

Abstract
Changes in mitochondrial matrix [Ca2+] evoked by trains of action potentials were studied in levator auris longus motor terminals using Ca2+-sensitive fluorescent indicator dyes (rhod-2, rhod-5F). During a 2500 impulse 50 Hz train, mitochondrial [Ca2+] in most wild-type terminals increased within 5–10 s to a plateau level that was sustained until stimulation ended. This plateau was not due to dye saturation, but rather reflects a powerful buffering system within the mitochondrial matrix. The amplitude of this plateau was similar for stimulation frequencies in the range 15–100 Hz. Plateau amplitude was sensitive to temperature, with no detectable stimulation-induced increase in fluorescence at temperatures below 17 °C, and increasing magnitudes as temperature was increased to near-physiological levels (38 °C). When stimulation ended, mitochondrial [Ca2+] decayed slowly back to prestimulation levels over a time course of hundreds of seconds. Similar measurements were also made in motor terminals of mice expressing the G93A mutation of human superoxide dismutase 1 (SOD1-G93A). In mice > 100 days old, all of whom exhibited hindlimb paralysis, some terminals continued to show wild-type mitochondrial [Ca2+] responses, but in other terminals mitochondrial [Ca2+] did not plateau, but rather continued to increase throughout most of the stimulus train. Thus mechanism(s) that limit stimulation-induced increases in mitochondrial [Ca2+] may be compromised in some SOD1-G93A terminals.
Mitochondrial uptake of Ca2+ contributes to buffering moderate-to-large cytosolic Ca2+ loads in neurons and other secretory cells (Friel & Tsien, 1994; Stuenkel, 1994; Werth & Thayer, 1994; White & Reynolds, 1995; Herrington et al. 1996; Tang & Zucker, 1997; Kaftan et al. 2000; Suzuki et al. 2002). Mitochondrial sequestration of Ca2+ may be especially significant in nerve terminals, which sustain large stimulation-induced Ca2+ influxes into a small volume. In lizard motor nerve terminals, measurements of cytosolic and mitochondrial matrix [Ca2+] demonstrated that mitochondrial Ca2+ sequestration is the major mechanism limiting the increase in spatially averaged cytosolic [Ca2+] during trains of 25 or more action potentials delivered at 50 Hz (David et al. 1998; David, 1999). Consistent with these findings, David & Barrett (2000) showed that in mouse motor nerve terminals stimulation-induced increases in cytosolic [Ca2+] are also greatly increased by drugs that prevent mitochondrial Ca2+ uptake. These cytosolic [Ca2+] responses are also increased by lowering the temperature, suggesting that mitochondrial Ca2+ uptake in mouse terminals might be temperature-dependent. Here we extend this work on mouse motor terminals by measuring [Ca2+] within the mitochondrial matrix. We report that during repetitive stimulation, mitochondrial [Ca2+] increases to a plateau whose amplitude increases with increasing temperature, but not with increasing stimulation frequency (15–100 Hz).
Stimulation-induced mitochondrial [Ca2+] responses were also measured in motor terminals of SOD1-G93A mice. SOD1 is a cytosolic, homodimeric, Cu2+/Zn2+-dependent metalloenzyme that catalyses the conversion of superoxide anion to hydrogen peroxide, thus contributing to cellular defence against oxidative stress. This enzyme is not required for motoneuron survival, but may be necessary for maintenance of normal hindlimb neuromuscular junctions (Flood et al. 1999). In humans the G93A mutation, as well as several other SOD1 mutations, cause some familial forms of amyotrophic lateral sclerosis (ALS, Aguirre et al. 1999; Gellera et al. 2001) by a toxic gain-of-function mechanism. ALS is characterized by adult-onset progressive motor weakness and paralysis accompanied by death of spinal motoneurons (reviewed in Rowland & Shneider, 2001). Mice expressing these mutant forms of human SOD1, but not wild-type human SOD1, also develop progressive motor weakness, paralysis and death of spinal motoneurons (G93A mutation: Gurney et al. 1994; Chiu et al. 1995; G37R, Wong et al. 1995; G85R mutation, Bruijn et al. 1997). In ALS, different motoneuron pools degenerate at different rates; for example motoneurons innervating leg muscles degenerate earlier than those innervating external eye muscles. This pattern of degeneration is at least partially reproduced in the SOD1-G93A mouse model (Chiu et al. 1995).
Although SOD1 is a cytosolic protein, both normal and mutant SOD1 are present in the mitochondrial fraction (Sturtz et al. 2001; Jaarsma et al. 2001; Higgins et al. 2002; Mattiazzi et al. 2002). Both in vivo and in vitro evidence suggests that calcium dysregulation and/or mitochondrial dysfunction contribute to the motoneuron cell death in SOD1 mutant mice (reviewed by Menzies et al. 2002). This evidence includes mitochondrial vacuolization and degeneration (Chiu et al. 1995; Wong et al. 1995; Mourelatos et al. 1996; Kong & Xu, 1998; Jaarsma et al. 2000), immunohistochemical signs of oxidative damage to mitochondrial DNA in spinal motoneurons (Warita et al. 2001), and decreased activity of components of the mitochondrial respiratory chain in spinal cord tissue (Borthwick et al. 1999; Jung et al. 2002). The survival of mice expressing mutant human SOD1 can be prolonged by creatine, cyclosporin A and minocycline (Klivenyi et al. 1999; Keep et al. 2001; Zhu et al. 2002), which inhibit opening of the mitochondrial permeability transition pore, and by overexpression of bcl-2, an anti-apoptotic protein (Kostic et al. 1997), which increases mitochondrial ability to accumulate Ca2+ (Murphy et al. 1996; Zhu et al. 1999). Cultured motoneurons expressing SOD1-G93A exhibit increased intracellular [Ca2+], decreased intramitochondrial [Ca2+] and several indices of mitochondrial dysfunction (Kruman et al. 1999), and their survival is prolonged by agents that block plasma membrane Ca2+ channels and by expression of a Ca2+-binding protein, calbindin-D28K (Roy et al. 1998). This latter finding complements evidence that the motoneuron pools that die first in ALS have lower concentrations of cytosolic Ca2+ buffers (parvalbumin and calbindin-D28K) than the more resistant motoneuron pools (Ince et al. 1993; Alexianu et al. 1994; Siklos et al. 1998).
