

Abstract
Sulfated steroids like pregnenolone sulfate (PS) are found endogenously in the central nervous system where they may modulate GABAA receptors. Understanding the mechanism of steroid inhibition is important for understanding the conditions under which endogenous steroids modulate GABAA receptor function, assessing their potential clinical utility, and for evaluating sulfated steroids as probes of receptor behaviour. Some previous studies suggest that sulfated steroid inhibition exhibits activation dependence, whilst other studies suggest only slow, time-dependent inhibition, perhaps reflecting slow PS association with receptors. We tested activation dependence in several ways. Steroid potency increased 2- to 3-fold with ≈10-fold change in GABA concentration. PS inhibition of saturating partial agonist responses suggested that the level of channel activation, rather than receptor occupancy by agonist, is important for PS inhibition. Inhibition by sulfated steroids exhibited weak or no voltage dependence. Responses to rapid applications of exogenous GABA differed little whether PS was pre-applied or simply co-applied with GABA, consistent with the hypothesis that the actions of PS are facilitated by receptor activation. PS applied during steady-state GABA responses exhibited slow onset and offset rate constants. The offset, rather than onset, was significantly slowed by elevated GABA concentration. At hippocampal synapses, large, multiquantal IPSCs were inhibited more effectively by a fixed concentration of PS than small quantal content IPSCs, consistent with known ‘pooling’ of transmitter following multiquantal release. Picrotoxinin, although superficially similar to PS in its activation dependence, was dissimilar from PS in a number of details. In summary, PS inhibition exhibits activation dependence that may be explained by activation-dependent binding and altered desensitization.
Steroids produced endogenously within the CNS are commonly referred to as neurosteroids (Mensah-Nyagan et al. 1999; Robel et al. 1999). One of the more common neurosteroids, pregnenolone sulfate (PS), is synthesized from cholesterol through the activity of mitochondrial side-chain cleavage enzyme, which produces pregnenolone, and a sulfotransferase, which produces PS. PS and other similar endogenous pregnane and androstane steroids inhibit GABAA receptor-mediated synaptic transmission (Majewska et al. 1988).
Recent data from our lab suggest that PS inhibition of GABAA receptor-mediated currents may in some way be activation dependent (Shen et al. 2000; Wang et al. 2002). However, other studies suggest that, within the margin of experimental resolution, PS does not bind more rapidly to open states of the receptor than to closed states (Akk et al. 2001). Understanding the mechanism of PS action is important for several reasons. Firstly, PS and other sulfated neuroactive steroids are present in the brain at high nanomolar concentrations (Baulieu & Robel, 1990; Corpechot et al. 1997; Wang et al. 1997). Acting separately or aggregately, these steroids may be endogenous modulators of GABAA receptor activity. The conditions under which steroids may modulate GABAA receptors will be dictated by the mechanisms by which the steroids bind to and modulate the receptor or channel. Secondly, the sulfated steroids may be important probes of GABAA receptor gating and function. Finally, we have shown that sulfated steroids can reverse the actions of GABAA receptor-potentiating steroids and the actions of other GABAA-potentiating drugs, such as barbiturates (Wang et al. 2002). This suggests that PS and similar drugs may be useful clinically at low concentrations for reversing anaesthesia, or treating drug intoxication.
In the present work, we examined PS modulation of GABAA receptors expressed in Xenopus oocytes and native receptors of cultured hippocampal neurons to examine properties of PS actions at GABAA receptors. We found that PS block is dependent on activation of receptors, but PS has several unique features compared with another state-dependent blocker, picrotoxinin. We found that many, but not all, features of PS block can be incorporated into a scheme by which PS alters receptor desensitization.
METHODS
Xenopus oocyte expression studies
Stage V–VI oocytes were harvested from sexually mature female Xenopus laevis (Xenopus One, Northland, MI, USA) under 0.1 % tricaine (3-aminobenzoic acid ethyl ester) anaesthesia, according to protocols approved by the Washington University Animal Studies Committee. All frogs were humanely killed under deep anaesthesia following the final oocyte collection. Oocytes were defolliculated by shaking for 20 min at 37 °C in collagenase (2 mg ml−1) dissolved in calcium-free solution containing (mm): 96 NaCl, 2 KCl, 1 MgCl2, and 5 Hepes at pH 7.4. Capped mRNA, encoding rat GABAA receptor α1, β2 and γ2L subunits was transcribed in vitro using the mMESSAGE mMACHINE Kit (Ambion, Austin, TX, USA) from linearized pBluescript vectors containing receptor coding regions. Subunit transcripts were injected in equal parts (20–40 ng total RNA) 8- 24 h following defolliculation. Oocytes were incubated for up to 5 days at 18 °C in ND96 medium containing (mm): 96 NaCl, 1 KCl, 1 MgCl2, 2 CaCl2 and 10 Hepes at pH 7.4, supplemented with pyruvate (5 mm), penicillin (100 i.u. ml−1), streptomycin (100 μg ml−1) and gentamycin (50 μg ml−1). The cDNAs for the rat GABAA receptor subunits were originally provided by A. Tobin (University of California, Los Angeles, USA; α1), P. Malherbe (Hoffman-La Roche, Switzerland; β2) and C. Fraser (National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, MD, USA; γ2L).
Oocyte electrophysiology
Two-electrode voltage-clamp experiments were performed with a Warner OC725 amplifier 2–5 days after RNA injection. The extracellular recording solution was ND96 medium with no supplements. Intracellular recording pipettes were filled with 3 M KCl and had open tip resistances of ≈1 MΩ. Drugs were applied from a common tip via a gravity-driven multibarrel drug-delivery system. Oocytes were not pre-equilibrated with antagonist before co-application of GABA. Cells were clamped at −70 mV for all experiments, and, unless otherwise noted, the current at the end of 20–30 s drug applications was measured for quantification of current amplitudes.
Because α1β2 subunits alone can form functional receptors, we confirmed expression of all three subunits pharmacologically. GABA responses showed benzodiazepine potentiation (≈2-fold at 1 μm lorazepam) and low Zn2+ sensitivity (< 10 % inhibition at 1 μm Zn2+), suggesting strong presence of the γ2 subunit.
Hippocampal microcultures
Primary microcultures of hippocampal cells were prepared from 1-to-3-day postnatal Sprague-Dawley rats as previously described (Mennerick et al. 1995). Halothane-anaesthetized rats were decapitated and the hippocampi removed. Hippocampi were cut into 500 μm-thick transverse slices. Single-cell suspensions were prepared with 1 mg ml−1 papain digestion in oxygenated Leibovitz L-15 medium, followed by mechanical trituration in modified Eagle's medium containing 5 % horse serum, 5 % fetal calf serum, 17 mm D-glucose, 400 μm glutamine, 50 i.u. ml−1 penicillin, and 50 μg ml−1 streptomycin. Cells were plated in the modified Eagle's medium at a density of 75 cells per mm2 onto 35 mm plastic culture dishes pre-coated with collagen microdroplets sprayed onto a layer of 0.15 % agarose. The anti-mitotic cytosine arabinoside (5–10 μm) was added on the third day after plating to halt glial proliferation. Electrophysiology was performed 8–15 days following plating for synaptic experiments (to allow synaptic maturation) and 4–6 days following plating to study exogenous GABA responses (to limit current amplitudes and associated clamp errors).
