

Abstract
Nitric oxide (NO) can function as either a pro-inflammatory or anti-inflammatory molecule, depending upon its concentration and the microenvironment in which it is produced. We tested whether muscle-derived NO affects muscle inflammation and membrane lysis that occur in modified muscle use. Transgenic mice with muscle-specific over-expression of neuronal NO synthase (nNOS) were generated in which transgene expression was driven by the human skeletal muscle actin promoter. Transgenic mice and non-transgenic littermates were subjected to hindlimb muscle unloading followed by reloading, which causes muscle inflammation and membrane lysis. NOS expression decreased in transgenic and non-transgenic mice during muscle unloading. Muscle inflammation was assessed by immunohistochemistry after 24 h of muscle reloading following 10 days of unloading. Soleus muscles of non-transgenic mice showed significant increases in the concentrations of neutrophils (4.8-fold) and macrophages (11.3-fold) during reloading, compared to mice that experienced unloading only. Muscles of transgenic mice showed 51 % fewer neutrophils in reloaded muscles than those of non-transgenic mice, but macrophage concentrations did not differ from non-transgenic mice. Muscle membrane damage was determined by measuring influx of an extracellular marker dye. Significantly more membrane damage occurred in muscles of non-transgenic mice experiencing reloading than in ambulatory controls. However, membrane damage in the reloaded muscles of transgenic mice did not differ from that in ambulatory mice. In vitro cytotoxicity assays confirmed that mouse neutrophils lyse muscle cell membranes, and showed that inhibition of NOS in muscle and neutrophil co-cultures significantly increased neutrophil-mediated lysis of muscle cells. Together, these data show that muscle-derived NO can function as an anti-inflammatory molecule in muscle that experiences modified loading, and that NO can prevent neutrophil-mediated damage of muscle cell membranes in vivo and in vitro.
Inflammatory cells have the capacity to cause muscle membrane lysis and muscle cell death both in vivo and in vitro. Muscle injury by inflammatory cells has been examined most thoroughly in experimental models of muscle ischaemia followed by reperfusion, in which neutrophils have been clearly demonstrated to promote muscle fibre damage during the reperfusion phase (Korthuis et al. 1988; Carden & Korthuis, 1989; Yokota et al. 1989; Walden et al. 1990). Much of the muscle damage in this injury model requires the production of superoxide that may directly or indirectly promote damage to the target muscle cell (Korthuis et al. 1985; Yokota et al. 1989; Kawasaki et al. 1993). Rat neutrophils can lyse muscle membranes in vitro by superoxide-dependent mechanisms (Nguyen & Tidball, 2003), which suggests that neutrophil-derived superoxide may contribute to muscle injury in vivo.
Recent findings have shown that muscle cells can influence the level of cytotoxicity of inflammatory cells. NOS inhibition in muscle and neutrophil co-cultures caused an increase in superoxide concentration in the co-cultures and an increase in muscle cell lysis that could be prevented by superoxide dismutase (Nguyen & Tidball, 2003). These results suggest that the release of nitric oxide (NO) can protect muscle from superoxide-mediated damage caused by neutrophils in vitro, although it was not shown definitively that muscle cells were the source of the cytoprotective NO in those in vitro assays. However, several observations concerning the pathophysiology of dystrophin-deficient muscle have indicated that muscle-derived NO can protect against muscle membrane lysis by inflammatory cells in vivo (Wehling et al. 2001). Firstly, dystrophin-deficiency results in loss of NOS from skeletal muscle as a secondary consequence of dystrophin-deficiency (Brenman et al. 1995; Chang et al. 1996). In addition, dystrophic muscle experiences extensive inflammation (reviewed by Spencer & Tidball, 2001), and the depletion of macrophages from dystrophic muscle greatly reduces muscle pathology and the occurrence of muscle membrane lesions (Wehling et al. 2001). Finally, the transgenic expression of NOS in dystrophin-deficient muscle can normalize levels of NO production in muscle, and results in a large reduction in muscle inflammation and muscle membrane damage (Wehling et al. 2001). Together, these findings indicate that the loss of normal NO generating capacity by dystrophic muscle results in an increase in muscle damage that is caused by inflammatory cells.
Modified muscle loading also produces muscle inflammation and membrane damage, although there are no definitive findings that show whether inflammatory cells contribute to membrane damage during modified muscle use. Rodent hindlimb muscle unloading followed by reloading through normal ambulation has been employed as a model for examining muscle inflammation and injury during modified muscle use (Krippendorf & Riley, 1993, 1994; St Pierre & Tidball, 1994). Much of the membrane damage that occurs during reloading in this model corresponds to the invasion into the muscle by neutrophils, which show significant increases in concentration in muscle within 2 h of reloading (Tidball et al. 1999a; Frenette et al. 2000). However, the time course of muscle membrane damage does not correspond to continued muscle loading or to the time course of macrophage invasion into the muscle (Tidball et al. 1999a). Although these observations are consistent with the possibility that neutrophils induce muscle membrane damage in this model, an alternative interpretation is that neutrophil invasion and muscle membrane damage are independent consequences of another occurrence.
Previous work has shown that unloading of rat hindlimb muscle for 10 days resulted in a large reduction in NOS expression (Tidball et al. 1998). If NO were to function in abrogating membrane damage, decreased NOS expression during muscle unloading could render muscle more vulnerable to damage by inflammatory cells during reloading. In the present investigation, we test the hypothesis that muscle-derived NO can protect muscle from inflammation and muscle membrane damage that result from modified muscle use. We use the rodent hindlimb unloading/reloading model because both inflammation and membrane lysis are well-characterized consequences of modified use in this model (Krippendorf & Riley, 1993, 1994; Kasper, 1995; Tidball et al. 1999a). Our findings show that the muscle-specific expression of a NOS transgene inhibits increases in neutrophil concentration in muscle that experiences modified loading, without affecting the invasion of the reloaded muscle by macrophages. In addition, expression of the transgene prevents increases in muscle membrane damage. These findings suggest that muscle-derived NO functions as an anti-inflammatory molecule, and support the view that neutrophils play an important role in muscle membrane damage during modified muscle use.
METHODS
All experimental protocols involving the use of animals were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the University of California Los Angeles Institutional Animal Care and Use Committee.