Motor nerve terminals contain abundant mitochondria, so we investigated whether expression of SOD1-G93A would alter mitochondrial handling of the large Ca2+ loads produced by repetitive nerve stimulation. We report that in SOD1-G93A mice > 100 days old the majority of levator auris longus motor terminals exhibit an abnormal, continuously ramping (rather than plateauing) increase in mitochondrial [Ca2+] during repetitive stimulation. Thus disrupted mitochondrial handling of Ca2+ loads may contribute to the motor terminal pathology that develops during this disease.
METHODS
Preparation and solutions
Neuromuscular preparations were dissected from mice killed with 100 % CO2, in accordance with guidelines of the University of Miami Animal Care and Use Committee. Experiments used SOD1-G93A mice 100–120 days old (all of which exhibited hindlimb paralysis) or wild-type littermates also 100–120 days old. A founder mouse expressing a high copy number of human SOD1-G93A, originally described by Gurney et al. (1994), was purchased from Jackson Labs. SOD1-G93A mice were identified by PCR of tail clips taken at 8–20 days. The touchdown PCR assay recommended by Jackson Labs used a mixture of two sets of primer pairs, one for mutant human SOD1 (5′-CAT CAG CCC TAA TCC ATC TGA-3′ and 5′-CGC GAC TAA CAA TCA AAG TGA-3′) and the other for wild-type mouse interleukin-2 (IL-2, 5′-CTA GGC CAC AGA ATT GAA AGA TCT-3′ and 5′-GTA GGT GGA AAT TCT AGC ATC ATC C-3′). There is a competition for nucleotides during the amplification of SOD1-G93A and IL-2 cDNAs (236 and 324 bp, respectively).
The levator auris longus muscle (Angaut-Petit et al. 1987; Erzen et al. 2000) was removed together with its attachment sites on the skin of the scalp and the midline muscles at the back of the head. This muscle is very thin (1–2 muscle layers), which facilitates imaging studies. Preparations were bathed in mammalian saline solution (mm: NaCl 145, NaHCO3 24, glucose 5.5–11, CaCl2 2, MgCl2 0.5, and Na2HPO4 0.1, pH 7.4, gassed with 95 % O2/5 % CO2) and pinned in a small chamber (0.75 ml volume) with a glass coverslip bottom and silicone rubber walls. The motor nerve was stimulated with brief, suprathreshold depolarizing pulses (e.g. 50 Hz for 50 s) delivered via a suction electrode; the resulting muscle contractions demonstrated the viability of the preparation. Contractions were then blocked using d-tubocurare (2 μm). During imaging an outer chamber surrounding the chamber containing the muscle was slowly infused with a 95 % O2/5 % CO2 gas mixture. The temperature of the preparation was monitored using a small thermocouple in the experimental chamber.
Loading of Ca2+ indicator dyes into mitochondria
Preparations were bath-loaded with the acetoxymethylester (AM) form of Ca2+ indicator dyes in the rhod family (rhod-2 [Kd ≈ 0.6 μm]; rhod-5F (Kd ≈ 1.9 μm); occasionally rhod-5FF (Kd ≈19 μm) or rhod-5N (Kd ≈320 μm)). These dyes show Ca2+-dependent fluorescence after the AM moiety is cleaved by esterases in the cytosol and mitochondrial matrix. A variety of loading protocols were tested to maximize intramitochondrial loading, including some in which tetraisopropylpyrophosphoramide (final concentration 1 μm), an irreversible blocker of non-specific esterases, was added 15 min prior to incubation with dye. The easiest protocol that yielded reliable intramitochondrial dye loading (see David et al. 2003) was to expose the preparation to a high concentration of dye (5–20 μm) for 2–4 min, and then rinse with dye-free solution for 30–60 min at room temperature in an atmosphere containing 95 % O2/5 % CO2.
The localization of indicator dye in motor terminal mitochondria was verified using three criteria (David, 1999). First, the resting fluorescence had a patchy distribution localized to the terminal region (not present in the motor axon), suggesting localization in terminal organelles rather than throughout the cytosol (e.g. Figs 3A and 5A). Second, the fluorescence increase was time-locked to the onset of nerve stimulation and had a slow post-stimulation decay. Third, the stimulation-induced fluorescence response was abolished by carbonyl cyanide m-chlorophenyl hydrazone (CCCP, 1–2 μm, e.g. Fig. 6). CCCP is a protonophore that collapses the transmembrane voltage gradient (and thus the driving force for Ca2+ entry) across the inner mitochondrial membrane. The latter two features distinguish mitochondrial [Ca2+] responses from cytosolic [Ca2+] responses, which have a rapid initial component of decay and are increased by CCCP (David & Barrett, 2000). Mitochondria also cluster within the end-plate region of the muscle fibre, but muscle mitochondria are unlikely to contribute significantly to the stimulation-induced responses recorded here because tubocurare blocks (and thereby prevents Ca2+ influx through) end-plate nicotinic receptors.
Figure 3. F/Frest responses evoked by 50 Hz stimulation in neighbouring wild-type terminals filled with rhod-5F and imaged using a Perkin-Elmer confocal system.
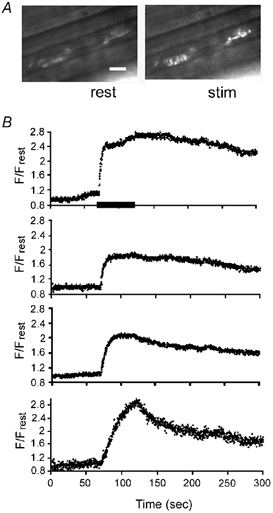
A, fluorescence micrograph showing two of the four terminals at rest and during stimulation (stim). Calibration, 50 μm. B, F/Frest responses evoked simultaneously in four adjacent terminals (including those shown in A). The horizontal bar in the upper record indicates the duration of stimulation for all four records. Images were sampled at 0.23 s image−1 at 26 °C.
Figure 5. Heterogeneity of stimulation-induced F/Frest responses recorded in various regions of a rhod-5F-filled SOD1-G93A motor terminal stimulated for 50 s at 50 Hz at 27 °C (duration indicated by bar).