Culture electrophysiology
Whole-cell recordings were performed on hippocampal microcultured neurons, using an Axopatch 1D amplifier (Axon Instruments, Foster City, CA, USA) interfaced to a Pentium III-based computer via a Digidata 1200 acquisition board (Axon Instruments). Recordings were at room temperature. Access resistance (5–10 MΩ) was electronically compensated 90–100 %.
At the time of experiments, culture medium was replaced with an extracellular recording solution consisting of (mm): 138 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 10 Hepes, 0.025 D(-)-2-amino-5-phosphonovaleric acid (D-APV), 0.001 1,2,3,4-tetrahydro-6-nitro-2,3-dioxobenzo[f]quinoxaline-7-sulfonamide (NBQX) (pH 7.25). For experiments examining the effect of the rapidly dissociating GABAA receptor antagonist 1,2,5,6-tetrahydropyridin-4-yl-methylphosphinic acid (TPMPA; Fig. 8C and D), the bath solution contained 5 μm CGP55845 to block GABAB agonist effects of TPMPA. The standard pipette solution for autaptic recordings contained (mm): 140 KCl, 4 NaCl, 0.5 CaCl2, 5 EGTA, 10 Hepes (pH 7.25). For exogenous applications of drugs, CsCl replaced KCl in the pipette solution. For ease of comparing IPSCs from different cells that varied in the complexity of decay kinetics (2–3 exponential components), we used the model-independent 10–90 % decay time of IPSCs as our primary measure of IPSC duration.
Figure 8. PS inhibition depends on quantal content of hippocampal synaptic responses.

A, autaptic IPSCs were either evoked in the presence of 4 mm Ca2+o (no added Mg2+o; top panel) or in the presence of 1 mm Ca2+o−3 mm Mg2+o (bottom panel). In interleaved trials, PS (5 μm) was pre-applied to synapses. Control IPSCs (PS absent) are shown as thin traces. IPSCs acquired in the presence of 5 μm PS are shown as thick traces. B, the effects of PS on IPSC peak amplitude and decay are summarized from 7 neurons stimulated under the conditions described in A. Because of the multi-exponential decay of IPSCs, 10–90 % decay time was used to quantify IPSC decays. Asterisks denote P < 0.05, two-tailed paired t test. C, TPMPA block of IPSCs is increased in low quantal content conditions. Ca2+ and Mg2+ concentrations were the same as for panel A. D, summary of TPMPA effects. (n = 5, P = 0.03). The control IPSC peak amplitude was 2.5 ± 0.7-fold larger in high Ca2+ than in low Ca2+.
Exogenous drugs were applied with a multi-barrel pipette coupled to miniature solenoid valves that allowed rapid switching (≈100 ms on whole cells; Fig. 5F) between solutions. Drugs were applied in synaptic studies with a slower flow rate. Autaptic release of neurotransmitter was evoked in voltage-clamped solitary neurons with a 2 ms voltage pulse to 0 mV from a holding potential of −70 mV. This stimulation protocol elicits an escaped action potential in the partially clamped axons that triggers transmitter release (Bekkers & Stevens, 1991; Mennerick et al. 1995). Whenever possible, at least three traces in each experimental condition were acquired for analysis. Release was evoked every 25 s for synaptic responses, and drug exposure was initiated approximately 20 s before stimulation. Control conditions were interleaved with experimental conditions to counterbalance any time-dependent changes. Data sampling frequency was 5–10 kHz.
Figure 5. GABA concentration dependence of inhibition onset and offset in hippocampal neurons.

A, 2 μm GABA was applied for 20 s, followed by 10 s of GABA + PS (5 μm), then returned to GABA alone. The lower panels (grey traces) show the onset and offset of PS block at higher resolution. Superimposed (continuous black lines) are single exponential fits of time constants 594 ms (PS onset) and 856 ms (PS offset). B, same protocol from the same cell as in A, but using 20 μm GABA as the agonist. The fits are exponentials with time constants 721 ms (onset) and 1.72 s (PS offset). C and D, summary of the time constants of onset and offset of PS effect and picrotoxinin effect (10 μm) at 2 μm GABA (black columns) and 20 μm GABA (open columns) determined from single exponential fits. For PS, there was no significant difference between onset time constants in 2 or 20 μm GABA (n = 16; P > 0.3). Offset time constants were slowed by the increased GABA concentration (n = 16; P < 0.01, asterisk). For picrotoxinin, both onset and offset were significantly slowed at 20 μm GABA (asterisks, P < 0.01, n = 16). E, degree of steady- state block at 2 and 20 μm GABA in the same cells represented in C and D. Asterisk denotes P < 0.01. Both antagonists yielded larger block with increased GABA concentration. F, to estimate the speed of solution exchanges, during GABA application the Cl−o concentration in the bath solution was decreased to 50 % of that in the normal saline. Cells were clamped (+20 mV) near the reversal potential predicted by the low Cl−o concentration. The average time constant from 6 solution exchanges from 2 cells was 91.4 ± 14.6 ms.
Data analysis
Data acquisition and analysis were performed with pCLAMP software (Axon Instruments). Data plotting and curve fitting were done with Sigma Plot software (SPSS Science, Chicago, IL, USA) or IGOR Pro (Lake Oswego, OR, USA). Data are presented in the text and figures as means ± standard deviation. Statistical differences were determined using Student's two-tailed t test or a one-way analysis of variance (ANOVA). Fitting of the dose-response relationships was performed using the Hill equation: I = Imax × Cn/(EC50n + Cn), where C is the concentration of steroid (or agonist, e.g. Fig. 1), Imax is the maximum current amplitude, EC50 is the concentration of steroid (or agonist) that produces 50 % of Imax, and n is the Hill coefficient. Simulations were performed with the NEURON simulation program (http://www.neuron.yale.edu/), as described in the Discussion.
Figure 1. PS inhibition of recombinant α1β2γ2L responses in Xenopus oocytes is associated with receptor activation.

A, response of a voltage-clamped oocyte to application of 2.5 μm GABA alone (thick trace) and simultaneously co-applied PS (1.0 μm; thin trace). Clamp potential was - 70 mV. Arrows in A-C indicate the time point at which measurements of block were made. B, response in another oocyte to 30 μm GABA in the absence and presence of 1.0 μm PS. C, similar protocol as in A and B, but using a high concentration of the partial agonist P4S. D, summary inhibition curves from six cells using 30 μm GABA (○) and six additional cells challenged with 2.5 μm GABA (•). Grey triangles represent three cells challenged with 1 mm P4S. Error bars represent standard deviations, and the continuous lines represent fits of the Hill equation with IC50 values of 8.0, 0.36, and 1.42 μm for 2.5 and 30 μm GABA, and 1 mm P4S, respectively. The Hill slopes for the three curves were 0.9, 1.0, and 0.8, respectively. E, agonist concentration-response curves for peak response to GABA (•) and P4S (grey triangles) on the same set of five oocytes. Hill fits yielded estimates for the EC50 of 15.3 μm for GABA and 47.2 μm for P4S and Hill slopes of 2.56 and 0.8. F, from the concentration-response curves in E, functionally equivalent concentrations of 10 μm GABA and 1 mm P4S were chosen at which to evaluate the effect of 3 μm PS. Similar inhibitory effects were observed at the functionally equivalent concentrations. Inhibition was calculated as: IPS/Icontrol - 1, where Icontrol is the current at the end of a 30 s application of agonist alone and IPS is the current at the end of a 30 s co-application of agonist and PS.