Cytotoxicity assays
C2C12 myotubes were used for assays to assess neutrophil-mediated cytotoxicity. C2C12 cells were cultured in 96-well plates in 10 % fetal bovine serum (FBS) in Dulbecco's modified Eagle's medium (DMEM) until they were confluent. The cells were then cultured in serum-free medium for approximately 14 h to promote their fusion to myotubes. Cultures were then returned to complete medium for approximately 4 days before use in cytotoxicity assays.
Myeloid cells were collected from the peritoneal spaces of C57 BL6 mice approximately 20 h after intraperitoneal injection of 12 % sodium caseinate. Cells from the peritoneal exudate were centrifuged at 500 g for 5 min and then resuspended in 0.85 % ammonium chloride to lyse erythrocytes. The cells were again pelleted and then resuspended in Hanks' balanced salt solution (HBSS). The suspension was overlaid onto Histopaque 1077 (Sigma, St Louis, MO, USA) and then centrifuged at 400 g for 45 min at room temperature. Neutrophils were then collected from pelleted cells. The purity of the neutrophil preparations exceeded 90 %, as assessed morphologically in haematoxylin-stained preparations of the isolated cells that were adhered to microscope slides by centrifugation (Cytospin, Shandon, USA).
C2C12 myotubes were incubated in HBSS containing 51Cr and 0.25 % FBS for 2 h, and then washed twice in HBSS before use in cytotoxicity assays. Myotubes were then co-cultured with neutrophils in HBSS containing 0.25 % FBS and 0.64 μM phorbol 12-myristate 13-acetate (PMA) for 24 h, after which the medium was collected and 51Cr release into the medium was assayed by scintillation counting. In addition, L-nitro-arginine methyl ester (L-NAME) was added to some cultures to inhibit NO production by NOS. Cytotoxicity was expressed as a percentage of total lysis by setting 0 % as chromium released spontaneously by C2C12 myotubes incubated for 24 h in the absence of neutrophils. 100 % cytotoxicity was set at the chromium release into the media by C2C12 myotubes incubated with 0.1 % Triton X-100 in HBSS. Neutrophil density in cytotoxicity cultures was expressed as the number of neutrophils per mm2 of surface area in the dish, as described previously (Nguyen & Tidball, 2003). Relative cell numbers were not expressed as effector-to-target-cell ratios because the variabilities in cell proliferation and fusion that occur as myoblasts differentiate into myotubes precludes knowledge of the actual number of target cells. Instead, relative target cell numbers were normalized to the surface area of the culture dish, in which they form an adherent and continuous monolayer. All cytotoxicity assays were performed at least three times, with six replicates of each condition in each assay, and each value was expressed as the mean with its standard error.
Production of NOS transgenic mice
Transgenic (Tg) mice with the muscle-specific expression of rat brain nNOS were generated as previously described (Wehling et al. 2001). Expression of the transgene was driven by the human skeletal actin promoter and the vp1 intron (provided by Dr Jeffrey Chamberlain, University of Washington, USA). Tg mice were produced at the University of California, Irvine Transgenic Mouse Facility by microinjection of purified plasmid into zygotes from C57Bl/6J × Balb/c parents. All comparisons were made with age-matched littermates that were not Tg. PCR of tail DNA was used to identify Tg mice, as described previously (Spencer et al. 2002). In addition, overexpression of the transgene was confirmed in each experimental animal by Western blot analysis using rabbit anti-nNOS (Transduction Labs, San Diego, USA).
Hindlimb muscle unloading and reloading
Muscle injury and inflammation were induced by subjecting mice to 10 days of muscle unloading of both hindlimbs, followed by reloading for 24 h by normal weight bearing. Unloading was achieved using an apparatus that was a modification of that described by Morey-Holton & Globus (2002). Modifications of the original apparatus, which was designed for rat hindlimb unloading, were made to accommodate the smaller body size of mice used in the present investigation and did not influence the unloading that was experienced by the animals. Muscle unloading through this manipulation has been shown previously to produce 40 % mass loss of the soleus muscle in a 10 day period (Thomason & Booth, 1990). Reloading for 24 h by return to normal ambulation has been shown previously to produce muscle inflammation, fibre injury and membrane lesions in soleus muscle fibres (Krippendorf & Riley, 1993, 1994; St Pierre & Tidball, 1994; Kasper, 1995; Tidball et al. 1999a). The ‘reloaded’ groups of animals consisted of six Tg mice and six non-Tg littermates that were subjected to hindlimb unloading followed by reloading for 24 h. The ‘unloaded only' groups consisted of six Tg and six non-Tg littermates that were subjected to hindimb unloading for 10 days and then immediately killed for tissue collection, without experiencing reloading. The ‘ambulatory control' groups consisted of six Tg and seven non-Tg control mice that experienced normal cage activity until killed for tissue collection. All mice were 2–3 months of age. Mice were monitored daily throughout the periods of hindlimb unloading and reloading, and they exhibited no signs of stress, such as vocalization, loss of appetite, or failure to groom. Following killing by intraperitoneal injection of an overdose of sodium pentabarbitol, soleus muscles were rapidly dissected from each animal. One soleus muscle from each animal was rapidly frozen in isopentane, and used for either immunohistochemical analysis or Western blot analysis. The second soleus from each animal was used for assessment of muscle membrane damage.
Immunohistochemistry
Cross-sections 10 μm thick were taken from the midbellies of soleus muscles and used for immunohistochemical analysis. Sections from each muscle were fixed by immersion in acetone, and then immunolabelled for neutrophils using rat anti-Ly6G (Pharmingen) or for macrophages using rat anti-F4/80. Anti-F4/80 was prepared by ammonium sulfate precipitation of immunoglobulins from F4/80 hybridoma cultures (American Type Culture Collection). The concentrations of precipitated immunoglobulins were assayed by ELISA, after which the samples were diluted to 0.1 to 0.2 μg ml−1 of anti-F4/80 for use in immunohistochemistry. Sections were processed as described previously (Wehling et al. 2001) and immunoreactive cells were identified using a biotinylated mouse anti-rat IgG secondary antibody and horseradish peroxidase-conjugated avidin before reaction with 3-amino-9-ethylcarbazole (Vector). Control sections were processed identically, except that incubation with primary antibody was eliminated.
