance with the University of Michigan Committee on the Use and Care of Animals. Anaesthesia was maintained during surgery by placing the head of the animal in a custom-made chamber through which the halothane–air mixture was administered. The depth of anaesthesia was monitored (abolition of the foot pinch withdrawal reflex) prior to and during surgery. An abdominal laparotomy was performed and crystals of the retrograde tracer 1,1′-dioctadecyl-3,3,3′,3′-tetramethylinodcarbocyanine perchlorate (DiI) were applied to the serosal surface of the major curvature of the gastric fundus or corpus, to the antrum/pylorus, to the duodenum (to the antimesenteric surface at the level of the bifurcation of the hepatic and pancreatico–duodenal junction) or to the caecum (at the level of the ileo–caecal junction). The dye was embedded in place with epoxy resin that was allowed to harden before the entire surgical area was flushed with warmed sterile saline and the wound closed with 5/0 suture. The animal was allowed to recover for 10–15 days prior to removal of the brainstem for electrophysiological study.
Electrophysiology
The method of the removal and slicing of the brainstem has been described already (Travagli et al. 1991; Browning et al. 1999). Briefly, the rats were anaesthetized deeply with halothane before being killed by induction of a bilateral pneumothorax. The brainstem was removed and placed in oxygenated physiological saline solution at 4 °C. Using a vibratome, six to eight coronal slices (200 μm thick) containing the DVC were cut. The brainstem slices were stored in oxygenated physiological saline at 35 ± 1 °C for at least 90 min prior to use.
A single slice was transferred to the oxygenated perfusion chamber on the stage of a Nikon E600FN microscope equipped with TRITC epifluorescence filters. The slice was held in place with a nylon mesh and maintained at 35 ± 1 °C by continuous perfusion with warmed Krebs solution. Once a DiI-filled neuron was identified, its identity was confirmed under bright field illumination using DIC optics. The brief (< 2 s) periods of illumination used to identify a DiI-filled neuron do not alter its electrophysiological properties (Mendelowitz & Kunze, 1991; Browning et al. 1999).
Whole-cell recordings were performed with patch pipettes of resistance 4–7 MΩ when filled with potassium gluconate solution (see below for composition) using a single electrode voltage clamp amplifier (Axoclamp 1D, Axon Instruments, Union City, CA, USA). Data were filtered at 2 kHz, digitized via a Digidata 1320 interface and stored on an IBM PC utilizing pCLAMP8 software (Axon Instruments). Only recordings with a series resistance (i.e. pipette + access resistance) < 15 MΩ were used. For a neuron to be considered acceptable for recording, it had to be stable at the holding potential, the action potential evoked following injection of DC had to overshoot and the membrane had to return to baseline after the after-hyperpolarization. Data analysis was performed using pCLAMP8 software.
Electrical stimulation
Excitatory postsynaptic currents (EPSCs) were recorded from neurons that were voltage clamped at −60 mV. Inhibitory postsynaptic currents (IPSCs) were recorded from neurons that were voltage clamped at −50 mV and bathed continuously with Krebs solution containing the non-selective glutamate antagonist kynurenic acid (1 mm). Bipolar tungsten electrodes (WPI Ltd, Sarasota, FL, USA) were used to stimulate electrically the NTS. Pairs of stimuli (0.1–1.0 ms, 10–500 μA) were applied using a Master 8 stimulator (AMPI, Jerusalem, Israel) every 20 s to evoke submaximal EPSCs (average 299 ± 10 pA) or submaximal IPSCs (average 307 ± 32 pA). Note that the variations in EPSC or IPSC amplitude in any one neuron were ±5 %. The value of each evoked current was thus taken as the average of at least three traces.
Drug application and statistical analysis
Drugs were applied to the bath via a series of manually operated valves. Results were compared before and after drug administration, with each neuron serving as its own control (Student's paired t test). When different concentrations of BIBP3226 were tested, the results were analysed using the group t test. Results are expressed as means ± s.e.m. with significance defined as P < 0.05.
Cells were classified as responders if perfusion with PPs (100 nm) induced at least a 10 % variation in the peak amplitude of the evoked EPSCs or IPSCs. Similarly, neurons were classified as responders if perfusion with PPs (100 nm) induced a membrane current larger than 15 pA. Comparisons of responsive vs. non-responsive neurons in the treatment groups were analysed using the χ2 test. Results are expressed as means ± s.e.m. with significance defined as P < 0.05.
The concentration–response curves for NPY and PYY (Fig. 1) were obtained using data from neurons on which at least three different concentrations of PPs were tested. The responses to perfusion with 0.3 and 1 μm NPY or PYY did not induce a significant decrease in the amplitude of the EPSCs. These data were not included because the small inhibition and the ±5 % variation in the EPSC amplitude generated unreliable measurements. The curves were generated using the regression analysis function in the Statistica software (Statsoft, Tulsa, OK, USA). The concentration of PPs that produced the half-maximum drug response (EC50) was estimated using a third order polynomial regression (Pitts et al. 1990).
Figure 1. NPY and PYY induced a concentration-dependent inhibition in evoked EPSC amplitude.

A, representative traces showing the concentration-dependent inhibition of the evoked EPSC amplitude by NPY. Full recovery was achieved between successive applications of NPY. Each trace is the average of 3 EPSCs. Holding potential, −60 mV. B, graphical representation of the concentration-dependent effects of NPY and PYY expressed as percentage reduction in EPSC amplitude. Note that both NPY and PYY induced inhibitions of similar magnitudes and had similar estimated IC50 values. (Each data point represents the average of 3–18 neurons for NPY, and 5–22 neurons for PYY.)
Drugs and solutions
Krebs solution (mm): 126 NaCl, 25 NaHCO3, 2.5 KCl, 1.2 MgCl, 2.4 CaCl2, 1.2 NaH2PO4 and 11 dextrose, maintained at pH 7.4 by bubbling with 95 % O2–5 % CO2. Intracellular solution (mm): 128 potassium gluconate; 10 KCl, 0.3 CaCl2, 1 MgCl2, 10 Hepes, 1 EGTA, 2 Na2ATP and 0.25 NaGTP, adjusted to pH 7.35 with KOH. DiI was purchased from Molecular Probes (Eugene, OR, USA); NPY, PYY, [Leu31,Pro34]NPY and NPY(3–36), diprotin-A and yohimbine were purchased from Bachem (King of Prussia, PA, USA). All other chemicals were purchased from Sigma Chemical Company (St Louis, MO, USA).
RESULTS
NPY, PYY, [Leu31,Pro34]NPY or NPY(3–36) was applied to a total of 350 GI-projecting DMV neurons (255 gastric, 95 intestinal); 86 of these neurons were tested with more than one agonist. The response to pharmacological treatments did not reveal any qualitative (i.e. percentage of responsive neurons and type of response) or quantitative (i.e. amplitude of the response) difference between gastric- and intestinal-projecting neurons; all the data were therefore pooled.
