

Abstract
We sought to quantify the contribution of cardiac output (Q) and total vascular conductance (TVC) to carotid baroreflex (CBR)-mediated changes in mean arterial pressure (MAP) during mild to heavy exercise. CBR function was determined in eight subjects (25 ± 1 years) at rest and during three cycle exercise trials at heart rates (HRs) of 90, 120 and 150 beats min−1 performed in random order. Acute changes in carotid sinus transmural pressure were evoked using 5 s pulses of neck pressure (NP) and neck suction (NS) from +40 to −80 Torr (+5.33 to −10.67 kPa). Beat-to-beat changes in HR and MAP were recorded throughout. In addition, stroke volume (SV) was estimated using the Modelflow method, which incorporates a non-linear, three-element model of the aortic input impedance to compute an aortic flow waveform from the arterial pressure wave. The application of NP and NS did not cause any significant changes in SV either at rest or during exercise. Thus, CBR-mediated alterations in Q were solely due to reflex changes in HR. In fact, a decrease in the carotid-HR response range from 26 ± 7 beats min−1 at rest to 7 ± 1 beats min−1 during heavy exercise (P = 0.001) reduced the contribution of Q to the CBR-mediated change in MAP. More importantly, at the time of the peak MAP response, the contribution of TVC to the CBR-mediated change in MAP was increased from 74 ± 14 % at rest to 118 ± 6 % (P = 0.017) during heavy exercise. Collectively, these findings indicate that alterations in vasomotion are the primary means by which the CBR regulates blood pressure during mild to heavy exercise.
Recently, we quantified the contribution of cardiac output (Q) and total vascular conductance (TVC) to the carotid baroreflex (CBR)-mediated change in mean arterial pressure (MAP) at rest and found that alterations in vasomotion are the primary means by which the CBR regulates blood pressure (Ogoh et al. 2002). Furthermore, since stroke volume (SV) was not significantly altered during CBR stimulation, we identified that the heart rate (HR) response was a true representation of the reflex-mediated changes in Q at rest (Ogoh et al. 2002). Whether the mechanisms by which the CBR regulates blood pressure differ during exercise remains unclear. This is an important question considering the significant cardiovascular and haemodynamic adjustments that occur with exercise.
Numerous investigations have demonstrated that CBR control of blood pressure remains operable during mild to heavy exercise without any change in reflex sensitivity (Potts et al. 1993; Papelier et al. 1994; Norton et al. 1999a). However, several studies suggest that the CBR responds to changes in MAP differently from rest to exercise. Previous work has demonstrated that the magnitude of the CBR-mediated increase in HR in response to neck pressure (NP) decreased during exercise, while the reflex bradycardia in response to neck suction (NS) was augmented in comparison to rest (Potts et al. 1993; Potts & Raven, 1995). More recently, Keller et al. (2003) identified that decreases in leg vascular conductance in response to NP were attenuated in the exercising leg compared with rest. Collectively, these studies suggest that the relative contribution of Q and TVC to baroreflex-mediated changes in MAP may be altered by exercise. The major unknown in determining the contribution of Q and TVC to the baroreflex-mediated changes in MAP during exercise is whether the baroreflex adjusts its ability to alter SV in comparison to rest. This is particularly important considering the large increases in SV that occur with exercise due to the muscle pump (Laughlin, 1987) and increases in mitral valve flow velocity (Turkevich et al. 1988).
Previously, we utilized pulsed-Doppler ultrasound recordings of ascending aortic blood flow velocity and aortic root dimension to quantify beat-to-beat changes in SV during the application of NP and NS at rest and quantified the relative contributions of Q and TVC to the CBR-mediated changes in MAP (Ogoh et al. 2002). However, movement artifacts and signal quality compromise the fidelity of the pulsed-Doppler ultrasound recordings of the aorta during exercise. Therefore, in the present investigation we used a three-element model (Modelflow) to estimate SV from the impedance changes of the arterial blood flow velocity curve obtained from beat-to-beat measurements of arterial blood pressure (Wesseling et al. 1993). This technique has been shown to track fast and brisk changes in stroke volume during various experimental protocols (Jansen et al. 1990; Gratz et al. 1992; Stok et al. 1993), including submaximal cycling exercise (Sugawara et al. 2001). Thus, we were able to measure beat-to-beat changes in SV during mild to heavy exercise.
The primary goal of this investigation was to determine the contribution of reflex changes in SV to the CBR-mediated changes in MAP during exercise. This allowed us to proportion the change in MAP between the reflex-mediated response of Q and TVC to each NP and NS stimulus. In addition, we examined the influence of workload-induced decreases in cardiac filling time on the SV response to the CBR-mediated changes in Q and TVC by comparing responses to NP and NS during mild, moderate and heavy exercise. We hypothesized that with increasing exercise workloads the SV contribution to the CBR-mediated changes in MAP would be unchanged from rest. Thus, alterations in TVC would be the primary means by which the CBR regulates blood pressure during mild to heavy exercise.