The total number of neutrophils or macrophages in each section was counted microscopically. The total volume of each section analysed was determined by measuring the area of each section using a stereological, point-counting technique (Spencer et al. 2001), and then multiplying that value by the section thickness (10 μm). The concentration of neutrophils or macrophages in soleus muscles of each experimental and control group was expressed as the mean and its standard error. Values were compared using Student's t test with the level of statistical significance set at P < 0.05.
Western analysis
Relative quantity of nNOS in soleus muscles was assayed for each animal analysed. After freezing soleus muscles in isopentane, a portion of each muscle was homogenized in reducing sample buffer (80 mM Tris, pH 6.8, containing 0.1 M dithiothreitol and 70 mM SDS), then boiled and finally centrifuged to remove particulates. Protein concentration in the supernatant was measured by the method of Minamide & Bamburg (1990) and then 30 μg of each sample were loaded on 10 % polyacrylamide gels that were prepared according to Laemmli (1970). The gels were then electrophoretically transferred onto nitrocellulose (Burnette, 1981), after which the blots were incubated with a rabbit antibody to nNOS (Transductions Labs) diluted 1:1000 in TBS (50 mM Tris, pH 7.6, containing 150 mM NaCl and 0.1 % NaN3) containing 0.05 % Tween 20 and 3 % bovine serum albumin. After extensive washing with TBS, the blots were incubated with secondary antibody conjugated to horseradish peroxidase and the bound antibody was detected by enhanced chemiluminescence (Amersham). Relative quantities of nNOS in experimental samples were compared by scanning densitometry (Alpha Innotech) of the reaction product at the molecular mass of nNOS (160 kDa) in Western blots. Values were normalized to the concentration of nNOS in ambulatory NOS non-Tg mice.
Assay of fibre membrane injury
Injuries to soleus muscle fibre membranes were assayed by measuring the relative quantities of the fluorescent extracellular tracer dye procion orange in the cytoplasm of fibres from muscles in each experimental and control group. Muscles that are incubated in procion orange solutions exclude the marker dye, unless membrane lesions are present. One soleus from each animal was incubated in 0.5 % procion orange dye solution in Krebs Ringer solution for 1 h followed by two 5-min washes with Krebs Ringer solution. The soleus muscles were then rapidly frozen in isopentane, and midbelly cross-sections of each muscle were cut. We incubated muscles in procion orange following dissection instead of injecting dye into the animals prior to dissection because the latter could feasibly cause an increase in inflammatory cell activation.
Two assessments of fibre membrane injury were used. First, we used an assay employed previously (e.g. Greelish et al. 1999; Hack et al. 2000; Wehling et al. 2001) in which the number of brightly-fluorescent, injured fibres in each section was expressed as a percentage of the total number of fibres in the section. However, we observed that there were many fibres that displayed levels of cytosolic fluorescence that appeared higher than those that were normally observed in healthy, ambulatory muscle fibres, although not as high as those in the fibres that are typically identified as injured in this assay. Injury to these less fluorescent fibres was assessed in an additional assay for injury, in which we measured the fluorescence of each fibre in every section of each mouse that was analysed, and assayed for shifts in fluorescence of the entire population of fibres.
In this second injury assay, all sections were viewed by epifluorescence, and the fluorescence intensity of each individual fibre was measured in a circular area of 8 μm diameter that was sampled at the centre of each fibre using a digital imaging system (Bioquant, Nashville, TN, USA). Photobleaching of fibres outside the field of signal acquisition was prevented by reducing the size of the field diaphragm. Fluorescence intensity values for each fibre were then corrected for background, by measuring background signal from an area of the slide that contained no tissue, and subtracting the background value from the cytosolic fluorescence measurements.
RESULTS
Neutrophil-mediated lysis of muscle cells in vitro is exacerbated by NOS inhibition
Cytotoxicity assays showed that mouse peritoneal neutrophils can lyse C2C12 myotubes with or without activation by PMA. Myotube lysis was significantly greater when neutrophils were added at 20 000 neutrophils per mm2 of cultured myotubes, compared to control cultures that did not contain neutrophils (Fig. 1). A positive relationship was seen between L-NAME concentration and the level of cytotoxicity (Fig. 2), which indicates that NO reduces neutrophil-mediated cytotoxicity of myotubes in vitro. Addition of L-NAME to myotube cultures in the absence of neutrophils did not affect cytotoxicity (Fig. 2).
Figure 1. Neutrophils lyse muscle cells in vitro.
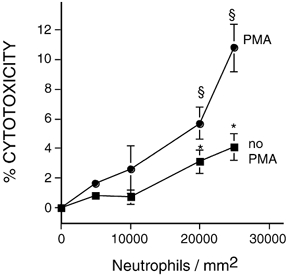
Peritoneal neutrophils were cultured on substrata covered with a confluent layer of C2C12 myotubes. Cytotoxicity was assayed by release of chromium from the myotubes. Myotubes were cultured without neutrophils or with 5000, 10 000, 20 000 or 25 000 neutrophils per mm2. Neutrophils in some cultures were activated by the addition of PMA. Release of chromium from myotubes was significantly greater than spontaneous release at P < 0.05, * or at P < 0.01, §. Mann–Whitney two-tailed test. n = 4 for each group. Bars = s.e.m.
Figure 2. NOS inhibition increases neutrophil-mediated lysis of myotubes in vitro.
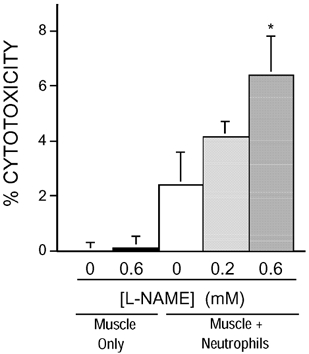
Neutrophils were cultured at 5000 neutrophils per mm2 on a confluent layer of myotubes, and cytolysis was measured by chromium release. Addition of L-NAME (0.6 mM) caused a significant increase of cytolysis compared to co-cultures without L-NAME (0 mM); * P < 0.01. Mann–Whitney two-tailed test. n = 5 for each group. Bars = s.e.m.
