

Abstract
In the heart, reperfusion following an ischaemic episode can result in a marked increase in [Ca2+]i and cause myocyte dysfunction and death. Although the Na+–Ca2+ exchanger has been implicated in this response, the ionic mechanisms that are responsible have not been identified. In this study, the hypothesis that the diastolic membrane potential can influence Na+–Ca2+ exchange and Ca2+ homeostasis during chemically induced hypoxia–reoxygenation has been tested using right ventricular myocytes isolated from adult rat hearts. Superfusion with selected [K+]o of 0.5, 2.5, 5, 7, 10 and 15 mm yielded the following resting membrane potentials: −27.6 ± 1.63 mV, −102.2 ± 1.89, −86.5 ± 1.03, −80.1 ± 1.25, −73.6 ± 1.02 and −66.4 ± 1.03, respectively. In a second set of experiments myocytes were subjected to chemically induced hypoxia–reoxygenation at these different [K+]o, while [Ca2+]i was monitored using fura-2. These results demonstrated that after chemically induced hypoxia–reoxygenation had caused a marked increase in [Ca2+]i, hyperpolarization of myocytes with 2.5 mm [K+]o significantly reduced [Ca2+]i (7.5 ± 0.32 vs. 16.9 ± 0.55 %); while depolarization (with either 0.5 or 15 mm [K+]o) significantly increased [Ca2+]i (31.8 ± 3.21 and 20.8 ± 0.36 vs. 16.9 ± 0.55 %, respectively). As expected, at depolarized membrane potentials myocyte hypercontracture and death increased in parallel with Ca2+ overload. The involvement of the Na+–Ca2+ exchanger in Ca2+ homeostasis was evaluated using the Na+–Ca2+ exchanger inhibitor KB-R7943. During reoxygenation KB-R7943 (5 μm) almost completely prevented the increase in [Ca2+]i both in control conditions (in 5 mm [K+]o: 2.2 ± 0.40 vs. 10.8 ± 0.14 %) and in depolarized myocytes (in 15 mm [K+]o: −2.1 ± 0.51 vs. 11.3 ± 0.05 %). These findings demonstrate that the resting membrane potential of ventricular myocytes is a critical determinant of [Ca2+]i during hypoxia–reoxygenation. This appears to be due mainly to an effect of diastolic membrane potential on the Na+–Ca2+ exchanger, since at depolarized potentials this exchanger mechanism operates in the reverse mode, causing a significant Ca2+ influx.
Although restoration of coronary blood flow is essential for the survival of ischaemic myocardium, reperfusion often results in enhanced cardiac dysfunction and/or irreversible cell damage (Park & Lucchesi, 1999). Rapid development of abnormal intracellular Ca2+ homeostasis represents a major pathway leading to this reperfusion injury (Tani, 1990), and changes in sarcolemmal Na+–Ca2+ exchange constitute a likely candidate for this abnormal Ca2+ regulation (Eigel & Hadley, 2001).
Under physiological conditions, the primary function of the sarcolemmal Na+–Ca2+ exchanger in mammalian heart is to remove Ca2+ from the myoplasm during diastole (Blaustein & Lederer, 1999). However, the Na+–Ca2+ exchanger can also work in reverse mode; as a result it can contribute to significant Ca2+ influx, and raise intracellular calcium during reperfusion (Schäfer et al. 2001; Eigel & Hadley, 2001). The resulting calcium overload has been shown to contribute to: (i) reperfusion injury as well as ventricular arrhythmias in isolated rabbit hearts (Elias et al. 2001), (ii) cardiac stunning in isolated perfused ferret hearts (Kusuoka et al. 1993) and (iii) hypercontracture and death of rat ventricular myocytes (Schäfer et al. 2001; Inserte et al. 2002).
Several different mechanisms can alter the direction and/or rate of Na+–Ca2+ exchange during hypoxia- reoxygenation. For example, hypoxia can lead to the acidification of the myoplasm, which activates the Na+-H+ exchanger upon reoxygenation (Park et al. 1999). The subsequent increase in [Na+]i may result in reverse mode Na+–Ca2+ exchanger activity. Previous studies have demonstrated that the activation of reverse mode Na+–Ca2+ exchanger activity upon reperfusion is a secondary result of [Na+]i accumulation, caused by stimulation of the Na+-H+ exchanger during ischaemia- reperfusion (Tani & Neely, 1989; Murphy et al. 1991), and block of the Na+-H+ exchanger can protect against ischaemia–reperfusion injury (Karmazyn, 1999; Buerke et al. 1999). The generation of oxygen-derived free radicals has also been implicated in enhancing Na+–Ca2+ exchange in isolated ventricular myocytes (Goldhaber, 1996; Zeitz et al. 2002).
The direction of the electrogenic Na+–Ca2+ exchanger current is dependent on the [Na+] and [Ca2+] electrochemical gradients, as well as the membrane potential. Under physiological conditions, the calculated reversal potential for a 3 Na+:1 Ca2+ exchange mechanism is approximately −40 mV in cardiac myocytes (Blaustein & Lederer, 1999). Ischaemia can lead to sustained increases of [K+] in the T-tubular and extracellular spaces as a result of significant K+ loss from ischaemic cells in the setting of impaired perfusion (Wilde & Aksnes, 1995). In a recent study, Cascio et al. (2001) showed that ischaemia causes extracellular K+ accumulation and depolarization of the membrane potential (Vm) (e.g. when [K+]o was 14 mm, Vm depolarized by 20 mV after 30 min of ischaemia), resulting in intermittent failure of conduction in coronary perfused rabbit papillary muscle. In this milieu, slowed conduction and altered refractoriness can cause re-entrant ventricular arrhythmias and may contribute to sudden cardiac death (Janse & Wit, 1989; Carmeliet, 1999). The Na+–Ca2+ exchanger inhibitor, KB-R7943, has been shown to reduce reperfusion injury and Ca2+ overload in ventricular myocytes (Mukai et al. 2000; Satoh et al. 2000). However, recent studies have not addressed how sustained depolarization of the resting membrane potential can alter calcium fluxes due to Na+–Ca2+ exchange during reperfusion.
In the present experiments, the hypothesis that the diastolic membrane potential of cardiac myocytes can significantly modulate the extent of hypoxia- reoxygenation-induced Ca2+ overload by altering mode-selective Na+–Ca2+ exchanger activity was evaluated. The results consistently show that hyperpolarization of rat ventricular myocytes can protect against (i) reoxygenation-induced Ca2+ overload and (ii) cellular hypercontracture. In contrast, depolarization of the diastolic potential augments Ca2+ overload, and increases the incidence of cell hypercontracture and death during reoxygenation. These findings and the observation that KB-R7943, an inhibitor of Na+–Ca2+ exchange, prevents Ca2+ overload during reoxygenation at both normal and depolarized resting membrane potentials confirm the involvement of the Na+–Ca2+ exchanger in this important aspect of diastolic function/dysfunction in mammalian ventricle (Zile & Brutsaert, 2002).
