

Abstract
A transient (Ipeak) and a sustained (Isus) outward K+ current were measured, using whole-cell voltage-clamp methods, in isolated rat ventricular myocytes obtained by enzymatic dispersion. A comparison was made between male and female rats following induction of (insulin-deficient) diabetes with streptozotocin (STZ). In control (non-diabetic) rats, both currents were smaller in cells obtained from females, as compared to males (P < 0.005). However, whereas inducing diabetes in male rats significantly attenuated both Ipeak and Isus (P < 0.005), Ipeak was unchanged in female diabetic rats. Isus was significantly (P < 0.005) reduced, but the extent of reduction was smaller (P < 0.02) than in males. The formation of angiotensin II (ATII) or endothelin-1 (ET-1) was blocked using inhibitors of angiotensin-converting enzyme (ACE) and endothelin-converting enzyme (ECE), respectively. In cells from diabetic males both inhibitors significantly (P < 0.005) enhanced K+ currents. In contrast, no effect was observed in cells from female diabetic rats. However, in ovariectomized (Ovx) diabetic females the in vitro inhibition of ATII and ET-1 formation augmented the two K+ currents, but not when oestradiol was administered in vivo prior to cell isolation. In cells from diabetic males, incubation with 100 nM 17β-oestradiol significantly (P < 0.005) enhanced both Ipeak and Isus. This effect was blocked if ATII or ET-1 was added to the medium. These results show that autocrine modulation of K+ currents by renin-angiotensin and endothelin systems is attenuated or absent in female diabetic rats. Oestradiol plays a key role in reducing this modulation. These results may underlie some of the sex differences associated with development of cardiac arrhythmias.
Sex-dependent differences in the development of cardiovascular disease are well established (Rossouw, 2002), although underlying mechanisms are only partly understood. Elevated levels of oestradiol have long been considered to have a protective role in females (Babiker et al. 2002); however, this has recently been called into question (Rossouw, 2002; Grady et al. 2002). In addition to differences in the development of coronary disease, there are sex differences in the propensity for various types of cardiac arrhythmias (Pham & Rosen, 2002). Some rhythm disorders are more common in males, as is sudden cardiac death (Larsen & Kadish, 1998). However, for some types of arrhythmias, females may be at greater risk (Pham & Rosen, 2002; Bailey & Curtis, 2002). Importantly, sex-related differences in the repolarization of the cardiac action potential and in underlying K+ currents have recently been established (Trepanier-Boulay et al. 2001).
Diabetes mellitus is an increasingly prevalent pathology (Nathan et al. 1997) with cardiovascular disease and associated arrhythmias recognized as major long-term, life-threatening complications (Nathan et al. 1997; Wild et al. 1999). Diabetes has been shown to counter the protective effects of female gender in the onset of coronary disease (Colhoun et al. 2000; Brown et al. 2001), possibly due to altered lipid profiles (Roeters van Lennep et al. 2002). Despite the prevalence of cardiac disease and diabetes, sex-dependent differences in the regulation of ion currents, which may underlie the development of cardiac arrhythmias, have not been extensively addressed in general, and in the setting of diabetes in particular.
Several pathological conditions such as diabetes and heart failure are associated with an increase in the activity of a local, cardiac renin-angiotensin system (RAS) (Dostal, 2000; Fiordaliso et al. 2000; Barlucchi et al. 2001). The effects of elevated angiotensin II (ATII) may be quite detrimental (Dostal, 2000; Fiordaliso et al. 2000), and indeed blocking formation of ATII with angiotensin-converting enzyme (ACE) inhibitors was shown to benefit diabetic patients (Zuanetti et al. 1997; Gerstein et al. 2000). We have recently shown that autocrine or paracrine release of angiotensin II contributes to the attenuation of repolarizing K+ currents in the setting of diabetes. These currents are augmented by inhibiting the formation of ATII, as well as by blocking ATII receptors (Shimoni, 2001). The expression of some of the channel proteins underlying these currents (Nerbonne, 2000) was also augmented by ACE inhibition (Shimoni & Liu, 2003). We also demonstrated that a paracrine or autocrine action of endothelin-1 contributes to cardiac K+ current attenuation in diabetes (Shimoni & Liu, 2003). This peptide is important for several reasons. It has been suggested that endothelin-1, which is synthesized, stored and released in the heart under pathological conditions (Russell & Molenaar, 2000), is involved in the onset of cardiac arrhythmias (Duru et al. 2001). Circulating endothelin-1 levels are increased in diabetes (Ferri et al. 1995; Saltevo et al. 2000), and long-term endothelin-1 receptor blockade was found to improve cardiovascular function in rats (Verma et al. 2001).
Some aspects of RAS activation are known to be sex dependent (Fischer et al. 2002). Recently, the abundance of ACE was shown to be significantly larger in male rat hearts, in comparison to females (Freshour et al. 2002). It is worth noting that oestradiol has been shown to interact with the RAS (Kuroski de Bold, 1999), preventing some of the consequences of RAS activation (Brosnihan et al. 1997; Gallagher et al. 1999). Furthermore, oestradiol (or its metabolites) inhibits both endothelin-1 binding (Duru et al. 2001) and endothelin-1 synthesis (Morey et al. 1998; Dubey et al. 2001).
It was therefore hypothesized that electrophysiological consequences of diabetes may show sex-dependent differences, particularly regarding regulation of K+ currents by angiotensin II and endothelin-1. This study was thus designed to answer the following questions. (1) Are K+ currents affected differently in (type 1) diabetic female rats, as compared to males? (2) Are there sex-related differences in the interaction of the angiotensin II or endothelin-1 systems with K+ currents (and channel proteins) in myocytes from diabetic rats? (3) Does oestradiol affect K+ currents in diabetic rat myocytes, and is this (at least partly) related to angiotensin II or endothelin-1?
METHODS
Experiments were performed in accordance with the guidelines of the Animal Care Committee of the University of Calgary.
Animals
Male and female Sprague-Dawley rats of comparable weight (200-250 g) were used. They were divided into control and diabetic groups. Diabetes was induced with a single I.V. injection of streptozotocin (STZ, 100 mg kg−1), and experiments were performed 1–2 weeks after injection. Glucose and insulin levels were determined in the clinical laboratory of the Foothills Hospital using standard assays, to confirm the diabetic status of the animals. In addition, ovariectomized female rats were used, divided into three groups. Group 1 remained untreated (Ovx), whereas diabetes was induced with STZ 2 weeks after ovariectomy (Ovx-STZ) in groups 2 and 3. In addition, group 3 received oestradiol replacement (0.5 μg ml−1 in the drinking water for 1 week before induction of diabetes, and for the subsequent 1–2 weeks until cell isolation).
Cell preparation
Rats were anaesthetized by CO2 inhalation and killed by cervical dislocation. Hearts were mounted on a Langendorff apparatus and the aortas cannulated for retrograde coronary perfusion. Calcium-containing, calcium-free and enzyme-containing (Yakult collagenase and Sigma protease type XVI) solutions were used to perfuse the hearts (for details see Shimoni et al. 1998; Shimoni, 2001). The free right ventricular wall was cut into pieces, and myocytes were isolated by further incubation in a shaker bath, with a solution containing collagenase and protease. Cells were then stored for up to 10 h in a storage solution containing 0.1 mM CaCl2 and 10 mg ml−1 albumin. Cells were divided into control (untreated) and treatment groups. Untreated cells maintained current densities for this duration.