METHODS
Seven men and one woman (means ±s.e.m.: age 25 ± 1 years; height 183 ± 3 cm; weight 74.0 ± 2.2 kg) were recruited for voluntary participation in the present study. All subjects were free of any known cardiovascular and pulmonary disorders, were normotensive and were not using prescribed or over the counter medications. Each subject provided written informed consent as approved by The Ethics Committee of Copenhagen (01–386/02). All experiments were performed in accordance with the Declaration of Helsinki. Subjects were requested to abstain from caffeinated beverages for 12 h and strenuous physical activity and alcohol for at least 24 h prior to arriving at the laboratory. Before any experiments were performed each subject visited the laboratory for familiarization with the techniques and procedures of the protocol.
Experimental measurements
Arterial blood pressure was measured by a Teflon catheter (1.1 mm i.d., 20 gauge) placed in the brachial artery of the non-dominant arm. The catheter was connected to a sterile disposable pressure transducer (Uden, The Netherlands) positioned at the level of the right atrium in the midaxillary line. HR was monitored using a standard lead II electrocardiogram (ECG). The arterial blood pressure and ECG signal outputs were connected to a Dialogue 2000 monitor (IBC-Danica, Copenhagen, Denmark) interfaced with a personal computer equipped with customized data acquisition software for the beat-to-beat measurement of cardiovascular variables. SV was calculated off-line from the blood pressure waveform using a Modelflow software program incorporated into BeatScope version 1.0 software (TNO-TPD, Biomedical Instrumentation, Amsterdam, The Netherlands). Modelflow is a three-element model that uses the arterial input impedance, including continuous correction for variations in the diameter, the compliance of the aorta, and total peripheral resistance, describing the relationship between aortic flow and pressure, and computing SV (Wesseling et al. 1993). This methodology has been shown to track fast and brisk changes in SV during various experimental protocols (Jansen et al. 1990; Gratz et al. 1992; Stok et al. 1993), including submaximal cycling exercise (Sugawara et al. 2001). Individual parameters of age, sex, height and weight are required for the model and were entered prior to the collection of data for each subject. Q was calculated from the product of SV and HR, and TVC was calculated beat-to-beat from the ratio of Q to MAP (TVC =Q/ MAP).
Experimental protocol
On the experimental day, the subjects arrived at the laboratory at least 2 h following a light meal. The subjects were seated in a semi-recumbent position on a hospital bed that was modified to allow the subject to perform seated upright cycling exercise (Galbo et al. 1987). After being instrumented for the measurement of blood pressure, SV (Modelflow) and HR, each subject was fitted with a malleable lead neck collar that encircled the anterior 2/3 of the neck for the application of neck pressure (NP) and neck suction (NS). Resting CBR function was determined using random-ordered 5 s pulses of NP and NS presented at +40, +20, 0, −20, −40, −60 and −80 Torr during a 10–15 s breath-hold at end-expiration. Under resting conditions, four to five pulses of NP and NS were applied at each pressure and were separated by a period of 45 s. Exercise CBR function was determined during three 20 min bouts of exercise at steady-state heart rates of 90, 120 and 150 beats min−1 representing mild, moderate and heavy exercise workloads. The reason for using three workloads was to evaluate whether the contribution of the Q and TVC response to NP and NS would be altered by the decreases in cardiac filling time induced by the progressive workload-dependent increases in HR. The exercise bouts were performed in random order and each bout was separated by a period of 30–40 min to enable sufficient recovery from the preceding exercise trial. Subjects began exercising at a workload of 10 W, which was then adjusted accordingly to reach the target HR for that particular exercise bout (Table 1). Once the target HR was achieved subjects exercised for 6–8 min to ensure steady-state conditions before CBR function was assessed. During exercise, NP and NS were applied without the presence of a breath-hold (Eckberg et al. 1980; Norton et al. 1999b). Only two to three 5 s pulses of NP and NS at each pressure were applied during exercise, as the time for CBR testing was limited to 12–14 min. This enabled the subjects to be at a steady-state before CBR testing began (6–8 min) and also minimized any confounding effects of cardiovascular drift on CBR function (Norton et al. 1999b). A minimum of 30 s was allotted between each NP and NS trial during exercise.
Table 1.
Haemodynamic and cardivascular responses to mild (EX90), moderate (Ex120) and heavy (EX150) exercise
| WL(W) | SV(ml) | HR(beats min−1) | MAP (mmHg) | Q (1 min−1) | TVC (1 min−1 mmHg−1) | |
|---|---|---|---|---|---|---|
| Rest | 0 | 99.9 ± 4.2 | 64.0 ± 4.6 | 90.3 ± 2.3 | 6.3 ± 0.3 | 0.07 ± 0.004 |
| EX90 | 48.8 ± 11.3 | 119.5 ± 7.5* | 89.8 ± 2.2* | 88.5 ± 2.8 | 107 ± 0.7* | 0.122 ± 0.009* |
| EX120 | 1163 ± 10.5 | 126.8 ± 7.6* | 121.3 ± 3.7*† | 94.8 ± 2.9 | 15.4 ± 1.1*† | 0.163 ± 0.0011*† |
| EX150 | 168.8 ± 12.6 | 135.5 ± 6.3*† | 156.7 ± 2.5*†‡ | 105.7 ± 3.9*†‡ | 21.2 ± 0.8*†‡ | 0.202 ± 0.010*†‡ |
Values are means ±s.e.m. at rest and during three exercise (EX) workloads (heart rate of 90, 120 and 150 beats min−1). WL, workloads: SV, stroke volume; HR, heart rate; MAP, mean arteial pressure; Q, cardiac output; TVC, total vascular conductance
Different from rest (P < 0.05)
different from EX90 (P < 0.05)
different from EX120 (P < 0.05).
