

Abstract
Glycine is a major inhibitory neurotransmitter in the spinal cord and brainstem. Here we report the novel finding that presynaptic glycine autoreceptors modulate release from terminals synapsing onto rat spinal sacral dorsal commissural nucleus (SDCN) neurons. In mechanically dissociated SDCN neurons, in which functional presynaptic nerve terminals remain adherent to the isolated neurons, exogenously applied glycine (3 μM) increased the frequency of glycinergic spontaneous inhibitory postsynaptic currents (sIPSCs) without affecting their amplitudes or decay times. This suggests that glycine acts presynaptically to increase glycine release probability. Picrotoxin, at a concentration that had little direct effect on sIPSC frequency and amplitude (30 μM), significantly attenuated glycine-induced presynaptic sIPSC facilitation. The glycine-induced sIPSC frequency facilitation was completely abolished either in a Ca2+-free external solution or in the presence of 100 μM Cd2+, suggesting the involvement of extracellular Ca2+ influx into the nerve terminals. The glycine action was also completely occluded in the presence of 300 nM tetrodotoxin. In recordings from SDCN neurons in spinal cord slices, glycine (10 μM) increased evoked IPSC (eIPSC) amplitude and decreased the extent of paired-pulse facilitation. In response to brief high frequency stimulus trains the eIPSCs displayed a profound frequency-dependent facilitation that was greatly reduced by picrotoxin (30 μM). These results indicate that glycine acts at presynaptic autoreceptors, causing depolarization of the glycinergic nerve terminals, the subsequent activation of voltage-dependent Na+ and Ca2+ channels, and facilitation of glycine release. Furthermore, this presynaptic facilitation was observed under more physiological conditions, suggesting that these glycinergic autoreceptors may contribute to the integration of local inhibitory inputs to SDCN neurons.
Presynaptic G-protein-coupled receptors modulate synaptic transmission in the mammalian central nervous system (CNS) through a variety of mechanisms, including the inhibition of presynaptic voltage-dependent Ca2+ channels, the activation of presynaptic K+ channels and the direct modulation of presynaptic release machinery (for review see Wu & Saggau, 1997). Ionotropic, ligand-gated ion channels on presynaptic nerve terminals can also modulate neurotransmitter release (for review see MacDermott et al. 1999). Some ‘classical’ neurotransmitters modulate their own release by acting on presynaptic ionotropic autoreceptors. For example, acetylcholine facilitates cholinergic transmission by the activation of presynaptic nicotinic receptors in several CNS regions, including the hippocampus, cerebellum and cortex (Heilbronn et al. 1981; Rowell & Winkler, 1984; Lapchak et al. 1989). Glutamate, via kainate receptors, also modulates glutamate release from hippocampal mossy fibre terminals (Kamiya & Ozawa, 2000; Schmitz et al. 2000).
The activation of presynaptic GABAA receptors has long been known to depolarize primary afferent fibres, leading to a presynaptic inhibition of electrically evoked glutamate release (Rudomin & Schmidt, 1999; Cattaert & El Manira, 1999). GABAA receptor-mediated presynaptic depolarization has also recently been shown to facilitate spontaneous glutamate or glycine release, by activating presynaptic voltage-dependent Na+ and Ca2+ channels (Jang et al. 2001, 2002). GABAC receptors, expressed at a high level in the terminals of retina bipolar neurons, also induce presynaptic inhibition of neurotransmitter release (Matthews et al. 1994; Lukasiewicz et al. 1994). These findings suggest that inhibitory neurotransmitters such as GABA, by acting on presynaptic ionotropic receptors, participate in the presynaptic modulation of neurotransmitter release.
Glycine is a major inhibitory neurotransmitter in the spinal cord and brainstem. Previous morphological studies have revealed that in the spinal cord, some presynaptic boutons terminating on glutamatergic terminals contain not only GABA but also glycine (Todd et al. 1995; Maxwell et al. 1997). These findings suggest the possibility that glycine, like GABA, contributes to synaptic modulation among synapses in the spinal cord. However, as glycine receptors and gephyrin distribution seem to be localized to synapses on postsynaptic soma and dendrites (Mitchell et al. 1993; Todd et al. 1995; Colin et al. 1998), it is far from clear whether presynaptic glycine receptors contribute to the modulation of synaptic transmission in the spinal cord. Activation of presynaptic glycine receptors has recently been shown to depolarize the large, excitatory, calyx of Held synaptic terminals in the rat auditory brainstem nucleus (Turecek & Trussell, 2001, 2002). While these terminals are not directly innervated by glycine-containing boutons (as found in the spinal cord), glycine ‘spillover’ from adjacent synapses was sufficient to elicit the presynaptic depolarization. Somewhat surprisingly, considering that GABAA receptor-mediated primary afferent depolarization causes presynaptic inhibition, the glycine receptor-mediated depolarization led to an increase in both spontaneous and evoked glutamate release (Turecek & Trussell, 2001, 2002). In the present study, we have investigated the presence and actions of presynaptic glycine receptors on small, glycinergic nerve terminals projecting onto rat spinal sacral dorsal commissural nucleus (SDCN) neurons. Our results clearly demonstrate, for the first time, functional presynaptic glycinergic ‘autoreceptors’ on spinal interneuron terminals and show that activation of these receptors also causes presynaptic depolarization and facilitation of glycine release.
METHODS
Preparation
Wistar rats (10–12 days old) were decapitated under pentobarbital anaesthesia (50 mg kg−1, I.P.). A segment of the lumbosacral (L6-S2) spinal cord was dissected and transversely sliced at a thickness of 350 μm by use of a microslicer (VT1000S; Leica, Nussloch, Germany). Slices containing the SDCN were kept in a control incubation medium (see below) saturated with 95 % O2 and 5 % CO2 at room temperature (21–24 °C) for at least 1 h before mechanical dissociation. Slices were then transferred into a 35 mm culture dish (Primaria 3801; Becton Dickinson, Rutherford, NJ, USA) containing the standard external solution (see below), and the region of the SDCN was identified under a binocular microscope (SMZ-1; Nikon, Tokyo, Japan). Details of the mechanical dissociation procedure used to isolate single neurons with adherent and functional presynaptic boutons have been given previously (Rhee et al. 1999; Akaike & Moorhouse, 2003). Briefly, mechanical dissociation was accomplished using a custom-built vibration device and a fire-polished glass pipette oscillating at about 50–60 Hz. The tip of the fire-polished glass pipette was lightly placed on the surface of the SDCN and was vibrated horizontally (0.1–0.2 mm displacement) for about 2 min. Slices were removed and the mechanically dissociated neurons allowed to settle and adhere to the bottom of the dish for at least 15 min before recordings commenced.