Drugs
All drugs were from Sigma (St Louis, MO, USA) except for (3α,5β)-3-hydroxypregnan-20-one sulfate (3α5βPS), which was from Steraloids (Newport, RI, USA).
RESULTS
PS is more potent against high vs. low GABA concentrations
Initial experiments were performed in Xenopus oocytes expressing α1β2γ2L GABAA receptors to provide a homogeneous population of receptors. If PS inhibition of GABAA receptor-mediated currents is activation-dependent, then PS may be more potent at higher concentrations of GABA, conditions that promote a larger fraction of liganded and open receptor channels. This hypothesis was tested by measuring PS concentration- response curves at two different GABA concentrations: 2.5 μm (Fig. 1A) and 30 μm GABA (Fig. 1B). These concentrations of GABA are lower and higher, respectively, than the EC50 of GABA (≈15 μm, Fig. 1E). GABA currents in the presence and absence of varied PS concentrations were measured at the end of drug co-application (arrows in Fig. 1A-C) in each of six oocytes. Responses were normalized to the control response and fitted to the Hill equation. As shown in Fig. 1D, the IC50 of PS using 30 μm GABA was 0.36 μm, whilst the IC50 using 2.5 μm GABA was 8.0 μm. Thus, PS more potently blocked responses to the higher concentration of GABA. The dependence of degree of PS block on GABA concentration is similar to that of other state-dependent blockers such as picrotoxin at GABAA receptors (Dibas et al. 2002) and memantine (Parsons et al. 1993) and ifenprodil (Kew et al. 1996) at NMDA receptors.
PS effect requires both receptor binding and activation
The above data support the hypothesis of activation dependence, but do not distinguish between GABA binding and receptor activation as the prerequisite for the PS effect. This distinction was explored by testing the efficacy of PS in the presence of piperidine-4-sulfonic acid (P4S). P4S is a partial GABA agonist that relatively potently activates GABAA receptors (EC50 ≈50 μm, Fig. 1E) but produces only 30–40 % of the maximal GABA current (Fig. 1E; Ebert et al. 1994). We used this partial agonist to distinguish whether liganded receptors or activated receptors contributed to the PS blocking effect. We activated GABA receptors with a near-saturating concentration of P4S (1 mm; Fig. 1E) and evaluated the effects of varied PS concentrations. As shown in Fig. 1D, the IC50 of PS using P4S was 1.42 μm, intermediate between the values measured using low and high concentrations of GABA. Given the potency of P4S, this result suggests that channel opening rather than ligand binding is important for PS block.
To more precisely determine whether the activation level of P4S predicts the degree of PS block, we examined the fractional block for a concentration of P4S that produces a similar proportion of activated GABA receptor channels. From the concentration-response curves of GABA and P4S (Fig. 1E), 10 μm GABA was chosen as a functionally equivalent concentration to 1 mm P4S. Figure 1F shows that the fractional block was similar at these two concentrations of agonist and partial agonist.
Voltage dependence of sulfated steroids
Many activation-dependent blockers of other channels exhibit voltage dependence (though see e.g. Inoue & Akaike, 1988; Newland & Cull-Candy, 1992) usually suggesting a binding site within the transmembrane electric field and within the channel pore. Our own work suggests that carboxylated analogues of sulfated steroids exhibit significant voltage dependence (Mennerick et al. 2001). However, as previously reported by others (Majewska et al. 1988; Akk et al. 2001), PS block exhibited very little voltage dependence. In three oocytes tested at 2 μm GABA, 2 μm PS yielded 42 ± 5 % block at −90 mV and 47 ± 1 % block at +90 mV (Fig. 2). Similarly, at 20 μm GABA, 2 μm PS yielded 88 ± 1 % and 91 ± 2 % block at −90 and + 90 mV, respectively (data not shown). We have previously found that pregnane steroids carboxylated at C-3 do exhibit detectable voltage dependence, despite an apparently similar overall mechanism to PS (Mennerick et al. 2001). Therefore, we examined a representative sulfated pregnane steroid (3α5βPS). Despite similar chemical structure and apparent mechanism compared with PS, 3α5βPS exhibited detectable voltage dependence (Fig. 2C, ○). A least-squares fit of the 2 μm GABA data to the Woodhull model (Donevan et al. 1992) suggests that the anionic group on 3α5βPS experiences 28 % of the membrane electric field whilst PS experiences at most 2.5 % of the electric field. These numbers should be considered an upper limit on the degree of voltage dependence of 3α5βPS, however, because GABA responses themselves exhibit significant rectification (Fig. 2A). Thus, some of the apparent voltage dependence observed may be related to the activation dependence of sulfated steroid block.
Figure 2. Lack of voltage dependence of PS effects in Xenopus oocytes.

A, voltage pulses to potentials between −90 and +90 mV from −70 mV (200 ms, 20 mV increments) were elicited in the absence and presence of 2 μm GABA. Currents in the absence of GABA were digitally subtracted from currents in the presence of GABA to yield the GABA responses shown. The dotted line indicates the zero current level. B, GABA responses from the same cell but in the presence of 2 μm PS. C, summary of the percentage inhibition of the steady-state GABA current at different membrane potentials. There was little change in inhibition over the potential range examined (•). Also shown is the block by a sulfated pregnane steroid, 3α5βPS. In contrast to PS, considerable voltage dependence was noted (○; n = 3 oocytes at 2 μm GABA and 30 μm 3α5βPS). Note that the missing point is at the GABA reversal potential, where there was no current to measure.
Pre-application of PS elicits similar kinetics of block to co-application with GABA
We noted from oocyte experiments that co-application of PS with GABA tended to yield less effect on the initial part of the GABA current than on the steady-state GABA current (Fig. 1A and B). This could be consistent with either slow association of PS with receptors or with the idea that PS block is facilitated by activation of the receptors. To explore this issue, we turned to experiments on cultured hippocampal neurons, where solution exchange could be more rapidly achieved. Also, because hippocampal cells probably express subunit combinations different from the α1β2γ2 subunit combination (e.g. Christie & De Blas, 2002), these experiments afforded an opportunity to examine whether the activation dependence of PS generalizes to other subunits. We attempted to distinguish between slow PS association with receptors and activation-dependent PS effects by comparing co-applications of PS and GABA with co-applications that were preceded by a period of PS pre-application. Similar to oocyte experiments, we found that simultaneous co-application of 2 μm PS and 20 μm GABA yielded less PS effect on peak GABA current than on the current at the end of the 10 s co-application period (Fig. 3). The time constant of GABA current fade during application was 1.0 ± 0.1 s in the presence of PS and 2.3 ± 0.4 s in the absence of PS (Fig. 3D)
Figure 3. Time course of PS effects in cultured hippocampal neurons.