Data analysis
The carotid-HR and the carotid-MAP responses were evaluated by plotting the peak changes in HR and MAP, respectively, against estimated carotid sinus pressure (ECSP), which was calculated as MAP minus neck chamber pressure. Each carotid baroreflex stimulus-response curve was fitted to the logistic model described by Kent et al. (1972). This function incorporates the following equation:
where HR or MAP is the dependent variable, A1 is the range of response of the dependent variable (maximum - minimum), A2 is the gain coefficient (i.e. slope), A3 is the carotid sinus pressure required to elicit equal pressor and depressor responses (centring point), and A4 is the minimum response of HR or MAP. Data were fitted to this model by non-linear least-squares regression (using a Marquardt-Levenberg algorithm), which minimized the sum of squares error term to predict a curve of ‘best fit’ for each set of raw data. The coefficient of variation for the overall fit of this model to the individual responses was 18 % (Potts et al. 1993). The gain was calculated from the first derivative of the logistic function and the maximal gain (Gmax) was applied as the index of carotid baroreflex responsiveness. Threshold (CSPthr), the point where no further increase in the dependent variable occurred despite reductions in ECSP, and saturation (CSPsat), the point where no further decrease in the dependent variable occurred despite increases in ECSP, were calculated as the maximum and minimum second derivatives, respectively, of the logistic function curve. For calculation of CSPthr and CSPsat, we applied equations described by Chen & Chang (1991): CSPthr=−2.0/A2+A3 and CSPsat= 2.0/A2+A3. These calculations of CSPthr and CSPsat are the carotid sinus pressure at which MAP or HR is within 5 % of its maximal or minimal response (Potts et al. 1993).
Since TVC is the sum of all regional vascular conductances, the percentage contributions of Q and TVC to CBR-mediated changes in MAP can be calculated. This was achieved by calculating the predicted change in MAP during CBR stimulation, if only the individual changes in Q or TVC occurred and all other parameters remained at control values (Ogoh et al. 2002). The percentage contributions of Q and TVC were calculated as follows:
 |
where MAPQ is the MAP response to carotid baroreceptor stimulation due to Q alone, MAPTVC is the MAP response to carotid baroreceptor stimulation due to TVC alone, MAPcontrol is the MAP value prior to NP/NS, Qcontrol is the Q value prior to NP/NS and TVCcontrol is the TVC value prior to NP/NS.
As demonstrated previously, there is a time delay between the peak changes in HR and MAP in response to NP and NS (Potts et al. 1993; Ogoh et al. 2002). The peak change in HR occurs within 2–3 s from the initiation of the neck pressure pulse and returns to baseline at the time the peak MAP response occurs, which is typically 6–8 s following the onset of NP or NS (Potts & Raven, 1995; Ogoh et al. 2002). Taking these response times into consideration, in the current study changes in the percentage contribution of Q and TVC to the CBR-mediated change in MAP were calculated at the MAP that occurs at the time of the peak HR response as well as at the time of the peak MAP response.
Statistical analysis
One-way analysis of variance (ANOVA) with repeated measures was used to assess the differences in haemodynamic and CBR responses between rest and the three exercise bouts. Repeated measures one-way ANOVA was also used to compare the contributions of Q and TVC at the time of the peak HR and MAP response between rest and the three exercise bouts. A Student-Newman-Keuls test was employed post hoc when main effects were significant. Statistical significance was set at P < 0.05. Data are expressed as means ±s.e.m. Analyses were conducted using SigmaStat (Jandel Scientific Software, SPSS Inc., Chicago, IL, USA) for Windows.
RESULTS
Cardiovascular and haemodynamic responses to exercise
As expected, increases in Q and TVC were proportional to the exercise intensities used to elicit HRs of 90, 120 and 150 beats min−1 (Table 1).
Carotid baroreflex control of SV
Under resting conditions, NP and NS did not cause any significant changes in SV, as we have previously demonstrated using measurements of pulsed-Doppler ultrasound to quantify SV (Ogoh et al. 2002). More importantly, although SV was increased by exercise, NP and NS did not significantly alter SV during any of the three exercise bouts (Table 2).
Table 2.