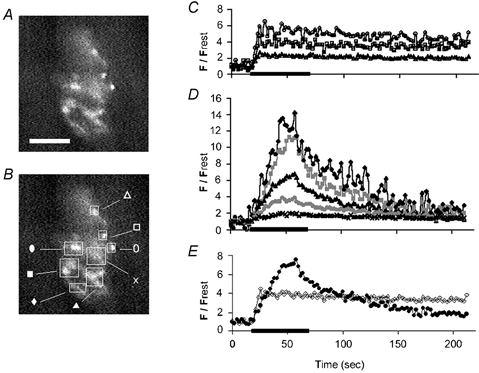
A and B show the same difference fluorescence image, with the location of regions of interest indicated by squares and labelled in B. The actual regions of interest used in the analysis were smaller, consisting of pixels within a circumference drawn around those regions whose fluorescence increased during stimulation. Calibration: 50 μm. C and D, F/Frest responses measured from subregions of this terminal, plotted using symbols shown in B. C, F/Frest responses from upper right regions of this terminal (open symbols) show plateaus. D, terminals from lower left regions of the terminal (filled symbols) exhibit ramping (see text). E, averages of the plateauing responses in C (open symbols) and of the ramping responses in D (filled symbols). The difference image in A was obtained by subtracting the average of 19 prestimulation images from the average of 16 images sampled during the stimulus train (2.13 s image−1). We were unable to determine whether the ramping and non-ramping regions of this terminal were innervated by the same or different motor axons. The observation that most of the responses in D started to decline before the end of the stimulation train raises the possibility of failure of axonal conduction and/or opening of the mitochondrial permeability transition pore.
Figure 6. Ramping F/Frest response in a SOD1-G93A terminal (♦) is inhibited by CCCP (○).
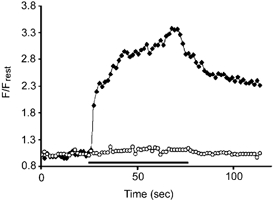
The horizontal bar indicates the duration of 50 Hz stimulation. The F/Frest response recovered partially following CCCP washout (not shown). Recordings were carried out at 28 °C.
Imaging and analysis of fluorescence responses
Most imaging experiments used an Odyssey XL confocal microscope (Noran Instruments, Middleton, WI, USA) equipped with a 20 mW diode-pumped green laser (Milles Griot) that provided the 532 nm excitation light for rhod dyes. A 550 nm barrier filter (Chroma) was used in the emission pathway. To reduce photodamage, we used lower laser intensities in combination with maximal gain on the photomultiplier tube and a pixel dwell time of 800 ns, which proved optimal for the Noran system. Each image was the average of eight consecutive exposures, to improve the signal-to-noise ratio. Usually a series of 100 images was collected during each trial, with the first 10–20 images used to determine the baseline before the onset of stimulation. To study responses evoked by different stimulation frequencies, the various frequencies were presented in mixed order and one standard frequency (usually 50 Hz) was retested one or more times during the experiment. Because mitochondrial [Ca2+] responses in motor terminals decay slowly, repeated stimulus trains were separated by intervals of at least 15 min.
Even with the adjustments indicated above, only a small percentage of motor terminals studied with the Noran system yielded multiple mitochondrial [Ca2+] responses with an acceptable signal-to-noise ratio. A Wallac Ultraview confocal microscope (Perkin-Elmer Life Sciences, Boston, MA, USA), tested during a brief demonstration at the University of Miami, yielded better results, as shown in Fig. 3. This system, which incorporates a Yokogawa spinning disk, reduces photodamage and improves the signal-to-noise ratio by sweeping the preparation with high-frequency, low-intensity excitation pulses.
Image data were analysed on a Pentium computer using V++ software (Digital Micro Optics, Auckland, NZ). To identify regions of interest (ROI) that responded to nerve stimulation, the 10–20 images collected prior to stimulation were averaged to yield a control (resting) image, which was subtracted from the average of 20 images collected during the maximal response to stimulation, yielding a difference image. ROIs defined on this difference image were copied to identical coordinates of each image in the image stack, and mean fluorescence values for the ROI on each of these images were calculated. Background fluorescence was estimated in regions surrounding the motor terminal. Stimulation-induced responses were plotted as F/Frest, where F is the (background-subtracted) fluorescence at a particular post-stimulation time, and Frest is the resting (control, background-subtracted) fluorescence. A 3-point moving bin average was used to reduce noise in the records collected with the Noran confocal.
All indicator dyes were from Molecular Probes (Eugene, OR, USA). Other reagents came from Sigma.
RESULTS
Stimulation-induced elevations of mitochondrial [Ca2+] are sensitive to temperature but not to stimulation frequency
The elevation of cytosolic [Ca2+] produced by 50–100 Hz stimulation of mouse motor terminals is greater at cool than at near-physiological temperatures (David & Barrett, 2000). To test whether this temperature dependence might be due to differences in mitochondrial Ca2+ uptake, we measured stimulation-induced elevations in mitochondrial [Ca2+] over a range of temperatures. Figure 1 illustrates F/Frest responses recorded in two experiments in which it was possible to record responses at three different temperatures from the same wild-type terminal. The indicator dye was rhod-2 in Fig. 1A, and the lower-affinity rhod-5F in Fig. 1B. The responses rose to a plateau, attained within 5–10 s following the onset of stimulation, and decayed slowly after stimulation stopped. Within each terminal the F/Frest responses were temperature-dependent, with no significant stimulation-related increase at 17 °C, and increasing amplitudes at higher temperatures. This result was not due to the temperature dependence of rhod dyes, since mitochondrial fluorescence responses recorded in lizard motor terminals using the same rhod dyes did not vary with temperature over this range (not shown). Most subsequent experiments were conducted at temperatures ranging from 25–30 °C, where responses were detectable and preparations remained more stable than at higher temperatures.
Figure 1. Temperature dependence of mitochondrial [Ca2+] responses (recorded as F/Frest) evoked by 50 Hz stimulation in two wild-type motor nerve terminals.
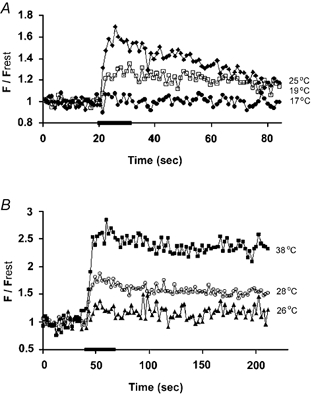
A, terminal loaded with rhod-2 and stimulated for 12 s at 17, 19 and 25 °C. B, terminal loaded with rhod-5F and stimulated for 30 s at 26, 28 and 38 °C. Horizontal bars indicate stimulation interval; note that A and B use different time and fluorescence axes.