Presynaptic effects of PPs – excitatory synaptic transmission
EPSCs were evoked upon stimulation of the adjacent NTS and recorded from identified DMV neurons. Perfusion with NPY and PYY (0.1–300 nm for both) induced a concentration-dependent inhibition in amplitude of evoked EPSCs in the majority of DMV neurons (94/145 or 65 % of neurons for NPY and 80/125 or 64 % of neurons for PYY; Fig. 1). The remaining neurons were unresponsive to NPY or PYY. The effects of NPY and PYY were qualitatively and quantitatively similar: both produced similar maximum decrease in EPSC amplitude at 100 nm (30 ± 1.7 % reduction of EPSCs peak amplitude for NPY and 32 ± 1.9 % reduction for PYY) and had similar estimated IC50 values (approximately 40 nm).
In neurons in which NPY (100 nm) or PYY (100 nm) decreased EPSC amplitude, the ratio of the EPSCs evoked by two identical electrical pulses delivered a few milliseconds apart increased from a control value of 0.82 ± 0.04 to 1.07 ± 0.05 (for NPY, P < 0.05; Fig. 2A and B) and from 0.80 ± 0.03 to 0.96 ± 0.04 (for PYY, P < 0.05). Such an alteration in the paired-pulse ratio is suggestive of a presynaptic site of action.
Figure 2. NPY and PYY decreased the EPSC amplitude via actions at presynaptic sites.

A, representative traces showing the inhibition in EPSC amplitude induced by NPY (100 nm) and alteration in the paired-pulse ratio (the paired-pulse ratio compares the amplitude of the second current (C2) with that of the first current (C1); EPSCs were evoked 40 ms apart). Each trace is the average of 3 EPSCs. B, the evoked EPSCs obtained in the presence of NPY (100 nm) were normalised to the amplitude of the control trace. The changes in the paired pulse ratio are more visible as a result. C, application of PYY (100 nm) does not affect the membrane responses to hyperpolarizing voltage commands between −50 and −60 mV.
The presynaptic site of action of NPY and PYY was further confirmed by monitoring the input resistance of the DMV neuron before and during PP application. In 19 neurons in which NPY (n = 9) or PYY (n = 10) decreased the amplitude of the EPSCs, the value of the input resistance was 462 ± 59 MΩ. In the presence of NPY or PYY the input resistance was 447 ± 69 MΩ (P > 0.05; Fig. 2C). In these cells, the EPSC rise time was 1.53 ± 0.11 ms in control and 1.48 ± 0.12 ms following NPY or PYY (P > 0.05). Similarly, the EPSC decay time was 2.81 ± 0.22 ms in control and 2.84 ± 0.22 ms following NPY or PYY (P > 0.05).
The response to NPY or PYY did not show tachyphylaxis. In fact, repeated superfusions of the preparation with 100 nm NPY or PYY 15 min apart gave similar results (Fig. 3A). In detail, the first superfusion of NPY decreased the EPSC amplitude by 24 ± 4.3 % (from 163 ± 26 pA in control to 122 ± 17 pA in NPY; P < 0.05vs. control; n = 4) while the second superfusion of NPY decreased the EPSC amplitude by 27 ± 5.7 % (from 163 ± 33 pA in control to 115 ± 19 pA in NPY; P < 0.05vs. control; P > 0.05vs. first application). Similarly, the first superfusion of PYY decreased the EPSC amplitude by 30 ± 5.1 % (from 219 ± 31 pA in control to 156 ± 33.3 pA in PYY; P < 0.05vs. control; n = 4) while the second superfusion of PYY decreased the EPSC amplitude by 31 ± 7.5 % (from 192 ± 20.3 pA in control to 130 ± 14 pA in PYY; P < 0.05vs. control; P > 0.05vs. first application).
Figure 3. The presynaptic inhibitory actions of NPY and PYY were mimicked by Y1 and Y2 receptor selective agonists.

A1, perfusion with NPY (100 nm) decreased the amplitude of the evoked EPSCs; A2, following a 10 min washout, NPY perfusion induced a similar inhibition of the evoked EPSCs. B, representative traces showing that the Y1 receptor selective agonist [Leu31,Pro34]NPY (100 nm) inhibited the amplitude of the evoked EPSCs. Pretreatment with the Y1 receptor selective antagonist BIBP3226 (0.1 μm) prevented the inhibition induced by [Leu31,Pro34]NPY. C, in another neuron, EPSCs were evoked as in A. The Y2 receptor selective agonist NPY(3–36) (100 nm) induced an inhibition in the amplitude of the evoked EPSCs that was unaffected by the Y1 receptor selective antagonist BIBP3226 (1 μm). Each trace is the average of 3 EPSCs. Holding potential, −60 mV.
Y1 and Y2 receptor selective agonists mimicked the inhibitory effects of NPY and PYY
The Y1 receptor selective agonist [Leu31,Pro34]NPY was applied to 89 neurons (64 gastric- and 25 intestinal-projecting) and induced an inhibition in amplitude of evoked EPSCs in approximately 53 % of the neurons (55 % gastric- and 48 % intestinal-projecting neurons). [Leu31,Pro34]NPY (100 nm) induced a 23 ± 1.8 % inhibition in EPSC amplitude (P < 0.05) that was accompanied by an increase in the paired-pulse ratio (from 0.72 ± 0.04 to 0.91 ± 0.06; P < 0.05). Such inhibitory effects were prevented by pretreatment with the Y1 receptor selective antagonist, BIBP3226 (Rudolf et al. 1994; Jacques et al. 1995). In four neurons, [Leu31,Pro34]NPY induced a 26 ± 7.4 % inhibition in evoked EPSC amplitude. Following wash out and 10 min perfusion with 0.1 μm BIBP3226, the inhibition induced by [Leu31,Pro34]NPY was reduced to 1 ± 2.6 % (P > 0.05 compared with control; P < 0.05 compared with inhibition in the absence of antagonist).
Perfusion with the Y2 receptor selective agonist NPY(3–36) (100 nm) was applied to 68 neurons (47 gastric- and 21 intestinal-projecting DMV neurons) and inhibited evoked EPSC amplitude in 41 % of neurons (66 % gastric, 48 % intestinal). The maximum inhibition in EPSC amplitude induced by NPY(3–36) (100 nm) was 26 ± 2.1 % (P < 0.05) and was accompanied by an increase in the paired-pulse ratio (0.87 ± 0.06 to 1.08 ± 0.08, P < 0.05). The presynaptic inhibitory effects of 100 nm NPY(3–36) were unaffected by pretreatment with the Y1 antagonist BIBP3226, even at a concentration (1 μm) that is 10-fold higher than that required to completely antagonize the effects of [Leu31,Pro34]NPY. Specifically, in four neurons, NPY(3–36) induced a 21 ± 6.0 % inhibition in evoked EPSC amplitude. Following wash out and 10 min perfusion with BIBP3226, NPY(3–36) induced a 24 ± 11.2 % inhibition (P < 0.05 compared with control; P > 0.05 compared with inhibition in the absence of antagonist; Fig. 3C).
