

Abstract
Five nicotinic acetylcholine receptor (nAChR) mutations are currently linked to autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE). The similarity of their clinical symptoms suggests that a common functional anomaly of the mutations underlies ADNFLE seizures. To identify this anomaly, we constructed rat orthologues (S252F, +L264, S256L, V262L, V262M) of the human ADNFLE mutations, expressed them in Xenopus oocytes with the appropriate wild-type (WT) subunit (α4 or β2), and studied the Ca2+ dependence of their ACh responses. All the mutations significantly reduced 2 mM Ca2+-induced increases in the 30 μM ACh response (P < 0.05). Consistent with a dominant mode of inheritance, this reduction persisted in oocytes injected with a 1:1 mixture of mutant and WT cRNA. BAPTA injections showed that the reduction was not due to a decrease in the secondary activation of Ca2+-activated Cl− currents. The S256L mutation also abolished 2 mM Ba2+ potentiation of the ACh response. The S256L, V262L and V262M mutations had complex effects on the ACh concentration-response relationship but all three mutations shifted the concentration-response relationship to the left at [ACh]≥ 30 μM. Co-expression of the V262M mutation with a mutation (E180Q) that abolished Ca2+ potentiation resulted in 2 mM Ca2+ block, rather than potentiation, of the 30 μM ACh response, suggesting that the ADNFLE mutations reduce Ca2+ potentiation by enhancing Ca2+ block of the α4β2 nAChR. Ca2+ modulation may prevent presynaptic α4β2 nAChRs from overstimulating glutamate release at central excitatory synapses during bouts of synchronous, repetitive activity. Reducing the Ca2+ dependence of the ACh response could trigger seizures by increasing α4β2-mediated glutamate release during such bouts.
Autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) patients suffer from brief, partial epileptic seizures that appear to originate in the frontal lobe and occur almost exclusively during phase 2 sleep (Scheffer et al. 1995). Three α4 (Steinlein et al. 1995, 1997; Hirose et al. 1999) and two β2 nicotinic acetylcholine receptor (nAChR) mutations (De Fusco et al. 2000; Phillips et al. 2001) are currently linked to ADNFLE. All five mutations lie within (or immediately adjacent to) M2, the putative pore-forming region of the nAChR subunits (reviewed in Karlin et al. 1995), and they produce similar clinical symptoms (Steinlein et al. 1995, 1997; Hirose et al. 1999; De Fusco et al. 2000; Ito et al. 2000; Phillips et al. 2001). Previous studies have shown that the ADNFLE mutations have a range of effects on the α4β2 ACh response (reviewed in Sutor et al. 2001). However, the similarity of their clinical symptoms suggests that a common functional anomaly of the mutations generates ADNFLE seizures.
Previous studies have reported that several ADNFLE mutations have common effects on the α4β2 ACh response. The human α4(S248F), α4(776ins3), α4(S252L) and β2(V287M) ADNFLE mutations shift the ACh concentration-response relationship to the left (Bertrand et al. 2002). Rat orthologues (S252F, +L264) of the human α4(S248F) and α4(776ins3) mutations induce use-dependent potentiation of the 100 nM ACh response, delay the rising phase of the 5–30 nM ACh response, and reduce 2.5 mM Ca2+-induced increases in the peak 30 μM ACh response (Figl et al. 1998). The human α4(S248F) mutation also displays use-dependent potentiation (Kuryatov et al. 1997) and the human α4(776ins3) mutation reduces 2.5 mM Ca2+ potentiation of the peak 30 μM ACh response (Steinlein et al. 1997). However, it is unclear whether use-dependent potentiation, delays in the rising phase of the 5–30 nM ACh response, and reductions in the Ca2 dependence of the ACh response are common features of all the ADNFLE mutations.
We constructed rat orthologues (S256L, V262L, V262M) of the human α4(S252L), β2(V287L) and β2(V287M) mutations (Fig. 1A and B) and co-expressed them in Xenopus oocytes with the appropriate wild-type (WT) subunit to determine whether use-dependent potentiation, delays in the rising phase of the 5–30 nM ACh response and reductions in Ca2+ potentiation were common features of the ADNFLE mutations. The rat (Goldman et al. 1987; Deneris et al. 1988) and human (Anand et al. 1990; Monteggia et al. 1995) α4 and β2 amino acid sequences share 89 % and 95 % identity, respectively. Their greatest divergence occurs in the intracellular cytoplasmic loop between the M3 and M4 transmembrane domains (Anand et al. 1990; Monteggia et al. 1995). Their β2 M2 sequences are identical (Fig. 1B). The α4 M2 sequences differ by a single conservative amino acid substitution (I ⇔ V) at position 1′ (Fig. 1A).
Figure 1. Locations of the autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) mutations.

The block diagram above A indicates the nicotinic subunit domains. A, the aligned amino acid sequences of the rat and human wild-type (WT), and the rat S252F, S256L and +L264 M2 domains. The rat and human WT α4 M2 sequences differ by a single amino acid at position 1. B, alignment of the rat and human WT, and rat V262L and V262M β2 M2 domains, as in A. The rat and human WT β2 M2 sequences are identical. The mutated residues are shown in bold. The primed numbers above the sequences indicate the position of the residue relative to the amino (N)-terminus (position 1′) of M2 (Charnet et al. 1990).
Our results show that the S256L mutation does not induce use-dependent potentiation or delay the rising phase of the 10 nM ACh response. Thus, these effects are not common features of the ADNFLE mutations. In contrast, all five ADNFLE mutations reduce 2 mM Ca2+-induced increases in the peak 30 μM ACh response and this reduction is consistent with a dominant mode of inheritance. During bouts of synchronous repetitive activity, reducing the Ca2+ dependence of presynaptic α4β2 nAChRs could induce seizures by increasing the relative amount of α4β2-mediated glutamate release at central excitatory synapses.
METHODS
Oocyte expression
Stage V-VI Xenopus oocytes were surgically isolated following previously published protocols (Quick et al. 1994a). All surgeries were in done in compliance with the methods approved by the Institutional Animal Care and Use Committee (IACUC) at the California Institute of Technology. Ovarian lobes were removed from female Xenopus laevis anaesthetized by immersion in 0.2 % tricaine methanesulphonate (pH 7.4; Sigma, St Louis, MO, USA) for 45–60 min. Xenopus were humanely killed after the final oocyte extraction. The oocyte follicular layer was removed using Type A collagenase (1–2 h in a 2 mg ml−1 collagenase solution; Boehringer Mannheim, Indianapolis, IN, USA). To increase receptor expression, the α4–1(Goldman et al. 1987) and β2 inserts (Deneris et al. 1988) were subcloned into a vector containing a 5′ untranslated region from the alfalfa mosaic virus that enhanced protein translation, and a long 3′ poly A tail (Figl et al. 1998). We used the Stratagene QuikChange kit (La Jolla, CA, USA) to construct the α4 and β2 mutations, and verified them by DNA sequencing. Capped cRNA was synthesized in vitro using the mMessage mMachine RNA transcription kit (Ambion, Austin, TX, USA). After a 24 h incubation in a modified Barth's solution containing (mM): NaCl 96, Hepes 5, sodium pyruvate 2.5, KCl 2, CaCl2 1.8 and MgCl2 1 with 2.5 μg ml−1 gentamicin (Sigma) and 5 % horse serum (pH 7.4, Irvine Scientific, Santa Ana, CA, USA), the isolated oocytes were injected with rat α4 and β2 cRNA, or mouse thyrotrophin-releasing hormone receptor (TRHr) cRNA. The injected oocytes were incubated for ≥ 24 h in the modified Barth's solution at 15 °C before electrophysiological recordings or [3H]epibatidine binding measurements were attempted.
Electrophysiological recordings
We voltage clamped the oocytes with two, 3 MΩ KCl-filled microelectodes (1.5–4 MΩ resistance) at −50 mV using a GeneClamp voltage clamp (Axon Instruments, Union City, CA, USA). During the voltage-clamp recordings, the oocytes were continually superfused with a nominally Ca2+-free saline solution (ND98) containing (mM): NaCl 98, MgCl2 1 and Hepes 5 (pH 7.4) at 20–23 °C, unless otherwise stated. We added 2 mM CaCl2 to the ND98 to measure the Ca2+ dependence of the α4β2 ACh response and 2 mM BaCl2 to measure its Ba2+ dependence. We used a concentration of 2 mM Ca2+ rather than the previously used 2.5 mM (Steinlein et al. 1997; Figl et al. 1998) because the extracellular Ca2+ concentration in the mammalian brain is 1.5–2 mM (reviewed in Egelman et al. 1999). ACh was applied to the oocytes using a U-tube microperfusion system (Cohen et al. 1995). The time constant for solution exchange was ≈0.5 s. The voltage-clamp currents were digitized with a personal computer equipped with a DigiData 1200 A/D interface and pCLAMP V.6.03 software (Axon Instruments). To avoid aliasing, the voltage-clamp currents were filtered at one-quarter to one-third of the sampling frequency with an 8-pole, low-pass Bessel filter. Unless otherwise stated, we used Student's unpaired t tests to determine whether two independent groups of measurements were significantly different, and the Student-Newman-Keuls test (SigmaStat V.1, Jandel Scientific) for multi-group comparisons. All the normalized ACh concentration-response data were fitted initially to the Hill equation. If the Hill coefficient from this initial fit was < 0.8, then the data were refitted to the sum of two hyperbolic binding functions (equivalent to the sum of two Hill equations with both Hill coefficients fixed to unity). If the Hill coefficient was 0.8–1.0, then the data were refitted to a single hyperbolic binding function.
BAPTA injections
The oocytes were injected with 50 nl of a BAPTA solution containing (mM): BAPTA 100, NaOH 85, KOH 2.5 and Hepes 10 (pH 7.4), 5 min before the recordings began, to prevent activation of the endogenous Ca2+-activated Cl− current by the TRHr or α4β2 nAChR (Haghighi et al. 2000). The BAPTA injections produced a final intracellular BAPTA concentration of ≈5 mM. Control oocytes were injected with 50 nl of sterile water.