METHODS
Resting membrane potential measurements
Adult rats were killed with pentobarbital (150 mg kg−1, I.P.) according to the University of Alberta Animal Policy and Welfare Committee and Canadian Council on Animal Care (CCAC) Guidelines. The hearts were removed and right ventricular myocytes were then obtained by enzymatic dissociation using standard protocols, which have been described previously (Bouchard et al. 1993; Light et al. 1998). The resting membrane potentials of these freshly isolated myocytes were measured using the perforated patch-clamp technique. Amphotericin B (Sigma Chemical Co., St Louis, MO, USA) was dissolved in dimethyl sulphoxide (40 mg ml−1) and diluted into the pipette solution immediately before use to give a final concentration of 80 μg ml−1. Pipettes were then back-filled with a solution containing the following (mm): KCl 10, potassium aspartate 130, Hepes 10, MgCl2 1.4, EGTA 1, glucose 10. The pH of the solution was adjusted to 7.2 with KOH. Patch pipettes were pulled using borosilicate glass shanks (G85150T, Warner Instrument Corp.) to yield pipettes with a resistance of 2–6 MΩ when filled with pipette solution. After a gigaohm seal was formed, series resistance was monitored to assess perforation; an access resistance of < 20 MΩ was deemed acceptable. Junction potentials were corrected before seal formation. Membrane potential recordings were made in current-clamp mode using an Axopatch 200B patch-clamp amplifier controlled with Clampex 8.0 software (Axon Instruments, Foster City, CA, USA) for data acquisition and analysis. Data were sampled at 200 Hz, filtered at 50 Hz, digitized (Digidata 1320A, Axon Instruments) and stored on computer. Cells were superfused with control solution (pH 7.4) containing (mm): NaCl 140, Hepes 10, CaCl2 1.0, MgCl2 1.4, glucose 10; KCl levels were adjusted to 0.5, 2.5, 5, 7, 10 and 15 mm to selectively alter membrane potential. Myocytes were directly exposed to each superfusate via a multi-input perfusion pipette. The time to change the solution at the tip of the recording pipette was approximately 2 s. All patch-clamp experiments were performed at room temperature (22 ± 1 °C).
Measurements of [Ca2+]i
A sustained increase in [Ca2+]i has been found to correlate closely with cell mortality in the setting of ischaemia–reperfusion (for a review, see Bolli & Marbán, 1999). Thus, continuous assessment of [Ca2+]i has been used as an indicator of Ca2+ homeostasis in a number of models of chemically induced hypoxia–reoxygenation (Jovanovic et al. 1998). Right ventricular myocytes from adult rats were loaded for 15 min with the esterified form of the Ca2+-sensitive fluorescent probe fura-2 AM (2 μm, dissolved in a 50 % dimethyl sulfoxide and 50 % v/v pluronic acid mixture; Molecular Probes, Eugene, OR, USA). After loading, cells were washed and placed on coverslips for observation at × 200 magnification with an inverted microscope (Olympus, CK40), while being superfused with the control solution containing (mm): NaCl 140, KCl 5, Hepes 10, CaCl2 2.0, MgCl2 1.4, glucose 10, or with selected test solutions. A Photon Technology International (PTI, Lawrenceville, NJ, USA) imaging system with PTI Imagemaster software (version 1.49) was used for data acquisition and analysis. Fura-2 was excited with alternating 340 and 380 nm wavelengths of light, and the emitted light intensity at 520 nm was digitized and stored. An estimate of [Ca2+]i was obtained from the calculated 340 nm/380 nm ratio. Normalized values were expressed as the ratio F(test)/F(control), where F(test) is the image ratio of 340 nm/380 nm at a given time point in test conditions and F(control) is the mean image ratio of 340 nm/380 nm measured during the stable 2 min normoxic period before the application of the solutions which resulted in hypoxic insult. In each successful protocol myocytes were subjected to 8 min of chemically induced hypoxia (5 mm 2-deoxy-glucose and 4 mm NaCN) and then 8 min of reoxygenation in the normal solution. All experimental solutions contained 100 μm probenecid, an inhibitor of the uric acid transporter system, to slow fura-2 AM leakage from the cells (Di Virgilio et al. 1990). Results from only those myocytes that did not develop hypercontracture during chemically induced hypoxia- reoxygenation are included in the figures which illustrate changes in [Ca2+]i during chemically induced hypoxia or reoxygenation.
Drugs and chemicals
KB-R7943 (Tocris, Ellisville, MO, USA) and probenecid (Sigma Chemical Co.) were made up as 5 and 100 mm stock solutions in dimethyl sulfoxide, respectively. Pluronic F-127 (Molecular Probes) was prepared as a 20 % w/v stock solution in dimethyl sulfoxide. Each stock solution was diluted to the required concentration immediately before use.
Statistics
Data are expressed as means ± s.e.m. Statistical significance was evaluated using Student's unpaired and paired t tests, as appropriate, with Origin 6.0 software. A level of P < 0.05 was considered to be statistically different.
RESULTS
Inhibition of Na+–Ca2+ exchange protects myocytes against reoxygenation-induced Ca2+ overload
Chemically induced hypoxia (8 min) caused a progressive increase in [Ca2+]i (6.1 ± 0.29 %). This was followed by a further, relatively large rise in [Ca2+]i upon reoxygenation (10.8 ± 0.14 %). Application of the Na+–Ca2+ exchanger inhibitor (Elias et al. 2001) KB-R7943 (5 μm) in the reoxygenation solution significantly reduced this reoxygenation-induced Ca2+ overload (2.2 ± 0.40 vs. 10.8 ± 0.14 % in controls, P < 0.05; Fig. 1). This finding is similar to the results of Schäfer et al. (2001). In a different group of cells, KB-R7943 (5 μm) was applied during both chemically induced hypoxia and reoxygenation to determine whether the observed increase in [Ca2+]i was also sensitive to the Na+–Ca2+ exchanger inhibitor during chemically induced hypoxia. The pattern of Ca2+ load in these experiments was virtually identical to that observed when KB-R7943 was applied only in the reoxygenation solution (Fig. 1). These results suggest that the Ca2+ load in chemically induced hypoxia and reoxygenation do not share the same mechanism and confirm the involvement of the Na+–Ca2+ exchanger in the reoxygenation-induced increase in [Ca2+]i. In these experiments some myocytes developed hypercontracture, which was preceded by a rapid increase in [Ca2+]i. These myocytes were no longer viable, thus they were excluded from evaluation of [Ca2+]i as a function of [K+]o (Fig. 4C). All solutions contained 5 mm [K+]o in the experiments shown in Fig. 1.
Figure 1. Chemically induced hypoxia followed by reoxygenation results in Ca2+ overload in rat ventricular myocytes.