In these wild-type terminals, mitochondrial F/Frest responses from different subregions of the same terminal were usually similar (not shown), and thus were averaged together. Wild-type cytosolic [Ca2+] responses also exhibited intra-terminal homogeneity (David & Barrett, 2000).
A few experiments were also attempted using lower-affinity rhod dyes. Stimulation-induced responses could occasionally be detected with rhod-5FF (Kd ≈ 19 μm), but not with rhod-5N (Kd ≈ 320 μm).
Figure 2 (left) shows that the plateau amplitude of F/Frest did not vary as stimulation frequency was increased from 50 to 100 Hz. This finding was confirmed by repeated measurements in 10 terminals, which exhibited no significant change in plateau amplitude as stimulation frequency was varied over 15–100 Hz. Little or no significant increase in F/Frest was detected at stimulation frequencies ≤ 10 Hz (not shown). The right portion of the trace in Fig. 2 was recorded from the same terminal after the plasma membrane was permeabilized with digitonin. When bath [Ca2+] was increased from near zero to 1 mm, mitochondrial F/Frest in the permeabilized terminal increased to values much higher than the plateau amplitude recorded during motor nerve stimulation prior to permeabilization, demonstrating that this plateau was not due to dye saturation.
Figure 2. F/Frest increases recorded in a rhod-2-filled terminal during stimulation at 50 Hz followed by 100 Hz (left), and when bath [Ca2+] was increased following permeabilization of the plasma membrane with digitonin (right, 40 μg ml−1 for 5 min).
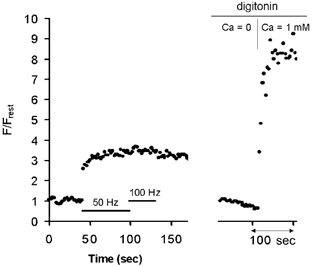
The finding that F/Frest was much greater in the Ca2+-exposed permeabilized terminal than in the intact stimulated terminal demonstrates that the plateau amplitude recorded during nerve stimulation was not due to rhod-2 saturation. The Ca2+-free solution contained 1 mm BAPTA. Recordings were carried out at 28 °C.
Sensitivity to photodamage necessitated the relatively low rates of image sampling employed in Figs 1 and 2, and made it difficult to measure the rise time of F/Frest responses and to follow the full extent of the slow post-stimulus decay of mitochondrial [Ca2+]. Figure 3B shows that a confocal imaging system that uses lower light intensities allowed collection of more images from a single terminal. Here mitochondrial F/Frest responses were obtained simultaneously from four neighbouring wild-type terminals loaded with rhod-5F. Figure 3A shows fluorescence micrographs of two of these terminals at rest and during 50 Hz stimulation. These F/Frest responses exhibited a higher signal-to-noise ratio than those in Figs 1 and 2, and appeared stable in spite of the higher rate of image collection. The shape of the upper three F/Frest responses resembled those in Figs 1 and 2. The plateau F/Frest values differed somewhat among the different terminals (see Discussion). The illustrated partial post-stimulus decays lasted several hundred seconds.
The lowest F/Frest response in Fig. 3B exhibited a somewhat different pattern, seen only rarely in wild-type terminals, in which F/Frest continued to increase throughout the stimulus train (ramping instead of plateauing). This F/Frest response also showed a faster initial decay than the F/Frest responses recorded in the other three terminals.
In some SOD1-G93A terminals mitochondrial [Ca2+] ramps rather than plateaus during repetitive stimulation
Fluorescence micrographs in Fig. 4 show motor terminals from SOD1-G93A mice stained with rhodamine 123, a positively charged dye whose accumulation within mitochondria depends on the potential gradient (internally negative) across the inner mitochondrial membrane. The staining patterns in Fig. 4 thus suggest that at least some mitochondria in these motor terminals retained a negative membrane potential. SOD1-G93A motor terminals displayed varying morphologies, some indistinguishable from wild-type (e.g. terminal at right in upper micrograph), others in various stages of apparent degeneration. Sprouting, thought to arise from surviving motor terminals expanding to innervate end-plates whose motoneurons have died, was observed frequently in SOD1-G93A (lower micrograph), but never in adult wild-type terminals. Some motor terminal areas visible with transmitted light failed to accumulate rhodamine 123 or rhod-2, suggesting that their mitochondria were depolarized or destroyed. Some myelinated axons (presumably motor) contained inclusion bodies (not shown). The presence of degenerating axons and terminals and sprouting terminals demonstrates that the levator auris longus motoneuron pool was affected by expression of SOD1-G93A.
Figure 4. Fluorescence micrographs of SOD1-G93A motor terminals loaded with rhodamine 123.
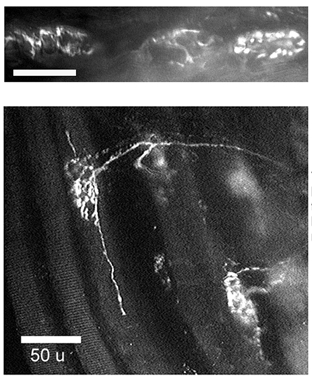
The upper micrograph illustrates three neighbouring terminals. The terminal at the right shows a wild-type staining pattern; the other two exhibit signs of degeneration (absence of bouton-like fluorescence clusters, irregular outline suggestive of dye uptake only into perisynaptic Schwann cells). The lower micrograph shows sprouting terminals. Rhodamine 123 was loaded using a 10 min exposure to 0.1 μm dye, followed by washout with dye-free saline solution. Calibration bars indicate 50 μm.
Stimulation-induced mitochondrial [Ca2+] responses could be elicited only in those SOD1-G93A terminals whose rhod staining patterns showed clusters of round, bouton-like structures arranged in a compact pattern like that seen in wild-type terminals (Fig. 4 top, right). Responses in mutant mice exhibited greater heterogeneity, both within and among terminals, than those in wild-type mice. The terminal in Fig. 5A displayed the spectrum of mitochondrial [Ca2+] responses measured in responsive SOD1-G93A terminals. Subregions of this terminal are indicated in Fig. 5B. Subregions in the upper right (marked by open symbols) yielded normal-looking F/Frest responses (plotted in Fig. 5C and averaged in Fig. 5E) that rose rapidly to a plateau and exhibited a slow post-stimulus decay. In contrast, the lower left regions of this terminal (filled symbols in Fig. 5B, D and E) displayed ramping behaviour, ranging from slower-than-normal attainment of a plateau during the stimulus train, to a higher-than-normal peak amplitude that decayed more rapidly than wild-type responses, and sometimes began decaying before the end of the stimulus train.