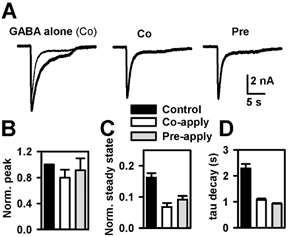
Experiments were performed on primary cultures of hippocampal neurons, to permit faster exchange time of solutions than achievable on oocytes. A, the currents represent responses to 20 μm GABA. In the left panel, the current in response to co-application (Co) of 20 μm GABA and 2 μm PS is superimposed (thin trace) on the response to GABA alone (thick trace). In the middle panel, the response to co-applied GABA plus PS is re-plotted in isolation. In the right panel is the response to 20 μm GABA co-applied with 2 μm PS following a 20 s pre-application (Pre) of PS alone. B, summary of the effect of PS pre-application (grey columns) and co-application alone (open columns) on peak GABA response (n = 3). Responses were normalized to the peak GABA current in the absence of PS (black columns). C, summary of effects of pre-applied and co-applied PS on the steady-state current level relative to the control (GABA alone) peak response (same cells as in B). D, summary of PS effects on the apparent desensitization time constant (same cells as in B).
If this effect of PS is because of time-dependent association of PS with receptors, pre-application for more than 12.5 s should produce nearly steady-state block (assuming a PS binding rate of 0.2 μm−1 s−1; Akk et al. 2001) and addition of GABA to the pre-equilibrated receptors should yield a current that rises to the steady-state level of block. In contrast, the peak GABA response to 20 μm GABA after pre-application of 2 μm PS for 20 s differed only slightly from the peak GABA response with co-application (Fig. 3B). The similarity in the degree and time course of PS block, whether or not receptors were pre-equilibrated with PS, is consistent with the idea that PS block is facilitated by receptor activation.
We compared the actions of PS to the known activation-dependent blocker of GABAA receptors, and probable channel blocker, picrotoxinin (Ffrench-Constant et al. 1993; Yoon et al. 1993; Zhang et al. 1994). We examined the effects of 5 μm GABA and 5 μm PS or 5 μm GABA and 10 μm picrotoxinin (Fig. 4). Results for PS were qualitatively similar to the results using 20 μm GABA (Fig. 3). PS block was associated with a more pronounced fade than responses to GABA alone, and this was true whether PS was pre-applied before co-application with GABA or solely co-applied with GABA (Fig. 4A and C). In this experimental protocol, 10 μm picrotoxinin elicited a similar profile of block to PS, including the larger effect on steady-state GABA response than peak, regardless of whether picrotoxinin was pre-applied (Fig. 4B and D).
Figure 4. Comparison of co- and pre- application of PS and picrotoxinin (PTXNIN) on hippocampal neurons.
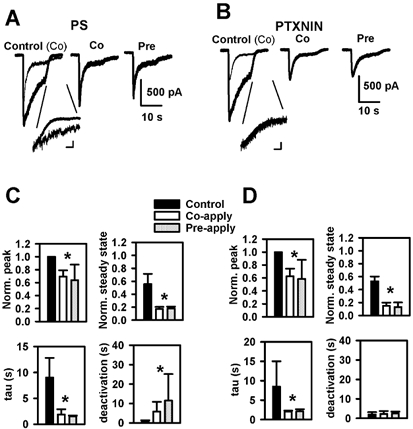
A and B, protocols were performed as in Fig. 3, except that 5 μm GABA and 5 μm PS (A) or 10 μm picrotoxinin (B) was used. The insets for each panel highlight the deactivation time course of responses by showing the deactivation of current in the presence of antagonist scaled to the deactivation in the presence of agonist alone. Time calibration bars for the insets represent 1 s. Current calibrations for A inset represent 100 pA (GABA alone) and 34 pA (GABA + PS). Current calibrations for B inset represent 200 pA (GABA alone) and 21 pA (GABA + picrotoxinin). Note that deactivation curves for picrotoxinin superimpose almost exactly. C and D, bars denote effects as in Fig. 3 B–D. Deactivation is the best exponential fit to the decay of the GABA current during washout of the GABA, antagonist combination. Summary data are from 5 cells treated with PS and 6 cells treated with picrotoxinin. Pre- and co-application conditions were different from control (asterisks) but did not differ from each other for each parameter. Note that PS and picrotoxinin had similar effects, except for effects on deactivation, where PS, but not picrotoxinin, significantly slowed deactivation.
Deactivation kinetics of GABA responses were markedly slowed in the presence of PS (Fig. 4A and C), an observation also made previously with prolonged application of low-affinity GABA agonists to nucleated membrane patches (Shen et al. 2000). Note that slowing of deactivation is selective for prolonged GABA applications. Deactivation to brief, synaptic-like pulses is speeded by PS (Shen et al. (2000) and Fig. 8). Interestingly, picrotoxinin did not exhibit the same slowing of deactivation following long pulses of GABA, implying mechanistic differences between the drugs (Shen et al. 1999), despite suggestions from binding experiments that PS and picrotoxinin may act at an overlapping site (Majewska & Schwartz, 1987; Majewska et al. 1990). Interestingly, ifenprodil, an activation-dependent NMDA antagonist, also slows deactivation (Kew et al. 1996).
Offset of PS block is slowed by increased GABA concentration
In single-channel recordings, the rate of PS association with GABA receptors has been found to be slow and independent of GABA concentration between 0.05 and 1 mm GABA (Akk et al. 2001). On the other hand, for picrotoxin (Yoon et al. 1993) and the NMDA antagonist ifenprodil (Kew et al. 1996), onset rate reportedly depends on agonist concentration. We explored the onset of PS block during steady-state responses to 2 and 20 μm GABA in whole-cell recordings. We found that the rate of development of PS block was slow and did not depend on the GABA concentration (Fig. 5). On the other hand, offset of block was significantly slowed at 20 μm GABA compared with 2 μm GABA. This slowing was approximately 2-fold (Fig. 5D), in reasonable agreement with the change in steady-state GABA current in the same cells at these two GABA concentrations (Fig. 5E). Therefore, slowing of recovery from block appears to account for the increased PS effects at high GABA concentration.
In contrast to a previous report (Yoon et al. 1993), we found that toxinin exhibited faster onset and offset kinetics at low GABA concentration than at high GABA concentration. Like PS, steady-state block using 10 μm toxinin increased roughly 2-fold with the 10-fold change in GABA concentration. This discrepancy with the older work may arise from the different GABA concentrations used, multiple effects of toxinin (Yoon et al. 1993), or the use of different experimental solutions designed to limit receptor desensitization in the previous work (Yoon et al. 1993).
Onset rate constant depends on PS and toxinin concentration
The rate of block development for PS is well below the estimates of diffusion-limited binding of drug. We therefore tested whether the onset time constant for PS block depends on PS concentration, expected if PS binding is rate limiting for current block (as opposed to a rate-limiting receptor conformational change promoted by PS). We found that across the range of 2.5–20 μm PS, the time constant of block development was dependent on PS concentration (Fig. 6B; P < 0.05, ANOVA). There was no significant relationship between PS concentration and the offset of PS block in the continued presence of GABA (P > 0.5, ANOVA; Fig. 6C). Similar to the relationship between PS concentration and time constant of block development, toxinin, at concentrations chosen to produce equivalent steady-state block of GABA receptors, exhibited a similar dependence of block rate on toxinin concentration (Fig. 6D-F; P < 0.05, ANOVA).
Figure 6. Effect of PS concentration on onset and offset of block in hippocampal neurons.