[Ca2+]i measurements based on the fura-2 AM ratiometric dye (i) in control conditions, (ii) during 8 min chemically induced hypoxia (CIH), and (iii) following 8 min reoxygenation. KB-R7943 (5 μm), a Na+–Ca2+ exchanger inhibitor, was applied during CIH and reoxygenation or during reoxygenation only. All solutions contained 5 mm [K+]o. * P < 0.05 vs. Control, n = 6 experiments, 2–5 cells per experiment.
Figure 4. Chemically induced hypoxia followed by reoxygenation results in changes in [Ca2+]i in rat ventricular myocytes at four different [K+]o levels.
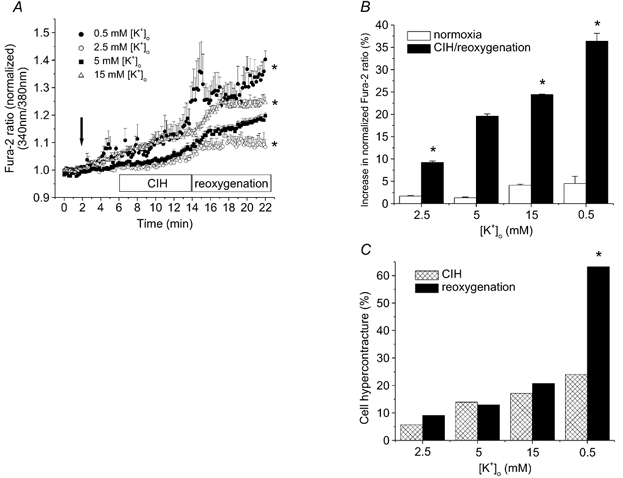
A, [Ca2+]i measurements (i) under control conditions (after 2 min in 5 mm [K+]o), (ii) after 4 min in each selected [K+]o, and (iii) during 8 min chemically induced hypoxia (CIH), (iv) followed by 8 min reoxygenation. The arrow denotes the time when [K+]o was changed. B, increase in [Ca2+]i load during the 4 min normoxic and 8 min CIH per 8 min reoxygenation period at selected [K+]o. The last three points of reoxygenation and of the period preceding CIH from panel A were used to construct panel B. C, effect of [K+]o on myocyte viability during hypoxia-reoxygenation. Hypercontracture was defined as a marked reduction in cell length. * P < 0.05 vs. control (5 mm [K+]o), n = 6 experiments and 2–6 cells per experiment.
Effect of selected [K+]o on resting membrane potentials of ventricular myocytes
To test whether intracellular Ca2+ homeostasis can be modulated significantly by diastolic membrane potential, a straightforward method of controlling membrane potential during Ca2+ imaging experiments was necessary. This was achieved based on the fact that the diastolic membrane potential of rat ventricular myocytes is controlled by an inwardly rectifying K+ conductance (Pandit et al. 2001). Thus, the diastolic potentials can be adjusted to predictable values using selected [K+]o in the experimental solutions. Furthermore, populations of myocytes can be depolarized in either high (e.g. 15 mm) or low (e.g. 0.5 mm) [K+]o thus removing [K+]oper se as an experimental variable. To confirm this rationale, the resting membrane potentials of isolated rat ventricular myocytes were measured at 0.5, 2.5, 5, 7, 10 and 15 mm [K+]o using the perforated patch-clamp technique. Figure 2 shows the effect of changing [K+]o on resting membrane potentials. As expected, superfusion of cells with 7, 10 and 15 mm [K+]o caused depolarization of the cell, while 2.5 mm [K+]o caused a marked hyperpolarization (Fig. 2A, Table 1).
Figure 2. Resting membrane potentials at selected [K+]o in rat ventricular myocytes.
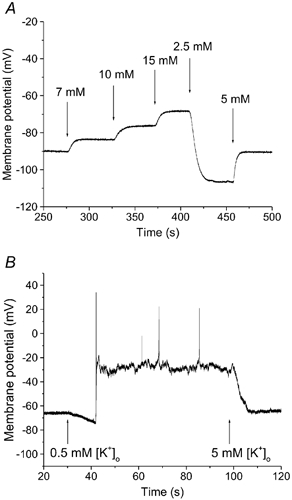
A, representative perforated patch recording from a quiescent right ventricular myocyte in response to selected changes in [K+]o, between 2.5 and 15 mm. Increases in [K+]o produced depolarization; while 2.5 mm [K+]o caused hyperpolarization of the resting membrane potential. B, representative recording from a rat right ventricular myocyte at 5 and 0.5 mm [K+]o. Note that, as expected, in 0.5 mm [K+]o after an initial hyperpolarization, the myocyte depolarized, and fired a number of action potentials. Arrows indicate the start of application of each [K+]o.
Table 1.
Resting membrane potentials at selected [k+]0 in isolated ventricular myocytes
| 0.5 mm | 2.5 mm | 5 mm | 7 mm | 10 mm | 15 mm | |
|---|---|---|---|---|---|---|
| Measured | −27.6 ± 1.63 | −102.2 ± 1.89 | −86.5 ± 1.03 | −80.1 ± 1.25 | −73.6 ± 1.02 | −66.4 ± 1.03 |
| Calculated | −143.2 | −102.3 | −84.7 | −76.1 | −67.0 | −56.7 |
For all groups, n = 7. Membrance patentials (mV) were determinded at 22°C while Ca2+ -imaging experiments were conducted at 35°C. The resting membrance potentials at 35°C, calculated using the Nernst equation, for the selected [K+]0 were: 0.5 mm, –149.5 mV; 2.5 mm, –168.8 mV; 5 mm, –88.4 mV; 7mM, –79.5 mV; 10 mm, –70.0 mV; 15 mm, –59.3 mV. The deviation of the measured resting membrance potential from the calculated value seen in 0.5 mm [K+]0 was expected due to the K+ -sensing function of the invwardly rectifying background K+ current, IK1 (Pandit et al. 2001). Based on these findings, in the Ca2+ -imaging experiments, diastolic membrance potential was altered by changing [K+]0.
Superfusion of cells with 0.5 mm [K+]o caused an initial hyperpolarization, as predicted. However, as [K+]o decreased progressively (e.g. in the T-tubules), the well known non-linearity in the inwardly rectifying background K+ current (IK1) and the fact that this [K+]o approached the Kd for the external K+ site of the Na+-K+ pump (Eisner & Lederer, 1979), caused marked membrane potential depolarization. This depolarization caused the myocytes to spontaneously fire action potentials (Fig. 2B). Table 1 shows the relationship between the measured resting membrane potentials and the equilibrium potentials calculated using the Nernst equation for K+ at selected [K+]o. Presumably the depolarization in 0.5 mm [K+]o is caused by (i) diminished turnover of the electrogenic Na+-K+ pump, and (ii) the well known ‘cross-over’ effect of the inwardly rectifying background K+ conductance in these ventricular myocytes (Pandit et al. 2001).