Current recording
Standard whole-cell voltage-clamp methods were used to record currents at room temperature (20–21 °C). A transient, inactivating current (Ipeak, measured as peak current) and a sustained current (Isus, measured at the end of 500 ms pulses; for details see Shimoni, 2001) were recorded and compared between groups. Cell capacitance was also measured. Current magnitudes were divided by cell capacitance and expressed as current density (pA pF−1). Series resistance was minimized with low resistance electrodes (2–3 MΩ).
Western blotting
Kv4.2 and Kv1.2 are two channel proteins that underlie the currents that we have studied (Nerbonne, 2000). We previously showed that ACE inhibition significantly augmented the expression of Kv4.2 and Kv1.2 in male diabetic hearts (Shimoni & Liu, 2003). In the present experiments we tested this in female rats.
The membrane fractions of ventricular tissue were used, in order to obtain sufficient protein. Since many of the effects observed on current magnitudes were only seen after 6 h, the following procedure was followed: aortas were cannulated and the hearts perfused using a Langendorff apparatus (at 37 °C, 70 cmH2O pressure) for at least 7 h in the absence or presence of the ACE inhibitor quinapril. After 7–9 h, during which the hearts maintained rhythmic spontaneous beating, both ventricles were dissected and frozen rapidly.
The ventricles were later thawed and (approximately 0.4 g tissue) homogenized in a 10 mM Tris buffer containing 0.5 M sucrose and a protease inhibitor cocktail tablet (Roche catalogue number, 1 836 153). The homogenate was centrifuged at 4 °C for 15 min at 1200 r.p.m. to remove debris and nuclei. The supernatant was removed and spun at 100 000 g for 1 h. The membrane pellet was re-suspended in Tris buffer containing 1 % Triton X-100. Trituration and vigorous vortexing were used to re-suspend the pellet until large particles were no longer visible. Protein concentration in the membrane fraction was determined (in duplicate for each sample) using the bicinchoninic acid assay (Pierce). Membrane proteins were separated by 10 % acrylamide SDS-PAGE and transferred to 0.45 μm nitrocellulose membranes prior to blocking with 5 % non-fat dried milk in a Tris-buffered solution (TBS). Membranes were labelled with rabbit polyclonal anti-Kv4.2 or anti-Kv1.2 (Alomone, diluted 1:400), washed with TBS and probed with 1:4000 horseradish peroxidase-conjugated goat anti-rabbit IgG. Detection was achieved with a chemiluminescent substrate (Pierce) using Biomax film. Identification of the correct bands corresponding to Kv4.2 and Kv1.2 was confirmed by using competing peptides (see Shimoni & Liu, 2003).
Chemicals
Drugs used were quinapril (Parke-Davis), to inhibit the angiotensin-converting enzyme, and a peptide fragment of big endothelin-1 (Sigma), which has been shown to inhibit the endothelin-converting enzyme (Zimmermann et al. 2001) The water-soluble form of 17β-oestradiol (Sigma) was also used.
Statistics
Unpaired t tests and one-way ANOVA were used to compare groups. The Student-Newman-Keuls multiple comparisons test was applied where appropriate. P < 0.05 was taken to indicate significant differences between experimental groups.
RESULTS
In the first series of experiments K+ currents in myocytes from control female rats were compared to currents in male rats. Several other laboratories have reported smaller repolarizing currents in myocytes from females, although some conflicting findings have been reported (Trepanier-Boulay et al. 2001; Wu & Anderson, 2002), even in the same species (mouse). Smaller repolarizing currents presumably contribute to the longer action potentials and QT intervals seen in the female electrocardiogram (Pham & Rosen, 2002). In our experiments, in 84 cells from males and 41 cells from females the mean densities (± s.e.m.) at +50 mV for Ipeak were 21.8 ± 0.9 and 18.7 ± 1.0 pA pF−1, respectively (P < 0.04), whereas for Isus the values were 8.4 ± 0.2 and 7.2 ± 0.3 pA pF−1, respectively, (P < 0.001).
We then proceeded to examine the effects of inducing diabetes in females. The injection of STZ produced a diabetic state in females comparable to the effect in males (Shimoni et al. 1998). Glucose and insulin levels were measured in a small sample. In our previous studies with male rats (Shimoni et al. 1998) we found that the STZ-induced diabetic state significantly enhanced plasma glucose levels, from 5.3 ± 0.3 (n = 3) to 27.2 ± 1.6 mM (n = 7). Insulin levels were significantly attenuated, from 107.2 ± 14.6 (n = 5) to 47.4 ± 4.1 pmol l−1 (n = 8). In the present experiments, plasma glucose concentration in diabetic females was also significantly augmented, to 40.3 ± 3.5 mM (n = 4). This was higher than in males for this small sample but was not considered to affect the results described below (see Discussion). The insulin level was significantly attenuated by STZ, to 46.4 ± 9.9 pmol l−1 (n = 4). As reported in our earlier work, insulin-deficient (type 1) diabetes attenuates both Ipeak and Isus in ventricular cells from male rats (Shimoni, 2001). In 80 cells from male rats, Ipeak (at +50 mV) was attenuated to 16.8 ± 0.7 pA pF−1 (P < 0.0001), and Isus was attenuated to 5.5 ± 0.2 pA pF−1 (P < 0.0001). However, in cells obtained from diabetic females, the effects on K+ currents were markedly different. In 40 cells, Ipeak (at +50 mV) was found to be unchanged, with a mean density of 20.1 ± 1.0 pA pF−1. Isus (at +50 mV) was significantly (P < 0.0001) attenuated, to 5.3 ± 0.2 pA pF−1. These results are shown in Fig. 1. Figure 1A shows sample current traces, while full current–voltage relationships for Ipeak and Isus are shown in Fig. 1B and C, respectively. Isus was significantly smaller (P < 0.02 to P < 0.0001) in the diabetic myocytes at membrane potentials in the range −30 to +50 mV.
Figure 1. Effects of STZ-induced diabetes on K+ currents in myocytes from female rats.
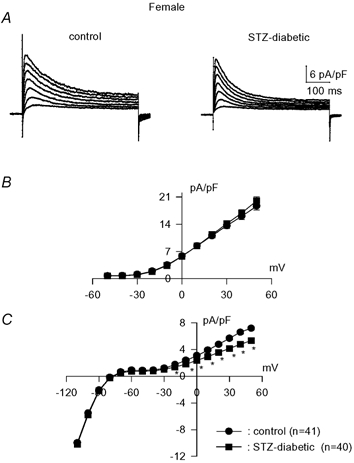
A, sample current traces obtained in response to 500 ms pulses from −80 mV to potentials in the range −10 to +50 mV in myocytes from untreated (left) and STZ-induced diabetic (right) females. B, mean (± s.e.m.) peak current densities plotted against membrane potentials obtained from control (n = 41, •) and diabetic (n = 40, ▪) rat cells. C, mean sustained current densities (measured at end of 500 ms pulses) from the same groups of cells, plotted against membrane potential. * Significant difference (P < 0.01 to P < 0.0001) in currents between −30 and +50 mV.