A-C, protocol was similar to that used in Fig. 5, except that GABA was kept constant at 5 μm, and PS concentration was varied from 2.5 to 20 μm. Statistical differences were evaluated with an ANOVA and are denoted by asterisks (P < 0.05). There was an effect of PS concentration on degree of block and a significant effect of PS on onset but not offset time constant (n = 14–22 cells at each concentration). D–F, picrotoxinin (PTXNIN; 10 μm) was substituted for PS in the experimental protocol. Note that there was a significant effect of picrotoxinin concentration on both the degree of block and the onset time constant (n = 4 cells).
PS and picrotoxinin actions on steady-state GABA responses at high GABA concentrations
PS and picrotoxinin exhibited similar blocking effects on GABA responses at low concentrations of GABA (Fig. 4 and Fig. 6). Although the mechanism(s) of picrotoxinin block have not been conclusively determined, some evidence favours a preferential interaction with open state of the receptor channel (Yoon et al. 1993). One might expect that such a preferential state-dependent association might produce a smaller effect on steady-state GABA responses at very high GABA concentrations, if interactions of the blocker with the open state buffer receptors away from desensitized states. We found a marked difference in the behaviour of PS and picrotoxinin at high (500 μm) GABA concentrations. Both drugs accelerated the fade in GABA current during the initial part of the co-application (Fig. 7), but while 5 μm PS markedly attenuated the steady-state GABA response, 10 μm picrotoxinin had little or no effect on the steady-state GABA response. The result was highlighted by examining the effect of PS or picrotoxinin application during the steady-state phase of the response to 500 μm GABA (Fig. 7C and D). PS application yielded a well-behaved block of the steady-state current, consistent with that observed at lower concentrations of GABA (Fig. 7C). By contrast, picrotoxinin, whilst producing similar block to PS at lower GABA concentrations (Fig. 5E), had a complex effect. Initially picrotoxinin reduced the steady-state current. The block was followed by slow relaxation back to near the steady-state level observed in the absence of picrotoxinin (Fig. 7D). The picrotoxinin results could be explained if open channels are preferentially blocked by picrotoxinin, and this open, blocked state absorbs channels away from the desensitized states. This result strengthens the conclusion that PS and picrotoxinin act through distinct mechanisms.
Figure 7. PS and picrotoxinin effects on responses to prolonged application of high GABA concentration in hippocampal neurons.
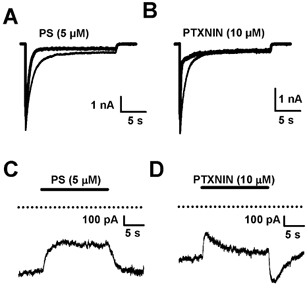
A, PS (5 μm) alters the time course of response to 500 μm GABA and inhibits the steady-state response when co-applied with GABA. B, in contrast, co-application of picrotoxinin (PTXNIN 10 μm; thick trace) with 500 μm GABA speeds the initial phase of the GABA response (control response shown as thin trace) but has little effect on the steady-state GABA response. C and D, responses to PS (C) and pictroxinin (D) application during the steady-state phase of the response to 500 μm GABA. GABA was pre-applied for 20 s before antagonist application. Note the inhibition of the steady-state GABA response by PS and complicated development of block followed by redevelopment of current during picrotoxinin application. Dotted lines indicate holding current level in the absence of GABA.
Effect of PS on IPSC decays
Given that our experiments suggest activation dependence on PS inhibition, we explored whether this activation dependence might be apparent in the course of synaptic transmission. Quantal GABA release results in a very brief transient of neurotransmitter (≈1 ms). When GABA is released with sufficiently large quantal content, spillover among synaptic release sites and/or rebinding of transmitter occurs, causing prolonged activation of GABA receptors (Korn & Dingledine, 1986; Isaacson et al. 1993). If PS actions are facilitated by receptor-channel activation, conditions that promote more or less receptor activation during synaptic release may influence the degree of PS inhibition. On the other hand, the slow actions of PS suggested by experiments like those in Fig. 5 and Fig. 6 might preclude any apparent activation dependence at synapses.
In microcultured autaptic synapses, we evoked GABA release under high and low quantal content conditions and examined PS inhibition. When high external [Ca2+] (4.0 mm) was used to evoke release, baseline IPSCs decayed 55 ± 38 % slower than IPSCs elicited in 1 mm Ca2+ (plus 3 mm Mg2+ to maintain total divalent cation concentration), presumably because of a briefer effective transmitter lifetime under low quantal content conditions (Isaacson et al. 1993). PS had a larger effect on both the peak and the decay of the IPSC when quantal content was elevated (Fig. 8). Decay was also differentially affected in the two conditions when a weighted time constant was used as a measure of decay (P < 0.05, data not shown). The effect did not result from a direct effect of divalent cations on PS block because neither peak nor steady-state PS inhibition of exogenous GABA responses (5 μm) was significantly different in 1 mm Ca2+-3 mm Mg2+versus 4 mm Ca2+-0 mm Mg2+ (n =7; P > 0.3).
To test the idea that GABA persists for a longer time following release under the high quantal content conditions, we performed experiments with a rapidly dissociating competitive antagonist, TPMPA, (Jones et al. 2001). Typically, competitive antagonists act non-competitively against PSCs because of the non-equilibrium conditions at the synapse. Thus, PSC amplitude in the presence of competitive antagonist is proportional to the number of receptors unoccupied by antagonist at the instant of transmitter release. However, previous experiments have shown that rapidly dissociating antagonists become less effective against synaptic currents during elevated transmitter output (Tong & Jahr, 1994; Wadiche & Jahr, 2001; Foster et al. 2002; Harrison & Jahr, 2003), consistent with the idea that transmitter persists long enough to displace some of the antagonist under these conditions. In our experiments we found that 500 μm TPMPA was less effective in high-Ca2+ conditions than in low-Ca2+ conditions, consistent with the idea of differing transmitter lifetimes in the two conditions (Fig. 8C and D). Our results do not distinguish whether the longer effective transmitter lifetime arises from multivesicular release from individual active zones (Wadiche & Jahr, 2001) or summation of transmitter spillover onto areas (possibly extrasynaptic) of receptors shared by multiple release sites (Isaacson et al. 1993; Diamond, 2001). Together, these results are consistent with the idea that PS is more effective when GABA lifetime is increased following synaptic release.
DISCUSSION
Several lines of evidence presented here are consistent with state-dependent block of GABAA receptors by PS. The IC50 for PS decreases in the presence of elevated GABA concentrations. Block of responses to high-efficacy agonists is greater than block of responses to partial agonists at saturating concentrations. Pre-equilibration with PS did not alter inhibition of peak GABA responses, in contrast to some previous results, which have been equivocal on the matter of activation dependence (Woodward et al. 1992; Zaman et al. 1992). These observations could be consistent with an interaction of PS with the open channel pore. This is the most typical mechanism of state-dependent blockers (e.g. Steinbach, 1968; Adams, 1976; Neher & Steinbach, 1978; Huettner & Bean, 1988; MacDonald & Nowak, 1990). Nevertheless, previous single-channel measurements have found no evidence for channel block (Mienville & Vicini, 1989; Akk et al. 2001).