Effect of chemically induced hypoxia–reoxygenation at 5 mm [K+]o on the resting membrane potential of ventricular myocytes
Eight myocytes from four different animals were subjected to 8 min of chemically induced hypoxia and 8 min reoxygenation, and their membrane potential was recorded using the perforated patch-clamp technique. Traces from three different myocytes shown in Fig. 3 indicate the range of responses that we observed. In all cases, the myocytes depolarized by a relatively small amount during chemically induced hypoxia (−69.1 ± 1.94 mV at 8 min of hypoxia vs. −74.3 ± 0.64 mV in 2 min control, n = 8, P < 0.05). In most cases, the characteristic ‘two stable membrane potential’ behaviour of a cell in which the resting potential is generated by a strong inward rectifier was observed. In some cases, provided that this spontaneous, mode switching of the resting potential was sufficiently slow, brief bursts of spontaneous action potentials were also recorded (Fig. 3).
Figure 3. Membrane potential during chemically induced hypoxia-reoxygenation in rat ventricular myocytes.

Representative whole cell (perforated patch) recordings from right ventricular myocytes at 5 mm [K+]o in control conditions, during 8 min chemically induced hypoxia (CIH), and following 8 min reoxygenation. Traces from three cells (A, B and C) indicate the range of responses observed. Note the 5 to 10 mV depolarization that occurred in all cells. The ‘two stable membrane potential’ behaviour of these ventricular myocytes was seen in most cases and is characteristic of cells expressing a strong inwardly rectifying K+ current. Bursts of spontaneous action potentials were seen in 4 out of 8 recordings.
Resting membrane potential and chemically induced hypoxia–reoxygenation Ca2+ overload
Our data describing the membrane potentials of normoxic myocytes at selected [K+]o levels provided the basis for assessing the role of diastolic membrane potential in reoxygenation-induced Ca2+ overload. In these experiments, the myocytes were first superfused with a control solution containing 5 mm [K+]o. The membrane potential of each population of cardiac myocytes was then changed by application (4 min) of one of the following [K+]o: 0.5, 2.5, 5 or 15 mm. These myocytes were then subjected to the chemically induced hypoxia- reoxygenation protocol (described above) at these selected [K+]o. Changing [K+]o did not significantly change [Ca2+]i during normoxic conditions (2.5 mm: 1.7 ± 0.08 %; 5 mm: 1.3 ± 0.17 %; 15 mm: 4.1 ± 0.24 %; 0.5 mm: 4.5 ± 1.61 %, P > 0.05; Fig. 4B). However, depolarization of the myocytes during superfusion with either 15 or 0.5 mm [K+]o significantly increased [Ca2+]i (11.8 ± 0.05 and 15.2 ± 1.92 % vs. 10.8 ± 0.14 % in controls, respectively, P < 0.05), while the hyperpolarization in 2.5 mm [K+]o significantly reduced Ca2+ overload following reoxygenation (4.2 ± 0.48 vs. 10.8 ± 0.14 % in controls, P < 0.05; Fig. 4A). The combined hypoxia–reoxygenation-induced increase in [Ca2+]i was significantly higher in myocytes superfused with either 15 or 0.5 mm [K+]o (20.8 ± 0.36 and 31.8 ± 3.21 % vs. 16.9 ± 0.55 % in controls, respectively, P < 0.05) and significantly lower in myocytes superfused with 2.5 mm [K+]o (7.5 ± 0.32 vs. 16.9 ± 0.55 % in controls, P < 0.05).
Effect of resting membrane potential and KB-R7943 on myocyte viability
As we expected, in this study there was a strong correlation between raised [Ca2+]i, hypercontracture and depolarization of the myocyte during both chemically induced hypoxia and reoxygenation. We use the term hypercontracture to describe myocytes exhibiting pathophysiological behaviour including dramatic cell shortening, rounding up or losing their rod-shaped cellular morphology, as well as to convey that this process was completely irreversible. Figure 4C shows the percentage of myocytes that hypercontracted during the hypoxia- reoxygenation protocol as a function of [K+]o. Application of the Na+–Ca2+ exchange inhibitor KB-R7943 (5 μm) (Iwamoto et al. 1996) markedly decreased the number of myocytes exhibiting hypercontracture. In fact, no myocytes hypercontracted during reoxygenation when KB-R7943 was included in the 5 mm [K+]o solution, and only 7 % of these myocytes hypercontracted during the depolarization caused by 15 mm [K+]o when KB-R7943 was present during reoxygenation.
KB-R7943 prevents reoxygenation-induced increase in [Ca2+]i at depolarized membrane potentials
As indicated, superfusion of myocytes with 15 mm [K+]o caused a marked depolarization of the resting membrane potential (Fig. 2A and Table 1) and an increase in [Ca2+]i. However, prior application of 5 μm KB-R7943 completely prevented the increase in [Ca2+]i induced by [K+]o-mediated membrane depolarization (Fig. 5). In fact, in the 5 μm KB-R7943-treated group the [Ca2+]i, as indicated by the fura-2 ratio, at the end of reoxygenation was lower than that at the end of the chemically induced hypoxia period. The change in the normalized fura-2 ratio in reoxygenation was −2.1 ± 0.51 vs. 11.8 ± 0.05 % in controls (P < 0.05). This result strongly suggests that Ca2+ influx due to reversal of Na+–Ca2+ exchange activity contributes to the increase in [Ca2+]i whenever the diastolic membrane potential depolarizes by, for example, 15–20 mV.
Figure 5. The Na+–Ca2+ exchange inhibitor KB-R7943 protects against chemically induced hypoxia and reoxygenation-induced Ca2+ overload in rat ventricular myocytes.
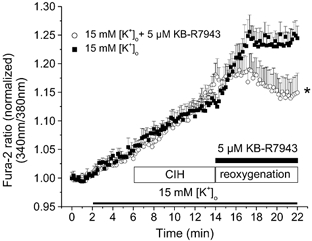
[Ca2+]i measurements in 15 mm [K+]o (i) under baseline conditions, and (ii) during 8 min chemically induced hypoxia, (iii) followed by 8 min reoxygenation. KB-R7943 (5 μm), a Na+–Ca2+ exchanger inhibitor, was applied during reoxygenation. * P < 0.05 vs. control (15 mm [K+]o), n = 6 experiments, 20 and 29 cells.
DISCUSSION
Main findings
Our results show that depolarization of the resting membrane potential of ventricular myocytes enhances the Ca2+ overload caused by reoxygenation, following chemically induced hypoxia. Importantly, hyperpolarization of the diastolic membrane potential reduces this effect, i.e. on reoxygenation, [Ca2+]i rises more slowly and reaches a lower steady-state level. Moreover, the Na+–Ca2+ exchange inhibitor KB-R7943 markedly attenuated the rise in [Ca2+]i during reoxygenation. We conclude that the magnitude of reoxygenation-induced Ca2+ overload is dependent on membrane potential, and that this effect is primarily mediated by Ca2+ influx due to the Na+–Ca2+ exchanger.