Stroke volume responses to NP and NS at rest and during mild (EX90), moderate (EX120) and heavy (EX150) exercise
| CP (Torr) | Rest(ml) | EX90(ml) | EX120(ml) | EX150(ml) |
|---|---|---|---|---|
| 40 | 98.0 ± 4.0 | 120.3 ± 7.7 | 129.8 ± 7.5 | 136.3 ± 6.5 |
| 20 | 99.4 ± 4.3 | 120.4 ± 8.0 | 127.2 ± 7.6 | 136.4 ± 6.7 |
| 0 | 99.9 ± 4.2 | 119.5 ± 7.5 | 126.8 ± 7.6 | 135.5 ± 6.3 |
| −20 | 101.8 ± 4.2 | 121.5 ± 7.6 | 128.3 ± 8.5 | 138.6 ± 6.5 |
| −40 | 100.8 ± 4.2 | 122.0 ± 7.1 | 131.3 ± 7.6 | 138.4 ± 6.8 |
| −60 | 101.0 ± 4.1 | 124.6 ± 7.1 | 127.6 ± 7.0 | 138.5 ± 6.3 |
| −80 | 99.2 ± 3.9 | 123.2 ± 7.3 | 129.8 ± 7.3 | 138.9 ± 6.1 |
Values are means ±s.e.m. at rest and during three excercise workloads. CP, chamber pressure.
Carotid baroreflex control of HR and MAP
CBR stimulus-response curves for both HR and MAP were relocated upward and rightward from rest during exercise in a workload-dependent manner (Fig. 1 and Fig. 2). This progressive resetting of the carotid-HR and carotid-MAP reflex function curves occurred without any changes in the maximal gain of the CBR. Although the carotid-MAP response range was unchanged from rest to exercise (P > 0.05), the carotid-HR response range decreased from 26 ± 7 beats min−1 at rest to 11 ± 2 and 7 ± 1 beats min−1 during moderate and heavy exercise, respectively (P < 0.01).
Figure 1.
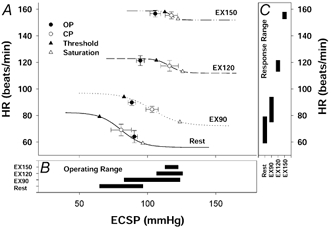
A, carotid-HR (cardiac) stimulus-response curves at rest and during mild (EX90), moderate (EX120) and heavy (EX150) exercise. Lines represent mean data fitted to the logistic function model. Symbols denote actual group data (means ±s.e.m.); OP, pre-stimulus operating point; CP, centring point; Threshold, carotid sinus threshold pressure; Saturation, carotid sinus saturation pressure; ECSP, estimated carotid sinus pressure. B, operating range of the carotid-HR stimulus-response curve derived from the logistic function model. C, response range (A1) of the carotid-HR stimulus-response curve derived from the logistic function model. The HR response curves were reset upward and rightward during exercise without a change in sensitivity. Although carotid sinus threshold pressure was augmented with the increasing exercise intensity, carotid sinus saturation pressure was not significantly increased during moderate and heavy exercise compared with mild exercise. Thus, the operating and response ranges of the carotid-HR baroreflex function curves were decreased during moderate and heavy exercise.
Figure 2.
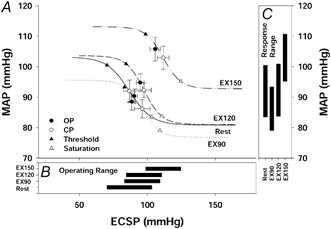
A, carotid-MAP (vasomotor) stimulus-response curves at rest and during mild (EX90), moderate (EX120) and heavy (EX150) exercise. Lines represent mean data fitted to the logistic function model. Symbols denote actual group data (means ±s.e.m.). B, operating range of the carotid-MAP stimulus-response curve derived from the logistic function model. C, response range (A1) of the carotid-MAP stimulus-response curve derived from the logistic function model. The MAP response curves were reset upward and rightward during exercise without a change in sensitivity. The operating and response ranges of the carotid-MAP baroreflex function curves were unchanged from rest to heavy exercise.
Carotid baroreflex control of Q and TVC
At rest and during mild and moderate exercise, the application of NP and NS produced CBR-mediated changes in Q with a similar range of response. However, during heavy exercise the range of response was slightly reduced (−25 ± 10 %, P = 0.190). Alterations in ECSP with NP and NS produced significant changes in TVC at rest and during all exercise workloads. In contrast to the Q response, the range of response of TVC to changes in ECSP during the three exercise workloads was increased from 0.019 ± 0.004 l min−1 mmHg−1 at rest to 0.043 ± 0.005, 0.038 ± 0.004 and 0.048 ± 0.003 l min−1 mmHg−1 during mild (EX90), moderate (EX120) and heavy (EX150) exercise, respectively (P < 0.05).
The contribution of Q and TVC to the CBR-mediated change in MAP
At rest, the contribution of Q to the CBR-mediated change in MAP was greatest at the time of the peak HR response during the 5 s period of NP or NS stimulation. However, after the peak HR response, the contribution of Q decreased, and the contribution of TVC peaked at approximately the same time as the peak MAP response (Fig. 3). Thus, under resting conditions the contribution of Q to the CBR-mediated change in MAP was greatest at the time of the peak HR response, whereas at the time of the peak MAP response changes in TVC were predominant. During exercise there was a significant decrease in the contribution of Q to the CBR-mediated change in MAP at the time of the peak HR response, with Q contributing only −8 ± 8 % during heavy exercise, which coincided with a greater contribution of TVC. Thus, as exercise intensity increased, the contribution of TVC became greater as the contribution of Q decreased at the time of the peak HR response. Similarly, at the time of the peak MAP response, the predominance of alterations in TVC in the CBR-mediated change in MAP at rest became even more profound during exercise.
Figure 3.