The PS onset rate constant also did not depend on GABA concentration between 2 and 20 μm GABA. Instead, a decrease in the rate of the PS offset was associated with the increased steady-state inhibition at high GABA concentration. This result is contrary to some forms of open channel block, where relief from block is often speeded by increased receptor activation (Gurney & Rang, 1984; Van Renterghem et al. 1987; Huettner & Bean, 1988; Inoue & Akaike, 1988). Picrotoxinin also behaved unexpectedly if channel opening is necessary to relieve block. Both onset and offset of block in the continuous presence of GABA were slowed at higher GABA concentrations. This was different from previous observations of picrotoxin's effects using higher GABA concentrations (Yoon et al. 1993). The behaviour we observed could be related to the presence of both use-dependent and use-independent mechanisms of picrotoxinin block (Yoon et al. 1993), suggesting that, if picrotoxinin acts as an open channel blocker, other effects are present as well.
Some forms of channel block require agonist presence to relieve the block (Lingle, 1983; Gurney & Rang, 1984; Van Renterghem et al. 1987; Huettner & Bean, 1988; Inoue & Akaike, 1988). We observed no evidence that agonist re-application was necessary for recovery from PS block. Following co-application and co-removal of GABA and PS, GABA responses returned quickly and without further agonist re-application to their full size (data not shown). In contrast we, like others (Inoue & Akaike, 1988; Yoon et al. 1993), observed that following simultaneous removal of GABA and picrotoxinin, GABA re-application was necessary for recovery of the response amplitude (data not shown).
One equivocal piece of evidence against channel block is that PS, although charged, fails to show voltage dependence (Fig. 2, Majewska & Schwartz, 1987; Akk et al. 2001). Although channel blockers often show voltage dependence (e.g. Donevan et al. 1992; Parsons et al. 1993), the premise of voltage dependence assumes that the charged moiety of the drug interacts with the channel deeply enough to sense the transmembrane field. It is possible that this is not true for PS, where the anionic group is at one end of the molecule (C-3). However, some sulfated (Fig. 2) and carboxylated (Mennerick et al. 2001) steroids, which share similar structure and apparent mechanism with PS, exhibit significant voltage dependence.
Our conclusions differ from those reached with single-channel cluster analysis, which suggest that, within the margin of measurement and estimation error, PS acts in a slow time-dependent fashion that is independent of whether the channel is open or closed (Akk et al. 2001). This apparent discrepancy may be a result of the differing experimental protocols. The whole-cell correlate of the channel clusters studied by Akk et al. (2001) is unclear. Also, the single-channel data readily estimate the onset rate of PS, but provide no information on the GABA or activation dependence of PS offset. Therefore, activation dependence resulting from a slowing of PS offset (Fig. 5) would not have been detected. Our data appear to agree with single-channel measurements that suggest PS association is slow and independent of GABA concentration.
Superficially, PS shares features with picrotoxinin, and past work has suggested that the two may share a binding site on the GABAA receptor (Majewska & Schwartz, 1987). Also, a PS-insensitive receptor was recently created by a point mutation of the rat α1 subunit in a position analogous to that of an invertebrate picrotoxin-insensitive receptor (Akk et al. 2001). The similarities between the antagonists observed in our work include failure of pre-incubation to substantially increase block (Fig. 4), dependence of steady-state block on agonist concentration (Fig. 1 and Fig. 5), and generally slow kinetics of onset and offset (Fig. 5 and Fig. 6). Dissimilarities include a difference in deactivation kinetics following prolonged GABA application (Fig. 4), a different dependence of onset and offset time constants on GABA concentration (Fig. 5), and qualitatively different behaviour at high GABA concentrations (Fig. 7).
To what extent can the observations made regarding the activation dependence of PS in the present work be incorporated into a mechanistic scheme? Previously, our group used a popular 7-state kinetic model (Jones & Westbrook, 1995; Jones et al. 1998; Mozrzymas et al. 1999; Fig. 9A) and proposed that PS may increase the rate of entry of receptors into a doubly liganded desensitized state (Shen et al. 2000). We explored whether the present results could be incorporated into the same scheme. In the older work (Shen et al. 2000), PS and GABA were present simultaneously, and therefore PS binding was not explicitly included. To account for the lack of difference between pre-applied PS and co-applied PS, we did not allow PS to bind the unliganded state of the receptor (Fig. 9A), but we allowed interaction with bound liganded and open states of the receptor (Fig. 9A). PS-bound states (grey states in Fig. 9A) obeyed the same kinetics as PS-unbound states (black states in Fig. 9A), except that the rate of entry into the doubly liganded desensitized state was speeded by 6-fold (asterisk in Fig. 9A), and the rate of the transition between the two desensitized states was altered to preserve microscopic reversibility.
Figure 9. Simulations of PS effects.

A, kinetic scheme. PS was allowed to bind singly and doubly liganded open and closed states. Following PS binding, entry into the D2 desensitized state (D2P) was speeded by 6-fold (*). The PS association rate constant was 1 μm−1 s−1, and the PS dissociation rate was 5 s−1. Faster rate constants produced fast kinetics and current relaxations that were not experimentally observed. The PS association rate constant is much slower than diffusion-limited rates, but is similar to the rate constant predicted from single-channel measurements (Akk et al. 2001). Other kinetic parameters were the same as previously reported (Mozrzymas et al. 1999; Shen et al. 2000). B, simulation of response to pulse application of 20 μm GABA in the absence and presence of PS for 10 s, beginning 6 s following the onset of the simulation. The thick line is the response to GABA alone. The thin line is the response to GABA plus 5 μm PS. C, magnified view of the deactivation phase of the responses shown in B. The PS deactivation trace has been scaled to the amplitude of the control deactivation trace. Note the slower deactivation of the current in the presence of PS. D, response to increasing PS concentrations applied during the steady-state phase of the response to 20 μm GABA. Responses to 5, 10, and 1000 μm PS (bottom, middle and top traces, respectively) are shown. The period of PS application is denoted by the horizontal bar. Note that the inhibition saturates at less than 100 % block. Peak GABA responses have been truncated to highlight effects on steady-state responses. E, degree of inhibition by 5 μm PS in response to 1 mm full agonist application (GABA) or 1 mm partial agonist (P4S). Partial agonist currents were simulated by slowing channel opening rates by 5—fold. Resulting simulated steady-state currents were approximately 3-fold smaller in response to the partial agonist. Note the simulations predict more inhibition of P4S-gated current than GABA-gated current, contrary to experimental observations. F–J, PS inhibition simulated by PS binding and inhibition of the doubly liganded open state of the receptor. Rate constants of PS binding and dissociation were 1 μm−1 s−1 and 0.5 s−1.
We should note some caveats of the model that we noticed at the outset. Although we used the 7-state scheme with previously published rate constants, a number of inadequacies in the simulations were unmasked by the long agonist applications designed to simulate the present experiments. The amount of macroscopic desensitization observed at low GABA concentrations was larger than experimentally observed, and the amount of macroscopic desensitization to high GABA concentrations was smaller than experimentally observed. Nevertheless, for discussion purposes, we retained the 7-state model because of its heuristic value and for comparison with previous work.
We found that the model accounted for a number of salient features observed in the experimental data. These include the apparent increase in macroscopic desensitization (Fig. 9B), the slowing of current deactivation kinetics upon simultaneous removal of both PS and agonist (Fig. 9C), the increase in steady-state block at elevated GABA concentration (data not shown), the increased onset rate with PS concentration (Fig. 9C), and the decreased offset rate constant with GABA concentration (data not shown). Thus, many major features observed in the experimental data were replicated in the simulations.