For the slice preparations, the lumbosacral spinal cord was dissected and transversely sliced at a thickness of 230 μm in a cold, low-Na+ medium (see below) using a microslicer (VT1000S, Leica). The slices were kept in an external bath solution (see below) saturated with 95 % O2 and 5 % CO2 at 34–35 °C for at least 1 h. They were then were transferred into a recording chamber, and the SDCN was identified under an upright microscope (DMLFSA, Leica). Once in the recording chamber, slices were continuously perfused at a rate of 3–4 ml min−1.
All experiments conformed to the guiding principles for the care and use of animals approved by the Council of the Physiological Society of Japan.
Electrical measurements
All electrical measurements were performed using the conventional whole-cell patch recording mode at a holding potential (VH) of −30 to −40 mV (CEZ-2300; Nihon Kohden, Tokyo, Japan). Patch pipettes were made from borosilicate capillary glass (1.5 mm outer diameter, 0.9 mm inner diameter; G-1.5; Narishige, Tokyo, Japan) in two stages on a vertical pipette puller (PB-7; Narishige). The resistance of the recording pipettes filled with internal solution was 5–6 MΩ. Isolated neurons were viewed under phase contrast on an inverted microscope (Diapot; Nikon). Current and voltage were continuously monitored on an oscilloscope (VC-6023; Hitachi) and a pen recorder (RECTI-HORIT-8K; Sanei, Tokyo, Japan), and recorded on digital-audio tape (RD-120TE; TEAC). Membrane currents were filtered at 1 kHz (E-3201A Decade Filter; NF Electronic Instruments, Tokyo, Japan), digitized at 4 kHz, and stored on a computer equipped with pCLAMP 8.0 (Axon Instruments). When recording, 10 mV hyperpolarizing step pulses (30 ms in duration) were periodically delivered to monitor the access resistance, and recordings were discontinued if the access resistance changed markedly. Experiments on dissociated neurons were performed at room temperature (21–24 °C) while slice experiments were performed at 34- 35 °C.
Evoked glycinergic IPSCs (eIPSCs) were elicited by applying short current pulses (100 μs, 30–50 μA) at 0.05 Hz through a glass pipette (7–8 μm inner diameter) placed around the central canal of the spinal cord, and filled with the bath solution. Electrical pulses were generated using a stimulator (SEN-7203, Nihon Kohden) attached to an isolator unit (SS-701J, Nihon Kohden). Data were acquired using pCLAMP 8 software, filtered at 3 kHz, digitized at 10 kHz and stored on a personal computer.
Data analysis
sIPSCs were counted and analysed using the MiniAnalysis program (Synaptosoft, NJ, USA), as described previously (Jang et al. 2002). Briefly, spontaneous events were screened automatically using an amplitude threshold of 5 pA and then visually accepted or rejected based upon their 10–90 % rise and 90–37 % decay times. The inter-event intervals and amplitudes of a large number of sIPSCs obtained from the same neuron were examined by constructing cumulative probability distributions, and these distributions were compared under different conditions using the Kolmogorov-Smirnov (K-S) test with Stat View software (SAS Institute, Inc.). The concentration-response data were fitted to the following modified Michaelis-Menten equation, using a least-squares fitting routine:
where I is the amplitude of the glycine-induced postsynaptic current and C is the corresponding agonist concentration. EC50 and nH denote the half-effective concentration and the Hill coefficient, respectively. Numerical values are provided as means ± s.e.m. using values normalized to the control. Differences in mean sIPSC amplitude and frequency were tested by Student's paired two-tailed t test using their absolute values, rather than the normalized ones. Values of P < 0.05 were considered significant. The responses to pairs and trains of stimulation were determined by averaging between 20 and 40 trials for each experimental condition. The amplitude of eIPSCs was measured using the Clampfit 8 program. Data are presented as means ± s.e.m. using values normalized to the control. Differences in mean eIPSC amplitude were tested by Student's paired two-tailed t test using their absolute values, rather than the normalized ones. Values of P < 0.05 were considered significant.
Solutions
The ionic composition of the incubation medium consisted of (mM): 124 NaCl, 5 KCl, 1.2 KH2PO4, 24 NaHCO3, 2.4 CaCl2, 1.3 MgSO4 and 10 glucose saturated with 95 % O2 and 5 % CO2. The pH was about 7.45. The low-Na+ medium consisted of (mM): 230 sucrose, 3 KCl, 1.5 KH2PO4, 10 MgSO4, 0.5 CaCl2, 26 NaHCO3 and 10 glucose saturated with 95 % O2 and 5 % CO2. The standard external solution used for recordings from isolated neurons contained (mM): 150 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose and 10 Hepes. The Ca2+-free external solution contained (mM): 150 NaCl, 5 KCl, 5 MgCl2, 2 EGTA, 10 glucose and 10 Hepes. These external solutions were adjusted to a pH of 7.4 with Tris-base. For recording sIPSCs, external solutions routinely contained 20 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 50 μM DL-2-amino-5-phosphonovaleric acid (AP5) to block glutamatergic currents. The ionic composition of the internal (patch-pipette) solution for the whole-cell patch recording was (mM): 145 caesium methanesulfonate, 5 TEA-Cl, 5 CsCl, 2 EGTA and 10 Hepes, pH adjusted to 7.2 with Tris-base. To reduce the postsynaptic GABAA response, all experiments were performed with the above ATP-free internal solution, which causes the postsynaptic GABAA response to run down and eventually disappear (Jang et al. 2002). In the slice experiments, the external bath solution consisted of (mM): 124 NaCl, 3 KCl, 1.5 KH2PO4, 26 NaHCO3, 2 CaCl2, 1 MgCl2 and 10 glucose saturated with 95 % O2 and 5 % CO2. The pH was about 7.5. To isolate glycinergic eIPSCs, this external solution routinely contained 20 μM CNQX + 50 μM AP5, 15 μM 2-[3-carboxypropyl]-3-amino-6-[4-methoxyphenyl]pyridazinium bromide (SR95531) and 3 μM (2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl] (phenylmethyl)phosphinic acid (CGP55845A) to block ionotropic glutamate, GABAA and GABAB receptors, respectively.