Although Isus in cells from diabetic females was significantly smaller than in control females, the relative attenuation at +50 mV (by 26 %) was significantly (P < 0.02) smaller than the attenuation of Isus in diabetic males (in which Isus was attenuated by 34 %). This was determined as follows: the individual current densities in all male and female diabetic cells were calculated as a ratio of the mean current in the respective controls (8.4 and 7.2 pA pF−1 in males and females, respectively). The mean values for these ratios were 0.66 ± 0.02 (n = 80) and 0.74 ± 0.03 (n = 54) in males and females, respectively, which was significantly different (P < 0.02, using a two-tailed t test).
These results show that the effects of insulin-deficient diabetes on K+ currents are markedly blunted in female rats. This blunted response in females is presumably due to multiple mechanisms. We attempted to investigate whether some of these are related to an altered interaction of the renin-angiotensin and endothelin-1 systems with repolarizing currents.
In subsequent experiments we exposed cells from diabetic female rats to the ACE inhibitor quinapril (1 μM in vitro) for 5–9 h. In cells from male rats this was shown (Shimoni, 2001) to significantly augment Ipeak and Isus. However, in striking contrast, quinapril had no effect on either current in ventricular myocytes from diabetic females. No significant differences (P > 0.05) were measured in current densities for either Isus or Ipeak, as illustrated in Fig. 2A and B. Figure 2C shows the increase in both currents obtained in cells from male diabetic rats, for the sake of comparison.
Figure 2. Effects of quinapril in myocytes from diabetic female rats.
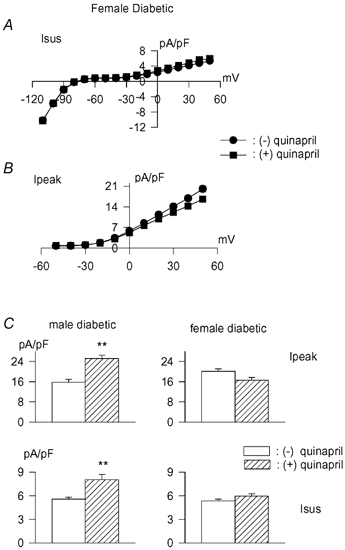
A, steady state current–voltage relationship (mean ± s.e.m. current densities) in the absence (n = 40, •) or following 5–9 h incubation with 1 μM quinapril (n = 30, ▪). B, peak current densities plotted against membrane potentials for the same cells. C, comparison of the effects of quinapril on current density at +50 mV in myocytes from diabetic males (left) and females (right). Ipeak values in top row, Isus in the bottom row. Open bars are values in the absence of drug, whereas hatched bars are values following 5–9 h in quinapril. ** Significant (P < 0.001) enhancement of both currents only in males, with no significant effects of quinapril in females.
In earlier work we found that a paracrine or autocrine effect of endothelin-1 (ET-1) is also involved in the attenuation of K+ currents in myocytes from male diabetic rats (Shimoni & Liu, 2003). Thus, the in vitro inhibition (for 5–9 h) of either the receptors for ET-1 or the endothelin-converting enzyme (ECE) leads to a significant augmentation of both Ipeak and Isus. At +50 mV, the values of Ipeak in the absence (n = 20 cells) or presence (n = 17) of the ECE inhibitor were 14.3 ± 1.7 and 22.0 ± 1.7 pA pF−1, respectively (P < 0.003). The values for Isus were 4.7 ± 0.3 and 7.0 ± 0.4 pA pF−1, respectively (P < 0.0001). In view of the lack of effect of the ACE inhibitor quinapril in cells from female diabetic rats, we also investigated the effects of ECE inhibition.
In marked contrast to the augmentation of both K+ currents in the diabetic male cells, there were no effects in cells from diabetic females. This result is shown in Fig. 3. Sample current traces from cells obtained from male diabetic rats are shown in Fig. 3A, in the absence (left) or presence for 6 h (right) of the ECE inhibitor. Figure 3B shows the mean current densities at different membrane potentials for Ipeak (left) and Isus (right). Both are significantly (P < 0.005) augmented between −20 and +50 mV. Figure 3C shows current traces obtained in cells from female diabetic rats, in the absence (left) or presence (right) of the ECE inhibitor, with the mean current densities plotted in Fig. 3D.
Figure 3. Sex differences in the effects of inhibiting the endothelin-converting enzyme (ECE).
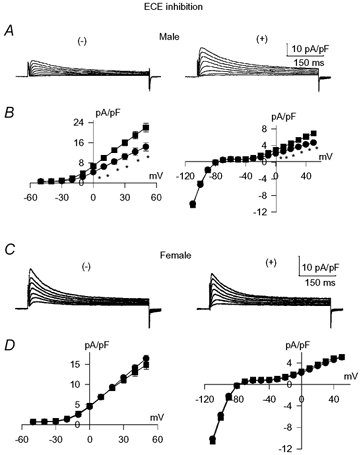
A and B, effects of incubation of myocytes obtained from male diabetic rats with 5 μM ECE inhibitor for 5–9 hours. A, sample current traces in cells without (left) and with (right) the ECE inhibitor. B, current–voltage relationships for Ipeak (left) and Isus (right) in the absence (•, n = 20) or presence (▪, n = 17) of the ECE inhibitor. * Significant increase (P < 0.005) in both currents between −20 and +50 mV. C and D, equivalent results in cells from female diabetic rats (n = 33 and n = 23 in the absence and presence of the ECE inhibitor, respectively). In contrast to the effects in cells from diabetic males, ECE inhibition did not augment either current in cells from diabetic females.
In our earlier work (Shimoni & Liu, 2003) we established that the augmentation of K+ currents by ACE and ECE inhibition in diabetic males is associated with an increased expression of Kv4.2 and Kv1.2, two of the channel proteins that underlie Ipeak and Isus, respectively (Nerbonne, 2000). In the present experiments we measured the effects of quinapril on Kv4.2 and Kv1.2 in diabetic female hearts. The hearts were perfused on a Langendorff apparatus for 7–8 h with or without quinapril, and then rapidly frozen. Subsequently, hearts were thawed and homogenized, and Western blots performed on membrane fractions using Kv4.2- and Kv1.2-specific antibodies (see Methods for details). Band intensity (normalized to a band on a Coomassie blue gel) was measured by densitometry. The results showed that Kv4.2 was unchanged in female diabetic hearts (n = 4) in comparison to controls (n = 4), and that quinapril did not augment Kv4.2 expression (n = 4). Kv1.2 consistently showed two bands, the lower of which was abolished by a competing peptide (Shimoni & Liu, 2003; and Fig. 4). The density of Kv1.2 was significantly decreased in the diabetic female hearts (n = 4), paralleling the reduction in Isus. However, quinapril did not augment its expression, (n = 4) in marked contrast to the effect in diabetic male hearts (Shimoni & Liu, 2003). These results are shown in Fig. 4.
Figure 4. Western blots obtained in membrane extracts from female hearts.