[3H]Epibatidine binding to immunoisolated receptors
We purchased [3H]epibatidine (specific activity of 30–50 Ci mmol−1) from Amersham Life Science, Inc. (Arlington Heights, IL, USA). The WT and S256L [3H]epibatidine concentration- binding relationships were measured as in Shafaee et al. (1999), except that the α4β2 nAChRs were incubated for 12 h in [3H]epibatidine in the current experiments. Oocytes expressing α4β2 nAChRs were solubilized in a lysis buffer containing (mM): NaCl 50, sodium phosphate buffer 50, EGTA 5 and EDTA 5 with 2 % Triton X-100 and the Complete protease inhibitor (1 tablet per 40 ml of lysis buffer, Boehringer Mannheim; Gerzanich et al. 1995). The solubilized receptors were immunoprecipitated onto EIA/RIA strip plates (Costar Corning Corp., Cambridge, MA, USA) coated with the anti-α4 antibody mAb 299 (Research Biochemicals, Natick, MA, USA). The wells were coated with antibody the previous day by adding 0.5 μg of mAb 299 to 100 μl of a 10 mM sodium bicarbonate solution (pH 8.8) for an overnight incubation at 4 °C. We blocked the antibody-coated wells with bovine serum albumin (3 %) in 200 μl of a PBS-Tween buffer containing (mM): NaCl 10 and sodium phosphate 100 with 0.05 % Tween 20 (pH 7.5) for 2 h at 4 °C. The blocked wells were rinsed three times with the PBS-Tween buffer. Aliquot parts of the solubilized receptor in lysis buffer (100 μl) were added to each well and incubated overnight at 4 °C. On the following day, the wells were rinsed three times with the PBS-Tween buffer and the appropriate [3H]epibatidine concentration was added to each well in PBS-Tween buffer for 12 h at 20–23 °C. This incubation time was ≥ 20 times longer than the [3H]epibatidine dissociation time constant for rat α4β2 receptors expressed in oocytes (Shafaee et al. 1999). To avoid radioligand depletion, we adjusted the amount of receptor in the wells to keep the bound [3H]epibatidine to within 10 % or less of the total [3H]epibatidine added. Incubation volumes of 4 ml were used for [3H]epibatidine concentrations of 0.001–0.3 nM. Volumes of 200 μl were used for [3H]epibatidine concentrations > 0.3 nM. The free [3H]epibatidine concentration was corrected for radioligand depletion. Non-specific binding was measured by adding 1 mM cold (−)nicotine to the wells. Non-specific binding was negligible (near background radiation levels) except for 30 nM [3H]epibatidine. Each measurement was repeated three times. We added 2–3 ml of liquid scintillation cocktail (Research Products International Corp., Mount Prospect, IL, USA) to each sample and measured the amount of [3H]epibatidine bound using a scintillation counter. To obtain the equilibrium dissociation constant (Kd) for [3H]epibatidine binding and the maximum bound [3H]epibatidine (Bmax), we fitted the [3H]epibatidine concentration-binding data to a hyperbolic binding function using the non-linear least squares regression routine in SigmaPlot V. 4 (SPSS, Chicago, IL, USA).
[3H]Epibatidine binding to intact oocytes
We used nearly saturating [3H]epibatidine (10 nM) and unlabelled ACh concentrations (1 mM) to measure WT and S256L surface receptor expression in intact oocytes. Half of the oocytes were used to measure total [3H]epibatidine binding and half of them were used to measure non-specific binding. To measure total [3H]epibatidine binding, we incubated individual oocytes in 200 μl of ND98 (see above) containing 10 nM [3H]epibatidine for 2 min at 20–23 °C. This incubation time minimized [3H]epibatidine uptake by the oocytes but was sufficient for 10 nM [3H]epibatidine binding to the receptors to approach equilibrium (Shafaee et al. 1999). To measure non-specific binding, we incubated individual oocytes in 200 μl of ND98 containing 10 nM [3H]epibatidine and 1 mM ACh for 2 min. ACh is a hydrophilic quaternary amine that cannot readily cross the cell membrane and, thus, selectively blocks [3H]epibatidine binding to surface nAChRs. After the incubations, each group of oocytes was rinsed three times in 10 ml ND98 and solubilized individually overnight in 200 μl concentrated nitric acid. We added 2 ml scintillation fluid to each sample and measured the amount of [3H]epibatidine bound using a scintillation counter.
RESULTS
S256L does not induce use-dependent potentiation or delay the rising phase of the ACh response
Previous experiments have shown that the rat S252F and +L264 mutations induce use-dependent potentiation of the 100 nM ACh response and prolong the rise time of the 5–30 nM ACh response (Figl et al. 1998). To determine whether the S256L mutation had similar effects on the ACh response, we used the same protocol and perfusion system (U-tube) for these experiments (Fig. 2B) that we used previously for the S252F and +L264 experiments (Figl et al. 1998). In contrast to the S252F and +L264 mutations (Figl et al. 1998), the S256L mutation failed to exhibit use-dependent potentiation during a series of ten 150 ms applications of 100 nM ACh spaced 5 s apart (Fig. 2A). These experiments were replicated three times. Likewise, the S256L mutation failed to delay the rising phase of the 10 nM ACh response (Fig. 2B). On the contrary, the S256L mutation accelerated the rising phase of the 10 nM ACh response, presumably because it increased the rate of ACh-induced desensitization (Bertrand et al. 2002; Matsushima et al. 2002). These experiments were replicated seven times. The superposition of the normalized WT and S256L responses after the ACh application ceased (Fig. 2B) shows that the difference between the rising phases of the WT and S256L responses was not the result of a difference in the rate of solution exchange. We did not carry out similar experiments on the V262L or V262M receptors because we were only interested in common effects of the five reported ADNFLE mutations.
Figure 2. The S256L mutation does not induce use-dependent potentiation of the 100 nM ACh response or delay the rising phase of the 10 nM ACh response.
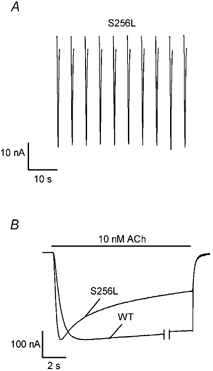
A, traces showing the inward currents evoked by applying ten, 150 ms pulses of 100 nM ACh spaced 5 s apart to an oocyte expressing S256L receptors. B, normalized WT and S256L 10 nM ACh responses on an expanded time scale. A portion of the WT response between the double vertical lines was omitted to align the beginning and end of the WT and S256L responses. The bars above the traces indicate the timing of the ACh application.
S256L, V262L and V262M reduce Ca2+ modulation of the 30 μM ACh response
Similar to the S252F and +L264 mutations (Figl et al. 1998), the S256L mutation also reduced Ca2+-induced increases in the peak 30 μM ACh response compared with WT (Fig. 3A and B). The effects of the S256L mutation on Ca2+ modulation of the ACh response depended on the agonist concentration (Fig. 3A and B). Removing 2 mM Ca2+ from the saline solution had little effect on the peak 30 μM ACh response of the S256L mutation but it had WT-like effects on the peak 10 nM ACh response of the mutation (Fig. 3A and B). The V262L and V262M mutations had similar effects on Ca2+ modulation of the ACh response (Fig. 3C-F). Thus, all three mutations dramatically reduced 2 mM Ca2+-induced increases in the 30 μM ACh response but they had little effect on 2 mM Ca2+-induced increases in the 10 nM ACh response.
Figure 3. S256L, V262L and V262M reduce the Ca2+ dependence of the 30 μM ACh response.
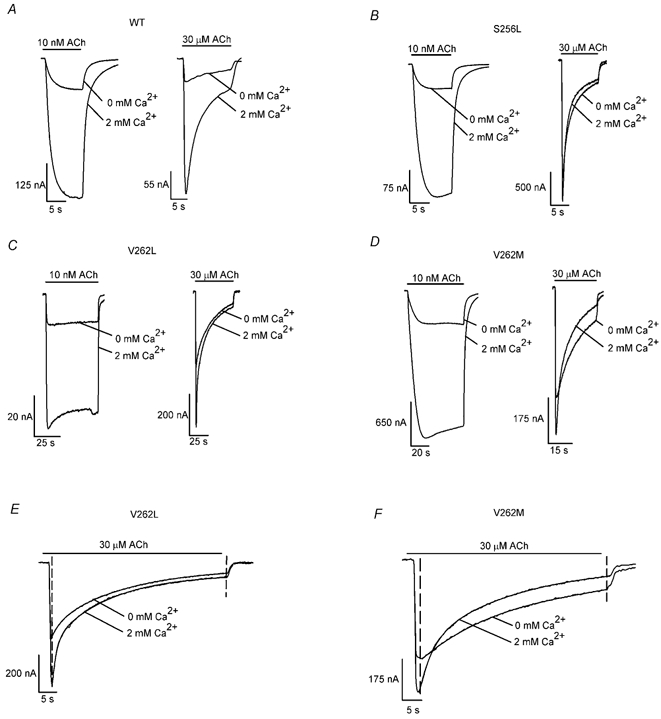
The traces show the WT (A), S256L (B), V262L (C) and V262M (D) 10 nM and 30 μM ACh responses in 0 and 2 mM added extracellular Ca2+ (n = 3–16 oocytes). Adding 2 mM Ca2+ to the saline solution increased the peak WT 10 nM and 30 μM ACh response by similar factors. It also increased the peak WT (A), S256L (B), V262L (C) and V262M (D) 10 nM ACh responses by similar factors. In contrast, adding 2 mM Ca2+ to the saline solution increased the peak S256L (B), V262L (C) and V262M (D) 30 μM ACh responses two- to 3-fold less than the WT response (A). The rise times of the ACh responses are somewhat slower than those in Fig. 2 because the rate of solution exchange was slower. The 10 nM and 30 μM ACh data in A-D were obtained from different oocytes. To avoid saturating the voltage clamp, we recorded the 30 μM ACh responses 1–2 days after cRNA injection and the 10 nM ACh responses 4–5 days after injection. E and F, the V262L (E) and V262M (F) 30 μM ACh responses are shown on an expanded time scale to facilitate comparison with the WT (A) and S256L (B) 30 μM ACh responses. Fits to the sum of two exponentials and a constant term (between the dashed lines) are superimposed on the responses. The mean ±s.e.m. fast (τf) and slow (τs) time constants, amplitudes of the fast (If) and slow (Is) exponential components, and amplitude of the steady-state component (Iss) for the V262L responses were 3.00 ± 0.04 s, 21.0 ± 0.1 s, 71 ± 1 nA 304 ± 1 nA, and 30 ± 1 nA in 0 mM Ca2+, and 1.00 ± 0.01 s, 16 ± 1 s, 231 ± 1 nA, 360 ± 1 nA and 63 ± 1 nA in 2 mM Ca2+. The corresponding V262M values were 18 ± 2 s, 50 ± 20 s, 190 ± 60 nA, 210 ± 40 nA and 40 ± 20 nA in 0 mM Ca2+, and 4.0 ± 0.01 s, 30 ± 1 s, 249 ± 5 nA, 332 ± 2 nA and 6 ± 6 nA in 2 mM Ca2+. The bars above the traces indicate the timing of the ACh applications.