The Y1 antagonist, BIBP3226, prevented the effects of NPY but not PYY
BIBP3226 attenuated the ability of NPY, but not PYY, to inhibit EPSC amplitude. In detail, 100 nm NPY induced a 30 ± 4.5 % inhibition in EPSC amplitude; following wash out and 10 min pretreatment with 0.1 μm BIBP3226, the NPY-induced inhibition was attenuated partially to 19 ± 3.3 % (n = 8; P < 0.05). In another set of neurons, 100 nm NPY induced a 20 ± 2.8 % inhibition in EPSC amplitude; following wash out and 10 min pretreatment with 1 μm BIBP3226, the NPY-induced inhibition was antagonized completely to 1 ± 4.1 % (n = 5; P > 0.05vs. control; P < 0.05vs. BIBP3226 0.1 μm; Fig. 3C). In contrast, even at 1 μm BIBP3226 had no effect on the inhibition of EPSC amplitude induced by 100 nm PYY. In detail, PYY perfusion induced a 22 ± 3.1 % inhibition in EPSC amplitude; in the presence of 1 μm BIBP3226, PYY induced a 19 ± 3.6 % inhibition (n = 5; P < 0.05 compared with control; P > 0.05 compared with inhibition in the absence of BIBP3226).
Both NPY and PYY are subject to enzymatic cleavage by endogenous diaminopeptidase IV (DAP IV) to form selective Y2 receptor agonists such as NPY(3–36) and PYY(3–36) (Grandt et al. 1993; Mentlein et al. 1993). Since the inhibitory actions of NPY were completely prevented by the Y1 receptor selective antagonist BIBP3226 (0.1 μm), one can assume that enzymatic degradation of NPY to form a Y2 receptor selective agonist does not occur to a significant degree. The actions of PYY, however, were unaffected by the Y1 antagonist, even at the higher concentration (1 μm), raising the possibility that PYY was cleaved to a Y2 receptor selective agonist. In order to ascertain whether, under the current experimental conditions, PYY was prevented from exerting any actions at presynaptic Y1 receptors because of enzymatic cleavage to a Y2 receptor selective ligand, we tested whether BIBP3226 attenuated the inhibitory effects of PYY in the presence of the DAP IV inhibitor diprotin-A (1 mm). In detail, in four neurons, PYY induced a 33 ± 4.5 % inhibition in evoked EPSC amplitude. Following 10 min superfusion with a combination of diprotin-A and 1 μm BIBP3226 (which per se had no effect on the EPSC amplitude), the PYY-induced inhibition was unchanged at 32 ± 5.3 % (P < 0.05 compared with control; P > 0.05 compared with PYY in the absence of peptidase inhibitor and antagonist).
Interactions of PPs with α adrenoceptors: studies with agonists
Since many reports have demonstrated that the effects of NPY are related to noradrenaline (NA) release (Wiley & Owyang, 1987; Martire & Pistritto, 1992; Khanna et al. 1993; Sawa et al. 1995; Kapoor & Sladek, 2001), we assessed the effects of pharmacological manipulations of adrenoceptors on the inhibition of EPSC amplitude induced by NPY and PYY.
In six neurons, NPY (100 nm) induced a 26 ± 3.9 % inhibition in EPSC amplitude (P < 0.05). In those same neurons, perfusion with the α2 adrenoceptor agonist UK14,304 (1 μm) induced 22 ± 2.5 % inhibition in EPSC amplitude (P < 0.05). In the presence of UK14,304, however, the inhibition induced by NPY was reduced to 12 ± 5.3 % (P < 0.05 compared with control, P < 0.05 compared with the inhibition in the absence of UK14,304). However, the occlusion of the inhibitory effects of NPY in the presence of α2 adrenoceptor activation was not due to a limitation in the maximum possible inhibition in EPSC amplitude; in fact, in three neurons, UK14,304 induced a 47 ± 11.0 % inhibition in EPSC amplitude (P < 0.05). Addition of methionine enkephalin (1 μm) caused an additional 35 ± 11.5 % reduction in EPSC amplitude reducing EPSC amplitude to 18 ± 11.5 % of control (P < 0.05 compared with inhibition in the absence of methionine enkephalin, P < 0.05 compared with control),
In contrast, perfusion with UK14,304 did not occlude the inhibition of EPSC amplitude induced by 100 nm PYY (29 ± 6.9 and 26 ± 7.4 % in PYY and in PYY + UK14,304, respectively; n = 6, P > 0.05) or by 100 nm [Leu31,Pro34]NPY (20 ± 3.5 and 28 ± 4.2 % in [Leu31,Pro34]NPY and in [Leu31,Pro34]NPY + UK14,304, respectively; n = 5, P > 0.05).
Interactions of PPs with α adrenoceptors: studies with antagonists
In four neurons, NPY (100 nm) induced a 25 ± 4.4 % inhibition in EPSC amplitude. Following wash out and 10 min superfusion with the α1 adrenoceptor agonist phenylephrine (10 μm), which itself had no effect on EPSC amplitude (98.6 ± 3.4 % of control; P > 0.05), the inhibition of EPSC amplitude induced by NPY was unchanged at 27.1 ± 1.7 % (P > 0.05 compared with inhibition in the absence of phenylephrine, P < 0.05 compared with control). Similarly, in four neurons, PYY (100 nm) induced a 24 ± 4.2 and a 25 ± 1.9 % inhibition in evoked EPSC amplitude in control and in the presence of phenylephrine, respectively (P > 0.05 compared with inhibition in the absence of phenylephrine, P < 0.05 compared with control; data not shown).
In five neurons, NPY induced a 32 ± 2.9 % inhibition in EPSC amplitude (P < 0.05) that was reduced to 19 ± 4.5 % following 10 min pretreatment with the α2 adrenoceptor antagonist yohimbine (10 μm; P < 0.05vs. control, P < 0.05 compared with inhibition in the absence of yohimbine; Fig. 5). In contrast, yohimbine pretreatment did not antagonize the inhibition induced by 100 nm PYY (29 ± 6.9 and 26 ± 7.4 % in PYY and in PYY + yohimbine, respectively; n = 6; P < 0.05vs. control; P > 0.05 compared with inhibition in the absence of yohimbine; Fig. 5) or by 100 nm[Leu31,Pro34]NPY (27 ± 7.7 and 32 ± 6.3 % in [Leu31,Pro34]NPY and in [Leu31,Pro34]NPY + yohimbine; n = 5, P > 0.05).