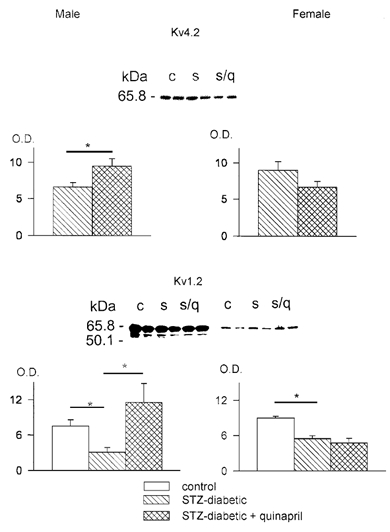
Top panel, blot obtained with an antibody directed against the channel protein Kv4.2 Two lanes each are shown from control (c), STZ-induced diabetic (s) and diabetic with quinapril (s/q) after 8 h perfusion. Below, summary data obtained by densitometry. Mean (± s.e.m.) optical density (O.D.) values are shown, normalized to bands in Coomassie blue gel stains (see Methods). On the left are results in males (blots shown in Shimon & Liu, 2003), in the absence of (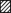 , n = 13) and following perfusion with quinapril (
, n = 13) and following perfusion with quinapril ( , n = 12). The increase in densitometry (P < 0.03) parallels the increase in Ipeak in males. On the right are the values obtained in females, showing no significant differences in the absence of (n = 4) or following perfusion with quinapril (n = 4), paralleling the lack of change in Ipeak. Bottom panel, blot obtained using an antibody against Kv1.2. The same samples as above were used. In addition, six lanes are shown (right) in which a competing peptide for Kv1.2 was used. The peptide blocked the lower of the two bands shown, which was the band measured by densitometry. The mean data in control (□), diabetic () and diabetic hearts perfused with quinapril () are shown (left) for males (blots shown in Shimon & Liu, 2003) and females (right). * Significant reduction (P < 0.02) in Kv1.2 expression in both panels in the diabetic hearts (n = 5 in males, n = 4 in females), but augmentation of channel protein was only seen in males (P < 0.02, n = 6), with no effect in females (n = 4).
, n = 12). The increase in densitometry (P < 0.03) parallels the increase in Ipeak in males. On the right are the values obtained in females, showing no significant differences in the absence of (n = 4) or following perfusion with quinapril (n = 4), paralleling the lack of change in Ipeak. Bottom panel, blot obtained using an antibody against Kv1.2. The same samples as above were used. In addition, six lanes are shown (right) in which a competing peptide for Kv1.2 was used. The peptide blocked the lower of the two bands shown, which was the band measured by densitometry. The mean data in control (□), diabetic () and diabetic hearts perfused with quinapril () are shown (left) for males (blots shown in Shimon & Liu, 2003) and females (right). * Significant reduction (P < 0.02) in Kv1.2 expression in both panels in the diabetic hearts (n = 5 in males, n = 4 in females), but augmentation of channel protein was only seen in males (P < 0.02, n = 6), with no effect in females (n = 4).
Thus, a major modulatory role of two different paracrine/autocrine factors is greatly blunted in cells from female rats. There is obviously a wide range of gender differences that could underlie this marked difference in sensitivity to the effects of inhibiting the formation of angiotensin II and endothelin-1. However, of major interest is the female sex hormone oestradiol. Oestradiol has been shown to down-regulate several components of the RAS (Kuroski de Bold, 1999; Fischer et al. 2002), and to reduce or inhibit several effects of angiotensin II (Brosnihan et al. 1997). Oestradiol was also shown to inhibit the production of endothelin-1 (Morey et al. 1998; Dubey et al. 2001). The next set of experiments examined the involvement of oestradiol. This was done by both in vitro and in vivo experiments.
It was hypothesized that if oestradiol blocks the RAS and endothelin system in females, preventing the autocrine modulation of K+ currents, this may be reversed by ovariectomy. Female rats were ovariectomized, (Ovx) and diabetes was induced with STZ 14 days after surgery. Ventricular myocytes were prepared 8–14 days after STZ injection. Current densities were compared in cells from Ovx and diabetic Ovx rats. Ovariectomy in itself augmented Ipeak (P < 0.02) but not Isus, with mean densities (at +50 mV) of 24.4 ± 2.4 and 7.8 ± 0.6 pA pF−1, respectively (n = 10). In diabetic cells (n = 21) from ovariectomized females both currents were significantly attenuated, to 18.6 ± 1.3 pA pF−1 (P < 0.03) and 4.9 ± 0.2 pA pF−1 (P < 0.0001). Sample current traces and full current–voltage relationships are shown in Fig. 5.
Figure 5. The effects of STZ-induced diabetes on K+ currents in ovariectomized (Ovx) female rats.
A, sample current traces obtained in a cell from an Ovx rat (left) and from an Ovx rat 10 days after induction of diabetes with STZ. Both peak and sustained currents are attenuated, in contrast to effects of STZ in normal females. B and C, mean current densities plotted against membrane potential for Ipeak (B) and Isus (C). * Significant reduction (P < 0.02 to P < 0.04) in peak currents between −20 and +50 mV, with Isus significantly (P < 0.0005) reduced between −50 and +50 mV.
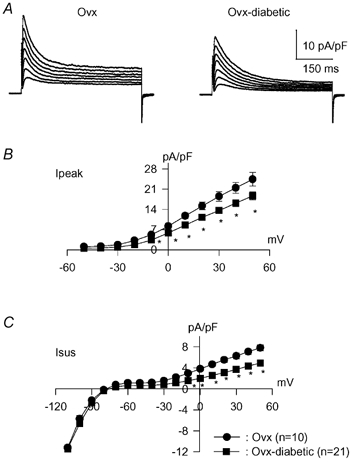
Thus, the attenuation of both currents under diabetic conditions is evident following ovariectomy (in contrast to normal females, as shown in Fig. 1). However, more striking was the restoration of the effects of ACE inhibition. In contrast to the absence of effect of quinapril on K+ currents in cells from diabetic females (as in Fig. 2), in vitro addition of quinapril (5–9 h) to cells from Ovx diabetic females markedly augmented both currents. The mean density of Ipeak at +50 mV in the absence and presence of quinapril was 18.6 ± 1.3 (n = 21) and 27.9 ± 1.3 pA pF−1 (n = 15) (P < 0.0001), respectively. The corresponding values for Isus were 4.9 ± 0.2 and 6.8 ± 0.4 pA pF−1 (P < 0.0001), respectively. Sample current traces and the current–voltage relationships are shown in Fig. 6.
Figure 6. The effects of ACE inhibition are restored in Ovx-diabetic rats.
A, sample current traces from cells obtained from Ovx-diabetic rats in the absence (left) or following 7 h incubation (right) with 1 μM quinapril. B and C, current–voltage relationships for Ipeak (B) and Isus (C). * Significant increase in Ipeak (P < 0.05 to P < 0.0001) between −10 and +50 mV, whereas Isus is augmented (P < 0.005) between −50 and +50 mV.
A similar restoration of the effects of the ECE inhibitor was found in the Ovx rats (see below). Our working hypothesis is that in females quinapril and the ECE inhibitor do not augment K+ currents since oestradiol already suppresses ACE and ECE. In Ovx rats, the removal of oestradiol presumably removes the inhibition of ACE and ECE, enabling the currents to be augmented by quinapril and the ECE inhibitor. This was tested in two ways (see below).
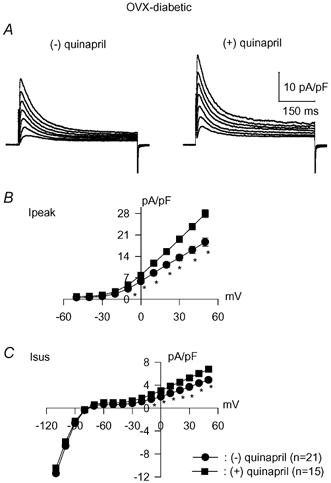
A direct role for oestradiol was investigated, to confirm the working hypothesis. This was done in experiments using oestradiol either in vivo or in vitro. First, Ovx rats received oestradiol (0.5 μg ml−1 in the drinking water). This was started 2 weeks after ovariectomy. One week later, diabetes was induced with STZ and cells were isolated after a further 8–10 days (oestradiol maintained throughout). The replacement of oestradiol in Ovx-diabetic rats was found to abolish the effects of quinapril on K+ currents, as in control female rats. This result is shown in Fig. 7, which shows current–voltage relationships in the absence and presence of quinapril for Ipeak (Fig. 7A) and Isus (Fig. 7B).