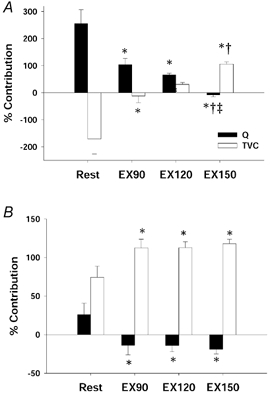
Percentage contribution of cardiac output (Q) and total vascular conductance (TVC) to the CBR-mediated changes in MAP at the time of the peak HR response (A) and at the time of the peak MAP response (B) at rest and during mild (EX90), moderate (EX120) and heavy (EX150) exercise. Bars represent the average contribution of all NP and NS stimuli for all subjects (means ±s.e.m.). *Different from Rest (P < 0.05); †different from EX90 (P < 0.05); ‡different from EX120 (P < 0.05). As exercise intensity increased, the contribution of Q progressively decreased at the time of the peak HR response. Similarly, at the time of the peak MAP response, the small contribution of Q seen at rest was completely eliminated with even mild exercise. The contribution of TVC at the time of both the peak HR response and the peak MAP response increased with exercise.
DISCUSSION
The major findings of the present investigation are threefold. First, similar to resting conditions, alterations in SV do not contribute to the CBR-mediated changes in MAP during exercise, indicating that CBR-mediated alterations in Q are solely due to reflex changes in HR. Second, a decrease in the carotid-HR response range during moderate to heavy exercise reduced the contribution of Q to the CBR-mediated change in MAP. Lastly, alterations in TVC predominate over Q in mediating CBR changes in MAP at rest and throughout mild to heavy exercise. These findings confirm our previous work indicating that at rest changes in MAP induced by CBR stimulation were primarily mediated by alterations in TVC, but more importantly we now extend these findings to dynamic exercise. Thus, we contend that alterations in vasomotion are the primary means by which the CBR regulates arterial blood pressure.
The data from the present investigation demonstrate that despite exercise intensity-induced increases in SV and decreases in cardiac filling time (Turkevich et al. 1988), SV was unchanged from pre-stimulus steady-state exercise values by NP and NS. Therefore, any CBR-mediated changes in Q were due to carotid-HR responses both at rest and during exercise. Due to exercise intensity-related decreases in the carotid-HR response range, the contribution of Q to the CBR-mediated change in MAP at the time of the peak HR and the peak MAP response was reduced from 256 ± 51 % and 26 ± 15 % at rest to −8 ± 8 % and −19 ± 6 % during heavy (EX150) exercise, respectively. In contrast, the contribution of TVC to the CBR-mediated change in MAP at the time of the peak HR and the peak MAP response was increased from −171 ± 55 % and 74 ± 14 % at rest to 106 ± 7 % and 118 ± 6 % during heavy (EX150) exercise, respectively. Thus, any contribution of Q to the CBR-mediated change in MAP at rest was progressively diminished by exercise, with the end result being no contribution of Q during heavy exercise. At the same time, the greater contribution of TVC to the CBR-mediated change in MAP at rest became even more profound throughout mild to heavy exercise.
It is important to recognize that the diminution of the contribution of Q with a progressive increase in the contribution of TVC from mild to heavy exercise primarily affected the MAP response at the time of the peak HR response when the CBR-mediated change in MAP was only ≈2–3 mmHg. However, at the time of the peak MAP response, which occurred ≈6–8 s following the onset of NP or NS, the small contribution of Q seen at rest was completely eliminated with even mild exercise as the contribution of TVC became significantly greater. Hence, the predominance of alterations in TVC in the CBR-mediated change in MAP observed at rest was maintained during exercise with a similar elevation in the contribution of TVC observed during mild, moderate and heavy exercise. Thus, the capacity of the carotid baroreflex to regulate arterial blood pressure during exercise depends critically on its ability to alter TVC. These findings are in accord with previous animal investigations reporting the importance of sympathetic control of vascular conductance in mediating CBR-induced changes in blood pressure during exercise (O'Leary et al. 1991; Collins et al. 2001). The similar pressor responses elicited by bilateral carotid occlusion at rest and during treadmill exercise in dogs were primarily dependent on changes in vascular conductance with the active skeletal muscle vascular bed becoming the primary site for carotid baroreflex-induced vasoconstriction during exercise. In agreement, Keller et al. (2003) measured CBR-mediated changes in leg blood flow and leg vascular conductance during unilateral leg exercise in humans and also identified that the active skeletal muscle is an essential site of the baroreflex-mediated vasomotor responses induced by NP and NS. Collectively, these studies along with the findings of the current investigation demonstrate conclusively that vasomotor control is essential to the regulation of arterial blood pressure by the carotid baroreflex during exercise.
Although sympathetic neural control of the vasculature is essential to the regulation of systemic blood pressure, sympathetic vasoconstriction during exercise could compromise blood flow to active muscle thereby limiting the delivery of oxygen to exercising muscle. As such, several studies have indicated that a metabolic modulation within active skeletal muscle attenuates sympathetically mediated vasoconstriction, consequently preserving oxygen delivery to meet the metabolic demands of the active muscles (Strange et al. 1990; Saltin et al. 1998; Hansen et al. 2000; Tschakovsky et al. 2002). Such attenuation, termed ‘functional sympatholysis’ by Remensnyder et al. (1962), appears to be exercise intensity dependent with a greater blunting of vasoconstriction during higher intensity exercise (Tschakovsky et al. 2002).