Drugs
The drugs and chemicals used in the present study were tetrodotoxin (TTX), strychnine, CNQX, AP5, EGTA, picrotoxin, glycine, SR95531 and ATP-Mg from Sigma (St Louis, MO, USA) and CGP55845A from Tocris (UK). For the isolated neuron experiments, solutions containing drugs were applied using the Y-tube system for rapid solution exchange (Akaike & Harata, 1994). For the slice experiments, drugs were applied by bath perfusion.
RESULTS
Most spinal neurons receive both GABAergic and glycinergic synaptic inputs (Todd & Sullivan, 1990; Schneider & Fyffe, 1992; Bohlhalter et al. 1994) and in most recordings a mixture of GABAergic and glycinergic sIPSCs were initially observed. To investigate whether exogenously applied glycine can modulate the release probability of glycine, we first isolated glycinergic sIPSCs. Rather than using the GABAA receptor antagonist bicuculline, which has been reported to affect glycine receptors at concentrations that fully block GABAA receptors (e.g. Shirasaki et al. 1991), we used ATP-free pipette solution, which induces a rapid rundown of the postsynaptic GABAA response (see also Jang et al. 2002). In all subsequent experiments, glycine was applied at least 50 min after membrane rupture in order to eliminate contamination by GABAergic sIPSCs.
Glycine acts presynaptically to facilitate spontaneous glycine release
Application of a low dose of glycine (3 μM) to single SDCN neurons induced a small, outward postsynaptic current at a VH of −30 mV that was associated with a very small change in input resistance, from 2.0 ± 0.2 GΩ in control conditions to 1.7 ± 0.3 GΩ in the presence of glycine (P = 0.08, n = 4; Fig. 1A and B). In just under half of the neurons tested (65/135), glycine also significantly increased sIPSC frequency. In 12 neurons in which this effect was fully analysed, glycine increased the mean sIPSC frequency to 244.7 ± 16.1 % of the control (P < 0.01) without affecting the mean sIPSC current amplitude (103.2 ± 9.1 % of control, P = 0.72; Fig. 1A and C). In the remaining neurons (70/135), glycine had no significant presynaptic effects. There was a trend for the glycine-induced increase in sIPSC frequency to subside somewhat during the 2 min application period and the residual sIPSC facilitation was rapidly reversed upon washout (Fig. 1Ab). The increase in mean frequency was also reflected in the cumulative frequency distributions, shifting the distribution of sIPSC inter-event intervals to the left without affecting the distribution of sIPSC amplitudes (data not shown). In cells in which glycine caused an increase in sIPSC frequency there was no concurrent effect on the kinetics of the sIPSCs, such as the rise time and the mean decay time, which was 13.8 ± 1.5 ms in control conditions and 13.3 ±1.7 ms in the presence of glycine (n = 10; Fig. 1C). To confirm that the observed sIPSCs were indeed glycinergic we investigated the effects of the potent glycine receptor antagonist strychnine. As shown in Fig. 1D, strychnine (0.5 μM) completely eliminated all sIPSCs, both in the absence and in the presence of applied glycine. Similar results were obtained in three other recordings. Taken together, these results suggest that glycine acts presynaptically to increase spontaneous glycine release.
Figure 1. Glycine facilitates glycinergic sIPSC frequency by presynaptic action.

Aa, typical trace of glycinergic sIPSCs before, during and after the application of 3 μM glycine. Insets represent the traces from each condition on an expanded time scale. b, the time course of sIPSC frequency during the application of glycine at a VH of −30 mV. The number of events in every 10 s period (•, presence of glycine; ○, absence of glycine) was summed and plotted. Each point is the mean from 12 neurons. B, current response to a 10 mV, 300 ms hyperpolarizing voltage step, with or without glycine. C, averaged sIPSCs in control conditions (n = 22 events) and in the presence of glycine (n = 28 events). The two averages are superimposed so as to demonstrate the lack of change in sIPSC kinetics. D, typical traces of glycinergic sIPSCs observed before, during and after the application of 3 μM glycine in a standard external solution (a) and in the presence of 0.5 μM strychnine (b). Note that strychnine completely eliminated all sIPSCs and application of glycine did not evoke any response.
Figure 2 shows the concentration-response relationship for both the glycine-induced increase in sIPSC frequency and the glycine-induced postsynaptic current (IGly). Glycine (3 μM) significantly facilitated the sIPSC frequency to 240.5 ± 18.5 % (n = 8, P < 0.01, Fig. 2A and Ba) without causing any marked postsynaptic current (Fig. 2A and C). A higher concentration of glycine (10 μM) evoked a small postsynaptic current but did not induce a further increase in sIPSC frequency (238.5 ± 34.8 % of the control, n = 8, P < 0.05). Glycine at even higher concentrations (30 μM and above) induced a large postsynaptic current, resulting in the occlusion of the glycinergic sIPSCs (Fig. 2A).
Figure 2. Concentration dependence of the pre- and postsynaptic actions of glycine.

A, typical traces of sIPSCs and sustained postsynaptic currents in the presence of various concentrations of glycine. The current response to 30 μM glycine (bottom trace) has been truncated. B, mean concentration–response relationship between glycine and sIPSC frequency (a) and sIPSC amplitude (b). Each column is the mean and s.e.m. of data from 8 neurons. Note that concentrations of glycine above 3 μM did not cause a further increase in sIPSC frequency. *P < 0.05, **P < 0.01. C, concentration—response relationship for postsynaptic currents induced by exogenous application of glycine. a, typical current responses induced by various concentrations of glycine. b, concentration–response curves in control conditions (•) and with co-application of glycine and 30 μM picrotoxin (○). All data were normalized to the current amplitude in response to 100 μM glycine. The smooth curves indicate the fit of the data to the Hill equation. The EC50 values were 57 μM (•) and 61 μM (○).
The abolition of sIPSCs in the presence of strychnine does not prove that applied glycine is activating presynaptic glycine receptors. Therefore, we investigated the effects of a low dose of picrotoxin on the glycine-induced increase in sIPSC frequency. Picrotoxin (30 μM), by itself, had no significant effect on either mean sIPSC frequency (112.5 ± 11.5 % of the control, n = 7) or mean sIPSC amplitude (93.8 ± 10.1 % of the control, n = 7, Fig. 3Ba). Furthermore, this dose of picrotoxin had no effect on the concentration-response relationship for the exogenous glycine-induced postsynaptic current (Fig. 2Cb). This insensitivity to low doses of picrotoxin confirms that the postsynaptic glycine receptors are heteromeric pentamers composed of α and β subunits (Pribilla et al. 1992; Handford et al. 1996). In contrast, however, picrotoxin (30 μM) significantly attenuated the glycine-induced increase in sIPSC frequency. In seven cells, the initial application of 3 μM glycine increased sIPSC frequency to 277.1 ± 22.4 % of the control frequency. In the same seven cells, subsequent co-application of glycine and picrotoxin facilitated sIPSC frequency to only 141.5 ± 11.9 % of the original control frequency (n = 7, Fig. 3A and Bb). This was significantly less facilitation than observed without picrotoxin (P < 0.01, Fig. 3Bb). It is unlikely that this reduction in the extent of glycine-induced facilitation was due to a run down of the facilitatory effects, as following picrotoxin washout, application of glycine again induced marked facilitation of sIPSC frequency (data not shown). Furthermore, in separate experiments, repetitive brief applications of glycine induced similar degrees of sIPSC facilitation. It was also noted that picrotoxin did not alter the sIPSC rise and decay time constants (data not shown).