Figure 7. Effects of ACE inhibition in Ovx-diabetic rats with oestrogen replacement.
Currents were recorded in cells obtained 10 days after induction of diabetes with STZ and following 17 days of oestradiol in the drinking water. Ovariectomy was performed 4 weeks before experiments. Mean current densities are plotted against membrane potential for Ipeak (A) and Isus (B) in the absence of (•) or following incubation (5–9 h) with 1 μM quinapril (▪). Restoring oestradiol to Ovx rats abolishes the effects of ACE inhibition on both currents, as in normal female rats.
At +50 mV the mean densities of Ipeak in the absence (n = 9) or presence (n = 13) of quinapril were 15.2 ± 1.9 and 13.9 ± 1.7 pA pF−1 (P > 0.05), respectively. The corresponding values for Isus were 5.5 ± 0.4 and 5.9 ± 0.4 pA pF−1 (P > 0.05), respectively.
Similar results were found with the ECE inhibitor. Whereas no effect on either K+ current was found in the diabetic female (in contrast to the male diabetic rats), current augmentation was restored in Ovx-diabetic rats. Oestradiol replacement in the Ovx-diabetic rats abolished the augmentation of both Ipeak and Isus by ECE inhibition. A summary of the results in these three conditions is shown in Fig. 8, with Ipeak shown in the top panel and Isus below. In Ovx rats ECE inhibition increased Ipeak and Isus from 18.2 ± 1.1 and 4.8 ± 0.2 pA pF−1 (n = 34), respectively, to 25.6 ± 1.3 and 7.6 ± 0.3 pA pF−1 (n = 25), respectively (P < 0.0001). In the oestradiol-replaced Ovx rats the values for Ipeak in the absence and presence of the ECE inhibitor were 15.0 ± 1.3 (n = 16) and 15.3 ± 1.3 pA pF−1 (n = 19), respectively (P > 0.05). The corresponding values for Isus were 5.6 ± 0.2 and 5.6 ± 0.4 pA pF−1, respectively.
Figure 8. The effects of ECE inhibition on K+ currents in STZ-induced diabetes depends on oestradiol.
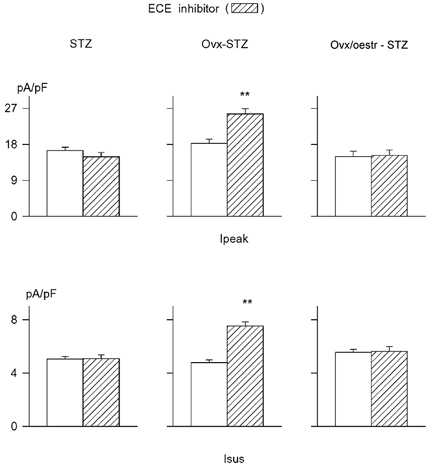
Mean current densities at +50 mV are plotted in the absence (□) or following 5–9 h incubation with 5 μM ECE inhibitor ( ). Ipeak values are in the top row, with Isus values in the bottom row. In cells from STZ-induced diabetic female rats (left) no effect of ECE inhibition is evident. In Ovx-diabetic rats ECE inhibition significantly augments both currents (middle), whereas restoring oestrogen to Ovx-diabetic rats abolishes the augmentation of both currents. ** Significant difference (P < 0.001).
). Ipeak values are in the top row, with Isus values in the bottom row. In cells from STZ-induced diabetic female rats (left) no effect of ECE inhibition is evident. In Ovx-diabetic rats ECE inhibition significantly augments both currents (middle), whereas restoring oestrogen to Ovx-diabetic rats abolishes the augmentation of both currents. ** Significant difference (P < 0.001).
Subsequently, in vitro experiments with oestradiol were performed, to test the hypothesis that the interaction of oestradiol with the autocrine systems modulates the K+ currents. Exposure of cells from male diabetic rats to 100 nM 17β-oestradiol for 5–9 h was found to induce effects comparable to those of quinapril. Both Ipeak and Isus were significantly augmented, as shown in Fig. 9. Sample current traces are shown in Fig. 9A, and current–voltage relationships for Ipeak and Isus are shown in Fig. 9B and C, respectively. Ipeak densities were significantly (P < 0.005) augmented at membrane potentials in the range −30 to +50 mV. Isus was significantly (P < 0.0005) increased at potentials from −40 to +50 mV.
Figure 9. Effects of oestradiol in myocytes from diabetic males.
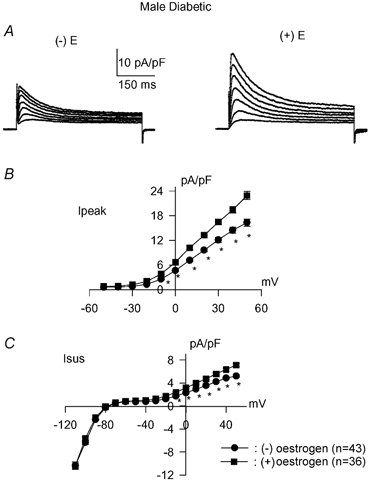
A, sample current traces (same voltage protocol as Fig. 1) from diabetic male rat myocytes in the absence (left, (−)E) or following (right (+)E) 6 h exposure to 100 nM 17β-oestradiol. B, current–voltage relationship for peak currents in the absence (•, n = 43 cells) or following administration of 17β-oestradiol (▪, n = 36). * Significant increase in Ipeak by oestradiol (P < 0.002 to P < 0.0008) between −40 and +50 mV. C, steady-state current–voltage relationship obtained in the same cells as in B. * Significant increase in Isus by oestradiol (P < 0.04 to P < 0.0001) between −60 and +50 mV.
The final set of experiments examined whether the augmentation of these currents by oestradiol is associated with angiotensin II and endothelin-1. It was assumed that if K+ current augmentation by oestradiol is (at least partly) mediated through an inhibition of the formation or action of angiotensin II or endothelin-1, it may be possible to antagonize the augmentation of K+ currents by oestradiol by including angiotensin II or endothelin-1 in the incubation medium. When cells from diabetic male rats were incubated for 5–9 h with both 17β-oestradiol (100 nM) and angiotensin II (100 nM), the augmentation of currents seen with oestradiol alone was significantly reduced. Figure 10 shows mean (± s.e.m.) current densities for Ipeak and Isus in the absence or presence of oestradiol, as well as with oestradiol and angiotensin II.
Figure 10. Angiotensin and endothelin-1 prevent K+ current augmentation by oestradiol in male diabetic rat myocytes.