On the other hand, at least two salient features of our experimental data were unaccounted for by the simulations. The accelerated desensitization model predicted saturation of inhibition at less than 100 % current block (Fig. 9C). We tested this prediction both in oocytes and at hippocampal autapses and found that inhibition could be driven to 100 % with sufficiently high PS concentration (data not shown). Another important feature unaccounted for by the accelerated desensitization model was the action of P4S (Fig. 9D). Because desensitization occurs from liganded closed states of the receptor in the 7-state scheme, saturating responses to partial agonists, modelled by 5-fold increases in the channel closing rates, were predicted to be more inhibited than saturating responses to full agonist (Fig. 9D), contrary to experimental data (Fig. 1).
Interestingly, we found that a simple interaction of blocker with the doubly liganded open state of the receptor (Fig. 9F) could also reasonably simulate all of the features explained by the accelerated desensitization model listed above (e.g. Fig. 9G and H, and data not shown). In addition, this mechanism accounted for both the observation of 100 % maximum block and the observed P4S effects (Fig. 9I and J). We should note, however, that this mechanism cannot satisfactorily account for the PS-induced kinetic changes previously observed with brief GABA applications (Shen et al. 2000), or apparently with single-channel data (Akk et al. 2001). For instance, the model shown in Fig. 9F predicts slowing of responses to synapse-like pulses of GABA, which is never experimentally observed (Fig. 8). Therefore, neither scheme simulates all PS actions. It is possible that alternative schemes of GABAA receptor gating, perhaps incorporating desensitized states proceeding directly from open states, might more satisfactorily incorporate all the past and present observations regarding PS effects.
In summary, we find that the endogenous steroid PS acts in an activation-dependent manner to block GABAA receptor-mediated currents. The activation dependence is distinct from that of picrotoxinin, which has been suggested to share a mechanism with PS. At present it is difficult to incorporate all past and present observations into a single mechanism of PS block.
Acknowledgments
We thank Gustav Akk, Doug Covey, and lab members for discussion, and particularly Joe Henry Steinbach for critical reading of an early version of the manuscript. We thank Ann Benz and Amanda Shute for technical support. This work was supported by GM47969, AA12951, and a gift from the Bantly Foundation (C.F.Z.) and by the Klingenstein Fund, NS40488 and AA12952 (S.M.).
REFERENCES
- Adams PR. Drug blockade of open end-plate channels. J Physiol. 1976;260:531–552. doi: 10.1113/jphysiol.1976.sp011530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Bracamontes J, Steinbach JH. Pregnenolone sulfate block of GABAA receptors: mechanism and involvement of a residue in the M2 region of the α subunit. J Physiol. 2001;532:673–684. doi: 10.1111/j.1469-7793.2001.0673e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulieu EE, Robel P. Neurosteroids: a new brain function. J Steroid Biochem Mol Biol. 1990;37:395–403. doi: 10.1016/0960-0760(90)90490-c. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci U S A. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie SB, De Blas AL. α5 Subunit-containing GABAA receptors form clusters at GABAergic synapses in hippocampal cultures. Neuroreport. 2002;13:2355–2358. doi: 10.1097/00001756-200212030-00037. [DOI] [PubMed] [Google Scholar]
- Corpechot C, Collins BE, Carey MP, Tsouros A, Robel P, Fry JP. Brain neurosteroids during the mouse oestrous cycle. Brain Res. 1997;766:276–280. doi: 10.1016/s0006-8993(97)00749-x. [DOI] [PubMed] [Google Scholar]
- Diamond JS. Neuronal glutamate transporters limit activation of NMDA receptors by neurotransmitter spillover on CA1 pyramidal cells. J Neurosci. 2001;21:8328–8338. doi: 10.1523/JNEUROSCI.21-21-08328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibas MI, Gonzales EB, Das P, Bell-Horner CL, Dillon GH. Identification of a novel residue within the second transmembrane domain that confers use-facilitated block by picrotoxin in glycine alpha 1 receptors. J Biol Chem. 2002;277:9112–9117. doi: 10.1074/jbc.M111356200. [DOI] [PubMed] [Google Scholar]
- Donevan SD, Jones SM, Rogawski MA. Arcaine blocks N-methyl-D-aspartate receptor responses by an open channel mechanism: whole-cell and single-channel recording studies in cultured hippocampal neurons. Mol Pharmacol. 1992;41:727–735. [PubMed] [Google Scholar]
- Ebert B, Wafford KA, Whiting PJ, Krogsgaard-Larsen P, Kemp JA. Molecular pharmacology of γ-aminobutyric acid type A receptor agonists and partial agonists in oocytes injected with different α, β, and γ receptor subunit combinations. Mol Pharmacol. 1994;46:957–963. [PubMed] [Google Scholar]
- Ffrench-Constant RH, Rocheleau TA, Steichen JC, Chalmers AE. A point mutation in a Drosophila GABA receptor confers insecticide resistance. Nature. 1993;363:449–451. doi: 10.1038/363449a0. [DOI] [PubMed] [Google Scholar]
- Foster KA, Kreitzer AC, Regehr WG. Interaction of postsynaptic receptor saturation with presynaptic mechanisms produces a reliable synapse. Neuron. 2002;36:1115–1126. doi: 10.1016/s0896-6273(02)01106-6. [DOI] [PubMed] [Google Scholar]
- Gurney AM, Rang HP. The channel-blocking action of methonium compounds on rat submandibular ganglion cells. Br J Pharmacol. 1984;82:623–642. doi: 10.1111/j.1476-5381.1984.tb10801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison J, Jahr CE. Receptor occupancy limits synaptic depression at climbing fiber synapses. J Neurosci. 2003;23:377–383. doi: 10.1523/JNEUROSCI.23-02-00377.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettner JE, Bean BP. Block of N-methyl-D-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proc Natl Acad Sci U S A. 1988;85:1307–1311. doi: 10.1073/pnas.85.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Akaike N. Blockade of γ-aminobutyric acid-gated chloride current in frog sensory neurons by picrotoxin. Neurosci Res. 1988;5:380–394. doi: 10.1016/0168-0102(88)90024-7. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Solis JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- Jones MV, Jonas P, Sahara Y, Westbrook GL. Microscopic kinetics and energetics distinguish GABA(A) receptor agonists from antagonists. Biophys J. 2001;81:2660–2670. doi: 10.1016/S0006-3495(01)75909-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Sahara Y, Dzubay JA, Westbrook GL. Defining affinity wth the GABAA receptor. J Neurosci. 1998;18:8590–8604. doi: 10.1523/JNEUROSCI.18-21-08590.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- Kew JN, Trube G, Kemp JA. A novel mechanism of activity-dependent NMDA receptor antagonism describes the effect of ifenprodil in rat cultured cortical neurones. J Physiol. 1996;497:761–772. doi: 10.1113/jphysiol.1996.sp021807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn SJ, Dingledine R. Inhibition of GABA uptake in the rat hippocampal slice. Brain Res. 1986;368:247–255. doi: 10.1016/0006-8993(86)90568-8. [DOI] [PubMed] [Google Scholar]