One possibility is that the ramping F/Frest response in SOD1-G93A terminals represents a combination of a normal, non-ramping mitochondrial response with a ramping response originating from non-mitochondrial compartments. This possibility can be tested by inhibiting mitochondrial Ca2+ uptake, which would be expected to increase the amplitude of any response component originating from cytosol or endoplasmic reticulum. Figure 6 shows that CCCP instead eliminated the ramping F/Frest response recorded in an SOD1-G93A terminal, indicating that this response originated mainly from the mitochondrial compartment.
Ramping was measured by dividing F/Frest measured at the end of the stimulus train by F/Frest measured midway through the train (1250 stimuli), with values exceeding 1.1 classified as ramping. By this criterion 62 % (13/21) of SOD1-G93A terminals ramped (average ratio 1.4), compared to only 3 % (2/60) of wild-type terminals. As illustrated in the averaged records of Fig. 5E, ramping terminals or subregions tended to reach higher peak F/Frest amplitudes than plateauing terminals/subregions. The average peak F/Frest value was 3.85 ± 0.88 (s.e.m.) for 13 ramping SOD1-G93A terminals, 2.56 ± 0.35 for 8 nonramping SOD1-G93A terminals, and 2.11 ± 0.16 for 60 wild-type terminals. The difference between peak F/Frest values in SOD1-G93A ramping terminals and wild-type terminals was significant (P < 0.05, two-tailed t test).
Limited attempts to ionophoretically inject a different indicator dye into SOD1-G93A motor axons, to enable imaging of cytosolic [Ca2+] responses, were unsuccessful, due in part to degeneration of motor axons in these late-stage mice.
DISCUSSION
Physiological importance of mitochondrial Ca2+ uptake in motor terminals
The stimulation-induced increases in mitochondrial [Ca2+] recorded here are consistent with David & Barrett's (2000) pharmacological evidence for mitochondrial Ca2+ uptake in mouse motor terminals, and with their hypothesis that this uptake is greater at physiological than at cooler temperatures. This mitochondrial Ca2+ uptake is important for maintaining evoked transmitter release (and thus neuromuscular transmission) during high-frequency stimulation. In the absence of curare, the end-plate potential is well maintained during 50 Hz stimulation, but exhibits accelerated and prolonged depression when mitochondrial Ca2+ uptake is inhibited pharmacologically (David & Barrett, 2003). This depression of phasic release is correlated with a large increase in both cytosolic [Ca2+] and asynchronous release. Billups & Forsythe (2002) found that agents that block mitochondrial Ca2+ uptake also slow recovery from depression at the calyx of Held synapse.
The increase in stimulation-induced mitochondrial [Ca2+] responses with increasing temperature is not simply a reflection of temperature-dependent changes in cytosolic [Ca2+], because the amplitude of cytosolic [Ca2+] responses decreases with increasing temperature (David & Barrett, 2000). Taken together, the cytosolic and mitochondrial [Ca2+] responses suggest instead that mammalian mitochondria take up Ca2+ less readily at cool temperatures than at temperatures in the physiological range. This temperature dependence might be intrinsic to the mitochondrial Ca2+ uniporter, or be mediated by temperature-dependent production of cytosolic factors controlling the Ca2+ affinity of the uniporter.
Cool temperatures not only increase the stimulation-induced elevation of cytosolic [Ca2+], but also accelerate depression of phasic transmitter release at some mammalian synapses (Moyer & van Lunteren, 2001; Li & Burke, 2001; Pyott & Rosenmund, 2002). These similarities between the effects of cooling and the effects of pharmacologically inhibiting mitochondrial Ca2+ uptake raise the possibility that inhibition of mitochondrial Ca2+ uptake contributes to the acclerated depression of phasic release at low temperature.
Limitation on stimulation-induced increases in mitochondrial matrix [Ca2+] in wild-type motor terminals
Mitochondrial [Ca2+] responses recorded during 50 Hz stimulation of wild-type mouse motor terminals resemble in several respects the responses recorded in lizard motor terminals (David, 1999): [Ca2+] rises to a plateau amplitude that persists until stimulation stops, and then falls slowly back towards baseline levels over a time course of hundreds of seconds. As in lizard terminals, the amplitude of the mitochondrial [Ca2+] plateau in mouse terminals does not vary with stimulation frequency. These stimulation-induced mitochondrial [Ca2+] responses thus differ in several respects from stimulation-induced cytosolic [Ca2+] responses recorded in mouse, lizard, frog and crayfish motor terminals, where the plateau amplitude increases with increasing stimulation frequency, and the decay is initially rapid (Wu & Betz, 1996; Ravin et al. 1997; David et al. 1998; David & Barrett, 2000).
The plateau of mitochondrial [Ca2+] is not an artifact of dye saturation, as demonstrated in Fig. 2 and by the finding that subregions of some SOD1-G93A terminals exhibit peak F/Frest values several-fold greater than those in adjacent plateauing subregions (Fig. 5C and D). David (1999) and David et al. (2003) estimated the plateau amplitude of mitochondrial [Ca2+] at ∼1 μm in lizard motor terminals. This magnitude is consistent with the Kd values of the dyes (rhod-2, rhod-5F) that proved most suitable for imaging mouse mitochondrial [Ca2+] responses. We could not detect stimulation-induced responses with lower-affinity dyes (e.g. rhod-5N), suggesting that in wild-type mouse terminals matrix [Ca2+] did not reach the millimolar levels reported during stimulation of bovine adrenal chromaffin cells (Montero et al. 2000).