Figure 5. The inhibitory effects of NPY, but not PYY, involve the evoked release of NA.

PYY (100 nm) induced an inhibition in evoked EPSC amplitude that was unaffected by pretreatment with the α2 adrenoceptor antagonist yohimbine (10 μm). Conversely NPY (100 nm) induced an inhibition of evoked EPSCs that was partially prevented by pretreatment with yohimbine. Following treatment with reserpine, however, the NPY-induced inhibition was unaffected by yohimbine and completely antagonized by 0.1 μm BIBP3226. *P < 0.05vs. NPY alone.
The Y1 antagonist BIBP3226 and the α2 antagonist yohimbine prevented the effects of NPY
Pretreatment with 0.1 μm BIBP3226 and 10 μm yohimbine completely antagonized the ability of NPY to inhibit EPSC amplitude. In detail, 100 nm NPY induced a 32 ± 7.2 % inhibition in EPSC amplitude; following wash out and 10 min pretreatment with BIBP3226, the NPY-induced inhibition was attenuated partially to 23 ± 5.2 % (P < 0.05vs. NPY alone). Following wash out of NPY and 10 min perfusion with BIBP and yohimbine, the NPY-induced inhibition was antagonized almost completely to 5 ± 7.4 % (n = 3; P > 0.05vs. control; P < 0.05vs. BIBP3226 0.1 μm; Fig. 4).
Figure 4. The inhibitory effects of NPY involve the activation of both Y1 and α2 presynaptic receptors.

NPY (100 nm) induced an inhibition in evoked EPSC amplitude that was antagonized partially by pretreatment with the Y1 receptor selective antagonist BIBP3226 (0.1 μm) but antagonized completely by a pretreatment with a combination of BIBP3225 (0.1 μm) and yohimbine (10 μm). *P < 0.05vs. NPY alone; ** P < 0.05vs. NPY + BIBP3226.
The effects of NPY were attenuated by pretreatment with reserpine
The data described above suggest that the inhibitory effects of NPY might involve the activation of inhibitory adrenoceptors. To ascertain whether this involves a direct interaction of NPY with presynaptic adrenoceptors or an indirect action via the release of NA, recordings were made from 10 neurons from eight rats which underwent chemical sympathectomy with reserpine (5 mg kg−1i.p.) at 48 and 24 h prior to experimentation (Tricklebank et al. 1984). In these neurons, NPY (100 nm) induced a 30 ± 3.2 % inhibition in evoked EPSC amplitude (P < 0.05vs. control). In the presence of yohimbine (10 μm), the inhibition induced by NPY was 27 ± 2.7 % (n = 6; P < 0.05vs. control; P > 0.05 compared with NPY in the absence of yohimbine), i.e. following pretreatment with reserpine, the ability of yohimbine to partially attenuate the actions of NPY was ablated (Fig. 5). Furthermore, in the four remaining neurons from reserpine-treated rats, perfusion with NPY induced a 29 ± 3.4 % inhibition in evoked EPSC amplitude (P < 0.05vs. control). In the presence of 0.1 μm BIBP3226, the NPY effects were completely antagonized (i.e. a 0 ± 3.5 % inhibition; Fig. 5).
Responses to multiple peptides
Out of 38 neurons in which the actions of both NPY and PYY were assessed, both peptides were effective in reducing the amplitude of evoked EPSCs in 10 neurons. In a further nine neurons, PYY (but not NPY) inhibited excitatory synaptic transmission while in the remaining 19 neurons, neither peptide had any effect.
In 28 neurons in which the actions of both the Y1 receptor selective agonist [Leu31,Pro34]NPY and the Y2 receptor selective agonist NPY(3–36) (100 nm for both) were assessed, in eight neurons both peptides inhibited EPSC amplitude. In three neurons, however, only [Leu31,Pro34]NPY had any effect while in five neurons, only NPY(3–36) reduced evoked EPSC amplitude. In the remaining 12 neurons, neither agonist had any effect.
In 6 of the 10 neurons in which the actions of both NPY and the Y1 receptor selective agonist [Leu31,Pro34]NPY (100 nm for both) were assessed, both peptides inhibited EPSC amplitude. In one neuron, however, only NPY had any effect. There were no occasions in which [Leu31,Pro34]NPY but not NPY was effective in decreasing the amplitude of evoked EPSCs.
In 5 of 10 neurons in which the actions of both PYY and the Y2 receptor selective agonist NPY(3–36) (100 nm for both) were assessed, both peptides inhibited EPSC amplitude. There were no occasions when PYY but not NPY(3–36) or vice versa, was effective in decreasing the amplitude of evoked EPSCs. In the remaining five neurons, neither agonist had any effect. Results are summarized in Table 1.
Table 1.
Response to multiple peptides
| NPY and PYY | [Leu31,Pro34] NPY and NPY(3–36) | NPY and [Leu31,Pro34]NPY | PYY and NPY (3–36) | |
|---|---|---|---|---|
| Both responsive | 10 | 8 | 6 | 5 |
| Neither responsive | 19 | 12 | 3 | 5 |
| NPY only | 0 | — | 1 | — |
| PYY only | 8 | — | — | 0 |
| [Leu31, Pro34]NPY only | — | 3 | 0 | — |
| NPY (3-36) only | — | 5 | — | 0 |
NPY, neuropeptide Y; PYY, peptide YY.
Presynaptic effects of PPs – inhibitory synaptic transmission
NPY (100–300 nm) and PYY (100–300 nm) were applied to 19 neurons. Neither NPY (n = 8) nor PYY (n = 11) had any effect on evoked IPSC amplitude. In detail, in control conditions the amplitude of evoked IPSCs was 307 ± 53 pA; in the presence of NPY (100 nm) the IPSCs amplitude was 311 ± 53 pA (P > 0.05). Similarly, in control the amplitude of evoked IPSCs was 307 ± 42 pA compared with 297 ± 39 pA in the presence of PYY (P > 0.05; Fig. 6).
Figure 6. NPY and PYY did not alter evoked IPSC amplitude.
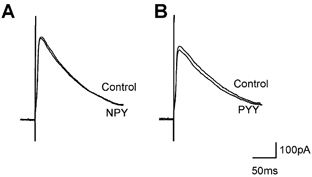
Neurons were voltage clamped at −50 mV and IPSCs evoked by electrical stimulation of the NTS. Superfusion with NPY (100 nm; A) or PYY (100 nm; B) did not have any effect on the amplitude of evoked IPSCs. Each trace is the average of 3 IPSCs.
Postynaptic effects of PPs
The postsynaptic response of NPY and PYY (both at 100 nm) did not differ either qualitatively or quantitatively in any neuronal type tested. An outward current of 28 ± 2.8 pA was induced by perfusion with NPY or PYY in 26 out of the 150 DMV neurons analysed, while an inward current of 27 ± 2.7 pA was induced in another 20 DMV neurons. The remaining 104 neurons either showed no response or responded with a change in current below the arbitrarily set limit of acceptance (15 pA). The low percentage of responsive neurons and the small amplitude of the postsynaptic currents prevented the pursuit of a thorough voltage clamp study.