- Lingle C. Blockade of cholinergic channels by chlorisondamine on a crustacean muscle. J Physiol. 1983;339:395–417. doi: 10.1113/jphysiol.1983.sp014723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JF, Nowak LM. Mechanisms of blockade of excitatory amino acid receptor channels. Trends Pharmacol Sci. 1990;11:167–172. doi: 10.1016/0165-6147(90)90070-O. [DOI] [PubMed] [Google Scholar]
- Majewska MD, Demirgoren S, London ED. Binding of pregnenolone sulfate to rat brain membranes suggests multiple sites of steroid action at the GABAA receptor. Eur J Pharmacol. 1990;189:307–315. doi: 10.1016/0922-4106(90)90124-g. [DOI] [PubMed] [Google Scholar]
- Majewska MD, Mienville JM, Vicini S. Neurosteroid pregnenolone sulfate antagonizes electrophysiological responses to GABA in neurons. Neurosci Lett. 1988;90:279–284. doi: 10.1016/0304-3940(88)90202-9. [DOI] [PubMed] [Google Scholar]
- Majewska MD, Schwartz RD. Pregnenolone-sulfate: an endogenous antagonist of the γ-aminobutyric acid receptor complex in brain. Brain Res. 1987;404:355–360. doi: 10.1016/0006-8993(87)91394-1. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Que J, Benz A, Zorumski CF. Passive and synaptic properties of neurons grown in microcultures and in mass cultures. J Neurophysiol. 1995;73:320–332. doi: 10.1152/jn.1995.73.1.320. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Zeng CM, Benz A, Shen W, Izumi Y, Evers AS, Covey DF, Zorumski CF. Effects on γ-aminobutyric acid (GABA)(A) receptors of a neuroactive steroid that negatively modulates glutamate neurotransmission and augments GABA neurotransmission. Mol Pharmacol. 2001;60:732–741. [PubMed] [Google Scholar]
- Mensah-Nyagan AG, Do-Rego JL, Beaujean D, Luu-The V, Pelletier G, Vaudry H. Neurosteroids: expression of steroidogenic enzymes and regulation of steroid biosynthesis in the central nervous system. Pharmacol Rev. 1999;51:63–81. [PubMed] [Google Scholar]
- Mienville JM, Vicini S. Pregnenolone sulfate antagonizes GABAA receptor-mediated currents via a reduction of channel opening frequency. Brain Res. 1989;489:190–194. doi: 10.1016/0006-8993(89)90024-3. [DOI] [PubMed] [Google Scholar]
- Mozrzymas JW, Barberis A, Michalak K, Cherubini E. Chlorpromazine inhibits miniature GABAergic currents by reducing the binding and by increasing the unbinding rate of GABAA receptors. J Neurosci. 1999;19:2474–2488. doi: 10.1523/JNEUROSCI.19-07-02474.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Steinbach JH. Local anaesthetics transiently block currents through single acetylcholine -receptor channels. J Physiol. 1978;277:153–176. doi: 10.1113/jphysiol.1978.sp012267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newland CF, Cull-Candy SG. On the mechanism of action of picrotoxin on GABA receptor channels in dissociated sympathetic neurones of the rat. J Physiol. 1992;447:191–213. doi: 10.1113/jphysiol.1992.sp018998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons CG, Gruner R, Rozental J, Millar J, Lodge D. Patch clamp studies on the kinetics and selectivity of N-methyl-D-aspartate receptor antagonism by memantine (1-amino-3,5-dimethyladamantan) Neuropharmacology. 1993;32:1337–1350. doi: 10.1016/0028-3908(93)90029-3. [DOI] [PubMed] [Google Scholar]
- Robel P, Schumacher M, Baulieu E-E. Neurosteroids: from definition and biochemistry to physiopathological function. In: Baulieu E-E, Robel P, Schumacher M, editors. Neurosteroids: A New Regulatory Function in the Nervous System. Totowa, NJ, USA: Humana Press; 1999. pp. 1–25. [Google Scholar]
- Shen W, Mennerick S, Covey DF, Zorumski CF. Pregnenolone sulfate modulates inhibitory synaptic transmission by enhancing GABAA receptor desensitization. J Neurosci. 2000;20:3571–3579. doi: 10.1523/JNEUROSCI.20-10-03571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen WX, Mennerick S, Zorumski EC, Covey DF, Zorumski CF. Pregnenolone sulfate and dehydroepiandrosterone sulfate inhibit GABA-gated chloride currents in Xenopus oocytes expressing picrotoxin-insensitive GABA(A) receptors. Neuropharmacology. 1999;38:267–271. doi: 10.1016/s0028-3908(98)00172-5. [DOI] [PubMed] [Google Scholar]
- Steinbach AB. A kinetic model for the action of xylocaine on receptors for acetylcholine. J Gen Physiol. 1968;52:162–180. doi: 10.1085/jgp.52.1.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong G, Jahr CE. Multivesicular release from excitatory synapses of cultured hippocampal neurons. Neuron. 1994;12:51–59. doi: 10.1016/0896-6273(94)90151-1. [DOI] [PubMed] [Google Scholar]
- Van Renterghem C, Bilbe G, Moss S, Smart TG, Constanti A, Brown DA, Barnard EA. GABA receptors induced in Xenopus oocytes by chick brain mRNA: evaluation of TBPS as a use-dependent channel-blocker. Brain Res. 1987;388:21–31. doi: 10.1016/0169-328x(87)90017-9. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE. Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron. 2001;32:301–313. doi: 10.1016/s0896-6273(01)00488-3. [DOI] [PubMed] [Google Scholar]
- Wang M, He Y, Eisenman LN, Fields C, Zeng CM, Mathews J, Benz A, Fu T, Zorumski E, Steinbach JH, Covey DF, Zorumski CF, Mennerick S. 3beta-hydroxypregnane steroids are pregnenolone sulfate-like GABA(A) receptor antagonists. J Neurosci. 2002;22:3366–3375. doi: 10.1523/JNEUROSCI.22-09-03366.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MD, Wahlstrom G, Backstrom T. The regional brain distribution of the neurosteroids pregnenolone and pregnenolone sulfate following intravenous infusion. J Steroid Biochem Mol Biol. 1997;62:299–306. doi: 10.1016/s0960-0760(97)00041-1. [DOI] [PubMed] [Google Scholar]
- Woodward RM, Polenzani L, Miledi R. Effects of steroids on γ-aminobutyric acid receptors expressed in Xenopus oocytes by poly(A)+ RNA from mammalian brain and retina. Mol Pharmacol. 1992;41:89–103. [PubMed] [Google Scholar]
- Yoon KW, Covey DF, Rothman SM. Multiple mechanisms of picrotoxin block of GABA-induced currents in rat hippocampal neurons. J Physiol. 1993;64:423–439. doi: 10.1113/jphysiol.1993.sp019643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman SH, Shingai R, Harvey RJ, Darlison MG, Barnard EA. Effects of subunit types of the recombinant GABAA receptor on the response to a neurosteroid. Eur J Pharmacol. 1992;225:321–330. doi: 10.1016/0922-4106(92)90106-6. [DOI] [PubMed] [Google Scholar]
- Zhang HG, Ffrench-Constant RH, Jackson MB. A unique amino acid of the Drosophila GABA receptor with influence on drug sensitivity by two mechanisms. J Physiol. 1994;479:65–75. doi: 10.1113/jphysiol.1994.sp020278. [DOI] [PMC free article] [PubMed] [Google Scholar]