The frequency-independent plateau amplitude and the slow post-stimulation decay of mouse and lizard mitochondrial [Ca2+] responses are consistent with the hypothesis that buffering of Ca2+ within the mitochondrial matrix includes reversible formation of an insoluble complex containing Ca and phosphate as well as mitochondrial import of phosphate (David, 1999). Kaftan et al. (2000) invoke a related idea with their hypothesis of ‘dynamic buffering’, in which the ability of the mitochondrial matrix to buffer incoming Ca2+ increases during stimulation. The ability of motor terminal mitochondria to limit the increase in matrix [Ca2+] probably contributes to their ability to keep taking up Ca2+ during prolonged stimulation, and also helps prevent opening of the permeability transition pore. Electron probe measurements of total Ca are consistent with the hypothesis that an insoluble complex contributes to matrix Ca2+ buffering: in sympathetic neurons stimulated with brief, high-[K+]-induced depolarizations and in dendrites of hippocampal neurons stimulated synaptically, total Ca2+ within mitochondria increases, and measurements of total Ca2+ within subregions of a given mitochondrion exhibit a pronounced heterogeneity, consistent with localized formation of Ca2+-containing complexes (Pivovarova et al. 1999, 2002). In contrast, measurements of total Ca2+ within endoplasmic reticulum are much more homogeneous (Hongpaisan et al. 2001).
According to the hypothesis mentioned above, the plateau amplitude of mitochondrial matrix [Ca2+] represents that [Ca2+] at which the solubility product of the hypothesized complex containing Ca2+ and phosphate is attained. The measured increase in this plateau amplitude with increasing temperature might arise if increasing temperature into the physiological range not only increases Ca2+ entry into mitochondria, but also decreases matrix [free inorganic phosphate], for example by stimulating mitochondrial ATP production. Another possibility is that the solubility product of the hypothesized complex increases with increasing temperature. Even at a constant temperature, the magnitude of plateau F/Frest values varied among experiments. Some of this variation probably resulted from the analytical technique. For example, calculated F/Frest values become smaller when more non-responsive pixels are included in the analysed region of interest, and are also sensitive to corrections for background fluorescence. Some of the variation in F/Frest might also reflect real differences in peak and/or resting matrix [Ca2+].
Evidence for mitochondrial Ca2+ dysregulation in some SOD1-G93A motor terminals
Figures 4 and 5 show that levator auris longus motor terminals in adult SOD1-G93A mice exhibit heterogeneous morphologies and mitochondrial [Ca2+] responses. Some terminals display stimulation-induced responses indistinguishable from those in wild-type littermates (Fig. 5C), suggesting that these terminals retained both a mitochondrial membrane potential sufficiently negative to take up Ca2+, and a normal ability to limit the increase in matrix [Ca2+]. Other SOD1-G93A terminals exhibit varying degrees of abnormality, including minimal accumulation of rhod dye, slower-than-normal attainment of the plateau amplitude, and marked ramping to higher-than-normal peak amplitudes. This latter abnormality suggests that mitochondria in some SOD1-G93A terminals are less able to limit stimulation-induced increases in matrix [Ca2+].
These ramping, high-amplitude matrix [Ca2+] responses may simply reflect a different, but harmless, mode of handling incoming Ca2+. Similarly shaped mitochondrial [Ca2+] responses were calculated in stimulated (wild-type) chromaffin cells by Montero et al. (2000) from light emitted by a mitochondrially targeted aequorin analogue.
Alternatively, the abnormally large stimulation-induced elevations in matrix [Ca2+] might reflect mitochondrial damage. The G93A mutation of SOD1 exhibits normal dismutase activity, but may lead to generation of excess hydroxyl radicals (Yim et al. 1996; Bogdanov et al. 1998; Andrus et al. 1998) that could damage mitochondria. Several aspects of mitochondrial function, including the phosphate transporter (Valenti et al. 2002), are especially sensitive to reactive oxygen species, and excessive [Ca2+] can itself increase mitochondrial free radical production (Dykens, 1994). Problems with mitochondrial Ca2+ handling might contribute to the findings of Roy et al. (1998) and Kruman et al. (1999) that SOD1-G93A spinal motoneurons in culture, as well as wild-type motoneurons transfected with mutant human SOD1, are more susceptible to a glutamate stress than spinal motoneurons expressing wild-type human SOD1. Dye-loaded mitochondria might also be damaged by the excitation light required for imaging. Perhaps excess endogenous generation of free radicals increases the sensitivity of SOD1-G93A mitochondria to such photodamage.
In summary, we present evidence that, during repetitive stimulation, mouse motor terminal mitochondria take up Ca2+ more readily at physiological than at cooler temperatures. These mitochondria contain a powerful buffering system that ‘caps’ the stimulation-induced elevation of matrix [Ca2+] at a value that does not vary with stimulation frequency over the range 15–100 Hz. We also show that in some motor terminals of SOD1-G93A mice, mitochondria are less able to limit this increase in matrix [Ca2+].
Acknowledgments
This work was supported by the Muscular Dystrophy Association, NIH grants RO1 NS 12404 and RO1 NS 12207, and an undergraduate summer research fellowship from the University of Miami for Ms Vila. We thank Dr Gavriel David for helpful comments concerning the manuscript, Baggi Somasundaram and Perkin-Elmer Life Sciences for their confocal microscope demonstration, and Edith Nonner and Myron Lutter for helping to maintain the SOD1-G93A mouse colony and for genotyping mice.