DISCUSSION
Using whole-cell patch clamp recordings from identified GI-projecting neurons of the rat DMV, we have demonstrated that the main effect of PPs in the DVC is to inhibit glutamatergic synaptic transmission between the NTS and the DMV rather than to alter GABAergic synaptic transmission or exert a direct effect on the DMV preganglionic motoneurons themselves. Our pharmacological evidence suggests that different receptors of the NPY family are involved. Specifically, activation of Y1, Y2 and another NPY-preferring receptor, possibly the putative Y3 receptor, inhibits excitatory synaptic transmission from the NTS to GI-projecting DMV neurons. The effects of the putative Y3 receptor activation are mediated by release of NA and its subsequent interaction with α2 adrenoceptors.
Even though the reduction in EPSC amplitude by PPs may appear relatively modest (∼30 %), it should be kept in mind that relatively small variations in synaptic inputs can have dramatic effects on the spontaneously active DMV neurons and on GI motility and secretion. In fact, a similar reduction in the GABAergic input from NTS to DMV (Browning & Travagli, 2001) is likely to underlie the severalfold increase in gastric motility observed upon administration of 5HT and TRH (McCann et al. 1988; Chi et al. 1996).
The present study indicates that NPY exerts its presynaptic inhibitory actions on glutamate currents via interactions with both the Y1 receptor and with another receptor, possibly the putative Y3 receptor, which, in turns, triggers the release of catecholamines with a subsequent α2 adrenoceptor-mediated decrease in EPSCs. Conversely, PYY appears to reduce EPSC amplitude via a non-Y1 and a non-Y3 receptor, presumably a Y2 receptor. Such conclusions are reached through several lines of evidence.
First, the presence of both Y1 and Y2 receptors on nerve terminals within the DVC was confirmed by the effectiveness of the receptor selective agonists, [Leu31,Pro34]NPY and NPY(3–36), respectively, in decreasing the evoked EPSC amplitude.
Second, the Y1 receptor selective antagonist BIBP3226, used at a concentration (0.1 μm) that prevented fully the presynaptic inhibitory actions of the Y1 selective agonist [Leu31,Pro34]NPY, attenuated, but did not completely antagonize, the presynaptic inhibitory action of NPY, but had no effect on the ability of PYY to inhibit evoked EPSC amplitude. These data, in conjunction with data showing that all the neurons that are inhibited by [Leu31,Pro34]NPY are also inhibited by NPY, support the conclusion that a portion of the inhibitory actions of NPY are mediated by Y1 receptors.
Conversely, PYY acts at sites other than Y1 and Y3 receptors. Our data show that all the neurons responding to PYY also respond to NPY(3–36) (Table 1) and that even at 1 μm, BIBP3226 does not antagonize the inhibitory effects of PYY, supporting the conclusion that the receptors that PYY interacts with are neither Y1 nor Y3 receptors but are probably Y2 receptors.
The explanation for such unexpected Y1 and Y2 receptor selectivity is unclear, because NPY and PYY are supposed to have similar affinities for both Y1 and Y2 receptors (Michel et al. 1999). It is unlikely that the lack of effect of PYY at Y1 receptors is due to degradation of PYY to a Y2-preferring ligand. In fact, inhibition of the endogenous enzyme DAP IV, which cleaves the peptide to a Y2-preferring metabolite (Grandt et al. 1993; Mentlein et al. 1993), does not uncover any latent Y1 receptor activity of PYY. On the other hand, it is likely that activation of another receptor, such as the NPY-preferring Y3 receptor (Grundemar et al. 1991; Glaum et al. 1997; Lee & Miller, 1998), is involved in the presynaptic inhibitory actions of NPY since some of its effects had a pharmacological profile different from that of PYY.
The actions of NPY and PYY also differ in the involvement of NA in their presynaptic inhibitory effects. While neither NPY nor PYY effects are occluded by the α1 adrenoceptor agonist phenylephrine, the inhibitory effects of NPY, but not PYY, are attenuated partially by α2 adrenoceptor blockade and are occluded partially by α2 adrenoceptor activation. The fact that the inhibitory actions of [Leu31,Pro34]NPY or PYY were not affected by α2 adrenoceptor agonists or antagonists suggests that, in addition to acting at Y1 receptors to inhibit glutamatergic synaptic transmission directly, NPY also acts at other receptors, possibly the putative Y3 receptor. Activation of these receptors, in turn, would induce the release of NA and activation of presynaptic α2 adrenoceptors that then inhibit excitatory synaptic transmission (Bertolino et al. 1997).
The DVC receives a dense innervation of noradrenergic nerve terminals (Rea et al. 1982) principally from noradrenergic nerve cell bodies within the A2 cell group of the NTS (Fukuda et al. 1987) and from the locus coeruleus (Ter Horst et al. 1991). We would like to suggest that the NPY-mediated NA release and consequent inhibition of EPSCs is due to the presence of the putative Y3 receptor only on the terminals of the projections from the locus coeruleus and/or on catecholaminergic neurons of the A2 cell group. Our data would suggest that these noradrenergic terminals, or neurons, express the putative Y3 receptors only, which would explain the involvement of NA in the inhibitory actions of NPY but not PYY or [Leu31,Pro34] NPY. It has been established that these NA-mediated effects of NPY occur in other areas, both within the CNS and the periphery (Wiley & Owyang, 1987; Martire & Pistritto, 1992; Khanna et al. 1993; Sawa et al. 1995; Kapoor & Sladek, 2001).
Interestingly, following reserpine-induced chemical sympathectomy, pretreatment with yohimbine does not attenuate the inhibitory actions of NPY, which are, rather, antagonized completely by pretreatment with 0.1 μm BIBP3226. These data would suggest that the α2-mediated responses of NPY are due to release of NA rather that a direct interaction of NPY with the α2 adrenoceptor itself. In reserpinized animals the magnitude of EPSC inhibition induced by NPY is similar to the inhibition obtained in control rats, which might suggest that following chemical sympathectomy, there is efficiency of Y1 receptor–effector coupling and/or that the circuitry has undergone a receptor rearrangement with an increased expression of Y1 receptors.
The differences in the mechanisms of inhibition of excitatory transmission by NPY and PYY may be explained by considering differences in their functions.