S256L, V262L and V262M enhance steady-state desensitization
To compare desensitization of the WT and mutant receptors, we fitted the desensitizing phase of the WT, S256L, V262L and V262M 30 μM ACh responses to the sum of two exponentials and a constant term (Table 1, Fig. 3E and F). Table 1 gives the fast (τf) and slow (τs) desensitization time constants, the fractional amplitudes of fast (Af) and slow (As) desensitization components, and the fractional amplitude of the steady-state response (Ass) for the mutant and WT receptors. A two-way analysis of variance (using receptor type and Ca2+ concentration as factors) showed that the S256L, V262L and V262M mutations significantly (P < 0.05) increased steady-state desensitization of the 30 μM ACh response (i.e. reduced the Ass). The mutant Ass values were 51–72 % lower than the WT value in 0 mM Ca2+ and 62–97 % lower than the WT value in 2 mM Ca2+ (Table 1). However, S256L, V262L and V262M did not uniformly affect the other desensitization parameters (τf, τs, Af and As). S256L significantly affected the τf for desensitization (P < 0.05) but V262L and V262M did not. S256L reduced the τf by 74 % in 0 mM Ca2+ and 72 % in 2 mM Ca2+, compared to the WT. S256L and V262L significantly reduced the τs for desensitization (P < 0.05) but V262M did not. The S256L τs was 88 % lower than the WT value in 0 mM Ca2+ and 87 % lower in 2 mM Ca2+. The V262L τs was 44 % lower than the WT value in 0 mM Ca2+ but it was not significantly different from the WT value in 2 mM Ca2+. S256L increased the Af by ≈2-fold in 0 mM Ca2+ but had no significant effect in 2 mM Ca2+. In contrast, V262L increased the Af by ≈2-fold in 2 mM Ca2+ but had no significant effect in 0 mM Ca2+. None of the mutations significantly affected the As. Adding 2 mM Ca2+ to the saline solution also had divergent effects on desensitization. A two-way analysis of variance showed that adding 2 mM Ca2+ to the saline solution had a significant overall effect on the τf, Af, As and Ass (P < 0.05). Ca2+ significantly increased the WT, V262L and V262M Af values by 2- to 3-fold but did not significantly affect the S256L Af. However, post hoc comparisons between the τf, As and Ass in 0 and 2 mM Ca2+ failed to show any significant differences. Finally, Ca2+ did not significantly affect the τs. Thus, the only uniform effect of the mutations on desensitization of the 30 μM ACh response was an increase in steady-state desensitization.
Table 1.
Mean fractional amplitudes and mean τ values for WT, S256L, V262L and V262M receptors
| [Ca2+](mM) | Af (%) | As(%) | Ass(%) | τf(S) | τs(S) | |
|---|---|---|---|---|---|---|
| WT (n = 25) | 0 | 16 ± 1 | 48 ± 2 | 36 ± 3 | 3.4 ± 0.4 | 34 ± 4 |
| S256L (n = 7) | 0 | 37 ± 3* | 53 ± 2 | 10 ± 1* | 0.9 ± 0.1* | 4 ± 1* |
| V262M (n = 14) | 0 | 19 ± 3 | 66 ± 5 | 14 ± 4* | 3 ± 1 | 19 ± 2* |
| V262M (n = 6) | 0 | 23 ± 7 | 64 ± 8 | 13 ± 3* | 5 ± 3 | 24 ± 6 |
| WT (n = 14) | 2 | 33 ± 6† | 38 ± 4 | 29 ± 4 | 1.8 ± 0.3 | 24 ± 4 |
| S256L (n = 4) | 2 | 46 ± 4 | 43 ± 3 | 11 ± 2* | 0.5 ± 0.1* | 3.2 ± 0.3* |
| V262L (n = 10) | 2 | 58 ± 5*† | 40 ± 4 | 1 ± 3* | 1.8 ± 0.3 | 21 ± 4 |
| V262M (n = 3) | 2 | 50 ± 5† | 47 ± 5 | 3 ± 1* | 2 ± 1 | 20 ± 5 |
All values are means ±s.e.m.
P < 0.05, significantly different from the corresponding WT value.
P < 0.05, significantly different from the corresponding value in 0 mM Ca2+.
Not all the mutations affect Ca2+ modulation in an ACh-dependent manner
To determine whether all five ADNFLE mutations reduced 2 mM Ca2+ modulation in an ACh concentration-dependent manner, we measured the ratio (ICa2+/I0) between the peak ACh response in 2 and 0 mM added Ca2+ for the WT, S252F, +L264, S256L, V262L and V262M receptors at three ACh concentrations (10 nM, 50 nM, 30 μM) (Fig. 4). All five mutations significantly reduced the ICa2+/I0 ratio of the 30 μM ACh response (P < 0.05). The WT ICa2+/I0 ratio at 30 μM ACh was 4.9 ± 0.4 (n = 16). (This value was greater than the previously reported ICa2+/I0 ratio of 2.9 ± 0.4 between 0 and 2.5 mM Ca2+ (Figl et al. 1998), probably because the ACh responses in the nominally Ca2+-free saline solution in the previous experiments were potentiated by residual Ca2+ in the water.) The mutant ICa2+/I0 ratios at 30 μM ACh ranged from 1.4 ± 0.1 (n = 4) to 2.5 ± 0.1 (n = 5). They were 50–72 % lower than the WT value but were not significantly different from each other. The S252F and +L264 mutations significantly reduced the ICa2+/I0 ratio at 10 nM and 50 nM ACh (P < 0.05). In contrast, the S256L, V262L and V262M mutations did not, although the S256L and V262M ICa2+/I0 ratios at 50 nM ACh were smaller than the corresponding WT value (Fig. 4). Thus, all five ADNFLE mutations reduced 2 mM Ca2+-induced increases in the peak 30 μM ACh response but only the S252F and +L264 mutations significantly reduced 2 mM Ca2+-induced increases in the 10 and 30 nM ACh responses.
Figure 4. All the rat ADNFLE orthologues significantly reduce the Ca2+ dependence of the 30 μM ACh response.
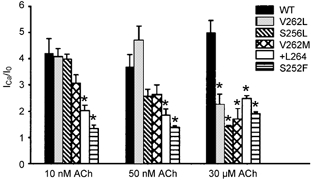
The bars denote the ratio (ICa2+/I0) of the peak 10 nM, 50 nM and 30 μM ACh response in 2 mM added Ca2+ to that in 0 mM added Ca2+. The error bars are ±s.e.m. (n = 3–16 oocytes). The asterisks denote a significant difference from the corresponding WT values (P < 0.05). To normalize the ICa2+/I0 distribution, we ranked the ratios and took the square roots of the ranks. Post hoc comparisons were carried out on the square root of the ranks using the Student-Newman-Keuls test.
Ba2+ blocks the S256L mutation
Ba2+ also acts as a positive allosteric modulator of neuronal nAChRs (Mulle et al. 1992). However, it is far less effective than Ca2+ at activating Cl− currents in Xenopus oocytes (Miledi et al. 1984). To determine whether the S256L mutation affected Ba2+ modulation of the α4β2 ACh response, we measured the WT and S256L 30 μM ACh response in zero and 2 mM extracellular Ba2+. Adding 2 mM Ba2+ to the saline solution increased the peak amplitude of the WT 30 μM ACh response by a factor of 1.20 ± 0.06 (Fig. 5A). In contrast, it reduced that of the S256L receptor by 16 ± 3 % (n = 7; Fig. 5A and B). The ratio between the peak WT 30 μM ACh response in 2 mM and 0 mM Ba2+ (1.20 ± 0.06, n = 5) was significantly greater (P < 0.01) than that of the S256L receptor (0.84 ± 0.03, n = 7). Thus, the S256L mutation not only eliminated the 2 mM Ba2+-induced increase in the peak 30 μM ACh response but also resulted in Ba2+ block of the receptor. Ba2+ is not a physiological cation. Thus, we did not carry out similar experiments with the other ADNFLE mutations.
Figure 5. Ba2+ (2 mM) blocks the S256L 30 μM ACh response.
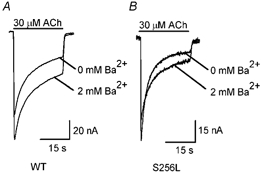
WT (A) and S256L (B) responses to 30 μM ACh in 0 and 2 mM Ba2+. Bars above the traces show the timing of the ACh applications.