At first our finding that exercise increased the contribution of TVC to the CBR-mediated pressor response may not appear consistent with previous studies indicating functional sympatholysis. However, based on our previous work demonstrating that during one-legged knee extension exercise CBR control of MAP was well maintained despite an attenuated CBR-mediated percentage reduction in exercising leg vascular conductance (LVC) in response to NP (Keller et al. 2003), we contend that a metabolic modulation of CBR vascular control is present within active skeletal muscle. Nevertheless it is important to note that in our previous study when CBR changes in LVC were expressed in absolute units of conductance, it was apparent that the CBR-mediated reduction in LVC in the exercising leg was greater than in the non-exercising leg, as well as that observed at rest. This is an important distinction, which can be explained by the fact that skeletal muscle blood flow increases dramatically with exercise compared to rest. As such, its contribution to systemic blood pressure regulation can remain high even when the relative (%) effects of vasomotor activity are blunted. In other words, a smaller percentage reduction in vascular conductance within exercising muscle may constitute an overall greater effect on systemic blood pressure due to the augmentation of blood flow in comparison to rest. Thus, despite an attenuated percentage reduction in vascular conductance in active muscle, the contribution of exercising muscle to CBR-induced vasomotor responses remains critical for blood pressure control along with vasoconstrictor responses in non-exercising muscle and visceral organs. Physiologically these alterations assist in redirecting cardiac output to active muscles to meet the metabolic demands of the exercising muscle and at the same time allow for the continued regulation of blood pressure during exercise.
Previously, we demonstrated that at rest SV was unchanged by the application of NP and NS and, therefore, the carotid-HR reflex primarily represented the contribution of Q to CBR-mediated changes in MAP. In addition, we demonstrated that the carotid-MAP reflex mainly represented the contribution of TVC to the CBR-mediated changes in MAP. The data from our current investigation confirm these previous findings and more importantly extend these observations to exercise. Thus, the carotid-HR reflex primarily represented the contribution of Q and the carotid-MAP reflex accurately reflected the CBR control of TVC not only at rest but also throughout mild to heavy exercise. Although previous studies (Potts et al. 1993; Norton et al. 1999a) have referred to the carotid-MAP reflex as the carotid-vasomotor reflex, this relationship was only speculative. The findings of the current investigation validate the appropriateness of using the carotid-MAP response as a measure of carotid-vasomotor responsiveness both at rest and during exercise.
Peak changes in HR and MAP in response to each NP and NS stimulus were used in developing the carotid-HR and carotid-MAP reflex function curves, respectively, at rest and during the three exercise intensities. Both the CBR-HR and CBR-MAP function curves were reset to operate around the prevailing arterial blood pressure of the exercise without an alteration in the maximal gain or sensitivity of the reflex (Potts et al. 1993; Raven et al. 1997, 2002; Norton et al. 1999a). In addition, as previously described, the resetting of the carotid baroreflex occurred in relation to the intensity of exercise, and the operating point was relocated away from the centring point and towards the threshold pressure of the reflex (Papelier et al. 1994; Norton et al. 1999a). Furthermore, we observed that the response range of the CBR-HR function curve was reduced during moderate (EX120) and heavy (EX150) exercise (Fig. 1). With no change in the maximal gain, it would be expected that reductions in the response range of the carotid-HR reflex would be associated with proportionate decreases in the operating range of the reflex. Indeed, an increase in the carotid sinus pressure at threshold occurred during EX120 and EX150 without a change in the carotid sinus pressure at saturation, thereby reducing the operating range. Re-analysis of the data developed by Norton et al. (1999a) in which CBR-HR function curves were calculated at rest and during exercise workloads of 50 %, 75 % and 100 % VO2,max identified a similar intensity-dependent reduction in the operating range of the CBR-HR reflex due to increases in the threshold without any changes in the saturation. We suggest that a possible mechanism by which the threshold of the CBR-HR reflex is affected and yet the saturation remains constant with increasing exercise intensity may be the withdrawal of vagal activity associated with the higher exercise intensities. In agreement with this idea, Potts and colleagues (Potts et al. 1993; Potts & Raven, 1995) indicated that the reduced tachycardic response to NP during 50 % VO2, peak compared to rest and 25 % VO2, peak exercise was due to the reduced vagal tone associated with the higher exercise intensity of 50 % VO2, peak.