Muscle unloading reduces the concentration of nNOS in mouse muscles
Western analysis of soleus muscles from mice experiencing hindlimb unloading for 10 days showed that there was a significant reduction in nNOS relative quantity during the unloading period (Fig. 3). Soleus muscles of NOS Tg mice also showed a significant decrease in nNOS relative quantity during a 10 day period of unloading, although the nNOS relative quantity remained much higher than the NOS relative quantity in ambulatory, non-Tg muscles. Scanning densitometry of nNOS Western blots of soleus muscle extracts from six animals from each treatment showed the following mean relative quantities of nNOS: ambulatory NOS non-Tg (1.0 arbitrary unit; s.e.m. = 0.14); ambulatory NOS Tg (51.0 arbitrary units; s.e.m. = 2.9); unloaded NOS non-Tg (0.52 arbitrary units; s.e.m. = 0.5); unloaded NOS Tg (40.0 arbitrary units; s.e.m. = 0.8).
Figure 3. Western blot analysis of nNOS in soleus muscles of ambulatory (AMB) and hindlimb-unloaded (UNL) mice.
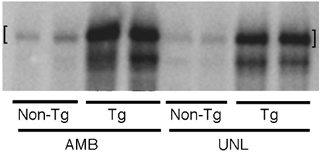
Lanes containing extracts of NOS Tg muscles (Tg) were loaded with 10 % of the sample mass used in lanes containing extracts of non-Tg littermates' muscles (non-Tg). Muscle extracts from two animals are shown for each experimental condition. The upper band (indicated by [ ]) is the intact, full-length nNOS at 160 kDa. The lower band is one of several NOS fragments that are detectable in muscle extracts from transgenic animals. Neither the 160 kDa band nor the lower-mass immunoreactive band is observed in nNOS Western blots of nNOS null mutant muscle extracts (data not shown).
Expression of a NOS transgene in muscle reduces the concentration of neutrophils, but not macrophages in reloaded soleus muscles
Previous investigations have shown that 24 h of hindlimb reloading that followed 10 or more days of muscle unloading in rats (Krippendorf & Riley, 1993; St Pierre & Tidball, 1994) and mice (Frenette et al. 2000) produced a large, significant increase in muscle invasion by both neutrophils and macrophages. In the present study, expression of the NOS transgene did not affect the concentration of either neutrophils or macrophages in ambulatory or unloaded muscles (Fig. 4 and Fig. 5). However, muscle unloading for 10 days caused small but significant increases in the concentrations of neutrophils and macrophages in soleus muscle of both Tg and non-Tg mice, when compared to ambulatory controls (Fig. 4 and Fig. 5). Muscle reloading for 24 h caused further increases in the concentrations of neutrophils and macrophages in the soleus muscles of Tg and non-Tg mice. However, expression of the NOS transgene resulted in 51 % fewer neutrophils in the solei of reloaded Tg mice than observed in non-Tg mice. The concentration of macrophages in the solei of reloaded mice was not affected by expression of the transgene.
Figure 4. Expression of a NOS transgene reduces the concentration of neutrophils in reloaded soleus muscles.
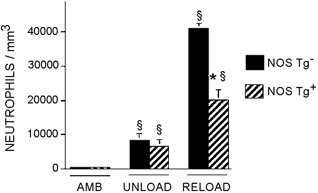
AMB, samples from ambulatory animals; UNLOAD, samples from animals experiencing muscle unloading only; RELOAD, samples from animals experiencing unloading followed by reloading. §Significantly different from ambulatory animals of same genotype, P < 0.05. * Significantly different from non-Tg animal experiencing the same experimental treatment, P < 0.05. Student's two-tailed t test. Bars = s.e.m.n = 6 for each group.
Figure 5. Expression of a NOS transgene does not affect the concentration of macrophages in soleus muscles.
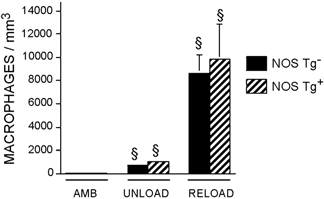
AMB, samples from ambulatory animals; UNLOAD, samples from animals experiencing muscle unloading only; RELOAD, samples from animals experiencing unloading followed by reloading. § Significantly different from ambulatory animals of same genotype, P < 0.05. Student's two-tailed t test. Bars = s.e.m.n = 6 for each group.
Expression of a NOS transgene in muscle reduces muscle membrane lesions during muscle reloading
Muscle fibres possessing membrane lesions were identified by the presence of the extracellular marker dye procion orange within the fibre cytoplasm when observed by epifluorescence (Figs 6–8). Some muscle fibres that contained high concentrations of procion orange were also observed to lie adjacent to neutrophils in the extracellular space (Fig. 6). Approximately 0.4 % of the fibres in midbelly cross-sections of muscle from ambulatory mice were intensely fluorescent, regardless of the expression of the transgene (Tg = 0.3; s.e.m. = 0.1; non-Tg = 0.4; s.e.m. = 0.1). Hindlimb unloading for 10 days produced a significant decrease in the number of injured, intensely fluorescent fibres in soleus muscles. Again, the proportion of injured fibres in soleus muscles did not differ between Tg mice and non-Tg littermates that experienced hindlimb unloading. However, muscle reloading for 24 h produced an increase in muscle fibre injuries to levels that were over five times greater in non-Tg mice than in Tg muscles, which indicates a strong, protective effect of the NOS transgene against membrane lesions associated with muscle reloading.
Figure 6. Micrographs showing relative distributions of neutrophils and brightly fluorescent fibres in procion orange-treated muscles.
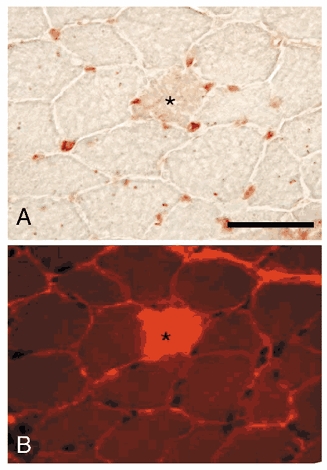
A, cross-section of soleus muscle from non-transgenic mouse subjected to hindlimb unloading, followed by 24 h reloading. The section was stained for neutrophils (red), some of which lie near the surface of a muscle fibre (*). B, cross-section taken adjacent to the section shown in Fig. 6A, but observed by indirect fluorescence microscopy to visualize fibres that contain cytosolic procion orange. The brightly fluorescent fibre (*) is the same as that indicated (*) in Fig. 6A. Bar = 50 μm.