PYY, for example, is a hormone released into the circulation from enterochromaffin cells following ingestion of a fatty meal or infusion of lipids into the duodenum (Pappas et al. 1985; Sheikh, 1991; Chen et al. 1997). PYY then acts centrally to inhibit gastric emptying and motility by a vagally mediated mechanism, the so-called ileal brake phenomenon. It is unlikely, however, that in vivo PYY itself exerts any actions within the DVC, rather, it is more likely that PYY is cleaved by the circulating endogenous enzyme, DAP IV, to form selective Y2 receptor agonists such as PYY(3–36) (Grandt et al. 1993; Mentlein et al. 1993). Previous studies have indeed demonstrated that the PYY-mediated inhibitory actions on GI motility result from interactions with Y2 receptors in the brainstem (Chen & Rogers, 1995, 1997; Chen et al. 1996, 1997). Our data suggest that PYY interaction with Y2 receptors induces mainly a decrease in the excitatory transmission between NTS and DMV.
NPY, in contrast, is contained within neurons of the dorsomedial NTS and within nerve terminals throughout the DVC (Harfstrand et al. 1987) and it is released as a neurotransmitter/neuromodulator. Previous reports have suggested that the central, vagally mediated effects of NPY on the GI tract depend upon the basal conditions of the GI tract at the time of application. Under conditions of increased levels of GI motility, which would engage an increased glutamatergic transmission, NPY decreases motility or gastric acid secretion (Chen et al. 1997). Under these conditions, neuronal release of NPY was suggested to activate only Y1 receptors (Chen et al. 1997). The present study, however, would indicate that NPY activates not only Y1 but also the putative Y3 receptors, even if Y2 receptors are present. While the vast majority of DMV neurons are cholinergic (Armstrong et al. 1990), at least two opposing vagal neuronal pathways exist which can be differentiated based on the neurochemical phenotype of the postganglionic enteric neurons they innervate. One circuit is a cholinergic excitatory pathway, inhibition of which would cause a decrease in gastric motility and secretion. The other circuit is a non-adrenergic, non-cholinergic (NANC) pathway, inhibition of which causes an increase of gastric tone, most commonly via nitric oxide release (for reviews see Travagli & Rogers, 2001; Travagli et al. 2003). Since activation of Y receptors on excitatory glutamatergic nerve terminals caused a decrease in glutamate release with a consequent inhibition of the DMV neuronal output, the overall GI response following application of PPs in the DVC would be a withdrawal of the cholinergic tone modulated by brainstem glutamatergic pathways.
It is interesting to note that only a subpopulation of DMV neurons appeared to be modulated by PPs. This observation further support the hypotheses proposed by us and other groups that there is a high degree of integration within the DMV that is reflected in discrete pharmacological properties of the neurons themselves as well as by the modulation of their synaptic inputs. Unfortunately, the correlation between these physiological/pharmacological differences and the function(s) subserved by the vagal motoneurons is far from being resolved.
In conclusion, we have provided strong evidence indicating that, under our experimental conditions, the effects of PPs are due mainly to inhibition of excitatory synaptic transmission between NTS and DMV. These effects are due to interactions with different receptors of the NPY family. NPY directly activates Y1 receptors located on NTS glutamatergic terminals impinging on DMV neurons. NPY also activates an NPY-preferring receptor, possibly the putative Y3 receptor, which releases NA to decrease the amplitude of EPSCs via α2 adrenoceptors. Conversely, PYY inhibits NTS–DMV excitatory transmission via activation of a non-Y1, non-Y3 receptor, possibly the Y2 receptor.
Acknowledgments
This manuscript was supported by NIH grant DK55530. We would like to thank Drs Moises Hermann and Owyang for critical comments on earlier versions of the manuscript.
References
- Adrian TE, Savage AP, Sagor GR, Allen JM, Bacarese-Hamilton AJ, Tatemoto K, Polak JM, Bloom SR. Effect of peptide YY on gastric, pancreatic, and biliary function in humans. Gastroenterology. 1985;89:494–499. doi: 10.1016/0016-5085(85)90442-1. [DOI] [PubMed] [Google Scholar]
- Armstrong DM, Manley L, Haycock JW, Hersh LB. Co-localization of choline acetyltransferase and tyrosine hydroxylase within neurons of the dorsal motor nucleus of the vagus. J Chem Neuroanat. 1990;3:133–140. [PubMed] [Google Scholar]
- Bertolino M, Vicini S, Gillis RA, Travagli RA. Presynaptic alpha-2 adrenoceptors inhibit excitatory synaptic transmission in rat brain stem. Am J of Physiol. 1997;272:G654–661. doi: 10.1152/ajpgi.1997.272.3.G654. [DOI] [PubMed] [Google Scholar]
- Browning KN, Renehan WE, Travagli RA. Electrophysiological and morphological heterogeneity of rat dorsal vagal neurons which project to specific areas of the gastrointestinal tract. J Physiol. 1999;517:521–532. doi: 10.1111/j.1469-7793.1999.0521t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning KN, Travagli RA. The peptide TRH uncovers the presence of presynaptic 5-HT1A receptors via activation of a second messenger pathway in the rat dorsal vagal complex. J Physiol. 2001;531:425–435. doi: 10.1111/j.1469-7793.2001.0425i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buell MG, Harding RK. Effects of peptide YY on intestinal blood flow distribution and motility in the dog. Regul Pept. 1989;24:195–208. doi: 10.1016/0167-0115(89)90238-3. [DOI] [PubMed] [Google Scholar]
- Chen CH, Rogers RC. Central inhibitory action of peptide YY on gastric motility in rats. Am J Physiol. 1995;269:R787–792. doi: 10.1152/ajpregu.1995.269.4.R787. [DOI] [PubMed] [Google Scholar]
- Chen CH, Rogers RC. Peptide YY and the Y2 agonist PYY-(13–36) inhibit neurons of the dorsal motor nucleus of the vagus. Am J Physiol. 1997;273:R213–218. doi: 10.1152/ajpregu.1997.273.1.R213. [DOI] [PubMed] [Google Scholar]