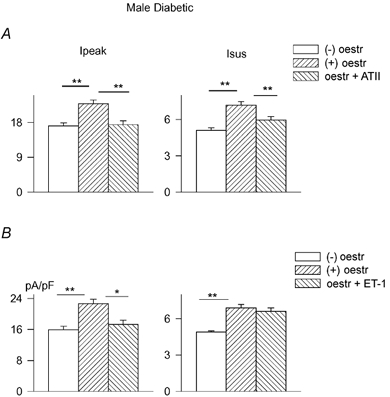
A, current densities at +50 mV (Ipeak, left; Isus, right) for currents in untreated cells (□, n = 42), in cells treated for 5–9 h with 100 nM 17β-oestradiol (, n = 36) and in cells treated with 17β-oestradiol and 100 nM angiotensin II (, n = 32). ** Significant reduction in Ipeak and Isus in the presence of oestradiol + ATII (in comparison to oestradiol alone) (P < 0.001). B, ET-1 also prevents augmentation of Ipeak by oestradiol. Mean densities (at +50 mV) for Ipeak (left) and Isus (right) are shown in the absence of drugs (□), following 5–9 h in the presence of 100 nM oestradiol () and in the presence of 100 nM oestradiol + 400 nM ET-1 (). * P < 0.05; * P < 0.005.
The combined drugs reduced Ipeak at membrane potentials ranging from 0 to +50 mV (P < 0.03 at 0 mV, P < 0.005 from +10 to +50 mV). Isus was significantly reduced between +20 and +50 mV (P < 0.05 between +20 and +40 mV, P < 0.003 at +50 mV (not shown)).
In control experiments, angiotensin II alone did not reduce Ipeak or Isus in cells from diabetic male rats. The values of Ipeak and Isus were 14.3 ± 0.8 and 5.1 ± 0.2 pA pF−1, respectively, (n = 33) in the absence, and 16.1 ± 1.0 and 5.7 ± 0.2 pA pF−1, respectively, (n = 30) following 5–8 h treatment with 100 nM angiotensin II.
Endothelin-1 was also added in vitro in the presence of oestradiol to see whether it could blunt the augmentation of currents by oestradiol. Inclusion of ET-1 in the incubation medium together with oestradiol was less effective than ATII in blunting the action of oestradiol. Whereas 100 nM ET-1 was ineffective, 200 nM was partially effective. Only when 400 nM ET-1 was used was there a significant (P < 0.01) reduction in the augmentation of Ipeak by oestradiol. Mean Ipeak densities at +50 mV were 15.9 ± 1.0, 22.7 ± 1.1 and 17.3 ± 1.1 pA pF−1 with no oestradiol (n = 34), with oestradiol alone (n = 34) and with oestradiol plus 400 nM ET-1 (n = 35), respectively. The effect of oestradiol on Isus was still maintained with ET-1 present. The corresponding values were 4.9 ± 0.1, 6.85 ± 0.3 and 6.6 ± 0.3 pA pF−1, respectively. These results are shown in Fig. 10B.
Addition of 400 nM endothelin-1 to cells (5–10 h) from male diabetic rats had no significant effects on either current. The values obtained in these experiments for Ipeak were 15.9 ± 1.1 (n = 23) and 16.4 ± 1.0 pA pF−1 (n = 21) in the absence of and following incubation with endothelin, respectively (P > 0.05). The corresponding values for Isus were 5.2 ± 0.2 and 6.1 ± 0.4 pA pF−1 (P > 0.05), respectively.
DISCUSSION
Summary of findings
In the present work, we report three novel findings: (1) insulin-deficient diabetes produces a significantly weaker attenuation of two repolarizing K+ currents in ventricular myocytes from female rats, as compared to males. (2) The augmentation of these currents by an ACE or an ECE inhibitor, observed in cells from diabetic males, is absent in cells from diabetic female rats. (3) This augmentation is restored following ovariectomy.
A key modulatory role of oestradiol involving interaction with the renin-angiotensin system is suggested by in vivo and in vitro experiments. Giving oestradiol to ovariectomized rats abolished the enhancement of K+ currents by ACE and ECE inhibition in isolated cells. Furthermore, both K+ currents were enhanced in cells from diabetic males, following addition of 17β-oestradiol (> 5 h). This effect was blunted or abolished if angiotensin II or endothelin-1 were included in the incubation medium.
Interpretation and significance
Although the overall protective effects of oestradiol, particularly with regard to development of coronary disease, have lately been questioned (Rossouw, 2002; Grady et al. 2002), there is abundant evidence for beneficial effects of oestradiol. Thus, oestradiol has been shown to attenuate the pressor response to angiotensin II (Brosnihan et al. 1997), to attenuate the development of cardiac hypertrophy (Van Eickels et al. 2001) and to reduce infarct size, the occurrence of ischaemia and reperfusion-induced arrhythmias (Node et al. 1997). Since inhibition of the renin-angiotensin system is considered to be cardio-protective in general (Fischer et al. 2002) and in diabetes in particular (Zuanetti et al. 1997; Gerstein et al. 2000), it is presumably this aspect of the action of oestradiol that mediates some of its protective effects. In our experiments, the working hypothesis was that a chronic elevated level of oestradiol in female rats blunts the effects of STZ-induced diabetes on the two K+ currents (Fig. 1). This would occur if the activation of the RAS, which normally mediates some of the attenuation of currents, is reduced or inhibited by oestradiol.
This is supported by direct evidence showing that ACE expression is lower in female than in male mice (Freshour et al. 2002). In their work androgens were shown to be a mediating component of this disparity, although our work, as well as earlier work by Brosnihan et al. (1997) and by Gallagher et al. (1999) suggests that oestradiol also regulates the renin-angiotensin system. The reported effects of oestradiol include a down-regulation of ACE related to reduced ACE mRNA levels (Gallagher et al. 1999), as well as a reduction in ACE activity (Kuroski de Bold, 1999). AT1 receptor mRNA and protein expression are also reduced by oestradiol (Nickenig et al. 2000). Oestradiol also inhibits endothelin-1 binding and synthesis (Duru et al. 2001; Dubey et al. 2001). It is interesting that there are also reported gender differences in the effects of oestradiol on basal and ATII-induced protein kinase C (PKC) activity (Lachowicz & Rebas, 2002). Thus, sex differences in PKC may also be of importance. This will be addressed in future studies.
Our working hypothesis is that the RAS is already inhibited by oestradiol in the females, with no further augmentation of currents by addition of the ACE or ECE inhibitors (Fig. 2 and Fig. 3). A direct role of oestrogen was suggested by the combined findings that showed that in ovariectomized rats ACE and ECE inhibitors could augment the two currents, as in males (Fig. 5 and Fig. 6) but that this was again lacking if oestradiol was given to these Ovx rats prior to cell isolation (Fig. 7 and Fig. 8).
Obviously, the actions of oestradiol on repolarizing currents are more complex, and probably involve actions unrelated to the autocrine systems. Some repolarizing currents in females are smaller, as reported by Trepanier-Boulay et al. (2001). In our experiments ovariectomy reversed the attenuation of Ipeak but not of Isus. However, the major effect of oestradiol, at least in the setting of diabetes, was to modulate these currents by interacting with angiotensin. The sex differences in the action of quinapril were reflected in changes in channel expression as well as in current magnitude, suggesting (but not proving) that the interaction occurs at the level of transcription. The sex differences in plasma glucose level which was observed in the small samples was not considered to affect the results, since despite the higher glucose levels in females the effects of diabetes on the K+ currents were smaller than in males.