Figure 3. Effects of picrotoxin on glycine-induced sIPSC frequency facilitation.

A, typical traces of glycinergic sIPSCs before, during and after the application of 3 μM glycine in standard external solution (a) and in the presence of 30 μM picrotoxin (b). The 2 traces were obtained from the same neuron. Ba, effect of 30 μM picrotoxin on glycinergic sIPSC frequency and amplitude. b, effect of picrotoxin on the facilitation of sIPSCs by glycine. In both a and b, data have been normalized relative to the original control sIPSC frequency and amplitude. Each column represents the mean and s.e.m. of data from 6 neurons. **P < 0.01.
Cellular mechanism underlying the glycine-induced increase in sIPSC frequency
We have recently described how the activation of presynaptic GABAA receptors on glycinergic nerve terminals projecting to isolated SDCN neurons facilitates spontaneous glycine release secondary to a presynaptic depolarization and external Ca2+ influx (Jang et al. 2002). The depolarization occurs as a result of a high intraterminal Cl− concentration, which is maintained by a bumetanide-sensitive transporter (Jang et al. 2002). Thus, we tested whether a similar glycine receptor-mediated presynaptic depolarization is responsible for the increase in sIPSC frequency observed in the present study. Firstly, we tested whether the glycine-induced increase in sIPSC frequency requires Ca2+ influx from the extracellular sites. In Ca2+-free external solution, sIPSC frequency was greatly reduced, to 46.1 ± 8.8 % of the control frequency (n = 7, P < 0.01) while mean sIPSC amplitude was unaffected (87.5 ± 6.5 % of control, n = 7, P = 0.15; Fig. 4A and B). Hence, under our recording conditions, Ca2+ influx from the extracellular sites plays an important role in spontaneous glycine release under control conditions. In the absence of external Ca2+, the facilitatory effect of glycine on sIPSC frequency was completely abolished, with sIPSC frequency in the presence of glycine being 103.2 ± 3.6 % of the Ca2+-free control condition (n = 7, P = 0.41, Fig. 4Ab and B). The application of 100 μM Cd2+, a non-selective blocker of voltage-dependent Ca2+ channels (VDCCs), also decreased the basal sIPSC frequency, to 47.5 ± 5.6 % of the control frequency (n = 6, P < 0.01), without affecting the mean sIPSC current amplitude (86.6 ± 6.9 %, n = 6, P = 0.28; Fig. 4A and C). In the continued presence of Cd2+, glycine application failed to affect sIPSC frequency, which was 96.2 ± 7.9 % of the Cd2+ condition (n = 6, P = 0.45; Fig. 4Ac and C). These results suggest that the increase in sIPSC frequency by glycine is due to extracellular Ca2+ influx via VDCCs.
Figure 4. Glycine-induced sIPSC frequency facilitation is mediated by Ca2+ influx through VDCCs.
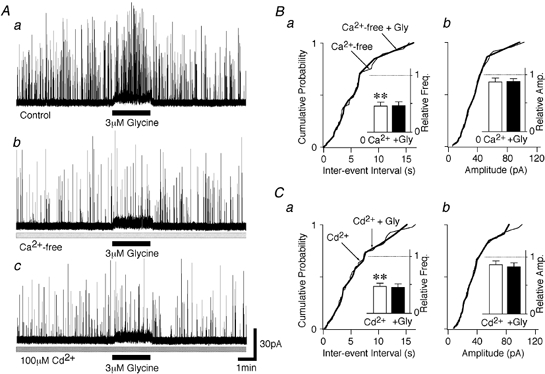
A, typical traces of glycinergic sIPSCs before, during and after the application of 3 μM glycine in standard external solution (a), in Ca2+-free external solution (b) and in external solution containing 100 μM Cd2+ (c). All traces were obtained from the same neuron. B, cumulative probability plots for sIPSC inter-event interval (a; P = 0.67, Ca2+ free vs. Ca2+ free + glycine) and sIPSC amplitude (b; P = 0.40) obtained from the trace shown in Ab (102 events for the Ca2+-free condition and 21 events for the Ca2+-free + glycine condition). The insets show the effects of the various manipulations on the mean sIPSC frequency and amplitude. Each column represents mean and s.e.m. data from 7 neurons, with the values all being normalized to the initial control values. **P < 0.01. C, cumulative probability plots for sIPSC inter-event interval (a; P = 0.23, Cd2+vs. Cd2+ + glycine) and sIPSC amplitude (b; P = 0.45) obtained from the trace shown in Ac (225 events for the Cd2+ condition and 29 events for the Cd2+ + glycine condition). As in B, insets show the mean and s.e.m., normalized to the initial control values (n = 6). ** P < 0.01.
Activation of presynaptic glycine receptors may depolarize the terminals sufficiently to directly activate VDCCs, or it may elicit action potentials and thereby indirectly activate VDCCs. In the presence of 300 nM TTX, which blocks voltage-dependent Na+ channels, glycine failed to increase sIPSC frequency, which was 115.6 ± 10.5 % of the TTX-containing control condition (n = 11, P = 0.21; Fig. 5A and B). These results suggest that the activation of glycine receptors depolarizes the glycinergic presynaptic nerve terminals and elicits TTX-sensitive action potentials, which subsequently result in the activation of VDCCs and facilitation of release. TTX, by itself, also significantly decreased sIPSC frequency to 64.9 ± 7.0 % of the control (n = 11, P < 0.01; Fig. 5B), without affecting mean sIPSC amplitude (95.1 ± 3.2 % of the control, P = 0.15), indicating that TTX-sensitive Na+ channels also contribute to glycine release under control conditions.
Figure 5. Glycine-induced sIPSC frequency facilitation requires TTX-sensitive Na+ channels.