Potential limitation
We have previously used pulsed-Doppler ultrasound measurements of aortic blood flow velocity to determine whether the 5 s periods of NP and NS stimulation induced changes in SV at rest (Ogoh et al. 2002). However, in the current study, using pulsed-Doppler to measure beat-to-beat SV responses to NP and NS during moderate to heavy semi-recumbent cycling exercise proved too difficult due to measurement artifacts related to thoracic cage movement and the quality of signal acquisition. Therefore, to eliminate these problems, we used the Modelflow method for SV measurements. This methodology has been shown to provide accurate estimates of SV from the intra-arterial blood pressure waveform under various haemodynamic conditions (Jansen et al. 1990; Gratz et al. 1992; Stok et al. 1993). In addition, Sugawara et al. (2001) compared the Modelflow method with pulsed-Doppler cardiography at rest and during exercise, and demonstrated that Modelflow could provide an accurate estimation of SV and Q during submaximal exercise. Although others have suggested that Modelflow is not an accurate method for estimating SV and Q during moderate exercise, these measurements were made from non-invasive blood pressure data collected by Portapres (Houtman et al. 1999). This probably minimized the quality of the blood pressure waveform recordings, thereby reducing the accuracy of the Modelflow methodology. Thus, we contend that the Modelflow method provides a suitable methodology for obtaining beat-to-beat measurements of SV during the application of NP and NS both at rest and during mild to heavy cycling exercise. Moreover, given that the CBR-mediated HR response does not significantly contribute to the peak MAP response induced by NP and NS, there would have to be a substantial change in SV in order for cardiac output to contribute significantly to the peak CBR-mediated MAP response. It is unlikely that such changes would have occurred without any changes in the Modelflow SV measurements. This is particularly true considering that the results on the contribution of SV and Q obtained at rest in the current study are nearly identical to those obtained in our previous study using pulsed-Doppler ultrasound measurements to assess SV (Ogoh et al. 2002).
We conclude that alterations in SV do not contribute to CBR-mediated changes in MAP during exercise, indicating that CBR-mediated alterations in Q are solely due to reflex changes in HR. More importantly, similar to resting conditions, alterations in TVC predominate over Q in mediating CBR changes in MAP during exercise. Thus, alterations in vasomotion are the primary means by which the CBR regulates arterial blood pressure during mild to heavy exercise.
Acknowledgments
The authors appreciate the time and effort expended by all the volunteer subjects. We thank Lisa Marquez for her assistance in preparing the document. This study was supported in part by NIH grant no. HL-045547 and by a grant from the Copenhagen Muscle Research Centre (the Danish National Research Foundation 504–14).
REFERENCES
- Chen HI, Chang KC. Assessment of threshold and saturation pressure in the baroreflex function curve: a new mathematical analysis. Jpn J Physiol. 1991;41:861–877. doi: 10.2170/jjphysiol.41.861. [DOI] [PubMed] [Google Scholar]
- Collins HL, Augustyniak RA, Ansorge EJ, O'Leary DS. Carotid baroreflex pressor responses at rest and during exercise: cardiac output vs. regional vasoconstriction. Am J Physiol Heart Circ Physiol. 2001;280:H642–648. doi: 10.1152/ajpheart.2001.280.2.H642. [DOI] [PubMed] [Google Scholar]
- Eckberg DL, Kifle YT, Roberts VL. Phase relationship between normal human respiration and baroreflex responsiveness. J Physiol. 1980;304:489–502. doi: 10.1113/jphysiol.1980.sp013338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbo H, Kjaer M, Secher NH. Cardiovascular, ventilatory and catecholamine responses to maximal dynamic exercise in partially curarized man. J Physiol. 1987;389:557–568. doi: 10.1113/jphysiol.1987.sp016672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratz I, Kraidin J, Jacobi AG, de Castro NG, Spagna P, Larijani GE. Continuous noninvasive cardiac output as estimated from the pulse contour curve. J Clin Monit. 1992;8:20–27. doi: 10.1007/BF01618083. [DOI] [PubMed] [Google Scholar]
- Hansen J, Sander M, Thomas GD. Metabolic modulation of sympathetic vasoconstriction in exercising skeletal muscle. Acta Physiol Scand. 2000;168:489–503. doi: 10.1046/j.1365-201x.2000.00701.x. [DOI] [PubMed] [Google Scholar]
- Houtman S, Oeseburg B, Hopman MT. Non-invasive cardiac output assessment during moderate exercise: pulse contour compared with CO2 rebreathing. Clin Physiol. 1999;19:230–237. doi: 10.1046/j.1365-2281.1999.00166.x. [DOI] [PubMed] [Google Scholar]