- Chen CH, Rogers RC, Stephens RL., Jr Intracisternal injection of peptide YY inhibits gastric emptying in rats. Regul Pept. 1996;61:95–98. doi: 10.1016/0167-0115(95)00143-3. [DOI] [PubMed] [Google Scholar]
- Chen CH, Stephens RL, Jr, Rogers RC. PYY and NPY control of gastric motility via action on Y1 and Y2 receptors in the DVC. Neurogastroenterol Motil. 1997;9:109–116. doi: 10.1046/j.1365-2982.1997.d01-26.x. [DOI] [PubMed] [Google Scholar]
- Chi J, Kemerer J, Stephens RL., Jr 5-HT in DVC: disparate effects on TRH analogue-stimulated gastric acid secretion, motility, and cytoprotection. Am J Physiol. 1996;271:R368–372. doi: 10.1152/ajpregu.1996.271.2.R368. [DOI] [PubMed] [Google Scholar]
- Dumont Y, Fournier A, St Pierre S, Schwartz TW, Quirion R. Differential distribution of neuropeptide Y1 and Y2 receptors in the rat brain. Eur J Pharmacol. 1990;191:501–503. doi: 10.1016/0014-2999(90)94189-5. [DOI] [PubMed] [Google Scholar]
- Fujimiya M, Inui A. Peptidergic regulation of gastrointestinal motility in rodents. Peptides. 2000;21:1565–1582. doi: 10.1016/s0196-9781(00)00313-2. [DOI] [PubMed] [Google Scholar]
- Fujimiya M, Itoh E, Kihara N, Yamamoto I, Fujimura M, Inui A. Neuropeptide Y induces fasted pattern of duodenal motility via Y2 receptors in conscious fed rats. Am J Physiol Gastrointest Liver Physiol. 2000;278:G32–38. doi: 10.1152/ajpgi.2000.278.1.G32. [DOI] [PubMed] [Google Scholar]
- Fukuda A, Minami T, Nabekura J, Oomura Y. The effects of noradrenaline on neurones in the rat dorsal motor nucleus of the vagus, in vitro. J Physiol. 1987;393:213–231. doi: 10.1113/jphysiol.1987.sp016820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan JG, Lawson DC, Cheng CA, Opara E, Taylor IL, Pappas TN. Intracerebroventricular neuropeptide Y increases gastric and pancreatic secretion in the dog. Gastroenterology. 1993;105:1069–1077. doi: 10.1016/0016-5085(93)90951-8. [DOI] [PubMed] [Google Scholar]
- Gillis RA, Quest JA, Pagani FD, Norman WP. Control centers in the central nervous system for regulating gastrointestinal motility. In: Wood JD, editor. Handbook of Physiology, section 6, The Gastrointestinal System, vol. I, Motility and Circulation. Bethesda, MD, USA: The American Physiological Society; 1989. pp. 621–683. part 2. [Google Scholar]
- Glaum SR, Miller RJ, Rhim H, Maclean D, Georgic LM, MacKenzie RG, Grundemar L. Characterization of Y3 receptor-mediated synaptic inhibition by chimeric neuropeptide Y-peptide YY peptides in the rat brainstem. Br J Pharmacol. 1997;120:481–487. doi: 10.1038/sj.bjp.0700883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandt D, Dahms P, Schimiczek M, Eysselein VE, Reeve JR, Mentlein R. Proteolytic processing by dipeptidyl aminopeptidase IV generates receptor selectivity for peptide YY. Med Klin. 1993;88:143–145. [PubMed] [Google Scholar]
- Grundemar L, Wahlestedt C, Reis DJ. Long-lasting inhibition of the cardiovascular responses to glutamate and the baroreceptor reflex elicited by neuropeptide Y injected into the nucleus tractus solitarius of the rat. Neurosci Lett. 1991;122:135–139. doi: 10.1016/0304-3940(91)90211-b. [DOI] [PubMed] [Google Scholar]
- Harfstrand A, Fuxe K, Terenius L, Kalia M. Neuropeptide Y-immunoreactive perikarya and nerve terminals in the rat medulla oblongata: relationship to cytoarchitecture and catecholaminergic cell groups. J Comp Neurol. 1987;260:20–35. doi: 10.1002/cne.902600103. [DOI] [PubMed] [Google Scholar]
- Hornby PJ. Receptors and transmission in the brain-gut axis. II Excitatory amino acid receptors in the brain-gut axis. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1055–1060. doi: 10.1152/ajpgi.2001.280.6.G1055. [DOI] [PubMed] [Google Scholar]
- Ishiguchi T, Amano T, Matsubayashi H, Tada H, Fujita M, Takahashi T. Centrally administered neuropeptide Y delays gastric emptying via Y2 receptors in rats. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1522–1530. doi: 10.1152/ajpregu.2001.281.5.R1522. [DOI] [PubMed] [Google Scholar]
- Jacques D, Cadieux A, Dumont Y, Quirion R. Apparent affinity and potency of BIBP3226, a non-peptide neuropeptide Y receptor antagonist, on purported neuropeptide Y Y1, Y2 and Y3 receptors. Eur J Pharmacol. 1995;278:R3–5. doi: 10.1016/0014-2999(95)00179-o. [DOI] [PubMed] [Google Scholar]
- Kapoor JR, Sladek CD. Substance P and NPY differentially potentiate ATP and adrenergic stimulated vasopressin and oxytocin release. Am J Physiol Regul Integr Comp Physiol. 2001;280:R69–78. doi: 10.1152/ajpregu.2001.280.1.R69. [DOI] [PubMed] [Google Scholar]
- Kawakubo K, Yang H, Tache Y. Gastric protective effect of peripheral PYY through PYY preferring receptors in anesthetized rats. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1035–1041. doi: 10.1152/ajpgi.00154.2002. [DOI] [PubMed] [Google Scholar]
- Khanna S, Sibbald JR, Day TA. Neuropeptide Y modulation of A1 noradrenergic neuron input to supraoptic vasopressin cells. Neurosci Lett. 1993;161:60–64. doi: 10.1016/0304-3940(93)90140-g. [DOI] [PubMed] [Google Scholar]
- Lee CC, Miller RJ. Is there really an NPY Y3 receptor? Regul Pept. 1998:75–76. 71–78. doi: 10.1016/s0167-0115(98)00054-8. [DOI] [PubMed] [Google Scholar]
- Leslie RA, McDonald TJ, Robertson HA. Autoradiographic localization of peptide YY and neuropeptide Y binding sites in the medulla oblongata. Peptides. 1988;9:1071–1076. doi: 10.1016/0196-9781(88)90091-5. [DOI] [PubMed] [Google Scholar]
- Lynch DR, Walker MW, Miller RJ, Snyder SH. Neuropeptide Y receptor binding sites in rat brain: differential autoradiographic localizations with 125I-peptide YY and 125I-neuropeptide Y imply receptor heterogeneity. J Neurosci. 1989;9:2607–2619. doi: 10.1523/JNEUROSCI.09-08-02607.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann MJ, Hermann GE, Rogers RC. Dorsal medullary serotonin and gastric motility: enhancement of effects by thyrotropin-releasing hormone. J Auton Nerv Syst. 1988;25:35–40. doi: 10.1016/0165-1838(88)90005-7. [DOI] [PubMed] [Google Scholar]