Ca2+-induced increases in the ACh response are not an artifact of Cl− currents
Ca2+ influx through the WT and mutant α4β2 nAChRs could potentially activate endogenous Ca2+-activated Cl− currents in Xenopus oocytes. Therefore, we used BAPTA injections (Kavanaugh et al. 1991; Wadiche et al. 1995; Haghighi et al. 2000) to determine whether the secondary activation of Ca2+-activated Cl− currents made a significant contribution to the 2 mM Ca2+-induced increases in the WT and mutant receptor ACh responses. Mouse TRHrs expressed in oocytes activate Cl− currents by triggering intracellular Ca2+ release (Quick et al. 1994b). To test the effectiveness of the BAPTA injections, we expressed mouse TRHrs in oocytes and examined the effects of BAPTA injections on the TRH response. After verifying that BAPTA injections completely blocked the TRH response (Fig. 6A and B), we examined their effects on 2 mM Ca2+-induced increases in the WT and V262M ACh responses (Fig. 7A-D). BAPTA injections reduced 2 mM Ca2+-induced increases in the WT response by a small, but insignificant, amount (Fig. 7A and B). The ICa2+/I0 ratio of the WT 30 μM ACh response was 5.0 ± 0.7 (n = 4) for the water-injected controls (Fig. 7A) and 3.8 ± 0.3 for the BAPTA-injected oocytes (n = 5; Fig. 7B). The BAPTA injections also failed to significantly affect Ca2+-induced increases in the V262M ACh response (Fig. 7C and D). The ICa2+/I0 ratio of the V262M 30 μM ACh response was 1.3 ± 0.1 (n = 4) for the water-injected controls (Fig. 7C) and 1.1 ± 0.1 (n = 5) for the BAPTA-injected oocytes (Fig. 7D). Thus, the secondary activation of Ca2+-activated Cl− currents by α4β2-mediated Ca2+ influx did not contribute significantly to 2 mM Ca2+-induced increases in the WT and V262M 30 μM ACh responses.
Figure 6. BAPTA injections completely eliminate the response of the TRHr to 100 nM TRH.
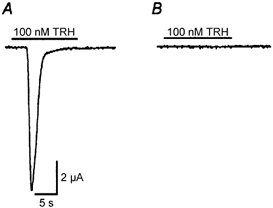
A, the application of 100 nM TRH evoked a peak inward current of 12.2 ± 2.5 μA (mean +s.e.m., n = 3 oocytes) in water-injected oocytes (see Methods). B, BAPTA-injected oocytes did not respond to 100 nM TRH (n = 6 oocytes). The bars above the traces indicate the timing of the TRH application.
Figure 7. BAPTA injections do not significantly affect 2 mM Ca2+-induced increases in the peak WT and V262M 30 μM ACh response.
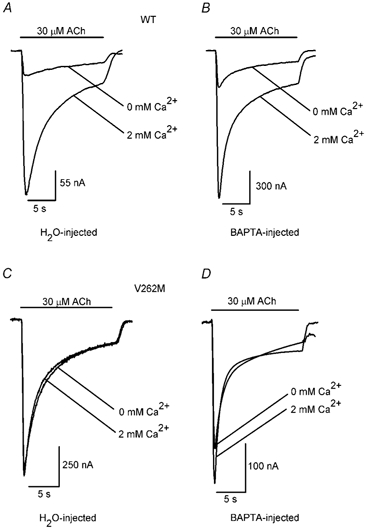
The traces denote the WT (A and B) and V262M 30 μM ACh responses (C and D) in 0 and 2 mM added extracellular Ca2+. The oocytes were injected with 50 nl of water (A and C) or 5 mM BAPTA (B and D) before the recordings. The bars above the traces indicate the timing of the ACh application.
The mutant effects on Ca2+ modulation are consistent with dominant inheritance
ADNFLE segregates in an autosomal dominant fashion (reviewed in Hirose et al. 2000). To determine whether the ADNFLE mutations reduced the Ca2+ dependence of the α4β2 ACh response in a dominant fashion, we measured the 2 mM Ca2+-induced increases in the peak WT, S256L, V262M and mock heterozygous receptor 1 mM ACh responses. We did not test the V262L, S252F and +L264 mutants. We used 1 mM ACh in these experiments to ensure maximum receptor activation. Since the α4:β2 subunit stoichiometry is expected to be 2:3 in central nicotinic receptors, we examined heterozygous conditions using an ADNFLE mutation present in the α4 subunit and one present in the β2 subunit. To mimic the homozygous condition, the oocytes were injected with S256L α4 and WT β2 cRNA in a 2:3 α4:β2 ratio, or with WT α4 and V262M β2 cRNA in the same α4:β2 ratio. To mimic the heterozygous condition, oocytes were injected with WT α4, S256L α4, and WT β2 cRNA in a 1:1:3 α4:α4:β2 ratio or, with WT α4, WT β2 and V262M β2 cRNA in a 2:1.5:1.5 α4:β2:β2 ratio. The ICa2+/I0 ratio of the WT 1 mM ACh response (3.2 + 0.3, n = 6; Fig. 8A) was within the range of the ICa2+/I0 ratios for the 10 nM, 50 nM and 30 μM ACh WT responses (from 3.6 ± 0.4 to 4.9 ± 0.4). The S256L and V262M mutations reduced the ICa2+/I0 ratio by comparable amounts in the heterozygous and homozygous conditions (Fig. 8B-E). The heterozygous S256L ICa2+/I0 ratio (1.5 ± 0.5, n = 4; Fig. 8B) was not significantly different from the homozygous ratio (1.0 ± 0.03, n = 3; Fig. 8C). Similarly, the heterozygous V262M ICa2+/I0 ratio (1.9 ± 0.2, n = 4; Fig. 8D) was not significantly different from the V262M homozygous ratio (1.8 ± 0.2, n = 3; Fig. 8E). Thus, the S256L and V262M mutations reduced the Ca2+ dependence of the ACh response in a manner consistent with dominant inheritance.
Figure 8. Heterozygous expression of the S256L and V262M mutations reduces the Ca2+ dependence of the 1 mM ACh response by nearly the same amount as homozygous expression.
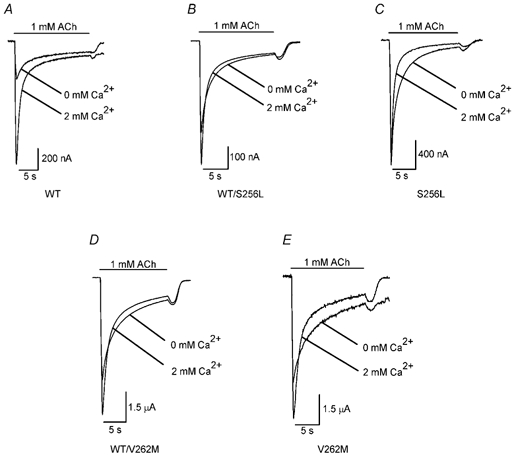
WT (A), heterozygous S256L (WT/S256L, B), homozygous S256L (C), heterozygous V262M (WT/V262M, D), and homozygous V262M (E) responses to 1 mM ACh in 0 and 2 mM added extracellular Ca2+. The bar above the traces shows the timing of the ACh application. The small rebound currents at the end of the ACh applications suggest that, at a concentration of 1 mM, ACh has a slight open-channel blocking effect on the WT and mutant channels.
S256L, V262L and V262M mutations increase ACh sensitivity at [ACh]≥ 30 μM
To determine whether the S256L, V262L or V262M mutations affected ACh sensitivity, we measured the WT and mutant ACh concentration-response relationships at −50 mV (Fig. 9). Consistent with previous studies (Covernton et al. 2000; Buisson & Bertrand, 2001), the WT, S256L and V262L ACh concentration-response data were best fitted by the sum of two hyperbolic binding functions. In contrast, the V262M data were adequately fitted by a single hyperbolic binding function. Visual inspection of the data in Fig. 9G shows that all three mutations appear to shift the concentration-response relationship to the left at ACh concentrations ≥ 30 μM. The high-affinity EC50, low-affinity EC50 and percentage of high-affinity receptors for the WT ACh concentration- response relationship were 1.5 ± 0.2 μM, 300 ± 130 μM and 67 ± 3 % (means ± S.E., degrees of freedom (d.f.) = 6), respectively (Fig. 9E and G). These values were similar to those (1.6 ± 0.2 μM, 62 ± 8.3 μM, 62 ± 3 %) reported for the human WT α4β2 ACh concentration- response relationship at −100 mV (Bertrand et al. 2002). The V262L mutation significantly reduced the high- and low-affinity EC50 values (0.42 ± 0.16 μM, 25 ± 20 μM, d.f. = 4) but it did not significantly affect the percentage of high-affinity receptors (59 ± 9 %; Fig. 9F and G). The S256L mutation did not significantly affect the high-affinity EC50, low-affinity EC50, or percentage of high-affinity receptors (1.2 ± 0.2 μM, 80 ± 30 μM and 59 ± 4 %, d.f. = 4; Fig. 9E and G). The EC50 of the V262M concentration-response relationship (1.8 ± 0.1 μM, d.f. = 7) was not significantly different from the high-affinity WT or S256L EC50 values. However, the V262M concentration-response relationship lacked a noticeable low-affinity component (Fig. 9F and G). Thus, the S256L, V262L and V262M mutations had divergent effects on the ACh concentration-response relationship but all three mutations appeared to increase the fractional response to non-saturating ACh concentrations ≥ 30 μM. Previous results (Fig. 4D in Figl et al. 1998) have shown that the S252F and +L264 mutations also shift the ACh concentration- response relationship to the left at ACh concentrations ≥ 10 μM ACh, although these mutations only marginally affect the EC50.
Figure 9. The WT, S256L, V262L and V262M ACh concentration-response relationships.