Na+–Ca2+ exchange and Ca2+ overload
The Na+–Ca2+ exchanger is an essential regulator of intracellular Ca2+ homeostasis in ventricular myocytes. Under physiological conditions, the primary function of this antiporter is the extrusion of Ca2+ during diastole, and it plays a significant role in excitation-contraction coupling in cardiac myocytes (Blaustein & Lederer, 1999). However, in the setting of hypoxia-reoxygenation the raised [Na+]i and depolarization combine to cause the Na+–Ca2+ exchanger to work in the reverse mode, thus increasing [Ca2+]i and contributing to cell injury and myocyte hypercontracture (Schäfer et al. 2001).
Insight into the extent to which the Na+–Ca2+ exchanger can contribute to either physiological or pathophysiological Ca2+ homeostasis requires knowledge of the exact stoichiometry of the cardiac Na+–Ca2+ exchanger. The majority of available evidence favours a stoichiometry of 3:1 for the Na+–Ca2+ exchanger (for a review see Blaustein & Lederer, 1999). However, results from a recent study based on electrophysiological measurements in HEK cells transfected with the rat cardiac Na+–Ca2+ exchanger NCX 1.1, have been interpreted in terms of a 4:1 stoichiometry (Dong et al. 2002). If this is the case, the Na+–Ca2+ exchanger may generate larger currents (operating in either forward or reverse mode), since the assumed current-voltage (I-V) relationship (a symmetrical hyperbolic function) is more curvilinear. (We note, however, that a recent publication by Hinata et al. (2002) calls into question the conclusion drawn by Dong et al. (2002).) Under either of these assumed stoichiometries even a small depolarization during ischaemia could lead to a significant outward Na+–Ca2+ current, i.e. an increased Ca2+ influx (see Fig. 6). However, since ENa/Ca = 4ENa - 2ECa in the 4:1 model (where E is the equilibrium potential), and ENa/Ca = 3ENa - 2ECa in the 3:1 model, the reversal potential of the Na+–Ca2+ exchanger would be more positive in the case of a 4:1 stoichiometry.
Figure 6. Mathematical simulation of Na+–Ca2+ exchange current as a function of membrane potential at selected [Na+]i and [K+]o.
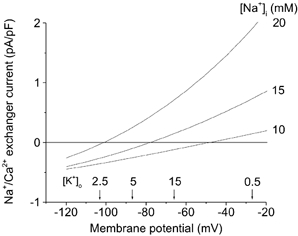
Na+–Ca2+ exchange current is shown assuming control (10 mm) and elevated [Na+]i. Arrows denote our experimental measurements of diastolic membrane potentials resulting from superfusion of ventricular myocytes with a solution containing selected [K+]o (mm). This simulation was done using the software described in Puglisi & Bers (2001).
A hypothetical relationship between diastolic membrane potential and Na+–Ca2+ exchange current and therefore reoxygenation-induced Ca2+ overload is illustrated in Fig. 6. The superimposed curves, which we computed using a conventional mathematical model of the Na+–Ca2+ exchanger assuming 3:1 stoichiometry, illustrate the steady-state Na+–Ca2+ exchanger current as the function of membrane potential at selected [Na+]i levels. These simulations are based on a recent publication by Puglisi & Bers (2001). In rat cardiac myocytes, measurements of resting [Na+]i yield values of between 10 and 16 mm, compared with approximately 4–5 mm in rabbit (Shattock & Bers, 1989; Despa et al. 2002). Ischaemia causes a time-dependent and marked (2- to 3-fold) increase in [Na+]i (Tani & Neely, 1989; Imahashi et al. 1999) as a consequence of inhibition of the Na+-K+ pump. Figure 6 shows the I-V relationship for the Na+–Ca2+ exchanger at selected (normal and elevated) [Na+]i. The Na+–Ca2+ exchange current measured in the resting state in rat ventricular myocytes with a higher [Na+]i causes an altered shape of the I-V curve in normoxic conditions as well as shifting the reversal potential to more hyperpolarized Vm (Despa et al. 2002). The steeper I-V curve could cause the exchanger to be more sensitive to smaller changes in membrane potential in the diastolic range: between −40 and −90 mV. The arrows denote the membrane potentials measured in our perforated patch-clamp experiments (Fig. 2) when myocytes were superfused with the [K+]o levels indicated. Our findings confirm that relative changes in [K+]o of electrically quiescent myocardial cells change membrane potential in the manner predicted for an excitable cell with the resting potential dominated by an inwardly rectifying K+ current.
The small amount of depolarization (5–10 mV) observed during chemically induced hypoxia-reoxygenation in the presence of 5 mm [K+]o (Fig. 3) could not be solely responsible for the induction of reverse mode Na+–Ca2+ exchanger activity. The activity of the Na+–Ca2+ exchanger is strongly dependent on both intra- and extracellular [Na+] and [Ca2+]. In our experiments extracellular [Na+] and [Ca2+] were set at 140 and 2 mm, respectively. However, as [Na+]i increases with the development of ischaemia, the I-V curve for the Na+–Ca2+ exchanger becomes steeper and shifts to the left. Therefore, in the presence of elevated [Na+]i smaller depolarizations of resting membrane potential are sufficient to cause Ca2+ entry via reverse mode Na+–Ca2+ exchanger activity. In addition, the significant increase in [K+] in the extracellular space during ischaemia (Rodríguez et al. 2002; modelled in our experiments by application of 15 mm [K+]o) can markedly depolarize the myocyte (Carmeliet, 1999). These changes favour the Na+–Ca2+ exchanger working in reverse mode generating an outward current and substantial Ca2+ entry. This can lead to Ca2+ overload in the myocyte.
Our results also demonstrate that this change in Ca2+ overload following depolarization is due to the change in resting membrane potential and not to a change in [K+]o itself. Thus, and as expected (Pandit et al. 2001), due to the K+-sensing property, and highly non-linear I-V curve of the K+ currents that control the resting membrane potential in mammalian ventricular myocytes, both 15 and 0.5 mm [K+]o caused a marked depolarization of the myocyte (Table 1), and these groups of myocytes showed the largest increase in [Ca2+]i during reoxygenation.
Pharmacological inhibition of the Na+–Ca2+ exchanger (5 μm KB-R7943) reduced increases in [Ca2+]i during reoxygenation only (Fig. 1). This may suggest that different mechanisms are responsible for elevation of [Ca2+]i during hypoxia, e.g. inhibition of ATP-dependent Ca2+ extrusion mechanisms, such as the sarcolemmal Ca2+-ATPase.