Figure 8. Frequency distribution of fluorescence intensities of all fibres in muscle cross-sections.
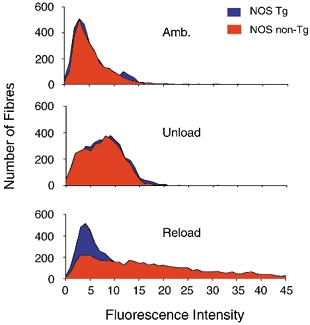
Shifts of peaks to the right reflect higher concentrations of intracellular procion orange, which result from increased membrane damage. The data shown in each graph are the aggregates of intracellular fluorescence measurements for all fibres in cross-sections of entire soleus muscles from each of six mice from each experimental condition.
Figure 7. Expression of a NOS transgene reduces the proportion of injured muscle fibres in reloaded soleus muscles.
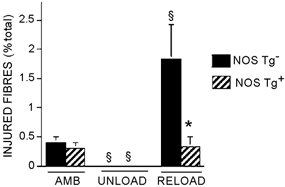
Data are the percentages of the total number of fibres per entire cross-section of soleus muscle that were brightly fluorescent following incubation in procion orange solution. AMB, samples from ambulatory animals; UNLOAD, samples from animals experiencing muscle unloading only; RELOAD, samples from animals experiencing unloading followed by reloading. §Significantly different from ambulatory animals of same genotype, P < 0.05. * Significantly different from non-Tg animal experiencing the same experimental treatment, P < 0.05. Student's two-tailed t test. Bars = s.e.m.n = 6 for each group.
We also assayed for effects of NOS transgene expression on muscle membrane injury by assaying for fluorescence of all fibres in each mid-belly cross-section of every muscle treated with the extracellular marker dye, procion orange. This second analysis of injury shows that the distribution of fluorescent intensities in fibres is continuous (Fig. 8). Thus, the selection and quantification of brightly fluorescent fibres as an index of injury, as performed in the routinely employed assay for fibre injury reported above, is subjective. In addition, this second analysis of injury shows that a large proportion of the muscle fibre population in reloaded, non-transgenic muscle shows an increase in fluorescence, relative to the mean fluorescence of healthy controls. This indicates that the typically employed technique of counting the proportion of the fibre population that is brightly fluorescent underestimates muscle membrane damage.
Soleus muscle fibres from ambulatory Tg and non-Tg muscles showed nearly identical levels of intracellular fluorescence (ambulatory Tg = 5.59 arbitrary units; s.e.m. = 0.80; ambulatory non-Tg = 5.7; s.e.m. = 1.04). In addition, the frequency distributions of fluorescence intensities for fibres in all ambulatory, NOS Tg soleus fibres and all ambulatory, non-Tg fibres were almost identical (Fig. 8). Similarly, there were no significant differences in the mean, fluorescence intensities of NOS Tg soleus fibres and non-Tg fibres after 10 days of hindlimb unloading (Tg = 8.31 arbitrary units; s.e.m. = 1.26; non-Tg = 7.5; s.e.m. = 0.58). The frequency distribution of fluorescence intensities for fibres in all unloaded, NOS-Tg soleus fibres and unloaded, non-Tg fibres did not differ (Fig. 8). However, reloading soleus muscles for 24 h following unloading produced a significant increase in intracellular, fibre fluorescence (reloaded Tg = 16.3 arbitrary units; s.e.m. = 1.09; non-Tg = 23.7; s.e.m. = 0.78). In addition, the variance of the data was substantially larger in the reloaded, non-Tg soleus muscles (variance = 298.3) than in reloaded, Tg muscles (variance = 177.4). The larger variance is reflected in the broadened and flattened frequency distribution of fluorescence intensities in fibres in the non-Tg muscles (Fig. 8).
DISCUSSION
The results of the present investigation show that the muscle-specific expression of an nNOS transgene in mice can prevent increases in neutrophil concentration and muscle membrane damage that result from modified muscle use. In addition, our findings show that mouse neutrophils can cause muscle membrane damage in vitro and indicate that NOS inhibition increases this damage. Together, these findings support the hypothesis that muscle-derived NO can protect against muscle membrane lysis by neutrophils both in vivo and in vitro, and they indicate that much of the damage to muscle membranes that occurs in modified muscle use may be a result of neutrophil-mediated events.
NO may protect against neutrophil-induced damage to muscle membranes through several possible mechanisms. Previous investigations showed that neutrophils can damage muscle cell membranes in vitro through superoxide-dependent events (Nguyen & Tidball, 2003). NO can protect against superoxide-mediated cytotoxicity by scavenging superoxide radicals (Rubanyi et al. 1991), or by inhibiting the activity of NADPH oxidase, which generates superoxide in neutrophils (Clancy et al. 1992). Either of these NO-dependent protective mechanisms may occur in the in vitro or in vivo models of injury that we use in the present study. However, NO may also protect against neutrophil cytotoxicity in vivo during modified muscle use through other mechanisms. For example, NO can inhibit the expression of molecules such as ICAM, VCAM and P-selectin (Niu et al. 1994; Gauthier et al. 1994; Adams et al. 1997), which are important in leukocyte diapedesis. Reduced expression of these adhesion molecules would reduce the concentration of inflammatory cells in the reloaded muscle.
The reductions in the relative quantities of nNOS that were observed in unloaded muscles of mice (present study) and rats (Tidball et al. 1998) indicate that unloaded muscle may be especially susceptible to neutrophil-mediated damage at the onset of reloading. Previous studies (Tidball et al. 1998) showed that levels of nNOS expression in muscle may not return to normal until approximately 2 days after return to ambulation following periods of muscle unloading. During this period, any protective roles of muscle-derived NO would be diminished, which may leave muscle membranes especially vulnerable to damage by inflammatory cells. Recent work (Wehling et al. 2001) indicated that loss of normal anti-inflammatory functions of muscle-derived NO can also promote muscle inflammation and membrane damage in dystrophin-deficient muscular dystrophy. Loss of dystrophin expression in muscle causes a secondary loss of nNOS from muscle (Brenman et al. 1995; Chang et al. 1996), and when NO production is normalized in dystrophic muscle, there is a large reduction in inflammation and muscle membrane damage (Wehling et al. 2001).