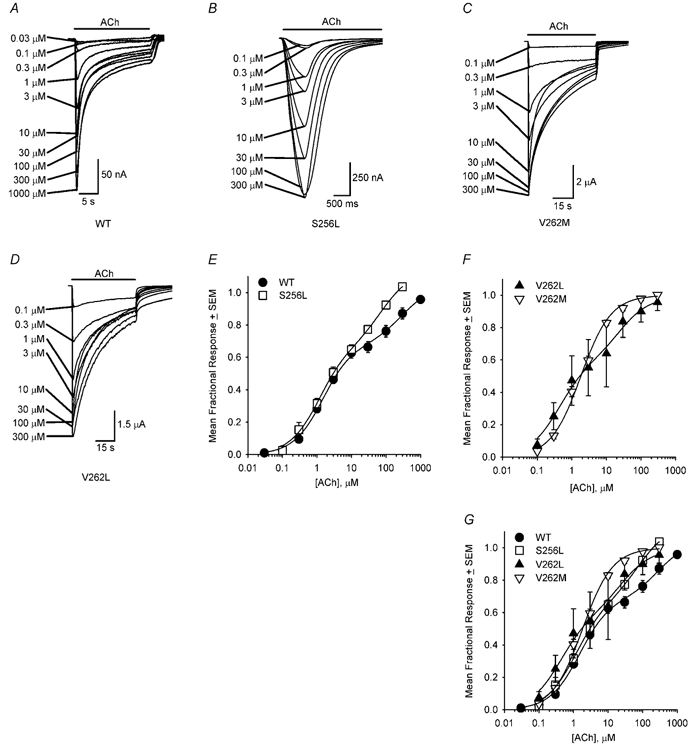
WT (A), S256L (B), V262M (C) and V262L (D) responses to 0.03–1000 μM ACh (0 mM added Ca2+). E, normalized WT and S256L ACh concentration-response relationships. F, same for V262L and V262M. The lines are fits to one, or the sum of two, hyperbolic binding function(s). G, normalized WT, S256L, V262L, and V262M ACh concentration-response relationships plotted together. The symbols and error bars (obscured in some cases) are the mean fractional ACh responses (normalized to the fitted maximum response for each oocyte) ±s.e.m. for 3–8 oocytes. See Results for the values of the fitted parameters.
S256L has little effect on [3 H]epibatidine affinity or surface receptor expression
To determine whether the S256L mutation affected the Kd or Bmax of [3H]epibatidine binding, we injected oocytes with equal amounts of receptor cRNA, immunoisolated the expressed WT or S256L receptors with mAb 299 and measured their [3H]epibatidine concentration-binding relationships (see Methods). Consistent with previous results for the S252F and +L264 mutations (Figl et al. 1998), the S256L mutation did not have a major effect on the [3H]epibatidine affinity of desensitized α4β2 nAChRs. The normalized WT and S256L [3H]epibatidine concentration-binding relationships were nearly superposed (Fig. 10A). The WT and S256L Kd values were 17 ± 2 pM and 11 ± 1 pM (d.f. = 7), respectively. The WT Kd was somewhat smaller than the value (30 ± 6 pM) previously measured for high-affinity rat WT α4β2 receptors expressed in Xenopus oocytes (Shafaee et al. 1999). The S256L Bmax (581 ± 12 c.p.m., d.f. = 7) was larger than the WT value (296 ± 5 c.p.m., d.f. = 7), suggesting that the S256L mutation increased total receptor expression ≈2-fold. However, the S256L mutation did not significantly affect surface receptor expression (Fig. 10B). The median [3H]epibatidine bound to surface WT receptors in intact oocytes was 3.4 fmol(oocyte)−1 (2.7 and 5.8 fmol (oocyte)−1 for 25th and 75th percentiles, respectively) and that bound to surface S256L receptors was 3.1 fmol (oocyte)−1 (2.4 and 3.5 fmol (oocyte)−1 for 25th and 75th percentiles, respectively). Because changes in surface expression or [3H]epibatidine affinity were not common features of the ADNFLE mutations, we did not perform similar experiments with the V262M and V262L mutations.
Figure 10. The S256L mutation has little effect on the [3H]epibatidine affinity of α4β2 nAChRs or their surface expression.
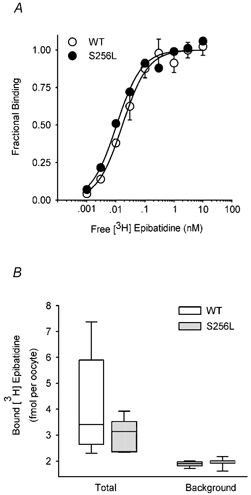
A, normalized WT and S256L [3H]epibatidine concentration-binding relationships. The symbols and error bars (obscured in some cases) are the mean fractional binding (normalized to the fitted Bmax) ±s.e.m. (n = 3 replicates). The smooth lines are fits to a simple hyperbolic binding function. See Results for the values of the fitted parameters. B, box plot showing the total and background [3H]epibatidine bound to intact oocytes expressing WT or S256L receptors. The total [3H]epibatidine bound to the WT and S256L nAChRs was significantly greater than background binding (P < 0.05, Kruskal-Wallis one-way analysis of variance on ranks). The lines inside the boxes are medians (n = 9–11 oocytes per group). The box boundaries closest to the median are the 25th percentiles. Those farthest away are the 75th percentiles. The error bars below and above the box indicate the 10th and 90th percentiles, respectively.
V262M enhances Ca2+ block of the α4β2 nAChR
Previous experiments show that a mutation (E172Q) in the N-terminal domain of α7 nAChRs completely eliminates Ca2+ potentiation of the ACh response (Galzi et al. 1996) and that 10–20 mM Ca2+ blocks the human α4β2 ACh response (Buisson et al. 1996). To determine whether the ADNFLE mutations reduced the Ca2+ dependence of the α4β2 ACh response by enhancing Ca2+ block, we made an α4 mutation (E180Q) orthologous to E172Q, co-expressed it with the WT β2 subunit or the V262M mutation, and measured the effects of 2 mM Ca2+ on the peak 30 μM ACh responses of the single E180Q and double E180Q/V262M mutations. Consistent with the effects of the orthologous α7 mutation (Galzi et al. 1996), co-expression of the E180Q mutation with the WT β2 subunit completely eliminated 2 mM Ca2+-induced increases in the 30 μM ACh response (Fig. 11A). The ICa2+/I0 of the E180Q 30 μM ACh response was 0.85 ± 0.05 (n = 7). Moreover, co-expression of E180Q and V262M resulted in Ca2+ block, rather than potentiation, of the 30 μM ACh response (Fig. 11B). The ICa2+/I0 of the E180Q/V262M 30 μM ACh response was 0.60 ± 0.01 (mean ±s.e.m., n = 4 oocytes). This value was significantly less than the corresponding E180Q ICa2+/I0 ratio (P < 0.01). Thus, adding 2 mM Ca2+ to the saline solution reduced the peak E180Q/V262M 30 μM ACh response by 40 %. However, neither the E180Q mutation itself, nor the combination of the E180Q and V262M mutations, affected the ACh concentration- response relationship significantly (Fig. 11C-E). The E180Q and E180Q/V262M ACh concentration-response data were best fitted by the sum of two hyperbolic binding functions. The high-affinity EC50, low-affinity EC50 and percentage of high-affinity receptors for the E180Q ACh concentration-response relationship were 1.6 ± 0.2 μM, 128 ± 20 μM and 60 ± 2 % (mean ± S.E., d.f. = 6), respectively. The corresponding values for the E180Q/V262M double mutation were 1.1 ± 0.2 μM, 260 ± 65 μM and 57 ± 4 %. Thus, the V262M mutation reduced the Ca2+ dependence of the α4β2 ACh response by enhancing Ca2+ block of the receptor. The effects of Ca2+ on combinations of the E180Q and other ADNFLE mutations (S252F, +L264, S256L, V262M) were not tested.
Figure 11. Ca2+ (2 mM) blocks the E180Q/V262M 30 μM ACh response.
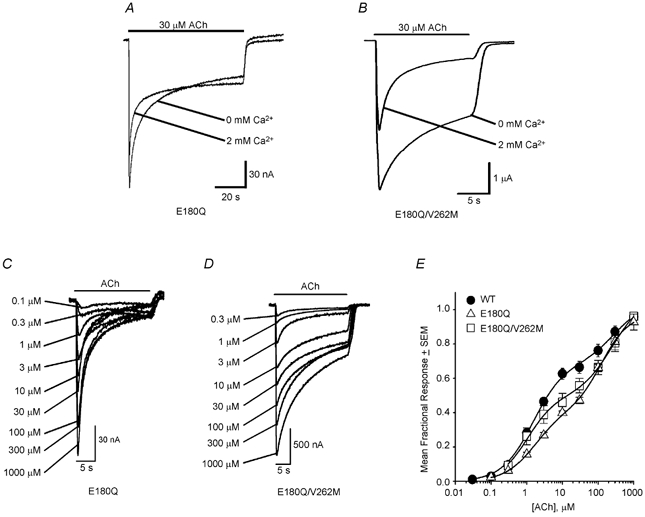
A and B, the E180Q response (A) and the E180Q/V262M response (B) to 30 μM ACh in the presence and absence of 2 mM added Ca2+. The oocytes were injected with 5 mM BAPTA prior to recording. C and D, the E180Q (C) and E180Q/V262M (D) responses to 0.1–1000 μM ACh. The bar above the traces in A-D indicates the timing of the ACh application. E, normalized WT, E180Q and E180Q/V262M ACh concentration- response relationships. The lines are fits to the sum of two hyperbolic binding functions using non-linear least-squares regression. The symbols and error bars (obscured in some cases) are the mean fractional ACh responses (normalized to the fitted maximum response for each oocyte) ±s.e.m. for 3–4 oocytes. See Results for the values of the fitted parameters.
DISCUSSION
The five currently reported ADNFLE mutations have at least one common effect on the α4β2 ACh response. They all reduce 2 mM Ca2+-induced increases in the peak 30 μM ACh response. The mutant ratios between the 30 μM ACh response in 2 and 0 mM Ca2+ are 50–72 % less than the WT value. Consistent with these results, previous experiments show that the human α4(776ins3) mutation also reduces the ratio between the peak 30 μM ACh response in 2.5 and 0 mM Ca2+ by a similar amount (Steinlein et al. 1997). The effects of the ADNFLE mutations on the Ca2+ dependence of the α4β2 ACh response are consistent with a dominant mode of inheritance and they appear to be mediated by an enhancement of Ca2+ block of the receptor. Similar to +L264 and S256L mutations (Figl et al. 1998), the S256L mutation has little effect on the [3H]epibatidine affinity of desensitized receptors. Moreover, the S256L mutation does not affect surface receptor expression.