A, typical traces of glycinergic sIPSCs before, during and after the application of 3 μM glycine in standard external solution (a) and in the presence of 300 nM TTX (b). The 2 traces were obtained from the same neuron. B, cumulative probability plots for sIPSC inter-event interval (a; P = 0.29, TTX vs. TTX + glycine) and current amplitude (b; P = 0.76) for the data shown in Ab (183 events for the TTX condition and 23 events for the TTX + glycine condition). The insets show mean and s.e.m. for sIPSC frequency and amplitude from 11 neurons, with data normalized to the original control conditions. **P < 0.01.
Effects of glycine and picrotoxin on electrically evoked IPSCs in spinal cord slices
We next examined whether the activation of presynaptic glycine receptors can modulate electrically evoked glycine release under more physiological conditions. When recording from SDCN neurons in these slice preparations, however, GABAergic IPSCs did not completely run down, even during prolonged recordings using ATP-free pipette solution (data not shown). This presumably reflects poor cell dialysis in these more intact neurons. Furthermore, since presynaptic GABAA and GABAB receptors are known to modulate glycinergic transmission at these synapses (Jang et al. 2002), glycinergic eIPSCs were recorded in the presence of 15 μM SR95531 and 3 μM CGP55845A, to block GABAA and GABAB receptors, respectively. Bath solutions also contained 20 μM CNQX and 50 μM AP5 to block glutamate receptors. In response to pairs of stimulus pulses, significant paired-pulse facilitation was observed, with the amplitude of the second eIPSC being increased to 137.2 ± 7.8 % of the first eIPSC amplitude (n = 5, P < 0.01; Fig. 6A and Bb). Bath application of 10 μM glycine significantly increased the first eIPSC amplitude, to 145.7 ± 11.5 % of the control amplitude (n = 5, P < 0.05, Fig. 6A and Ba), and abolished paired-pulse facilitation. The amplitude of the second eIPSC in the presence of glycine (10 μM) was now 98.4 ± 7.5 % of the amplitude of the first eIPSC (n = 5, P < 0.01; Fig. 6Bb). In the presence of 10 μM glycine, sIPSC frequency was again increased (data not shown). These results provide further evidence that glycine acts presynaptically to facilitate glycine release at these synapses.
Figure 6. Effect of glycine on IPSCs evoked by paired pulses.

A, typical traces of glycinergic eIPSCs obtained in response to paired-pulse stimulation (inter-stimulus interval = 50 ms), in control conditions (left trace) and again in the presence of 10 μM glycine (middle trace). Each trace represents the average of 20 trials. The right trace shows the superimposition of these two recordings. Ba, bar graph showing the averaged glycine-induced increase in the first eIPSC (eIPSC1) amplitude (n = 5). Error bars represent s.e.m.b, effects of glycine on the paired-pulse facilitation ratio (eIPSC2/eIPSC1). Connections between open circles represent the glycine-induced change in the paired-pulse facilitation ratio in each individual experiment, whereas filled circles represent the mean ±s.e.m. from 5 neurons. *P < 0.05.
Finally, to elucidate whether presynaptic glycine receptors may be activated by synaptically released glycine, and hence act like traditional neurotransmitter autoreceptors, we investigated the eIPSC response to brief high frequency stimulus trains, delivered at a variety of inter-stimulus intervals. During the higher frequency (33.3 and 100 Hz) trains, there was profound frequency facilitation of the glycinergic eIPSCs. In about half of the neurons examined, this frequency facilitation was markedly reduced in the presence of picrotoxin (30 μM). Figure 7A and B (left and middle panels) shows the averaged frequency facilitation at 33.3 and 100 Hz in neurons that were responsive to picrotoxin (defined as a 20 % or greater change in the second IPSC amplitude in the presence of picrotoxin; 5 of 9 neurons tested at 33.3 Hz and 7 of 12 neurons tested at 100 Hz). Picrotoxin (30 μM) had no effect on the amplitude of the first IPSC in the train, indicating again its lack of action on postsynaptic glycine receptors and that basal, circulating levels of glycine in the slice are not sufficient to activate these presynaptic receptors. At a train frequency of 10 Hz, there was no apparent frequency facilitation and picrotoxin did not affect the amplitude of eIPSCs evoked at this frequency (Fig. 7A and B, right panel). These results clearly indicate that synaptically released glycine can activate presynaptic autoreceptors during moderate and high frequency trains and cause a potentiation of glycinergic transmission in the spinal cord.
Figure 7. Picrotoxin reduces frequency-dependent facilitation of glycinergic eIPSCs.

A, typical averaged traces of eIPSCs in the absence and presence of 30 μM picrotoxin during a brief stimulation train presented at a frequency of 100 Hz (left), 33.3 Hz (middle) and 10 Hz (right). All traces represent the average of 20–40 trials, which were then superimposed in each experimental condition. B, plots of the normalized mean peak amplitudes of each eIPSC during brief stimulus trains presented at 100 Hz (left, n = 7 neurons), 33.3 Hz (middle, 5 neurons) and 10 Hz (right, 4 neurons). Each point represents the mean and s.e.m. ○, data recorded in control conditions; •, data recorded in the presence of picrotoxin. *P < 0.05,**P < 0.01.
DISCUSSION
Ionotropic glycine receptors are present on glycinergic nerve terminals
The present results demonstrate the existence of functional presynaptic glycine ‘autoreceptors’ on the terminals of inhibitory, glycinergic interneurons in the deep lamina of the dorsal horn. Application of glycine increased spontaneous glycine release by a mechanism that was sensitive to low doses of picrotoxin. This indicates that glycine was acting through picrotoxin-sensitive glycine receptors, rather than eliciting effects secondary to currents arising from presynaptic glycine transport. Furthermore, since all experiments were carried out in the presence of glutamate receptor antagonists, the glycine effect was not due to binding to the glycine site on the NMDA receptor. Some previous reports have suggested the existence of strychnine-sensitive glycine receptors on presynaptic terminals in various non-spinal cord preparations. Glycine was shown to inhibit K+-elicited neurotransmitter release in both cultured cerebellar granule cells (Wahl et al. 1994) and purified neurohypophysial nerve terminals (Hussy et al. 2001). Furthermore, Turecek & Trussell (2001, 2002) have recently provided direct evidence for presynaptic ionotropic glycine receptors on the large, glutamatergic, calyx of Held synaptic terminals in the rat brainstem. This study provides direct evidence that presynaptic glycine receptors can exist in more typical central synaptic terminals, and provides another example of presynaptic autoreceptors for classical, fast neurotransmitters.