Although the cytotoxicity data presented here show that nitric oxide can protect against muscle membrane damage caused by neutrophils in vitro, other NO-mediated mechanisms may also be present in vivo, which could reduce muscle membrane damage during modified loading. NO has been shown to be able to participate in several signalling pathways, especially signalling through cGMP and protein kinase G. The protective role of NO against muscle membrane damage in vivo could feasibly involve modifications in unidentified transcriptional or translational activities that are regulated by cGMP or protein kinase G. In particular, increased production of NO by muscle has been shown to increase the expression of membrane-associated, cytoskeletal proteins (Tidball et al. 1999b) that may protect against membrane damage during modified muscle use.
Previous investigations (Pizza et al. 1998) have shown that the systemic inhibition of NO production in rats by administering L-NAME via drinking water could reduce inflammation and muscle necrosis following hindlimb unloading and reloading. The findings that reduction of NO production by L-NAME treatment (Pizza et al. 1998) and that increase in NO production by expression of a NOS transgene in muscle can both reduce muscle inflammation in the same model appear contradictory. However, L-NAME treatments would inhibit all NOS isoforms, including nNOS in muscle, eNOS in endothelial cells and iNOS in inflammatory cells, whereas expression of the nNOS transgene driven by the skeletal muscle actin promoter would specifically modify muscle production of NO. Several recent investigations have shown that perturbations of NO production by different NOS isoforms in the same tissue can have opposite physiological effects. For example, NO that is produced by eNOS in cardiac myocytes inhibits contractility (Barouch et al. 2002) while NO derived from nNOS in cardiac myocytes increases contractility (Xu et al. 1998; Barouch et al. 2002).
The findings that the systemic inhibition of NO production (Pizza et al. 1998) and the muscle-specific increase in NO production (present investigation) both reduce muscle inflammation also illustrate the dichotomy of NO function, in which it can act as either a pro-inflammatory or anti-inflammatory molecule. For example, NO can activate cyclooxygenases (Salvemini et al. 1993), which can increase prostaglandin production and inflammation. NO can also function as a chemoattractant to inflammatory cells (Beauvais et al. 1995). However, NO can also induce apoptosis in inflammatory cells (Albina et al. 1993; von Knethen & Brune, 1997) and inhibit diapedesis (Akimitsu et al. 1995; Fukumura et al. 1997). In the present model, muscle-derived NO functions as an anti-inflammatory molecule, although NO that is derived from other sources appears to have a pro-inflammatory function (Pizza et al. 1998). NO derived from endothelial cell NOS (eNOS) may indirectly promote muscle inflammation in this unloading/ reloading model by causing an increase in vasodilatation, which could increase the delivery of inflammatory cells to the muscle. L-NAME administration has been shown previously to reduce blood flow to muscle, presumably through the inhibition of eNOS (Wilson & Kapoor, 1993; Hirai et al. 1994; Hickner et al. 1997). In some situations, muscle-derived NO can also increase vasodilatation by opposing alpha-adrenergic vasoconstriction (Thomas et al. 1998).
The influence of muscle-derived NO on neutrophil concentrations in muscle, without having an effect on macrophage concentrations, shows that macrophage invasion can occur at normal levels in the absence of normal neutrophil invasion. This may indicate that macrophage invasion is independent of neutrophil invasion, or that the reduced levels of neutrophil invasion in NOS Tg animals was sufficient for subsequent invasion by macrophages. In addition, the effect of expression of the NOS transgene on neutrophil, but not on macrophage, invasion may suggest that neutrophil diapedesis is NO sensitive, while macrophage diapedesis is not affected by NO in this model. NO has been shown to be capable of preventing cytokine-induced increases in the expression of P-selectin and E-selectin on endothelial cells in vivo and in vitro, which are associated with reductions in leukocyte rolling and adhesion to the endothelium in vivo and consequent reduction in diapedesis. However, in at least some in vivo models of inflammation, F4/80 macrophage invasion into tissue can occur at diminished levels in the absence of selectins. For example, F4/80 macrophages invade renal tissue during obstructive nephropathy in mice that are triple null mutants for P-, E-, and L-selectin, although the levels of invasion are attenuated (Lange-Sperandio et al. 2002). Thus, it is feasible that the absence of a NO effect on macrophage concentrations in reloaded muscle may reflect macrophage invasion by selectin-independent mechanisms. Alternatively, the increase in F4/80 macrophages in inflamed kidneys of triple-null selectin mutants (Lange-Sperandio et al. 2002) or in the reloaded muscles of NOS Tg mice (present study) may reflect the proliferation of resident macrophages in the tissue, rather than macrophage invasion. This possibility is under current investigation.
We conclude that muscle-derived NO can function as an anti-inflammatory molecule in muscle that experiences modified loading and that its release from muscle can reduce damage to the muscle cell membrane. Furthermore, these beneficial effects of muscle-derived NO may be mechanistically related. Our in vitro findings show that neutrophils lyse muscle cell membranes, which suggests that the reduction of muscle membrane damage in reloaded, NOS Tg muscles may result from a reduction in neutrophil-mediated lysis. In our continuing studies we are assessing the identity of the specific, neutrophil-derived, cytolytic molecules, so that we can test whether disruption of their synthesis can reduce muscle damage during modified muscle loading.
Acknowledgments
This work was supported by grants from the NIH (AR/HD47721, AR47855), NASA (NAG5–4837) and the Muscular Dystrophy Association, USA. Jackie Parente provided valuable technical assistance.