A reduction in α4β2 Ca2+ permeability cannot explain the effects of the ADNFLE mutations on Ca2+-induced increases in the ACh response. First, not all the ADNFLE mutations reduce Ca2+ permeability. The human α4(S248F) mutation reduces α4β2 Ca2+ permeability (Kuryatov et al. 1997) but the α4(776ins3) does not (Bertrand et al. 2002). Second, BAPTA injections show that Ca2+-activated Cl− currents do not make a significant contribution to 2 mM Ca2+-induced increases in the WT ACh response. Thus, even a total loss of α4β2 Ca2+ permeability cannot explain the effects of the ADNFLE mutations on 2 mM Ca2+-induced increases in the 30 μM ACh response.
A gain of function generates ADNFLE seizures
The absence of seizures in α4 (Marubio et al. 1999) and β2 knock-out mice (Picciotto et al. 1998), and the ability of carbamazepine (an α4β2 antagonist) to block ADNFLE seizures (Picard et al. 1999), suggests that a gain of α4β2 function underlies ADNFLE seizures. Postsynaptic Ca2+-permeable glutamate channels at central excitatory synapses appear to act as Ca2+ sinks that deplete Ca2+ from the limited extracellular space surrounding the synapse during bouts of synchronous repetitive firing (Vassilev et al. 1997; Rusakov et al. 2003). Under these circumstances, the reduced Ca2+ sensitivity of the ADNFLE mutations could produce a net gain of α4β2 function. The resulting increase in α4β2-stimulated glutamate release could generate seizures.
The effects of the α4(S248F) mutation on ACh sensitivity (at constant Ca2+ concentrations) are still not settled. Bertrand et al. (2002) report that the human α4(S248F), α4(776ins3), α4(S252L) and β2(V287M) ADNFLE mutations increase ACh sensitivity. However, Weiland et al. (1996) and Bertrand et al. (2002) report only a slight leftward shift in the α4(S248F) ACh concentration- response relationship relative to the WT, and Kuryatov et al. (1997) report a shift in the opposite direction. Also, our results and those of a previous study (Matsushima et al. 2002) show that the rat S256L mutation shifts the ACh concentration-response relationship only marginally to the left. Despite such differences, our results and those of Figl et al. (1998) show that all five rat ADNFLE orthologues produce a leftward shift in the ACh concentration-response relationship at ACh concentrations ≥ 30 μM.
It is unclear whether a gain of function based solely on an increase in ACh sensitivity could account for the ADNFLE seizures. If central cholinergic nerve terminals release nearly saturating ACh concentrations (as other types of central synapses apparently do (reviewed in Clements, 1996)), then increasing the ACh sensitivity of α4β2 nAChRs may only slightly increase the amplitude of nicotinic synaptic potentials. Another problem is that α4(S248F)-mediated reductions in the maximum ACh response and single-channel conductance (Kuryatov et al. 1997; Bertrand et al. 1998; Figl et al. 1998) could negate any gain in the synaptic potential amplitude produced by greater ACh sensitivity. Interestingly, a knock-in mouse with an α4 mutation that increases ACh sensitivity reduces the threshold for nicotine-induced seizures (Fonck et al. 2003), but it is unclear whether this mouse exhibits spontaneous seizures.
The ADNFLE mutations have a number of other pharmacological and biophysical effects on α4β2 nAChRs not shared by all the mutations. For example, some mutations induce use-dependent potentiation of the 100 nM ACh response, delay the rising phase of the 5–30 nM ACh response (Figl et al. 1998), increase the apparent rate of ACh-induced desensitization (Weiland et al. 1996; Kuryatov et al. 1997; Figl et al. 1998; Bertrand et al. 2002; Matsushima et al. 2002), reduce the maximum ACh response (Kuryatov et al. 1997; Figl et al. 1998), increase choline sensitivity (Bertrand et al. 2002), reduce Ca2+ permeability (Kuryatov et al. 1997), reduce the single-channel conductance (Kuryatov et al. 1997; Figl et al. 1998), and alter the voltage-jump relaxation time constants (Figl et al. 1998). Also, the α4(S248F), α4(776ins3) and α4(S252L) mutations reduce the half-maximal inhibitory concentration (IC50) of ACh-induced desensitization (Bertrand et al. 1998, 2002). However, similar effects have not been reported for the other ADNFLE mutations, and ACh-induced desensitization is probably too slow to affect nicotinic synaptic transmission under normal physiological conditions (Edmonds et al. 1995).
Ca2+ both potentiates and blocks neuronal AChRs
Extracellular Ca2+ has a dual effect on neuronal nAChRs. At concentrations of 1–8 mM, it potentiates the ACh response (Mulle et al. 1992; Vernino et al. 1992; Galzi et al. 1996), but at concentrations of 10–20 mM, it blocks the human α4β2 ACh response (Buisson et al. 1996). Consistent with previous results for α7 nAChRs (Galzi et al. 1996), our results show that a single mutation in the α4 N-terminal domain (E180Q) abolishes 2 mM Ca2+-induced increases in the α4β2 ACh response. Thus, the residues that mediate Ca2+-induced increases in the α4β2 ACh response apparently lie in the N-terminal domain. Because the residues that mediate the positive allosteric effects of Ca2+ and the presently known ADNFLE mutations are in different α4β2 domains, the ADNFLE mutations probably do not reduce Ca2+-induced increases in the ACh response by sterically hindering Ca2+ binding to its potentiation site.
Alternatively, the ADNFLE mutations could reduce Ca2+-induced increases in the α4β2 ACh response by enhancing Ca2+ block of the receptor. The effects of the E180Q/V262M mutation on Ca2+ modulation of the α4β2 ACh response and the effects of the S256L mutation on Ba2+ modulation support this alternative. Also, a couple of ADNFLE mutations (S248F, S256L) are located at positions that mediate the open-channel block of muscle nAChRs by QX-222 (Leonard et al. 1988; Charnet et al. 1990). Ca2+ could block α4β2 nAChRs by binding to a site in the channel pore (channel block) or at some other site. The ACh concentration dependence of the S256L-, V262L- and V262M-mediated reductions in Ca2+ potentiation and the locations of the ADNFLE mutations in and around M2 suggest that the mutations enhance channel block by Ca2+.
Open-channel block is a form of uncompetitive inhibition. Uncompetitive antagonists produce little inhibition near the foot of the agonist concentration- response relationship, but as the agonist concentration increases, they inhibit the agonist response more effectively. Thus, uncompetitive (open-channel) Ca2+ block could explain the ability of the S256L, V262L and V262M mutations to significantly reduce Ca2+ dependence at high ACh concentrations (30 μM), but not at the foot (10 nM, 50 nM) of the ACh concentration-response relationship. In contrast to the S256L, V262L, and V262M mutations, the S252F and +L264 mutations reduce Ca2+ dependence equally well at high (30 μM) and low (10 nM, 50 nM) ACh concentrations. Thus, Ca2+ may be able to reach its pore-blocking site in these mutations equally well in both the closed and open states. Molecular modelling also suggests that the human α4(S248F) mutation should enhance Ca2+ channel block of the α4β2 nAChR (Ortells et al. 2002).
The apparent voltage independence of the S252F- and +L264-mediated reductions in Ca2+ dependence (Figl et al. 1998) and Ca2+ block of the WT human α4β2 receptors (Buisson et al. 1996) suggests that the α4β2 divalent cation binding site lies near the extracellular boundary of the membrane electric field. A glutamate residue (αE262) located near the putative extracellular end of M2 (position 20′ in Fig. 1) mediates the block of muscle nAChRs by extracellular Mg2+ (Imoto et al. 1988). Thus, the aligning α4 glutamate (Fig. 1A) could participate in an α4β2 divalent cation blocking site. All five presently reported ADNFLE mutations reduce side-chain polarity. Such a reduction could draw the M2 helices closer together and constrict the channel pore. Bringing the negatively charged glutamates at position 20 in α4 closer together could enhance their affinity for divalent cations (Ortells et al. 2002).
If Ca2+ blocks open S256L, V262L and V262M channels, does one expect smaller synaptic potentials? Yes, if block proceeds quickly enough. At 0 mV, the bimolecular binding forward rate constant for open-channel blockers to muscle nAChRs is 0.5–5.0 (× 107) M−1 s−1 (Sanchez et al. 1986). Assuming a similar rate constant for Ca2+ binding to its α4β2 blocking site, open-channel block by Ca2+ would be established within < 0.1 ms and therefore could reduce synaptic currents. If central cholinergic nerve terminals release nearly saturating neurotransmitter concentrations (as GABAergic and glutamatergic terminals do (reviewed by Clements, 1996)), then the effects of the ADNFLE mutations on Ca2+ modulation at 30 μM ACh are the ones most relevant to synaptic transmission.
How could a change in Ca+ modulation lead to nocturnal frontal lobe seizures?