Na+-K+ pump activity and Ca2+-overload during hypoxia-reoxygenation
It is possible that altered Na+-K+ pump activity could indirectly influence Na+–Ca2+ exchanger activity by changing [Na+]i in ischaemic ventricular myocytes. However, if this was the case and if the Na+-K+ pump was the major cause of the changes in [Na+]i then during reoxygenation a significant hyperpolarization of the membrane potential would be expected, i.e. the equivalent of the classical ‘overdrive suppression response’ (Bhattacharyya & Vassalle, 1980). This was not observed. Furthermore, Hasin et al. (1984) found that the combination of cyanide and 2-deoxy-glucose (inhibitors of oxidative phosphorylation and glycolysis, respectively) did not exert its depolarizing effect through the inhibition of the Na+-K+ pump. Changes in intracellular pH (pHi) during metabolic inhibition could also influence the activity of the Na+-K+ pump and, therefore, Na+–Ca2+ exchanger function indirectly. Breitwieser et al. (1987) found that a change of the pHi in the acidic direction caused a reversible decrease in the maximum velocity of Na+-K+ pump current without affecting the Kd for [K+]o in the squid giant axon. Russell et al. (1983) found that the ouabain-sensitive Na+ efflux was sharply inhibited by acidic pHi and completely absent at pHi below 6.8 in giant barnacle muscle fibres. However, the data of Gao et al. (1995) from quiescent guinea-pig myocytes suggest that changes in pHi between 6.0 and 7.2 do not have any effect on the Na+-K+ pump. Thus, the pHi sensitivity of the Na+-K+ pump function appears to be isoform specific, with the α-subunit which is expressed in mammalian myocytes being relatively insensitive to changes in pHi.
Na+-H+ exchanger activity and reoxygenation-induced Ca2+ overload
The activity of the Na+–Ca2+ exchanger is strongly dependent on [Na+]i (Blaustein & Lederer, 1999) as indicated in Fig. 6. One of the key mechanisms leading to increased [Na+]i in ventricular myocytes upon reoxygenation is the activation of the Na+-H+ exchanger due to the acidification of the intracellular milieu during hypoxia (Park et al. 1999). This leads to an increase in [Na+]i and can result in reverse mode Na+–Ca2+ exchanger activity in reoxygenation (Tani & Neely, 1989; Murphy et al. 1991). Accordingly, inhibition of the Na+-H+ exchanger in the setting of ischaemia-reperfusion has been shown to protect the heart from reperfusion injury (Karmazyn, 1999; Buerke et al. 1999). Therefore, it is expected that the contribution of the Na+-H+ exchanger to elevations of [Na+]i and consequent reverse mode Na+–Ca2+ exchanger activity-induced Ca2+ overload upon reoxygenation was similar in all groups in this study.
Implications for cardioplegia
During cardiac surgery, the heart must be subjected to elective global ischaemia. Hyperkalaemic cardioplegic solutions are currently used to arrest the heart in an attempt to preserve its function. Although using cardioplegic solutions is essential, their application can also lead to postoperative cardiac dysfunction. Our results show that the elevated [K+]o used in hyperkalaemic cardioplegic solutions (23 mm for induction and 13 mm for maintenance; Fukuhiro et al. 2000) will depolarize the myocytes and suggest that this diastolic depolarization can result in (i) calcium overload due to reverse mode Na+–Ca2+ exchange (ii) cell hypercontracture on reperfusion. Several strategies have been developed to reduce Ca2+ overload in the setting of cardioplegia (Chambers & Hearse, 1999; Fukuhiro et al. 2000). Lowered [Ca2+]o in a Mg2+-containing cardioplegic solution has been shown to have beneficial effects on post-ischaemic function in isolated rat hearts (Fukuhiro et al. 2000). However, the precise control of free [Ca2+]o is technically difficult in blood cardioplegia and the use of citrate to chelate Ca2+ was found to be detrimental in the same study. The addition of a sodium channel blocker, tetrodotoxin (Snabaitis et al. 1997), or a Na+-H+ exchange inhibitor, HOE-642 (Fukuhiro et al. 2000), to the cardioplegic solution has been shown to improve post-ischaemic recovery. Application of ATP-sensitive K+ (KATP) channel openers has yielded inconsistent results. Pinacidil has been shown to enhance cardioprotection compared with the cardioplegic solution alone (Hosoda et al. 1994). However, the KATP opener lemakalim was cardioprotective when given alone, but did not improve post-ischaemic recovery when added to the hyperkalaemic cardioplegic solution (Galiñanes et al. 1992). Based on our results, it seems likely that the protective effect offered by lemakalim would be diminished markedly due to the depolarization of the ventricular myocardium in the hyperkalaemic milieu. Our findings (Fig. 4) show that hyperpolarization of the membrane potential significantly reduced reoxygenation-induced Ca2+ overload. Indeed, results from recent studies on cardioplegia are in agreement: solutions that hyperpolarize the maximum diastolic potential are more effective during cardioplegia (Chambers & Hearse, 1999).
Possible contribution of Ca2+ influx through L-type Ca2+ channels
In our protocol, which involved depolarization of ventricular myocytes with [K+]o, Ca2+ influx could have increased by opening L-type Ca2+ channels. However, all Ca2+ imaging experiments were performed on myocytes that were quiescent for most (≈80 %) of the experiment. Moreover, although some myocytes did fire action potentials (Fig. 3), they became quiescent within 1–2 s possibly due to voltage-dependent inactivation of INa and ICa at depolarized potentials. That is, even in these myocytes the observed brief bursts of spontaneous action potentials accounted for only a small percentage of the total length of the experiment. Also, we found that application of 10 μm nifedipine, a putative L-type Ca2+ channel blocker, did not change the reoxygenation-induced increase in [Ca2+]i in our experimental settings (increase in normalized fura-2 ratio in reoxygenation was 9.78 ± 0.51 vs. 9.96 ± 0.26 % in controls, n = 6, P > 0.05). Thus, a significant contribution to Ca2+ entry mediated by L-type Ca2+ channels during reoxygenation is very unlikely.
Summary
Our results provide a clear demonstration that the resting membrane potential of cardiac myocytes strongly modulates the changes in [Ca2+]i resulting from reoxygenation. This effect arises from the intrinsic voltage-dependent properties of the cardiac Na+–Ca2+ exchanger and the presence of an inwardly rectifying background K+ current. Depolarization-induced reverse-mode Na+–Ca2+ exchanger activity during reoxygenation can result in a marked elevation of [Ca2+]i thus increasing the possibility of hypercontracture and/or cell death. These findings have important implications for the understanding of factors that control myocyte viability during reoxygenation. For example, strategies aimed at maintaining an approximately normal membrane potential of ventricular myocytes would be expected to improve the rate and extent of recovery of excitability and contractility in the post-ischaemic myocardium.