The suggestion of an interaction between oestradiol and the two autocrine/paracrine systems was supported by the in vitro experiments on cells from diabetic males. In these cells, addition of 17β-oestradiol augments K+ currents (Fig. 9) at least partly by inhibiting angiotensin II and endothelin-1 formation or action. This is based on the finding that combining angiotensin II or endothelin-1 with 17β-oestradiol prevents current augmentation (Fig. 10). The presence of ATII and ET-1 presumably overcomes the suppression of RAS and the endothelin system by oestradiol and thus prevents current augmentation by oestradiol, although with ET-1 the effect of oestradiol on Isus was maintained (Fig. 10). The reason for this is not known, but may reflect the fact that Isus is a combination of several currents that may be subject to differential regulation.
Our results may be of significance in that they provide a mechanistic link between the protective actions of oestradiol and changes in repolarizing K+ currents. This may be associated with the reduction in some types of arrhythmias in females.
Limitations
One limitation is the uncertainty regarding other effects of oestradiol on K+ currents, and the mechanisms underlying the reduction in baseline current density in control female rats. If the major effect of oestradiol in control conditions was to suppress the RAS, these K+ currents should be larger in females and not smaller. However, it is very likely that the interaction of oestradiol with the RAS is mainly operational under pathological conditions, as has been suggested by others (Fischer et al. 2002). Thus, for example, there is a profound sexual dimorphism in hypertensive rats (Bachmann et al. 1993; Brosnihan et al. 1997), which is associated with the interaction of oestradiol and the RAS. It is important that the overall effect of diabetes is to reduce the female advantage in the development of cardiovascular disease (Colhoun et al. 2000; Brown et al. 2001). However, this may relate to coronary disease and altered lipid profiles associated with diabetes. It is possible that other mechanisms of arrhythmia development, such as changes in K+ currents as described here, are differentially regulated in diabetes, giving females an advantage. Further studies will be required to elucidate these issues.
In summary, the data presented here present a novel way in which K+ currents may be modulated in a sex-selective manner. Sex differences in propensity for cardiac arrhythmias are well recognized. Changes in patterns of repolarization, largely determined by changes in K+ currents are prevalent in several cardiac disease states such as heart failure (Nabauer & Kaab, 1998). Since changes in repolarization underlie various types of arrhythmias (e.g. Surawicz, 1997), the present results may have a wide implication for various cardiac pathologies.
Acknowledgments
This work was supported by the Canadian Institutes of Health and by a grant from the Canadian Diabetes Association in honour of the late Celeste DePaoli.
REFERENCES
- Babiker FA, De Windt LJ, Van Eickels M, Grohe C, Meyer R, Doevendans PA. Estrogenic hormone action in the heart: regulatory network and function. Cardiovasc Res. 2002;53:709–719. doi: 10.1016/s0008-6363(01)00526-0. [DOI] [PubMed] [Google Scholar]
- Bachmann J, Wagner J, Haufe C, Wystrychowski A, Ciechanowicz A, Ganten D. Modulation of blood pressure and the renin-angiotensin system in transgenic and spontaneously hypertensive rats after ovariectomy. J Hypertens. 1993;11(Suppl. 5):S226–S227. [PubMed] [Google Scholar]
- Bailey MS, Curtis AB. The effects of hormones on arrhythmias in women. Curr Womens Health Rep. 2002;2:83–88. [PubMed] [Google Scholar]
- Barlucchi L, Leri A, Dostal DE, Fiordaliso F, Tada H, Hintze TH, Kajstura J, Nadal-Ginard B, Anversa P. Canine ventricular myocytes possess a renin-angiotensin system that is upregulated with heart failure. Circ Res. 2001;88:298–304. doi: 10.1161/01.res.88.3.298. [DOI] [PubMed] [Google Scholar]
- Brosnihan KB, Li P, Ganten D, Ferrario CM. Estrogen protects transgenic hypertensive rats by shifting the vasoconstrictor-vasodilator balance of RAS. Am J Physiol. 1997;273:R1908–1915. doi: 10.1152/ajpregu.1997.273.6.R1908. [DOI] [PubMed] [Google Scholar]
- Brown RA, Walsh MF, Ren J. Influence of gender and diabetes on vascular and myocardial contractile function. Endocr Res. 2001;27:399–408. doi: 10.1081/erc-100107864. [DOI] [PubMed] [Google Scholar]
- Colhoun HM, Rubens MB, Underwood SR, Fuller JH. The effect of type 1 diabetes mellitus on the gender difference in coronary artery calcification. J Am Coll Cardiol. 2000;36:2160–2167. doi: 10.1016/s0735-1097(00)00986-4. [DOI] [PubMed] [Google Scholar]
- Dostal DE. The cardiac renin-angiotensin system: novel signalling mechanisms related to cardiac growth and function. Regul Pept. 2000;91:1–11. doi: 10.1016/s0167-0115(99)00123-8. [DOI] [PubMed] [Google Scholar]
- Dubey RK, Jackson EK, Keller PJ, Imthurn B, Rosselli M. Estradiol metabolites inhibit endothelin synthesis by an oestrogen receptor-independent mechanism. Hypertens. 2001;37:640–644. doi: 10.1161/01.hyp.37.2.640. [DOI] [PubMed] [Google Scholar]
- Duru F, Barton M, Luscher TF, Candinas R. Endothelins and cardiac arrhythmias: do endothelin antagonists have a therapeutic potential as antiarrhythmic drugs. Cardiovasc Res. 2001;49:272–280. doi: 10.1016/s0008-6363(00)00263-7. [DOI] [PubMed] [Google Scholar]
- Ferri C, Laurenti O, Bellini C, Faldetta MRC, Properzi G, Santucci A, De Mattia G. Circulating endothelin-1 levels in lean noninsulin-dependent diabetic patients. Influence of ACE inhibition. Am J Hypertens. 1995;8:40–47. doi: 10.1016/0895-7061(94)00180-J. [DOI] [PubMed] [Google Scholar]
- Fiordaliso F, Li B, Latini R, Sonnenblick EH, Anversa P, Leri A, Kajstura J. Myocyte death in steptozotocin-induced diabetes in rats is angiotensin II-dependent. Lab Invest. 2000;80:513–527. doi: 10.1038/labinvest.3780057. [DOI] [PubMed] [Google Scholar]
- Fischer M, Baessler A, Schunkert H. Renin angiotensin system and gender differences in the cardiovascular system. Cardiovasc Res. 2002;53:672–677. doi: 10.1016/s0008-6363(01)00479-5. [DOI] [PubMed] [Google Scholar]
- Freshour JR, Chase SE, Vikstrom KL. Gender differences in cardiac ACE expression are normalized in androgen-deprived male mice. Am J Physiol Heart Circ Physiol. 2002;283:H1997–2002. doi: 10.1152/ajpheart.01054.2001. [DOI] [PubMed] [Google Scholar]