- Jansen JR, Wesseling KH, Settels JJ, Schreuder JJ. Continuous cardiac output monitoring by pulse contour during cardiac surgery. Eur Heart J. 1990;11(suppl. I):26–32. doi: 10.1093/eurheartj/11.suppl_i.26. [DOI] [PubMed] [Google Scholar]
- Keller DM, Wasmund WL, Wray DW, Ogoh S, Fadel PJ, Smith ML, Raven PB. Carotid baroreflex control of leg vascular conductance at rest and during exercise. J Appl Physiol. 2003;94:542–548. doi: 10.1152/japplphysiol.00817.2002. [DOI] [PubMed] [Google Scholar]
- Kent BB, Drane JW, Blumenstein B, Manning JW. A mathematical model to assess changes in the baroreceptor reflex. Cardiology. 1972;57:295–310. doi: 10.1159/000169528. [DOI] [PubMed] [Google Scholar]
- Laughlin MH. Skeletal muscle blood flow capacity: role of muscle pump in exercise hyperemia. Am J Physiol Heart Circ Physiol. 1987;253:H993–1004. doi: 10.1152/ajpheart.1987.253.5.H993. [DOI] [PubMed] [Google Scholar]
- Norton KH, Boushel R, Strange S, Saltin B, Raven PB. Resetting of the carotid arterial baroreflex during dynamic exercise in humans. J Appl Physiol. 1999a;87:332–338. doi: 10.1152/jappl.1999.87.1.332. [DOI] [PubMed] [Google Scholar]
- Norton KH, Gallagher KM, Smith SA, Querry RG, Welch-O'Connor RM, Raven PB. Carotid baroreflex function during prolonged exercise. J Appl Physiol. 1999b;87:339–347. doi: 10.1152/jappl.1999.87.1.339. [DOI] [PubMed] [Google Scholar]
- Ogoh S, Fadel PJ, Monteiro F, Wasmund WL, Raven PB. Haemodynamic changes during neck pressure and suction in seated and supine positions. J Physiol. 2002;540:707–716. doi: 10.1113/jphysiol.2001.013259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary DS, Rowell LB, Scher AM. Baroreflex-induced vasoconstriction in active skeletal muscle of conscious dogs. Am J Physiol Heart Circ Physiol. 1991;260:H37–41. doi: 10.1152/ajpheart.1991.260.1.H37. [DOI] [PubMed] [Google Scholar]
- Papelier Y, Escourrou P, Gauthier JP, Rowell LB. Carotid baroreflex control of blood pressure and heart rate in men during dynamic exercise. J Appl Physiol. 1994;77:502–506. doi: 10.1152/jappl.1994.77.2.502. [DOI] [PubMed] [Google Scholar]
- Potts JT, Raven PB. Effect of dynamic exercise on human carotid-cardiac baroreflex latency. Am J Physiol Heart Circ Physiol. 1995;268:H1208–1214. doi: 10.1152/ajpheart.1995.268.3.H1208. [DOI] [PubMed] [Google Scholar]
- Potts JT, Shi XR, Raven PB. Carotid baroreflex responsiveness during dynamic exercise in humans. Am J Physiol Heart Circ Physiol. 1993;265:H1928–1938. doi: 10.1152/ajpheart.1993.265.6.H1928. [DOI] [PubMed] [Google Scholar]
- Raven PB, Fadel PJ, Smith SA. The influence of central command on baroreflex resetting during exercise. Exerc Sport Sci Rev. 2002;30:39–44. doi: 10.1097/00003677-200201000-00008. [DOI] [PubMed] [Google Scholar]
- Raven PB, Potts JT, Shi X. Baroreflex regulation of blood pressure during dynamic exercise. Exerc Sport Sci Rev. 1997;25:365–389. [PubMed] [Google Scholar]
- Remensnyder JP, Mitchell JH, Sarnoff SJ. Functional sympatholysis during muscular activity. Circ Res. 1962;11:370–380. doi: 10.1161/01.res.11.3.370. [DOI] [PubMed] [Google Scholar]
- Saltin B, Radegran G, Koskolou MD, Roach RC. Skeletal muscle blood flow in humans and its regulation during exercise. Acta Physiol Scand. 1998;162:421–436. doi: 10.1046/j.1365-201X.1998.0293e.x. [DOI] [PubMed] [Google Scholar]
- Stok WJ, Baisch F, Hillebrecht A, Schulz H, Meyer M, Karemaker JM. Noninvasive cardiac output measurement by arterial pulse analysis compared with inert gas rebreathing. J Appl Physiol. 1993;74:2687–2693. doi: 10.1152/jappl.1993.74.6.2687. [DOI] [PubMed] [Google Scholar]
- Strange S, Rowell LB, Christensen NJ, Saltin B. Cardiovascular responses to carotid sinus baroreceptor stimulation during moderate to severe exercise in man. Acta Physiol Scand. 1990;138:145–153. doi: 10.1111/j.1748-1716.1990.tb08826.x. [DOI] [PubMed] [Google Scholar]
- Sugawara J, Miyachi M, Tanabe T, Maeda S, Ajisaka R, Matsuda M. Non-invasive assessment of cardiac output during exercise: comparison between Modelflow method and Doppler echography method. Adv Exerc Sports Physiol. 2001;7:150. doi: 10.1046/j.0001-6772.2003.01211.x. [DOI] [PubMed] [Google Scholar]
- Tschakovsky ME, Sujirattanawimol K, Ruble SB, Valic Z, Joyner MJ. Is sympathetic neural vasoconstriction blunted in the vascular bed of exercising human muscle? J Physiol. 2002;541:623–635. doi: 10.1113/jphysiol.2001.014431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkevich D, Micco A, Reeves JT. Noninvasive measurement of the decrease in left ventricular filling time during maximal exercise in normal subjects. Am J Cardiol. 1988;62:650–652. doi: 10.1016/0002-9149(88)90676-5. [DOI] [PubMed] [Google Scholar]
- Wesseling KH, Jansen JR, Settels JJ, Schreuder JJ. Computation of aortic flow from pressure in humans using a nonlinear, three-element model. J Appl Physiol. 1993;74:2566–2573. doi: 10.1152/jappl.1993.74.5.2566. [DOI] [PubMed] [Google Scholar]