- Martire M, Pistritto G. Neuropeptide Y interaction with the adrenergic transmission line: a study of its effect on alpha-2 adrenergic receptors. Pharmacol Res. 1992;25:203–215. doi: 10.1016/s1043-6618(05)80069-6. [DOI] [PubMed] [Google Scholar]
- Masuda M, Tomita H, Okubo K, Miyasaka K. Vagal efferent nerve-dependent inhibitory action on pancreatic polypeptide and peptide YY in conscious rats: comparison with somatostatin. J Auton Nerv Syst. 1994;50:131–138. doi: 10.1016/0165-1838(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Matsuda M, Aono M, Moriga M, Okuma M. Centrally administered NPY inhibits gastric emptying and intestinal transit in the rat. Dig Dis Sci. 1993;38:845–850. doi: 10.1007/BF01295910. [DOI] [PubMed] [Google Scholar]
- Mendelowitz D, Kunze DL. Identification and dissociation of cardiovascular neurons from the medulla for patch clamp analysis. Neurosci Lett. 1991;132:217–221. doi: 10.1016/0304-3940(91)90305-d. [DOI] [PubMed] [Google Scholar]
- Mentlein R, Dahms P, Grandt D, Kruger R. Proteolytic processing of neuropeptide Y and peptide YY by dipeptidyl peptidase IV. Regul Pept. 1993;49:133–144. doi: 10.1016/0167-0115(93)90435-b. [DOI] [PubMed] [Google Scholar]
- Michel MC, Beck-Sickinger A, Cox H, Doods HN, Herzog H, Larhammar D, Quirion R, Schwartz T, Westfall TC. XVI International union of pharmacology reccommendations for the nomenclature of neuropeptide Y, peptide YY, and pancreatic polypeptide receptors. Pharmacol Rev. 1999;50:143–150. [PubMed] [Google Scholar]
- Naruse S, Kitagawa M, Ishiguro H, Hayakawa T. Feedback regulation of pancreatic secretion by peptide YY. Peptides. 2002;23:359–365. doi: 10.1016/s0196-9781(01)00612-x. [DOI] [PubMed] [Google Scholar]
- Pappas TN, Tache Y, Debas HT. Opposing central and peripheral actions of brain-gut peptides: a basis for regulation of gastric function. Surgery. 1985;98:183–190. [PubMed] [Google Scholar]
- Pitts DK, Kelland MD, Shen RY, Freeman AS, Chiodo LA. Statistical analysis of dose-response curves in extracellular electrophysiological studies of single neurons. Synapse. 1990;5:281–293. doi: 10.1002/syn.890050405. [DOI] [PubMed] [Google Scholar]
- Rea MA, Aprison MH, Felten DL. Catecholamines and serotonin in the caudal medulla of the rat: combined neurochemical-histofluorescence study. Brain Res Bull. 1982;9:227–236. doi: 10.1016/0361-9230(82)90135-6. [DOI] [PubMed] [Google Scholar]
- Rogers RC, Hermann GE, Travagli RA. Brainstem pathways responsible for oesophageal control of gastric motility and tone in the rat. Journal of Physiology. 1999;514:369–383. doi: 10.1111/j.1469-7793.1999.369ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolf K, Eberlein W, Engel W, Wieland HA, Willim KD, Entzeroth M, Wienen W, Beck-Sickinger AG, Doods HN. The first highly potent and selective non-peptide neuropeptide Y Y1 receptor antagonist: BIBP3226. Eur J Pharmacol. 1994;271:R11–13. doi: 10.1016/0014-2999(94)90822-2. [DOI] [PubMed] [Google Scholar]
- Sawa T, Mameya S, Yoshimura M, Itsuno M, Makiyama K, Niwa M, Taniyama K. Differential mechanism of peptide YY and neuropeptide Y in inhibiting motility of guinea-pig colon. Eur J Pharmacol. 1995;276:223–230. doi: 10.1016/0014-2999(95)00024-f. [DOI] [PubMed] [Google Scholar]
- Sheikh SP. Neuropeptide Y and peptide YY: major modulators of gastrointestinal blood flow and function. Am J Physiol. 1991;261:G701–715. doi: 10.1152/ajpgi.1991.261.5.G701. [DOI] [PubMed] [Google Scholar]
- Ter Horst GJ, Toes GJ, Van Willigen JD. Locus coeruleus projections to the dorsal motor vagus nucleus in the rat. Neuroscience. 1991;45:153–160. doi: 10.1016/0306-4522(91)90111-z. [DOI] [PubMed] [Google Scholar]
- Travagli RA, Gillis RA, Rossiter CD, Vicini S. Glutamate and GABA-mediated synaptic currents in neurons of the rat dorsal motor nucleus of the vagus. Am J Physiol. 1991;260:G531–536. doi: 10.1152/ajpgi.1991.260.3.G531. [DOI] [PubMed] [Google Scholar]
- Travagli RA, Hermann GE, Browning KN, Rogers RC. Musings on the wanderer: what's new in our understanding of vago-vagal reflexes?: III. Activity-dependent plasticity in vago-vagal reflexes controlling the stomach. Am J Physiol Gastrointest Liver Physiol. 2003;284:G180–187. doi: 10.1152/ajpgi.00413.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travagli RA, Rogers RC. Receptors and transmission in the brain-gut axis: potential for novel therapies. V. Fast and slow extrinsic modulation of dorsal vagal complex circuits. Am J Physiol Gastrointest Liver Physiol. 2001;281:G595–601. doi: 10.1152/ajpgi.2001.281.3.G595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tricklebank MD, Forler C, Fozard JR. The involvement of subtypes of the 5-HT1 receptor and of catecholaminergic systems in the behavioural response to 8-hydroxy-2-(di-n-propylamino)tetralin in the rat. Eur J Pharmacol. 1984;106:271–282. doi: 10.1016/0014-2999(84)90714-3. [DOI] [PubMed] [Google Scholar]
- Wiley J, Owyang C. Neuropeptide Y inhibits cholinergic transmission in the isolated guinea pig colon: mediation through alpha-adrenergic receptors. Proc Natl Acad Sci U S A. 1987;84:2047–2051. doi: 10.1073/pnas.84.7.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H. Central and peripheral regulation of gastric acid secretion by peptide YY. Peptides. 2002;23:349–358. doi: 10.1016/s0196-9781(01)00611-8. [DOI] [PubMed] [Google Scholar]
- Yang H, Kawakubo K, Wong H, Ohning G, Walsh J, Tache Y. Peripheral PYY inhibits intracisternal TRH-induced gastric acid secretion by acting in the brain. Am J Physiol Gastrointest Liver Physiol. 2000;279:G575–581. doi: 10.1152/ajpgi.2000.279.3.G575. [DOI] [PubMed] [Google Scholar]
- Yang H, Li WP, Reeve JR, Rivier J, Tache Y. PYY-preferring receptor in the dorsal vagal complex and its involvement in PYY stimulation in gastric acid secretion in rats. Br J Pharmacol. 1998;123:1549–1554. doi: 10.1038/sj.bjp.0701767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda M, Yokohama S, Tamori K, Sato Y, Nakamura K, Makino I. Neuropeptide Y in the dorsal vagal complex stimulates bicarbonate-dependent bile secretion in rats. Gastroenterology. 1997;112:1673–1680. doi: 10.1016/s0016-5085(97)70050-7. [DOI] [PubMed] [Google Scholar]