REFERENCES
- Adams MR, Jessup W, Hailstones D, Celermajer DS. L-arginine reduces human monocyte adhesion to vascular endothelium and endothelial expression of cell adhesion molecules. Circulation. 1997;95:662–668. doi: 10.1161/01.cir.95.3.662. [DOI] [PubMed] [Google Scholar]
- Akimitsu T, Gute DC, Korthuis RJ. Leukocyte adhesion induced by inhibition of nitric oxide production in skeletal muscle. J Appl Physiol. 1995;78:1725–1732. doi: 10.1152/jappl.1995.78.5.1725. [DOI] [PubMed] [Google Scholar]
- Albina JE, Cui S, Mateo RB, Reichner JS. Nitric oxide-mediated apoptosis in murine peritoneal macrophages. J Immunol. 1993;150:5080–5085. [PubMed] [Google Scholar]
- Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, Hobai IA, Lemmon CA, Burnett AL, O'Rourke B, Rodriguez ER, Huang PL, Lima JA, Berkowitz DE, Hare JM. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–339. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- Beauvais F, Michel L, Dubertret L. Exogenous nitric oxide elicits chemotaxis of neutrophils in vitro. J Cell Physiol. 1995;165:610–614. doi: 10.1002/jcp.1041650319. [DOI] [PubMed] [Google Scholar]
- Brenman HE, Chao DS, Sia H, Aldape K, Bredt DS. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995;82:743–752. doi: 10.1016/0092-8674(95)90471-9. [DOI] [PubMed] [Google Scholar]
- Burnette WN. 'Western blotting': electrophoretic transfer of proteins from sodium dodecyl sulfate–polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem. 1981;112:195–203. doi: 10.1016/0003-2697(81)90281-5. [DOI] [PubMed] [Google Scholar]
- Carden DL, Korthuis RJ. Mechanisms of postischemic vascular dysfunction in skeletal muscle: implications for therapeutic intervention. Microcirc Endothelium Lymphatics. 1989;5:277–298. [PubMed] [Google Scholar]
- Chang W-J, Iannaccone ST, Lau KS, Masters BSS, McCabe TJ, McMillan K, Padre RC, Spencer MJ, Tidball JG, Stull JT. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc Natl Acad Sci U S A. 1996;93:9142–9147. doi: 10.1073/pnas.93.17.9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy RM, Leszczynska-Piziak J, Abramson SB. Nitric oxide, an endothelial cell relaxation factor, inhibits neutrophil superoxide anion production via a direct action on the NADPH oxidase. J Clin Invest. 1992;90:1116–1121. doi: 10.1172/JCI115929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenette J, Cai B, Tidball JG. Complement activation promotes muscle inflammation during modified muscle use. Am J Pathol. 2000;156:2103–2110. doi: 10.1016/S0002-9440(10)65081-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumura D, Yuan F, Endo M, Jain RK. Role of nitric oxide in tumor microcirculation. Blood flow, vascular permeability, and leukocyte-endothelial interactions. Am J Pathol. 1997;150:713–725. [PMC free article] [PubMed] [Google Scholar]
- Gauthier TW, Davenpeck KL, Lefer AM. Nitric oxide attenuates leukocyte-endothelial interaction via P-selectin in splanchnic ischemia-reperfusion. Am J Physiol. 1994;267:G562–568. doi: 10.1152/ajpgi.1994.267.4.G562. [DOI] [PubMed] [Google Scholar]
- Greelish JP, Su LT, Lankford EB, Burkman JM, Chen H, Konig SK, Mercier IM, Desjardins PR, Mitchell MA, Zheng XG, Leferovich J, Gao GP, Balice-Gordon RJ, Wilson JM, Stedman HH. Stable restoration of the sarcoglycan complex in dystrophic muscle perfused with histamine and a recombinant adeno-associated viral vector. Nat Med. 1999;5:439–443. doi: 10.1038/7439. [DOI] [PubMed] [Google Scholar]
- Hack AA, Lam MJ, Cordier L, Shoturma DI, Ly CT, Hadhazy MA, Hadhazy HR, Sweeney HL, McNally EM. Differential requirement for individual sarcoglycans and dystrophin in the assembly and function of the dystrophin-glycoprotein complex. J Cell Sci. 2000;113:2535–2544. doi: 10.1242/jcs.113.14.2535. [DOI] [PubMed] [Google Scholar]
- Hickner RC, Fisher JS, Ehsani AA, Kohrt WM. Role of nitric oxide in skeletal muscle blood flow at rest and during dynamic exercise in humans. Am J Physiol. 1997;273:H405–410. doi: 10.1152/ajpheart.1997.273.1.H405. [DOI] [PubMed] [Google Scholar]
- Hirai T, Visneski MD, Kearns KJ, Zelis R, Musch TI. Effects of NO synthase inhibition on the muscular blood flow response to treadmill exercise in rats. J Appl Physiol. 1994;77:1288–1293. doi: 10.1152/jappl.1994.77.3.1288. [DOI] [PubMed] [Google Scholar]
- Kasper CE. Sarcolemmal disruption in reloaded atrophic skeletal muscle. J Appl Physiol. 1995;79:607–614. doi: 10.1152/jappl.1995.79.2.607. [DOI] [PubMed] [Google Scholar]
- Kawasaki S, Sugiyama S, Ishiguro N, Ozawa T, Miura T. Implication of superoxide radicals on ischemia-reperfusion-induced skeletal muscle injury in rats. Eur Surg Res. 1993;25:129–136. doi: 10.1159/000129267. [DOI] [PubMed] [Google Scholar]
- Korthuis RJ, Franger DN, Townsley MI, Taylor AE. The role of oxygen-derived free radicals in ischemia-induced increases in canine skeletal muscle vascular permeability. Circ Res. 1985;57:599–609. doi: 10.1161/01.res.57.4.599. [DOI] [PubMed] [Google Scholar]
- Korthuis RJ, Grisham MB, Granger DN. Leukocyte depletion attenuates vascular injury in postischemic skeletal muscle. Am J Physiol. 1988;254:H823–827. doi: 10.1152/ajpheart.1988.254.5.H823. [DOI] [PubMed] [Google Scholar]
- Krippendorf BB, Riley DA. Distinguishing unloading- versus reloading-induced changes in rat soleus muscle. Muscle Nerve. 1993;16:99–108. doi: 10.1002/mus.880160116. [DOI] [PubMed] [Google Scholar]