At low concentrations (≤ 10 μM), glycine significantly increased sIPSC frequency without eliciting any observable postsynaptic response, with an EC50 value of about 60 μM. The high sensitivity of the presynaptic response to glycine may be related to the presumably high input resistance of the tiny, dissociated presynaptic boutons. The opening of a single presynaptic glycine receptor may be sufficient to significantly depolarize the terminals and elicit an action potential, as has been observed in small, high input resistance cells (e.g. rat olfactory receptor neurons; Lynch & Barry, 1989). Alternatively, the presynaptic glycine receptors might have a higher sensitivity to glycine. Expression studies have suggested that recombinant α-homomeric glycine receptors may be slightly more sensitive to glycine than are αβ-heteromeric glycine receptors (Bormann et al. 1993; Handford et al. 1996) but this has not been consistently observed (Pribilla et al. 1992; Pistis et al. 1997). Picrotoxin, however, has been widely used to distinguish between α-homomeric and αβ-heteromeric glycine receptors as it shows a 20- to 400-fold increased sensitivity for block of homomeric glycine receptors (Pribilla et al. 1992; Handford et al. 1996; Pistis et al. 1997) and a picrotoxin concentration of about 30 μM appears optimal for this discrimination. At this concentration, the facilitation of sIPSC frequency induced by low glycine concentrations was almost abolished, while the somatic responses to applied glycine and the sIPSC amplitude were unaffected. This suggests that at least some of the presynaptic glycine receptors may have been α-homomers, although this should be further evaluated with more extensive pharmacological and immunohistochemical analysis. This conclusion is also consistent with a recent study suggesting that presynaptic glycine receptors in calyx terminals in the rat brainstem are α-homomers, based on their relatively large single channel conductances (about 90 pS; Turecek & Trussell, 2002).
Glycine depolarizes glycinergic nerve terminals
Reichling et al. (1994) have reported that in cultured rat dorsal horn neurons, glycine increases the intracellular Ca2+ concentration through the activation of VDCCs secondary to membrane depolarization. The glycine receptor-mediated Cl− conductance is excitatory and depolarizing due to an outwardly directed Cl− driving force. A similar, outwardly directed Cl− driving force is present in many presynaptic terminals, resulting in, for example, GABAA receptor-mediated terminal depolarization. We suggest that presynaptic glycine receptor activation, observed in the present study, also causes a presynaptic depolarization based on the following evidence: (1) the glycine-induced increase in sIPSC frequency was completely occluded in Ca2+-free external solution, or by the addition of Cd2+, suggesting the activation of VDCCs; and (2) the sIPSC facilitation was completely attenuated in the presence of TTX, implicating the activation of voltage-dependent Na+ channels. It was somewhat surprising that TTX totally abolished the effects on spontaneous release as presynaptic terminal depolarization, by itself, may be expected to increase basal Ca2+ levels and hence spontaneous glycine release. It may be that any such changes in Ca2+ were small and not sufficient to cause marked increases in glycine release, or that the presynaptic glycine receptors are located relatively far from the VDCCs in the boutons (i.e. a pre-terminal distribution) so that their activation cannot effectively and directly activate these Ca2+ channels.
In summary, presynaptic glycine receptor activation causes Cl− efflux down its electrochemical gradient, membrane depolarization and the subsequent activation of TTX-sensitive Na+ channels, causing action potential initiation and subsequent Ca2+ influx through VDCCs. We have recently reported a similar sequence of events following activation of presynaptic GABAA receptors on these glycinergic terminals, with the depolarizing Cl− gradient being maintained by a bumetanide-sensitive Na+-K+-Cl− cotransporter (Jang et al. 2002). Furthermore, in glutamatergic terminals in the rat calyx of Held, Turecek & Trussell (2001) directly recorded a presynaptic depolarization following glycine receptor activation (with intraterminal Cl− being left intact) and demonstrated that this led to an increase in intraterminal Ca2+ that was responsible for the facilitation of spontaneous and evoked glutamate release.
Physiological implications
In rat brainstem calyx synapses, synaptically released glycine seems to spill over to adjacent glutamatergic synapses and facilitate electrically stimulated glutamate release (Turecek & Trussell, 2001). By analogy, we found that synaptically released glycine acts on glycinergic terminals and facilitates action potential-dependent glycine release. The effects of picrotoxin on IPSC frequency facilitation were evident by the second IPSC within a train, indicating that synaptically released glycine activates presynaptic glycine receptors after even a single stimulus. A similar pattern of effects has been reported for the activation of presynaptic kainate receptors by synaptically released glutamate, i.e. their activation facilitates glutamate release from mossy fibre synapses, and LY382884, a selective kainate receptor antagonist, blocks high frequency-dependent facilitation of glutamatergic transmission (Lauri et al. 2001). Presynaptic GABAA receptors are directly activated by synaptically released GABA from axo-axonic synapses to cause primary afferent terminal depolarization and inhibition of action potential-evoked glutamate release (Levy, 1977; Rudomin & Schmidt, 1999). This effect is likely to be mediated by inactivating axonal Na+ channels and/or by shunting the presynaptic membrane (Segev, 1990; Graham & Redman, 1994; Cattaert & El Manira, 1999). Many axo-axonic synapses in the spinal cord contain glycine, as well as GABA, in their presynaptic terminals (Todd et al. 1995; Maxwell et al. 1997). Presynaptic glycine receptors, therefore, may be activated by synaptically released glycine from such axo-axonic synapses and/or by spillover of glycine from adjacent synapses.
It is rather interesting that the activation of presynaptic glycine receptors facilitated electrically stimulated glycine release onto SDCN neurons, while presynaptic GABAA receptors inhibit electrically evoked glycine release from the same group of glycinergic terminals (Jang et al. 2002). One explanation for this discrepancy is that the two receptor systems produce different presynaptic depolarizations. That is, activation of presynaptic glycine receptors causes a smaller presynaptic depolarization, sufficient to cause some increase in intraterminal [Ca2+] but not sufficient to significantly inactivate voltage-dependent Ca2+ and/or Na+ channels. In contrast, activation of presynaptic GABAA receptors leads to a larger depolarization, sufficient to inactivate these voltage-dependent channels. A larger depolarization could arise due to a higher density of GABAA receptors relative to glycine receptors. It should also be pointed out that our previous study on presynaptic GABAA receptors on glycinergic terminals synapsing onto SDCN neurons (Jang et al. 2002) used slightly different extracellular anion concentrations (e.g. 161 mM Cl−vs. 130 mM Cl− in the present study), although the effect of this difference in Cl− concentration would actually have reduced any differences in the extent of the GABA- and glycine-induced presynaptic depolarizations. Alternatively, the GABAA and glycine receptors may have different distributions; either differently distributed on the same terminals (e.g. bouton vs. axon) or present on different terminals that may, for example, have different intraterminal Cl− concentrations.