Acknowledgments
This work was supported by the Alberta Heritage Foundation for Medical Research (P.E.L., W.R.G.), Heart and Stroke Foundation of Canada (W.R.G.) and the Canadian Institutes for Health Research (P.E.L., W.R.G.). P.E.L. is a Scholar of the Alberta Heritage Foundation for Medical Research (AHFMR) and a Canadian Institutes of Health Research New Investigator. W.R.G. is a Medical Scientist of the Alberta Heritage Foundation for Medical Research. Salary support for Dr Baczkó was obtained from the Alberta Heart and Stroke Foundation Endowed Research Chair held by W.R.G.
REFERENCES
- Bhattacharyya M, Vassalle M. Metabolism dependence of overdrive-induced hyperpolarization. Arch Int Pharmacodyn Ther. 1980;246:28–37. [PubMed] [Google Scholar]
- Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- Bolli R, Marbán E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev. 1999;79:609–634. doi: 10.1152/physrev.1999.79.2.609. [DOI] [PubMed] [Google Scholar]
- Bouchard RA, Clark RB, Giles WR. Role of sodium-calcium exchange in activation of contraction in rat ventricle. J Physiol. 1993;472:391–413. doi: 10.1113/jphysiol.1993.sp019953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitwieser GE, Altamirano AA, Russell JM. Effects of pH changes on sodium pump fluxes in squid giant axon. Am J Physiol. 1987;253:C547–554. doi: 10.1152/ajpcell.1987.253.4.C547. [DOI] [PubMed] [Google Scholar]
- Buerke M, Rupprecht HJ, vom Dahl J, Terres W, Seyfarth M, Schultheiss HP, Richardt G, Sheehan FH, Drexler H. Sodium-hydrogen exchange inhibition: novel strategy to prevent myocardial injury following ischaemia and reperfusion. Am J Cardiol. 1999;83:19G–22G. doi: 10.1016/s0002-9149(99)00316-1. [DOI] [PubMed] [Google Scholar]
- Carmeliet E. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol Rev. 1999;79:917–1017. doi: 10.1152/physrev.1999.79.3.917. [DOI] [PubMed] [Google Scholar]
- Cascio WE, Yang H, Johnson TA, Muller-Borer BJ, Lemasters JJ. Electrical properties and conduction in reperfused papillary muscle. Circ Res. 2001;89:807–814. doi: 10.1161/hh2101.098612. [DOI] [PubMed] [Google Scholar]
- Chambers DJ, Hearse DJ. Developments in cardioprotection: ‘polarized’ arrest as an alternative to ‘depolarized’ arrest. Ann Thorac Surg. 1999;68:1960–1966. doi: 10.1016/s0003-4975(99)01020-6. [DOI] [PubMed] [Google Scholar]
- Despa S, Islam MA, Pogwizd SM, Bers DM. Intracellular [Na+] and Na+ pump rate in rat and rabbit ventricular myocytes. J Physiol. 2002;539:133–143. doi: 10.1113/jphysiol.2001.012940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio F, Steinberg TH, Silverstein SC. Inhibition of FURA-2 sequestration and secretion with organic anion transport blockers. Cell Calcium. 1990;11:57–62. doi: 10.1016/0143-4160(90)90059-4. [DOI] [PubMed] [Google Scholar]
- Dong H, Dunn J, Lytton J. Stoichiometry of the cardiac Na+/Ca2+ exchanger NCX1. 1 measured in transfected HEK cells. Biophys J. 2002;82:1943–1952. doi: 10.1016/S0006-3495(02)75543-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigel BN, Hadley RW. Antisense inhibition of Na+/Ca2+ exchange during anoxia/reoxygenation in ventricular myocytes. Am J Physiol Heart Circ Physiol. 2001;281:H2184–2190. doi: 10.1152/ajpheart.2001.281.5.H2184. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Lederer WJ. The role of the sodium pump in the effects of potassium-depleted solutions on mammalian cardiac muscle. J Physiol. 1979;294:279–301. doi: 10.1113/jphysiol.1979.sp012930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias CL, Lukas A, Shurraw S, Scott J, Omelchenko A, Gross GJ, Hnatowich M, Hryshko LV. Inhibition of Na+/Ca2+ exchange by KB-R7943: transport mode selectivity and antiarrhythmic consequences. Am J Physiol Heart Circ Physiol. 2001;281:H1334–1345. doi: 10.1152/ajpheart.2001.281.3.H1334. [DOI] [PubMed] [Google Scholar]
- Fukuhiro Y, Wowk M, Ou R, Rosenfeldt F, Pepe S. Cardioplegic strategies for calcium control. Low Ca2+, high Mg2+, citrate, or Na+/H+ exchange inhibitor HOE-642. Circulation. 2000;102(suppl. III):319–325. [PubMed] [Google Scholar]
- Galiñanes M, Shattock MJ, Hearse DJ. Effects of potassium channel modulation during global ischaemia in isolated rat heart with and without cardioplegia. Cardiovasc Res. 1992;26:1063–1068. doi: 10.1093/cvr/26.11.1063. [DOI] [PubMed] [Google Scholar]
- Gao J, Mathias RT, Cohen IS, Baldo GJ. Two functionally different Na/K pumps in cardiac ventricular myocytes. J Gen Physiol. 1995;106:995–1030. doi: 10.1085/jgp.106.5.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldhaber JI. Free radicals enhance Na+/Ca2+ exchange in ventricular myocytes. Am J Physiol. 1996;40:H823–833. doi: 10.1152/ajpheart.1996.271.3.H823. [DOI] [PubMed] [Google Scholar]
- Hasin Y, Barry WH. Myocardial metabolic inhibition and membrane potential, contraction, and potassium uptake. Am J Physiol. 1984;247:H322–329. doi: 10.1152/ajpheart.1984.247.2.H322. [DOI] [PubMed] [Google Scholar]
- Hinata M, Yamamura H, Li L, Watanabe Y, Watano T, Imaizumi Y, Kimura J. Stoichiometry of Na+–Ca2+ exchange is 3:1 in guinea-pig ventricular myocytes. J Physiol. 2002;545:453–461. doi: 10.1113/jphysiol.2002.025866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoda H, Sunamori M, Suzuki A. Effect of pinacidil on rat hearts undergoing hypothermic cardioplegia. Ann Thorac Surg. 1994;58:1631–1636. doi: 10.1016/0003-4975(94)91649-7. [DOI] [PubMed] [Google Scholar]
- Imahashi K, Kusuoka H, Hashimoto K, Yoshioka J, Yamaguchi H, Nishimura T. Intracellular sodium accumulation during ischaemia as the substrate for reperfusion injury. Circ Res. 1999;84:1401–1406. doi: 10.1161/01.res.84.12.1401. [DOI] [PubMed] [Google Scholar]