References
- Aguirre T, Matthijs G, Robberecht W, Tilkin P, Cassiman JJ. Mutational analysis of the Cu/Zn superoxide dismutase gene in 23 familial and 69 sporadic cases of amyotrophic lateral sclerosis in Belgium. Eur J Hum Genet. 1999;7:599–602. doi: 10.1038/sj.ejhg.5200337. [DOI] [PubMed] [Google Scholar]
- Alexianu ME, Ho B-K, Mohamed AH, La Bella V, Smith RG, Appel SH. The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann Neurol. 1994;36:846–858. doi: 10.1002/ana.410360608. [DOI] [PubMed] [Google Scholar]
- Andrus PK, Fleck TJ, Gurney ME, Hall ED. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 1998;71:2041–2048. doi: 10.1046/j.1471-4159.1998.71052041.x. [DOI] [PubMed] [Google Scholar]
- Angaut-Petit D, Molgo J, Connold AL, Faille L. The levator aruris longus muscle of the mouse: a convenient preparation for studies of short- and long-term presynaptic effects of drugs or toxins. Neurosci Lett. 1987;82:83–88. doi: 10.1016/0304-3940(87)90175-3. [DOI] [PubMed] [Google Scholar]
- Billups B, Forsythe ID. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci. 2002;22:5840–5847. doi: 10.1523/JNEUROSCI.22-14-05840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov MB, Ramos LE, Zu Z, Beal MF. Elevated ‘hydroxyl radical’ generation in vivo in an animal model of amyotrophic lateral sclerosis. J Neurochem. 1998;71:1321–1324. doi: 10.1046/j.1471-4159.1998.71031321.x. [DOI] [PubMed] [Google Scholar]
- Borthwick GM, Johnson MA, Ince PG, Shaw PJ, Turnbull DM. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Ann Neurol. 1999;46:787–790. doi: 10.1002/1531-8249(199911)46:5<787::aid-ana17>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Chiu AY, Zhai P, Dal Canto MC, Peters TM, Kwon YW, Prattis SM, Gurney ME. Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci. 1995;6:349–362. doi: 10.1006/mcne.1995.1027. [DOI] [PubMed] [Google Scholar]
- David G. Mitochondrial clearance of cytosolic Ca2+ in stimulated lizard motor nerve terminals proceeds without progressive elevation of mitochondrial matrix [Ca2+] J Neurosci. 1999;19:7495–7506. doi: 10.1523/JNEUROSCI.19-17-07495.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Barrett EF. Stimulation-evoked increases in cytosolic [Ca2+] in mouse motor nerve terminals are limited by mitochondrial uptake and are temperature-dependent. J Neurosci. 2000;20:7290–7296. doi: 10.1523/JNEUROSCI.20-19-07290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Barrett EF. Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J Physiol. 2003;548:425–438. doi: 10.1113/jphysiol.2002.035196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Barrett JN, Barrett EF. Evidence that mitochondria buffer physiological Ca2+ loads in lizard motor nerve terminals. J Physiol. 1998;509:59–65. doi: 10.1111/j.1469-7793.1998.059bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Talbot J, Barrett EF. Quantitative estimate of mitochondrial [Ca2+] in stimulated motor nerve terminals. Cell Calcium. 2003;33:197–206. doi: 10.1016/s0143-4160(02)00229-4. [DOI] [PubMed] [Google Scholar]
- Dykens JA. Isolated cerebral and cerebellar mitochondria produce free radicals when exposed to elevated Ca2+ and Na+: implications for neurodegeneration. J Neurochem. 1994;63:584–591. doi: 10.1046/j.1471-4159.1994.63020584.x. [DOI] [PubMed] [Google Scholar]
- Erzen I, Cvetko E, Obreza S, Angaut-Petit D. Fiber types in the mouse levator auris longus muscle: a convenient preparation to study muscle and nerve plasticity. J Neurosci Res. 2000;59:692–697. doi: 10.1002/(SICI)1097-4547(20000301)59:5<692::AID-JNR13>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Flood DG, Reaume AG, Gruner JA, Hoffman EK, Hirsch JD, Lin YG, Dorfman KS, Scott RW. Hindlimb motor neurons require Cu/Zn superoxide dismutase for maintenance of neuromuscular junctions. Am J Pathol. 1999;155:663–672. doi: 10.1016/S0002-9440(10)65162-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friel DD, Tsien RW. An FCCP-sensitive Ca2+ store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [Ca2+]i. J Neurosci. 1994;14:4007–4024. doi: 10.1523/JNEUROSCI.14-07-04007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellera C, Castellotti B, Riggio MC, Silani V, Morandi L, Testa D, Casali C, Taroni F, Di Donato S, Zeviani M, Mariotti C. Superoxide dismutase gene mutations in Italian patients with familial and sporadic amyotrophic lateral sclerosis: identification of three novel missense mutations. Neuromuscul Disord. 2001;11:404–410. doi: 10.1016/s0960-8966(00)00215-7. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Herrington J, Park YB, Babcock DF, Hille B. Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron. 1996;16:219–228. doi: 10.1016/s0896-6273(00)80038-0. [DOI] [PubMed] [Google Scholar]
- Higgins CMJ, Jung C, Gatha N, Xu Z-S. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J Neurosci. 2002;22:1–6. doi: 10.1523/JNEUROSCI.22-06-j0001.2002. RC215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongpaisan J, Pivovarova NB, Colegrove SL, Leapman RD, Friel DD, Andrews SB. Multiple modes of calcium-induced calcium release in sympathetic neurons II: a [Ca2+]i- and location-dependent transition from endoplasmic reticulum Ca accumulation to net Ca release. J Gen Physiol. 2001;118:101–112. doi: 10.1085/jgp.118.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince P, Stout N, Shaw P, Slade J, Hunziker W, Heizmann CW, Baimbridge KG. Parvalbumin and calbindin D-28k in the human motor system and in motor neuron disease. Neuropathol Appl Neurobiol. 1993;19:291–299. doi: 10.1111/j.1365-2990.1993.tb00443.x. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Haasdijk ED, Grashorn JA, Hawkins R, van Duijn W, Verspaget HW, London J, Holstege JC. Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis. 2000;7:623–643. doi: 10.1006/nbdi.2000.0299. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Rognoni F, van Duijn W, Verspaget HW, Haasdijk ED, Holstege JC. CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol. 2001;102:293–305. doi: 10.1007/s004010100399. [DOI] [PubMed] [Google Scholar]
- Jung C, Higgins CM, Xu X. Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. J Neurochem. 2002;83:535–545. doi: 10.1046/j.1471-4159.2002.01112.x. [DOI] [PubMed] [Google Scholar]
- Kaftan EJ, Xu T, Abercrombie RF, Hille B. Mitochondria shape hormonally induced cytoplasmic calcium oscillations and modulate exocytosis. J Biol Chem. 2000;275:25465–25470. doi: 10.1074/jbc.M000903200. [DOI] [PubMed] [Google Scholar]
- Keep M, Elmer E, Fong KS, Csiszar K. Intrathecal cyclosporin prolongs survival of late-stage ALS mice. Brain Res. 2001;894:327–331. doi: 10.1016/s0006-8993(01)02012-1. [DOI] [PubMed] [Google Scholar]