The SDCN is known to be intimately involved in nociceptive transmission (Honda, 1985; Ding et al. 1994; Lu et al. 1995; Vizzard et al. 1995) and receives abundant afferent inputs from both visceral and somatic organs. The convergence of visceral and somatic inputs onto SDCN neurons has been demonstrated using both electrophysiological (Honda, 1985) and anatomical techniques (Lu et al. 1995). The SDCN also integrates regulatory influences from brainstem descending pathways (Jones & Light, 1990). Hence, glycine autoreceptor-mediated presynaptic modulation of glycinergic transmission may play an important role in the integration of local inhibitory inputs within the spinal cord.
Acknowledgments
We thank Dr M. Brodwick for his valuable comments on this manuscript. This work was supported by The Japan Health Sciences Foundation (no. 21279, Research on Brain Science to N.A.).
H.-J. Jeong and I.-S. Jang contributed equally to this work.
REFERENCES
- Akaike N, Harata N. Nystatin perforated patch recording and its application to analysis of intracellular mechanism. Jpn J Physiol. 1994;44:433–473. doi: 10.2170/jjphysiol.44.433. [DOI] [PubMed] [Google Scholar]
- Akaike N, Moorhouse AJ. Techniques: Applications of the nerve-bouton preparation in neuropharmacology. Trends Pharmacol Sci. 2003;24:44–47. doi: 10.1016/s0165-6147(02)00010-x. [DOI] [PubMed] [Google Scholar]
- Bohlhalter S, Mohler H, Fritschy JM. Inhibitory neurotransmission in rat spinal cord: co-localization of glycine- and GABAA-receptors at GABAergic synaptic contacts demonstrated by triple immunofluorescence staining. Brain Res. 1994;642:59–69. doi: 10.1016/0006-8993(94)90905-9. [DOI] [PubMed] [Google Scholar]
- Bormann J, Rundstrom N, Betz H, Langosch D. Residues within transmembrane segment M2 determine chloride conductance of glycine receptor homo- and hetero-oligomers. EMBO J. 1993;12:3729–3737. doi: 10.1002/j.1460-2075.1993.tb06050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaert D, El Manira A. Shunting versus inactivation: Analysis of presynaptic inhibitory mechanisms in primary afferents of the crayfish. J Neurosci. 1999;19:6079–6089. doi: 10.1523/JNEUROSCI.19-14-06079.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin I, Rostaing P, Augustin A, Triller A. Localisation of components of glycinergic synapses during rat spinal cord development. J Comp Neurol. 1998;398:359–372. [PubMed] [Google Scholar]
- Ding YQ, Qin BZ, Li JS, Mizuno N. Induction of c-fos-like protein in spinoparabrachial tract-neurons locating within the sacral parasympathetic nucleus in the rat. Brain Res. 1994;659:283–286. doi: 10.1016/0006-8993(94)90894-x. [DOI] [PubMed] [Google Scholar]
- Graham B, Redman S. A simulation of action potentials in synaptic boutons during presynaptic inhibition. J Neurophysiol. 1994;71:538–549. doi: 10.1152/jn.1994.71.2.538. [DOI] [PubMed] [Google Scholar]
- Handford CA, Lynch JW, Baker E, Webb GC, Ford JH, Sutherland GR, Schofield PR. The human glycine receptor beta subunit: primary structure, functional characterisation and chromosomal localisation of the human and murine genes. Mol Brain Res. 1996;35:211–219. [PubMed] [Google Scholar]
- Heilbronn E, Haggblad J, Kubat B. Antibodies to the nicotinic acetylcholine receptor, obtained from serum of myasthenic patients, may decrease acetylcholine release from rat hippocampal nerve endings in vitro. Ann NY Acad Sci. 1981;377:198–207. doi: 10.1111/j.1749-6632.1981.tb33733.x. [DOI] [PubMed] [Google Scholar]
- Honda CN. Visceral and somatic afferent convergence onto neurons near the central canal in the sacral spinal cord of the cat. J Neurophysiol. 1985;53:1059–1078. doi: 10.1152/jn.1985.53.4.1059. [DOI] [PubMed] [Google Scholar]
- Hussy N, Bres V, Rochette M, Duvoid A, Alonso G, Dayanithi G, Moos FC. Osmoregulation of vasopressin secretion via activation of neurohypophysial nerve terminals glycine receptors by glial taurine. J Neurosci. 2001;21:7110–7116. doi: 10.1523/JNEUROSCI.21-18-07110.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang IS, Jeong HJ, Akaike N. Contribution of the Na-K-Cl cotransporter on GABAA receptor-mediated presynaptic depolarization in excitatory nerve terminals. J Neurosci. 2001;21:5962–5972. doi: 10.1523/JNEUROSCI.21-16-05962.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang IS, Jeong HJ, Katsurabayashi S, Akaike N. Functional roles of presynaptic GABAA receptors on glycinergic nerve terminals in the rat spinal cord. J Physiol. 2002;541:423–434. doi: 10.1113/jphysiol.2001.016535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SL, Light AR. Termination patterns of serotoninergic medullary raphe spinal fibers in the rat lumbar spinal cord: an anterograde immunohistochemical study. J Comp Neurol. 1990;297:267–282. doi: 10.1002/cne.902970209. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Ozawa S. Kainate receptor-mediated presynaptic inhibition at the mouse hippocampal mossy fibre synapse. J Physiol. 2000;523:653–665. doi: 10.1111/j.1469-7793.2000.t01-1-00653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapchak PA, Araujo DM, Quirion R, Collier B. Presynaptic cholinergic mechanisms in the rat cerebellum: evidence for nicotinic, but not muscarinic autoreceptors. J Neurochem. 1989;53:1843–1851. doi: 10.1111/j.1471-4159.1989.tb09251.x. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Delany C, Clarke VRJ, Bortolotto ZA, Ornstein PL, Isaac JTR, Collingridge GL. Synaptic activation of a presynaptic kainate receptor facilitates AMPA receptor-mediated synaptic transmission at hippocampal mossy fibre synapses. Neuropharmacology. 2001;41:907–915. doi: 10.1016/s0028-3908(01)00152-6. [DOI] [PubMed] [Google Scholar]