How might a reduction in the Ca2+ dependence of the α4β2 ACh response lead to nocturnal frontal lobe seizures? Similar to other nocturnal epileptiform discharges (Nobili et al. 1999a,b; Beelke et al. 2000; Ferrillo et al. 2000; Kostopoulos, 2000; Nobili et al. 2000, 2001a,b), thalamocortical oscillations during phase 2 sleep termed sleep spindles (Steriade et al. 1993) could trigger ADNFLE seizures. Presynaptic nAChRs facilitate both excitatory and inhibitory transmitter release in the cortex (reviewed in Wonnacott, 1997). Ca2+ modulation may act as a negative feedback mechanism to prevent presynaptic α4β2 nAChRs at central excitatory synapses from over-stimulating glutamate release during repetitive synaptic firing (Amador et al. 1995). If the synchronous, repetitive firing associated with sleep spindles selectively depletes Ca2+ from the extracellular space around excitatory synapses (Vassilev et al. 1997; Rusakov et al. 2003), then presynaptic α4β2 nAChRs should enhance inhibitory transmitter release more than excitatory transmitter release during sleep spindles. The resulting enhancement of α4β2-mediated lateral inhibition over α4β2-mediated lateral excitation may prevent spindle firing from spreading laterally through the cortex and initiating a partial epileptic seizure in normal individuals. However, because the ADNFLE mutations reduce the Ca2+ dependence of α4β2 nAChRs, the difference between α4β2-mediated excitatory and inhibitory transmitter release during sleep spindles may be less pronounced in ADNFLE patients than in normal individuals. The resulting enhancement of α4β2-mediated excitatory transmitter release could allow sleep spindles to spread beyond their normal focus in ADNFLE patients and trigger an epileptic seizure. This hypothesis suggests that carbamazepine inhibits ADNFLE seizures by preventing presynaptic α4β2 nAChRs from enhancing either excitatory or inhibitory neurotransmitter release. However, only a minority of ADNFLE patients have been mapped and there may be additional ADNFLE mutations outside the M2 domain that could have functional characteristics distinct from those described here.
Acknowledgments
This research was supported by the California Tobacco-Related Disease Research Program (6KT-0208), the Epilepsy Foundation (EFA/COHEN-01) and the NIH (NS43800, NS11756). We thank Kira Kostenka for help with the oocytes and Cesar Labarca, Purnima Deshpande, Carlos Fonck, Raad Nashmi, Andrew Tapper, Johannes Schwarz and Jim Boulter for comments.
REFERENCES
- Amador M, Dani JA. Mechanism for modulation of nicotinic acetylcholine receptors that can influence synaptic transmission. J Neurosci. 1995;15:4525–4532. doi: 10.1523/JNEUROSCI.15-06-04525.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R, Lindstrom J. Nucleotide sequence of the human nicotinic acetylcholine receptor β2 subunit gene. Nucleic Acids Res. 1990;18:4272. doi: 10.1093/nar/18.14.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beelke M, Nobili L, Baglietto MG, De Carli F, Robert A, De Negri E, Ferrillo F. Relationship of sigma activity to sleep interictal epileptic discharges: a study in children affected by benign epilepsy with occipital paroxysms. Epilepsy Res. 2000;40:179–186. doi: 10.1016/s0920-1211(00)00131-5. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Picard F, Le Hellard S, Weiland S, Favre I, Phillips H, Bertrand S, Berkovic SF, Malafosse A, Mulley J. How mutations in the nAChRs can cause ADNFLE epilepsy. Epilepsia. 2002;43:112–122. doi: 10.1046/j.1528-1157.43.s.5.16.x. [DOI] [PubMed] [Google Scholar]
- Bertrand S, Weiland S, Berkovic SF, Steinlein OK, Bertrand D. Properties of neuronal nicotinic acetylcholine receptor mutants from humans suffering from autosomal dominant nocturnal frontal lobe epilepsy. Br J Pharmacol. 1998;125:751–760. doi: 10.1038/sj.bjp.0702154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson B, Bertrand D. Chronic exposure to nicotine upregulates the human α4β2 nicotinic acetylcholine receptor function. J Neurosci. 2001;21:1819–1829. doi: 10.1523/JNEUROSCI.21-06-01819.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson B, Gopalakrishnan M, Arneric SP, Sullivan JP, Bertrand D. Human α4β2 neuronal nicotinic acetylcholine receptor in HEK 293 cells: a patch-clamp study. J Neurosci. 1996;16:7880–7891. doi: 10.1523/JNEUROSCI.16-24-07880.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charnet P, Labarca C, Leonard RJ, Vogelaar NJ, Czyzyk L, Gouin A, Davidson N, Lester HA. An open-channel blocker interacts with adjacent turns of α-helices in the nicotinic acetylcholine receptor. Neuron. 1990;4:87–95. doi: 10.1016/0896-6273(90)90445-l. [DOI] [PubMed] [Google Scholar]
- Clements JD. Transmitter timecourse in the synaptic cleft: its role in central synaptic function. Trends Neurosci. 1996;19:163–171. doi: 10.1016/s0166-2236(96)10024-2. [DOI] [PubMed] [Google Scholar]
- Cohen BN, Figl A, Quick MW, Labarca C, Davidson N, Lester HA. Regions of β2 and β4 responsible for differences between the steady state dose-response relationships of the α3β2 and α3β4 neuronal nicotinic receptors. J Gen Physiol. 1995;105:745–764. doi: 10.1085/jgp.105.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covernton PJ, Connolly JG. Multiple components in the agonist concentration-response relationships of neuronal nicotinic acetylcholine receptors. J Neurosci Methods. 2000;96:63–70. doi: 10.1016/s0165-0270(99)00185-5. [DOI] [PubMed] [Google Scholar]
- De Fusco M, Becchetti A, Patrignani A, Annesi G, Gambardella A, Quattrone A, Ballabio A, Wanke E, Casari G. The nicotinic receptor β2 subunit is mutant in nocturnal frontal lobe epilepsy. Nature Genet. 2000;26:275–276. doi: 10.1038/81566. [DOI] [PubMed] [Google Scholar]
- Deneris ES, Connolly J, Boulter J, Wada E, Wada K, Swanson LW, Patrick J, Heinemann S. Primary structure and expression of β2: a novel subunit of neuronal acetylcholine receptors. Neuron. 1988;1:45–54. doi: 10.1016/0896-6273(88)90208-5. [DOI] [PubMed] [Google Scholar]
- Edmonds B, Gibb AJ, Colquhoun D. Mechanisms of activation of muscle nicotinic acetylcholine receptors and the time course of endplate currents. Annu Rev Physiol. 1995;57:469–493. doi: 10.1146/annurev.ph.57.030195.002345. [DOI] [PubMed] [Google Scholar]
- Egelman DM, Montague PR. Calcium dynamics in the extracellular space of mammalian neural tissue. Biophys J. 1999;76:1856–1867. doi: 10.1016/s0006-3495(99)77345-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrillo F, Beelke M, Nobili L. Sleep EEG synchronization mechanisms and activation of interictal epileptic spikes. Clin Neurophysiol. 2000;111:S65–73. doi: 10.1016/s1388-2457(00)00404-1. [DOI] [PubMed] [Google Scholar]
- Figl A, Viseshakul N, Shafaee N, Forsayeth J, Cohen BN. Two mutations linked to nocturnal frontal lobe epilepsy cause use-dependent potentiation of the nicotinic ACh response. J Physiol. 1998;513:655–670. doi: 10.1111/j.1469-7793.1998.655ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonck C, Nashmi R, Deshpande P, Damaj MI, Marks MJ, Riedel A, Schwarz J, Collins AC, Labarca C, Lester HA. Increased sensitivity to agonist-induced seizures, straub tail, and hippocampal theta rhythm in knock-in mice carrying hypersensitive α4 nicotinic receptors. J Neurosci. 2003;23:2582–2590. doi: 10.1523/JNEUROSCI.23-07-02582.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galzi JL, Bertrand S, Corringer JP, Changeux JP, Bertrand D. Identification of calcium binding sites that regulate potentiation of a neuronal nicotinic acetylcholine receptor. EMBO J. 1996;15:5824–5832. [PMC free article] [PubMed] [Google Scholar]
- Gerzanich V, Peng X, Wang F, Wells G, Anand R, Fletcher S, Lindstrom J. Comparative pharmacology of epibatidine: a potent agonist for neuronal nicotinic acetylcholine receptors. Mol Pharmacol. 1995;48:774–782. [PubMed] [Google Scholar]
- Goldman D, Deneris E, Luyten W, Kochhar A, Patrick J, Heinemann S. Member of a nicotinic acetylcholine receptor gene family are expressed in different regions of the mammalian central nervous system. Cell. 1987;48:965–973. doi: 10.1016/0092-8674(87)90705-7. [DOI] [PubMed] [Google Scholar]
- Haghighi AP, Cooper E. A molecular link between inward rectification and calcium permeability of neuronal nicotinic acetylcholine α3β4 and α4β2 receptors. J Neurosci. 2000;20:529–541. doi: 10.1523/JNEUROSCI.20-02-00529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose S, Iwata H, Akiyoshi H, Kobayashi K, Ito M, Wada K, Kaneko S, Mitsudome A. A novel mutation of CHRNA4 responsible for autosomal dominant nocturnal frontal lobe epilepsy. Neurology. 1999;53:1749–1753. doi: 10.1212/wnl.53.8.1749. [DOI] [PubMed] [Google Scholar]
- Hirose S, Okada M, Kaneko S, Mitsudome A. Are some idiopathic epilepsies disorders of ion channels?: a working hypothesis. Epilepsy Res. 2000;41:191–204. doi: 10.1016/s0920-1211(00)00141-8. [DOI] [PubMed] [Google Scholar]
- Imoto K, Busch C, Sakmann B, Mishina M, Konno T, Nakai J, Bujo H, Mori Y, Fukuda K, Numa S. Rings of negatively charged amino acids determine the acetylcholine receptor channel conductance. Nature. 1988;335:645–648. doi: 10.1038/335645a0. [DOI] [PubMed] [Google Scholar]