- Gallagher PE, Li P, Lenhart JR, Chappell MC, Brosnihan KB. Estrogen regulation of angiotensin-converting enzyme mRNA. Hypertension. 1999;33:323–328. doi: 10.1161/01.hyp.33.1.323. [DOI] [PubMed] [Google Scholar]
- Gerstein HC, Yusuf S, Mann JFE, Hoogwerf B, Zinman B, Held C, Fisher M, Wolffenbuttel B, Bosch J, Richardson J, Pogue J, Halle JP. Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Lancet. 2000;335:253–259. [PubMed] [Google Scholar]
- Grady D, Herrington D, Bittner V, Blumenthal R, Davidson M, Hlatky M, Hsia J, Hulley S, Herd A, Khan S, Newby LK, Waters D, Vittinghoff E, Wenger N. Cardiovascular disease outcomes during 6. 8 years of hormone therapy. J Am Med Assoc. 2002;288:49–57. doi: 10.1001/jama.288.1.49. [DOI] [PubMed] [Google Scholar]
- Kuroski De Bold ML. Estrogen, natriuretic peptides and the renin-angiotensin system. Cardiovasc Res. 1999;41:524–531. doi: 10.1016/s0008-6363(98)00324-1. [DOI] [PubMed] [Google Scholar]
- Lachowicz A, Rebas E. Gender differences in steroid modulation of angiotensin II-induced protein kinase C activity in anterior pituitary of the rat. Biochem Biophys Res Commun. 2002;294:95–100. doi: 10.1016/S0006-291X(02)00433-3. [DOI] [PubMed] [Google Scholar]
- Larsen JA, Kadish AH. Effects of gender on cardiac arrhythmias. J Cardiovasc Electrophysiol. 1998;6:665–667. doi: 10.1111/j.1540-8167.1998.tb00950.x. [DOI] [PubMed] [Google Scholar]
- Morey AK, Razandi M, Pedram A, Hu RM, Prins BA, Levin AR. Oestrogens and progesterone inhibit the stimulated production of endothelin-1. Biochem J. 1998;330:1097–1105. doi: 10.1042/bj3301097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabauer M, Kaab S. Potassium channel down-regulation in heart failure. Cardiovasc Res. 1998;37:324–334. doi: 10.1016/s0008-6363(97)00274-5. [DOI] [PubMed] [Google Scholar]
- Nathan DM, Meigs J, Singer DE. The epidemiology of cardiovascular disease in type 2 diabetes mellitus: how sweet it is…or is it. Lancet. 1997;350(suppl. 1):4–9. doi: 10.1016/s0140-6736(97)90021-0. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM. Molecular basis of functional voltage-gated K+ channel diversity in the mammalian myocardium. J Physiol. 2000;525:285–298. doi: 10.1111/j.1469-7793.2000.t01-1-00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickenig G, Strehlow K, Wassermann S, Baumer AT, Albory K, Sauer H, Bohm M. Differential effects of estrogen and progesterone on AT (1) receptor gene expression in vascular smooth muscle cells. Circulation. 2000;102:1828–1833. doi: 10.1161/01.cir.102.15.1828. [DOI] [PubMed] [Google Scholar]
- Node K, Kitakaze M, Kosaka H, Minamino T, Funaya H, Hori M. Amelioration of ischemia-and reperfusion-induced myocardial injury by 17 beta-oestradiol: role of nitric oxide and calcium-activated potassium channels. Circulation. 1997;96:1953–1963. doi: 10.1161/01.cir.96.6.1953. [DOI] [PubMed] [Google Scholar]
- Pham TV, Rosen MR. Sex, hormones, and repolarization. Cardiovasc Res. 2002;53:740–751. doi: 10.1016/s0008-6363(01)00429-1. [DOI] [PubMed] [Google Scholar]
- Roeters van Lennep JER, Westerveld HT, Erkelens DW, Van der Wall EE. Risk factors for coronary heart disease: implications of gender. Cardiovasc Res. 2002;53:538–549. doi: 10.1016/s0008-6363(01)00388-1. [DOI] [PubMed] [Google Scholar]
- Rossouw JE. Hormones, genetic factors, and gender differences in cardiovascular disease. Cardiovasc Res. 2002;53:550–557. doi: 10.1016/s0008-6363(01)00478-3. [DOI] [PubMed] [Google Scholar]
- Russell FD, Molenaar P. The human heart endothelin system: ET-1 synthesis, storage, release and effect. Trends Pharmacol Sci. 2000;21:353–359. doi: 10.1016/s0165-6147(00)01524-8. [DOI] [PubMed] [Google Scholar]
- Saltevo J, Puolakka J, Ylikorkala O. Plasma endothelin in postmenopausal women with type 2 diabetes mellitus and metabolic syndrome: a comparison of oral combined and transdermal oestrogen-only treatment therapy. Diabetes Obes Metab. 2000;5:293–298. doi: 10.1046/j.1463-1326.2000.00087.x. [DOI] [PubMed] [Google Scholar]
- Shimoni Y. Inhibition of the formation or action of angiotensin II reverses attenuated K+ currents in type 1 and type 2 diabetes. J Physiol. 2001;537:83–92. doi: 10.1111/j.1469-7793.2001.0083k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoni Y, Ewart HS, Severson D. Type I and II models of diabetes produce different modifications of K+ currents in rat heart: role of insulin. J Physiol. 1998;507:485–496. doi: 10.1111/j.1469-7793.1998.485bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoni Y, Liu XF. Role of PKC in autocrine regulation of rat ventricular K+ currents by angiotensin and endothelin. Am J Physiol Heart Circ Physiol. 2003;284:H1168–1181. doi: 10.1152/ajpheart.00748.2002. [DOI] [PubMed] [Google Scholar]
- Surawicz B. Ventricular fibrillation and dispersion of repolarization. J Cardiovasc Electrophysiol. 1997;8:1009–1012. doi: 10.1111/j.1540-8167.1997.tb00624.x. [DOI] [PubMed] [Google Scholar]
- Trepanier-Boulay V, St-Michel C, Tremblay A, Fiset C. Gender-based differences in cardiac repolarization in mouse ventricle. Circ Res. 2001;89:437–444. doi: 10.1161/hh1701.095644. [DOI] [PubMed] [Google Scholar]
- Van Eickels M, Grohe C, Cleutjens JP, Janssen BJ, Wellens HJ, Doevendans PA. 17beta-estradiol attenuates the development of pressure-overload hypertrophy. Circulation. 2001;104:1419–1423. doi: 10.1161/hc3601.095577. [DOI] [PubMed] [Google Scholar]
- Verma S, Arikawa E, McNeill JH. Long-term endothelin receptor blockade improves cardiovascular function in diabetes. Am J Hypertens. 2001;14:679–687. doi: 10.1016/s0895-7061(01)01302-4. [DOI] [PubMed] [Google Scholar]
- Wild SH, Dunn CJ, McKeigue PM, Comte S. Glycemic control and cardiovascular disease in type 2 diabetes: a review. Diabetes Metab Res Rev. 1999;15:197–204. doi: 10.1002/(sici)1520-7560(199905/06)15:3<197::aid-dmrr28>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Wu Y, Anderson ME. Reduced repolarization reserve in ventricular myocytes from female mice. Cardiovasc Res. 2002;53:763–769. doi: 10.1016/s0008-6363(01)00387-x. [DOI] [PubMed] [Google Scholar]
- Zimmermann M, Jung C, Raabe A, Spanehl O, Fach K, Seifert V. Inhibition of endothelin-converting enzyme activity in the rabbit basilar artery. Neurosurgery. 2001;48:902–910. doi: 10.1097/00006123-200104000-00043. [DOI] [PubMed] [Google Scholar]
- Zuanetti G, Latini R, Maggioni AP, Franzosi M, Santoro L, Tognoni G. Effect of ACE inhibitor lisinopril on mortality in diabetic patients with acute myocardial infarction. Data from the GISSI-3 study. Circulation. 1997;96:4239–4245. doi: 10.1161/01.cir.96.12.4239. [DOI] [PubMed] [Google Scholar]