- Inserte J, Garcia-Dorado D, Ruiz-Meana M, Padilla F, Barrabes JA, Pina P, Agullo L, Piper HM, Soler-Soler J. Effect of inhibition of Na+/Ca2+ exchanger at the time of myocardial reperfusion on hypercontracture and cell death. Cardiovasc Res. 2002;55:739–748. doi: 10.1016/s0008-6363(02)00461-3. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Watano T, Shigekawa M. A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem. 1996;271:22391–22397. doi: 10.1074/jbc.271.37.22391. [DOI] [PubMed] [Google Scholar]
- Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischaemia and infarction. Physiol Rev. 1989;69:1049–1168. doi: 10.1152/physrev.1989.69.4.1049. [DOI] [PubMed] [Google Scholar]
- Jovanovic A, Jovanovic S, Lorenz E, Terzic A. Recombinant cardiac ATP-sensitive K+ channel subunits confer resistance to chemical hypoxia-reoxygenation injury. Circulation. 1998;98:1548–1555. doi: 10.1161/01.cir.98.15.1548. [DOI] [PubMed] [Google Scholar]
- Karmazyn M. The role of the myocardial sodium-hydrogen exchanger in mediating ischemic and reperfusion injury. From amiloride to cariporide. Ann N Y Acad Sci. 1999;874:326–334. doi: 10.1111/j.1749-6632.1999.tb09248.x. [DOI] [PubMed] [Google Scholar]
- Kusuoka H, Hurtado MCC, Marbán E. Role of sodium/calcium exchange in the mechanism of stunning: protective effect of reperfusion with high sodium solution. J Am Coll Cardiol. 1993;21:240–248. doi: 10.1016/0735-1097(93)90743-k. [DOI] [PubMed] [Google Scholar]
- Light PE, Shimoni Y, Harbison S, Giles WR, French RJ. Hypothyroidism decreases the ATP sensitivity of KATP channels from rat heart. J Membr Biol. 1998;162:217–223. doi: 10.1007/s002329900359. [DOI] [PubMed] [Google Scholar]
- Mukai M, Terada H, Sugiyama S, Satoh H, Hayashi H. Effects of a selective inhibitor of Na+/Ca2+ exchange, KB-R7943, on reoxygenation-induced injuries in guinea pig papillary muscles. J Cardiovasc Pharmacol. 2000;35:121–128. doi: 10.1097/00005344-200001000-00016. [DOI] [PubMed] [Google Scholar]
- Murphy E, Perlman M, London RE, Steenbergen C. Amiloride delays the ischemia-induced rise in cytosolic calcium. Circ Res. 1991;68:1250–1258. doi: 10.1161/01.res.68.5.1250. [DOI] [PubMed] [Google Scholar]
- Pandit SV, Clark RB, Giles WR, Demir SS. A mathematical model of action potential heterogeneity in adult rat left ventricular myocytes. Biophys J. 2001;81:3029–3051. doi: 10.1016/S0006-3495(01)75943-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C-O, Xiao X-H, Allen DG. Changes in intracellular Na+ and pH in rat heart during ischemia: role of Na+/H+ exchanger. Am J Physiol. 1999;45:H1581–1590. doi: 10.1152/ajpheart.1999.276.5.H1581. [DOI] [PubMed] [Google Scholar]
- Park JL, Lucchesi BR. Mechanisms of myocardial reperfusion injury. Ann Thorac Surg. 1999;68:1905–1912. doi: 10.1016/s0003-4975(99)01073-5. [DOI] [PubMed] [Google Scholar]
- Puglisi JL, Bers DM. LabHEART: an interactive computer model of rabbit ventricular myocyte ion channels and Ca transport. Am J Physiol Cell Physiol. 2001;281:C2049–2060. doi: 10.1152/ajpcell.2001.281.6.C2049. [DOI] [PubMed] [Google Scholar]
- Rodríguez B, Ferrero JM, Jr, Trénor B. Mechanistic investigation of extracellular K+ accumulation during acute myocardial ischemia: a simulation study. Am J Physiol Heart Circ Physiol. 2002;283:H490–500. doi: 10.1152/ajpheart.00625.2001. [DOI] [PubMed] [Google Scholar]
- Russell JM, Boron WF, Brodwick MS. Intracellular pH and Na fluxes in barnacle muscle with evidence for reversal of the ionic mechanism of intracellular pH regulation. J Gen Physiol. 1983;82:47–78. doi: 10.1085/jgp.82.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh H, Ginsburg KS, Qing K, Terada H, Hayashi H, Bers DM. KB-R7943 block of Ca2+ influx via Na+/Ca2+ exchange does not alter twitches or glycoside inotropy but prevents Ca2+ overload in rat ventricular myocytes. Circulation. 2000;101:1441–1446. doi: 10.1161/01.cir.101.12.1441. [DOI] [PubMed] [Google Scholar]
- Schäfer C, Ladilov Y, Inserte J, Schäfer M, Haffner S, Garcia-Dorado D, Piper HM. Role of the reverse mode of the Na+/Ca2+ exchanger in reoxygenation-induced cardiomyocyte injury. Cardiovasc Res. 2001;51:241–250. doi: 10.1016/s0008-6363(01)00282-6. [DOI] [PubMed] [Google Scholar]
- Shattock MJ, Bers DM. Ca flux and intracellular Na assessed by ion-selective microelectrodes. Am J Physiol. 1989;256:C813–822. doi: 10.1152/ajpcell.1989.256.4.C813. [DOI] [PubMed] [Google Scholar]
- Snabaitis AK, Shattock MJ, Chambers DJ. Comparison of polarized and depolarized arrest in the isolated rat heart for long-term preservation. Circulation. 1997;96:3148–3156. doi: 10.1161/01.cir.96.9.3148. [DOI] [PubMed] [Google Scholar]
- Tani M. Mechanisms of Ca2+ overload in reperfused ischemic myocardium. Annu Rev Physiol. 1990;52:543–559. doi: 10.1146/annurev.ph.52.030190.002551. [DOI] [PubMed] [Google Scholar]
- Tani M, Neely J. Role of intracellular Na+ in Ca2+ overload and depressed recovery of ventricular function of reperfused ischaemic rat hearts. Possible involvement of H+-Na+ and Na+–Ca2+ exchange. Circ Res. 1989;65:1045–1056. doi: 10.1161/01.res.65.4.1045. [DOI] [PubMed] [Google Scholar]
- Wilde A, Aksnes G. Myocardial potassium loss and cell depolarization in ischaemia and hypoxia. Cardiovasc Res. 1995;29:1–15. [PubMed] [Google Scholar]
- Zeitz O, Maass AE, Van Nguyen P, Hensmann G, Kogler H, Moller K, Hasenfuss G, Janssen PM. Hydroxyl radical-induced acute diastolic dysfunction is due to calcium overload via reverse-mode Na+–Ca2+ exchange. Circ Res. 2002;90:988–995. doi: 10.1161/01.res.0000018625.25212.1e. [DOI] [PubMed] [Google Scholar]
- Zile MR, Brutsaert DL. New concepts in diastolic dysfunction and diastolic heart failure: part II. Causal mechanisms and treatment. Circulation. 2002;105:1503–1508. doi: 10.1161/hc1202.105290. [DOI] [PubMed] [Google Scholar]