- Ito M, Kobayashi K, Fujii T, Okuno T, Hirose S, Iwata H, Mitsudome A, Kaneko S. Electroclinical picture of autosomal dominant nocturnal frontal lobe epilepsy in a Japanese family. Epilepsia. 2000;41:52–58. doi: 10.1111/j.1528-1157.2000.tb01505.x. [DOI] [PubMed] [Google Scholar]
- Karlin A, Akabas MH. Toward a structural basis for the function of nicotinic acetylcholine receptors and their cousins. Neuron. 1995;15:1231–1244. doi: 10.1016/0896-6273(95)90004-7. [DOI] [PubMed] [Google Scholar]
- Kavanaugh MP, Christie MJ, Osborne PB, Busch AE, Shen K-Z, Wu YN, Seeburg PH, Adelman JP, North RA. Transmitter regulation of voltage-dependent K+ channels expressed. Xenopus oocytes. Biochem J. 1991;277:899–902. doi: 10.1042/bj2770899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostopoulos GK. Spike-and-wave discharges of absence seizures as a transformation of sleep spindles: the continuing development of a hypothesis. Clin Neurophysiol. 2000;11:S27–S38. doi: 10.1016/s1388-2457(00)00399-0. [DOI] [PubMed] [Google Scholar]
- Kuryatov A, Gerzanich V, Nelson M, Olale F, Lindstrom J. Mutation causing autosomal dominant nocturnal frontal lobe epilepsy alters Ca2+ permeability, conductance, and gating of human α4β2 nicotinic acetylcholine receptors. J Neurosci. 1997;17:9035–9047. doi: 10.1523/JNEUROSCI.17-23-09035.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard RJ, Labarca CG, Charnet P, Davidson N, Lester HA. Evidence that the M2 membrane-spanning region lines the ion channel pore of the nicotinic receptor. Science. 1988;242:1578–1581. doi: 10.1126/science.2462281. [DOI] [PubMed] [Google Scholar]
- Marubio LM, del Mar Arroyo-Jimenez M, Cordero-Erausquin M, Léna C, Le Novère N, de Kerchove D'Exaerde A, Huchet M, Damaz MI, Changeux JP. Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature. 1999;398:805–810. doi: 10.1038/19756. [DOI] [PubMed] [Google Scholar]
- Matsushima N, Hirose S, Iwata H, Fukuma G, Yonetani M, Nagayama C, Hamanaka W, Matsunaka Y, Masatoshi I, Kaneko S, Mitsudome A, Sugiyama H. Mutation (Ser284Leu) of neuronal nicotinic acetylcholine receptor α4 subunit associated with frontal lobe epilepsy causes faster desensitization of the rat receptor expressed in oocytes. Epilepsy Res. 2002;48:181–186. doi: 10.1016/s0920-1211(01)00336-9. [DOI] [PubMed] [Google Scholar]
- Miledi R, Parker I. Chloride current induced by injection of calcium into Xenopus oocytes. J Physiol. 1984;357:173–183. doi: 10.1113/jphysiol.1984.sp015495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia LM, Gopalakrishnan M, Touma E, Idler KB, Nash N, Arneric SP, Sullivan JP, Giordano T. Cloning and transient expression of genes encoding the human α4 and β2 neuronal nicotinic acetylcholine receptor (nAChR) subunits. Gene. 1995;155:189–193. doi: 10.1016/0378-1119(94)00914-e. [DOI] [PubMed] [Google Scholar]
- Mulle C, Léna C, Changeux JP. Potentiation of nicotinic receptor response by external calcium in rat central neurons. Neuron. 1992;8:937–945. doi: 10.1016/0896-6273(92)90208-u. [DOI] [PubMed] [Google Scholar]
- Nobili L, Baglietto MG, Beelke M, De Carli F, De Negri E, Gaggero R, Rosadini G, Veneselli E, Ferrillo F. Distribution of epileptiform discharges during nREM sleep in the CSWSS syndrome: relationship with sigma and delta activities. Epilepsy Res. 2001a;44:119–128. doi: 10.1016/s0920-1211(01)00191-7. [DOI] [PubMed] [Google Scholar]
- Nobili L, Baglietto MG, Beelke M, De Carli F, De Negri E, Rosadini G, De Negri M, Ferrillo F. Modulation of sleep interictal epileptiform discharges in partial epilepsy of childhood. Clin Neurophysiol. 1999a;110:839–845. doi: 10.1016/s1388-2457(99)00021-8. [DOI] [PubMed] [Google Scholar]
- Nobili L, Baglietto MG, Beelke M, De Carli F, De Negri E, Tortorelli S, Ferrillo F. Spindles-inducing mechanism modulates sleep activation of interictal epileptiform discharges in the Landau-Kleffner syndrome. Epilepsia. 2000;41:201–206. doi: 10.1111/j.1528-1157.2000.tb00140.x. [DOI] [PubMed] [Google Scholar]
- Nobili L, Baglietto MG, Beelke M, De Carli F, Veneselli E, Ferrillo F. Temporal relationship of generalized epileptiform discharges to spindle frequency activity in childhood absence epilepsy. Clin Neurophysiol. 2001b;112:1912–1916. doi: 10.1016/s1388-2457(01)00624-1. [DOI] [PubMed] [Google Scholar]
- Nobili L, Ferrillo F, Baglietto MG, Beelke M, De Carli F, De Negri E, Schiavi G, Rosadini G, De Negri M. Relationship of sleep interictal epileptiform discharges to sigma activity (12–16 Hz) in benign epilepsy of childhood with rolandic spikes. Clin Neurophysiol. 1999b;110:39–46. doi: 10.1016/s0168-5597(98)00041-0. [DOI] [PubMed] [Google Scholar]
- Ortells MO, Barrantes GE. Molecular modelling of the interactions of carbamazepine and a nicotinic receptor involved in the autosomal dominant nocturnal frontal lobe epilepsy. Br J Pharmacol. 2002;136:883–895. doi: 10.1038/sj.bjp.0704786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HA, Favre I, Kirkpatrick M, Zuberi SM, Goudie D, Heron SE, Scheffer IE, Sutherland GR, Berkovic SF, Bertrand D, Mulley JC. CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am J Hum Genet. 2001;68:225–231. doi: 10.1086/316946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F, Bertrand S, Steinlein OK, Bertrand D. Mutated nicotinic receptors responsible for autosomal dominant nocturnal frontal lobe epilepsy are more sensitive to carbamazepine. Epilepsia. 1999;40:1198–1209. doi: 10.1111/j.1528-1157.1999.tb00848.x. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Rimondini R, Léna C, Marubio LM, Pich EM, Fuxe K, Changeux JP. Acetylcholine receptors containing the β2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–177. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- Quick MW, Lester HA. Methods of expression of excitability proteins in Xenopus oocytes. In: Conn PM, editor. Methods in Neurosciences. Vol. 19. Blackwell Science Inc; 1994a. pp. 261–279. [Google Scholar]
- Quick MW, Simon MI, Davidson N, Lester HA, Aragay AM. Differential coupling of G protein alpha subunits to seven-helix receptors expressed in Xenopus oocytes. J Biol Chem. 1994b;269:30164–30172. [PubMed] [Google Scholar]
- Rusakov DA, Fine A. Extracellular Ca2+ depletion contributes to fast activity-dependent modulation of synaptic transmission in the brain. Neuron. 2003;37:287–297. doi: 10.1016/s0896-6273(03)00025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez JA, Dani JA, Siemen D, Hille B. Slow permeation of organic cations in acetylcholine receptor channels. J Gen Physiol. 1986;87:985–1001. doi: 10.1085/jgp.87.6.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer IE, Bhatia KP, Lopes-Cendes I, Fish DR, Marsden CD, Andermann E, Andermann F, Desbiens R, Keene D, Cendes F. Autosomal dominant nocturnal frontal lobe epilepsy: a distinctive clinical disorder. Brain. 1995;118:61–73. doi: 10.1093/brain/118.1.61. [DOI] [PubMed] [Google Scholar]
- Shafaee N, Houng M, Truong A, Viseshakul N, Figl A, Sandhu S, Forsayeth JR, Dwoskin LP, Crooks PA, Cohen BN. Pharmacological similarities between native brain and heterologously expressed α4β2 nicotinic receptors. Br J Pharmacol. 1999;128:1291–1299. doi: 10.1038/sj.bjp.0702900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinlein OK, Magnusson A, Stoodt J, Betrand S, Weiland S, Berkovic SF, Nakken KO, Propping P, Bertrand D. An insertion mutation of the CHRNA4 gene in a family with autosomal dominant nocturnal frontal lobe epilepsy. Hum Mol Genet. 1997;6:943–947. doi: 10.1093/hmg/6.6.943. [DOI] [PubMed] [Google Scholar]
- Steinlein OK, Mulley JC, Propping P, Wallace RH, Phillips HA, Sutherland GR, Scheffer IE, Berkovic SF. A missense mutation in neuronal nicotinic acetylcholine receptor α4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nature Genet. 1995;11:201–203. doi: 10.1038/ng1095-201. [DOI] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Sutor B, Zolles G. Neuronal nicotinic acetylcholine receptors and autosomal dominant nocturnal frontal lobe epilepsy: a critical review. Eur J Physiol. 2001;442:642–651. doi: 10.1007/s004240100614. [DOI] [PubMed] [Google Scholar]
- Vassilev PM, Mitchel J, Vassilev M, Kanazirska M, Brown EM. Assessment of frequency-dependent alterations in the level of extracellular Ca2+ in the synaptic cleft. Biophys J. 1997;72:2103–2116. doi: 10.1016/S0006-3495(97)78853-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernino S, Amador M, Leutje CW, Patrick J, Dani JA. Calcium modulation and high calcium permeability of neuronal nicotinic acetylcholine receptors. Neuron. 1992;8:127–134. doi: 10.1016/0896-6273(92)90114-s. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Amara SG, Kavanaugh MP. Ion fluxes associated with excitatory amino acid transport. Neuron. 1995;15:721–728. doi: 10.1016/0896-6273(95)90159-0. [DOI] [PubMed] [Google Scholar]
- Weiland S, Witzemann V, Villarroel A, Propping P, Steinlein O. An amino acid exhange in the second transmembrane segment of a neuronal nicotinic receptor causes partial epilepsy by altering its desensitization kinetics. FEBS Lett. 1996;398:91–96. doi: 10.1016/s0014-5793(96)01215-x. [DOI] [PubMed] [Google Scholar]
- Wonnacott S. Presynaptic ACh receptors. Trends Neurosci. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]