- Klivenyi P, Ferrante RJ, Matthews RT, Bogdanov MB, Klein AM, Andreassen OA, Mueller G, Wermer M, Kaddurah-Daouk R, Beal MF. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nature Med. 1999;5:347–350. doi: 10.1038/6568. [DOI] [PubMed] [Google Scholar]
- Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic V, Jackson-Lewis V, De Bilbao F, Dubois-Dauphin M, Przedborski S. Bcl-2: prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science. 1997;277:559–562. doi: 10.1126/science.277.5325.559. [DOI] [PubMed] [Google Scholar]
- Kruman II, Pedersen WA, Springer JE, Mattson MP. ALS-linked Cu/Zn-SOD mutation increases vulnerability of motor neurons to excitotoxicity by a mechanism involving increased oxidative stress and perturbed calcium homeostasis. Exp Neurol. 1999;160:28–39. doi: 10.1006/exnr.1999.7190. [DOI] [PubMed] [Google Scholar]
- Li Y, Burke RE. Short-term synaptic depression in the neonatal mouse spinal cord: effects of calcium and temperature. J Neurophysiol. 2001;85:2047–2062. doi: 10.1152/jn.2001.85.5.2047. [DOI] [PubMed] [Google Scholar]
- Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- Menzies FM, Ince PG, Shaw PJ. Mitochondrial involvement in amyotrophic lateral sclerosis. Neurochem Int. 2002;40:543–551. doi: 10.1016/s0197-0186(01)00125-5. [DOI] [PubMed] [Google Scholar]
- Montero M, Alonso MT, Carnicero E, Cuchillo-Ibáñez I, Albillos A, García AG, García-Sancho J, Alvarez J. Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat Cell Biol. 2000;2:57–61. doi: 10.1038/35000001. [DOI] [PubMed] [Google Scholar]
- Mourelatos Z, Gonatas NK, Stieber A, Gurney ME, Dal Canto MC. The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu, Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc Natl Acad Sci U S A. 1996;93:5472–5477. doi: 10.1073/pnas.93.11.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer M, van Lunteren E. Effect of temperature on endplate potential rundown and recovery in rat diaphragm. J Neurophysiol. 2001;85:2070–2075. doi: 10.1152/jn.2001.85.5.2070. [DOI] [PubMed] [Google Scholar]
- Murphy AN, Bredesen DE, Cortopassi G, Wang E, Fiskum G. Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc Natl Acad Sci U S A. 1996;93:9893–9898. doi: 10.1073/pnas.93.18.9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivovarova NB, Hongpaisan J, Andrews SB, Friel DD. Depolarization-induced mitochondrial Ca accumulation in sympathetic neurons: spatial and temporal characteristics. J Neurosci. 1999;19:6372–6384. doi: 10.1523/JNEUROSCI.19-15-06372.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivovarova NB, Pozzo-Miller LD, Hongpaisan J, Andrews SB. Correlated calcium uptake and release by mitochondria and endoplasmic reticulum of CA3 hippocampal dendrites after afferent synaptic stimulation. J Neurosci. 2002;22:10653–10661. doi: 10.1523/JNEUROSCI.22-24-10653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyott SJ, Rosenmund C. The effects of temperature on vesicular supply and release in autaptic cultures of rat and mouse hippocampal neurons. J Physiol. 2002;539:523–535. doi: 10.1113/jphysiol.2001.013277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravin R, Spira ME, Parnas H, Parnas I. Simultaneous measurement of intracellular Ca2+ and asynchronous transmitter release from the same crayfish bouton. J Physiol. 1997;501:251–262. doi: 10.1111/j.1469-7793.1997.tb00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- Roy J, Minotti S, Dong L, Figlewicz DA, Durham HD. Glutamate potentiates the toxicity of mutant Cu/Zn superoxide dismutase in motor neurons by postsynaptic calcium-dependent mechanisms. J Neurosci. 1998;18:9673–9684. doi: 10.1523/JNEUROSCI.18-23-09673.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siklos L, Engelhardt JI, Alexianu ME, Gurney ME, Siddique T, Appel SH. Intracellular calcium parallels motoneuron degeneration in SOD-1 mutant mice. J Neuropathol Exp Neurol. 1998;57:571–598. doi: 10.1097/00005072-199806000-00005. [DOI] [PubMed] [Google Scholar]
- Stuenkel EL. Regulation of intracellular calcium and calcium buffering properties of rat isolated neurohypophysial nerve endings. J Physiol. 1994;481:251–271. doi: 10.1113/jphysiol.1994.sp020436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. A fraction of yeast Cu, Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J Biol Chem. 2001;276:38084–38089. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Osanai M, Mitsumoto N, Akita T, Narita K, Kijima H, Kuba K. Ca2+-dependent Ca2+ clearance via mitochondrial uptake and plasmalemmal extrusion in frog motor nerve terminals. J Neurophysiol. 2002;87:1816–1823. doi: 10.1152/jn.00456.2001. [DOI] [PubMed] [Google Scholar]
- Tang YG, Zucker RS. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18:483–491. doi: 10.1016/s0896-6273(00)81248-9. [DOI] [PubMed] [Google Scholar]
- Valenti D, Atlante A, Barile M, Passarella S. Inhibition of phosphate transport in rat heart mitochondria by 3′-azido-3′-deoxythymidine due to stimulation of superoxide anion mitochondrial production. Biochem Pharmacol. 2002;64:201–206. doi: 10.1016/s0006-2952(02)01065-1. [DOI] [PubMed] [Google Scholar]
- Warita H, Hayashi T, Murakami T, Manabe Y, Abe K. Oxidative damage to mitochondrial DNA in spinal motoneurons of transgenic ALS mice. Brain Res Mol Brain Res. 2001;89:147–152. doi: 10.1016/s0169-328x(01)00029-8. [DOI] [PubMed] [Google Scholar]
- Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured dorsal root ganglion neurons. J Neurosci. 1994;14:348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondria and Na+/Ca2+ exchange buffer glutamate-induced calcium loads in cultured cortical neurons. J Neurosci. 1995;15:1318–1328. doi: 10.1523/JNEUROSCI.15-02-01318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Wu L-G, Betz WJ. Nerve activity but not intracellular calcium determines the time course of endocytosis at the frog neuromuscular junction. Neuron. 1996;17:769–779. doi: 10.1016/s0896-6273(00)80208-1. [DOI] [PubMed] [Google Scholar]
- Yim MB, Kang JH, Yim HS, Kwak HS, Chock PB, Stadtman ER. A gain-of-function of an amyotrophic lateral sclerosis-associated Cu, Zn-superoxide dismutase mutant: An enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. Proc Natl Acad Sci U S A. 1996;93:5709–5714. doi: 10.1073/pnas.93.12.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Ling S, Yu X-D, Venkatesh LK, Subramanian T, Chinnadurai G, Kuo TH. Modulation of mitochondrial Ca2+ homeostasis by Bcl-2. J Biol Chem. 1999;274:33267–33273. doi: 10.1074/jbc.274.47.33267. [DOI] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu Du C, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]