- Krippendorf BB, Riley DA. Temporal changes in sarcomere lesions of rat adductor longus muscle during hindlimb reloading. Anat Rec. 1994;238:304–210. doi: 10.1002/ar.1092380304. [DOI] [PubMed] [Google Scholar]
- Laemmli K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lange-Sperandio B, Cachat F, Thornhill BA, Chevalier RL. Selectins mediate macrophage infiltration in obstructive nephropathy in newborn mice. Kidney Int. 2002;61:516–524. doi: 10.1046/j.1523-1755.2002.00162.x. [DOI] [PubMed] [Google Scholar]
- Minamide S, Bamburg JR. A filter paper dye-binding assay for quantitative determination of protein without interference from reducing agents or detergents. Anal Biochem. 1990;190:66–70. doi: 10.1016/0003-2697(90)90134-u. [DOI] [PubMed] [Google Scholar]
- Morey-Holton ER, Globus RK. Hindlimb unloading rodent model: technical aspects. J Appl Physiol. 2002;92:1367–1377. doi: 10.1152/japplphysiol.00969.2001. [DOI] [PubMed] [Google Scholar]
- Nguyen HX, Tidball JG. Interactions between neutrophils and macrophages promote macrophage killing of rat muscle cells in vitro. J Physiol. 2003;547:125–132. doi: 10.1113/jphysiol.2002.031450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu XF, Smith CW, Kubes P. Intracellular oxidative stress induced by nitric oxide synthesis inhibition increases endothelial cell adhesion to neutrophils. Circ Res. 1994;74:1133–40. doi: 10.1161/01.res.74.6.1133. [DOI] [PubMed] [Google Scholar]
- Pizza FX, Hernandez IJ, Tidball JG. Nitric oxide synthase inhibition reduces muscle inflammation and necrosis in modified muscle use. J Leukoc Biol. 1998;64:427–433. [PubMed] [Google Scholar]
- Rubanyi GM, Ho EH, Cantor EH, Lumma WC, Botelho LH. Cytoprotective function of nitric oxide: inactivation of superoxide radicals produced by human leukocytes. Biochem Biophys Res Commun. 1991;181:1392–1397. doi: 10.1016/0006-291x(91)92093-y. [DOI] [PubMed] [Google Scholar]
- St Pierre BA, Tidball JG. Differential response of macrophage subpopulations to soleus muscle reloading following rat hindlimb suspension. J Appl Physiol. 1994;77:290–297. doi: 10.1152/jappl.1994.77.1.290. [DOI] [PubMed] [Google Scholar]
- Salvemini D, Misko TP, Masferrer JL, Seibert K, Currie MG, Needleman P. Nitric oxide activates cyclooxygenase enzymes. Proc Natl Acad Sci U S A. 1993;90:7240–7244. doi: 10.1073/pnas.90.15.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer MJ, Guyon JR, Sorimachi H, Potts A, Richard I, Herasse M, Chamberlain J, Dalkilic I, Kunkel LM, Beckmann JS. Stable expression of calpain 3 from a muscle transgene in vivo: immature muscle in transgenic mice suggests a role for calpain 3 in muscle maturation. Proc Natl Acad Sci U S A. 2002;99:8874–8879. doi: 10.1073/pnas.132269299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer MJ, Montecino-Rodriguez E, Dorshkind K, Tidball JG. Helper (CD4+) and cytotoxic (CD8+) T cells promote the pathology of dystrophin-deficient muscle. Clin Immunol. 2001;98:235–243. doi: 10.1006/clim.2000.4966. [DOI] [PubMed] [Google Scholar]
- Spencer MJ, Tidball JG. Do immune cells promote the pathology of dystrophin-deficient myopathies. Neuromusc Disord. 2001;1:556–564. doi: 10.1016/s0960-8966(01)00198-5. [DOI] [PubMed] [Google Scholar]
- Thomason DB, Booth FW. Atrophy of the soleus muscle by hindlimb unweighting. J Appl Physiol. 1990;68:1–12. doi: 10.1152/jappl.1990.68.1.1. [DOI] [PubMed] [Google Scholar]
- Tidball JG, Berchenko E, Frenette J. Macrophage invasion does not contribute to muscle membrane injury during inflammation. J Leukoc Biol. 1999a;65:492–498. [PubMed] [Google Scholar]
- Tidball JG, Lavergne E, Lau KS, Spencer MJ, Stull JT, Wehling M. Mechanical loading regulates nitric oxide synthase expression and activity in developing and adult skeletal muscle. Am J Physiol. 1998;275:C260–266. doi: 10.1152/ajpcell.1998.275.1.C260. [DOI] [PubMed] [Google Scholar]
- Tidball JG, Spencer MJ, Wehling M, Lavergne E. Nitric-oxide synthase is a mechanical signal transducer that modulates talin and vinculin expression. J Biol Chem. 1999b;274:33155–33160. doi: 10.1074/jbc.274.46.33155. [DOI] [PubMed] [Google Scholar]
- von Knethen A, Brune B. Cyclooxygenase-2: an essential regulator of NO-mediated apoptosis. FASEB J. 1997;11:887–895. [PubMed] [Google Scholar]
- Walden DL, McCutchan HJ, Enquist EG, Schwappach JR, Shanley PF, Reiss OK, Terada LS, Leff JA, Repine JE. Neutrophils accumulate and contribute to skeletal muscle dysfunction after ischemia-reperfusion. Am J Physiol. 1990;259:H1809–1812. doi: 10.1152/ajpheart.1990.259.6.H1809. [DOI] [PubMed] [Google Scholar]
- Wehling M, Spencer MJ, Tidball JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol. 2001;155:123–131. doi: 10.1083/jcb.200105110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JR, Kapoor S. Contribution of endothelium-derived relaxing factor to exercise-induced vasodilation in humans. J Appl Physiol. 1993;75:2740–2744. doi: 10.1152/jappl.1993.75.6.2740. [DOI] [PubMed] [Google Scholar]
- Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor). by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- Yokota J, Minei JP, Fantini GA, Shires GT. Role of leukocytes in reperfusion injury of skeletal muscle after partial ischemia. Am J Physiol. 1989;257:H1068–1075. doi: 10.1152/ajpheart.1989.257.4.H1068. [DOI] [PubMed] [Google Scholar]