- Levy RA. The role of GABA in primary afferent depolarization. Prog Neurobiol. 1977;9:211–267. doi: 10.1016/0301-0082(77)90002-8. [DOI] [PubMed] [Google Scholar]
- Lu Y, Jin SX, Xu TL, Qin BZ, Li JS, Ding YQ, Shigemoto R, Mizuno N. Expression of c-fos protein in substance P receptor-like immunoreactive neurons in response to noxious stimuli on the urinary bladder: an observation in the lumbosacral cord segments of the rat. Neurosci Lett. 1995;198:139–142. doi: 10.1016/0304-3940(95)11991-5. [DOI] [PubMed] [Google Scholar]
- Lukasiewicz PD, Maple BR, Werblin FS. A novel GABA receptor on bipolar cell terminals in the tiger salamander retina. J Neurosci. 1994;14:1202–1212. doi: 10.1523/JNEUROSCI.14-03-01202.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch JW, Barry PH. Action potentials initiated by single channels opening in a small neuron (rat olfactory receptor) Biophys J. 1989;55:755–768. doi: 10.1016/S0006-3495(89)82874-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermott AB, Role LW, Siegelbaum SA. Presynaptic ionotropic receptors and the control of transmitter release. Ann Rev Neurosci. 1999;22:443–485. doi: 10.1146/annurev.neuro.22.1.443. [DOI] [PubMed] [Google Scholar]
- Matthews G, Ayoub GS, Heidelberger R. Presynaptic inhibition by GABA is mediated via two distinct GABA receptors with novel pharmacology. J Neurosci. 1994;14:1079–1090. doi: 10.1523/JNEUROSCI.14-03-01079.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell DJ, Kerr R, Jankowska E, Riddell JS. Synaptic connections of dorsal horn group II spinal interneurons: Synapses formed with the interneurons and by their axon collaterals. J Comp Neurol. 1997;380:51–69. [PubMed] [Google Scholar]
- Mitchell K, Spike RC, Todd AJ. An immunocytochemical study of glycine receptor and GABA in laminae I-III of rat spinal dorsal horn. J Neurosci. 1993;13:2371–2381. doi: 10.1523/JNEUROSCI.13-06-02371.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistis M, Belelli D, Peters JA, Lambert JJ. The interaction of general anaesthetics with recombinant GABAA and glycine receptors expressed in Xenopus laevis oocytes: a comparative study. Br J Pharmacol. 1997;122:1707–1719. doi: 10.1038/sj.bjp.0701563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribilla I, Takagi T, Langosch D, Bormann J, Betz H. The atypical M2 segment of the beta subunit confers picrotoxinin resistance to inhibitory glycine receptor channels. EMBO J. 1992;11:4305–4311. doi: 10.1002/j.1460-2075.1992.tb05529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichling DB, Kyrozis AK, Wang J, MacDermott AB. Mechanisms of GABA and glycine depolarization-induced calcium transients in rat dorsal horn neurons. J Physiol. 1994;476:411–421. doi: 10.1113/jphysiol.1994.sp020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee JS, Ishibashi H, Akaike N. Calcium channels in the GABAergic presynaptic nerve terminals projecting to Meynert neurons of the rat. J Neurochem. 1999;72:800–807. doi: 10.1046/j.1471-4159.1999.0720800.x. [DOI] [PubMed] [Google Scholar]
- Rowell PP, Winkler DL. Nicotinic stimulation of [3H]acetylcholine release from mouse cerebral cortical synaptosomes. J Neurochem. 1984;49:1593–1598. doi: 10.1111/j.1471-4159.1984.tb06083.x. [DOI] [PubMed] [Google Scholar]
- Rudomin P, Schmidt RF. Presynaptic inhibition in the vertebrate spinal cord revisited. Exp Brain Res. 1999;129:1–37. doi: 10.1007/s002210050933. [DOI] [PubMed] [Google Scholar]
- Schmitz D, Frerking M, Nicoll RA. Synaptic activation of presynaptic kainite receptors on hippocampal mossy fiber synapses. Neuron. 2000;27:327–338. doi: 10.1016/s0896-6273(00)00040-4. [DOI] [PubMed] [Google Scholar]
- Schneider SP, Fyffe RE. Involvement of GABA and glycine in recurrent inhibition of spinal motoneurons. J Neurophysiol. 1992;68:397–406. doi: 10.1152/jn.1992.68.2.397. [DOI] [PubMed] [Google Scholar]
- Segev I. Computer study of presynaptic inhibition controlling the spread of action potentials into axon terminals. J Neurophysiol. 1990;63:987–998. doi: 10.1152/jn.1990.63.5.987. [DOI] [PubMed] [Google Scholar]
- Shirasaki T, Klee MR, Nakaye T, Akaike N. Differential blockade of bicuculline and strychnine on GABA- and glycine-induced responses in dissociated rat hippocampal pyramidal cells. Brain Res. 1991;561:77–83. doi: 10.1016/0006-8993(91)90751-g. [DOI] [PubMed] [Google Scholar]
- Todd AJ, Spike RC, Chong D, Neilson M. The relationship between glycine and gephyrin in synapses of the rat spinal cord. Eur J Neurosci. 1995;7:1–11. doi: 10.1111/j.1460-9568.1995.tb01014.x. [DOI] [PubMed] [Google Scholar]
- Todd AJ, Sullivan AC. Light microscope study of the coexistence of GABA-like and glycine-like immunoreactivities in the spinal cord of the rat. J Comp Neurol. 1990;296:496–505. doi: 10.1002/cne.902960312. [DOI] [PubMed] [Google Scholar]
- Turecek R, Trussell LO. Presynaptic glycine receptors enhance transmitter release at a mammalian central synapse. Nature. 2001;411:587–590. doi: 10.1038/35079084. [DOI] [PubMed] [Google Scholar]
- Turecek R, Trussell LO. Reciprocal developmental regulation of presynaptic ionotropic receptors. Proc Natl Acad Sci U S A. 2002;99:13884–13889. doi: 10.1073/pnas.212419699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizzard MA, Erdman SL, De Groat WC. Increased expression of neuronal nitric oxide synthase (NOS) in visceral neurons after nerve injury. J Neurosci. 1995;15:4033–4045. doi: 10.1523/JNEUROSCI.15-05-04033.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl P, Elster L, Schousboe A. Identification and function of glycine receptors in cultured cerebellar granule cells. J Neurochem. 1994;62:2457–2463. doi: 10.1046/j.1471-4159.1994.62062457.x. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]