

Abstract
Stimulation of Calu-3 epithelia with 7,8-benzoquinoline, under short circuit current conditions, produced a current increase that was completely accounted for by the net flux of chloride, measured simultaneously with 36Cl−. Nevertheless the current stimulated by 7,8-benzoquinoline was sensitive to acetazolamide, which caused up to 50 % inhibition of the stimulated current, the remainder being sensitive to the Na+-K+-2Cl− cotransport inhibitor bumetanide. The effects of acetazolamide could be mimicked by either amiloride or by the di-sodium salt of 4,4′-dinitrostilbene-2,2′-disulphonic acid (DNDS) added to the basolateral side of the epithelium, but their actions were not additive. Amiloride was needed in sufficient concentration to inhibit the sodium-proton exchanger NHE1. DNDS blocks both the chloride-bicarbonate exchanger AE2 and the sodium-bicarbonate transporter NBC1. However, since 7,8-benzoquinoline activates basolateral K+ channels, causing hyperpolarisation, it is unlikely NBC1 is active after addition of 7,8-benzoquinoline. The effect of DNDS is, therefore, mainly on AE2. It is concluded that chloride enters the basolateral aspect of the cells using the Na+-K+-2Cl− cotransporter and a parallel arrangement of NHE1 with AE2, these latter two being sensitive to acetazolamide because of their association with the cytoplasmic form of carbonic anhydrase CAII. The effects of acetazolamide could be mimicked by removal of HCO3−/CO2 from the bathing medium, and furthermore showed that the NHE1-AE2 mechanism is particularly important when the transport rate is high. Thus part of the current stimulated by 7,8-benzoquinoline and inhibited by acetazolamide or HCO3−/CO2 removal can be said to represent bicarbonate-dependent chloride secretion.
The serous cells of the submucosal glands in the human lung are the richest source of the cystic fibrosis transmembrane conductance regulator (CFTR) in the airways (Engelhardt et al. 1992). These epithelial cells elaborate a fluid containing bicarbonate, antimicrobial peptides and enzymes, thought to be important in maintaining lung sterility (Basbaum et al. 1990), as well as adequate mucociliary clearance (Pilewski & Frizzell, 1999). Calu-3 cells, derived from a lung adenocarcinoma, have the properties of serous cells (Shen et al. 1994) and can be cultured as monolayers on permeable supports and exhibit transepithelial transport of ions (Moon et al. 1997).
There have been a number of studies in Calu-3 cells of the nature of the ions transported in response to various stimuli. In Calu-3 monolayers, the basal current was reduced by removal of bicarbonate ions; indeed removal of bicarbonate alone was as efficient at reducing the basal short circuit current (SCC) as removal of bicarbonate plus chloride ions (Singh et al. 1997). It was concluded that basal transport in Calu-3 cells was either bicarbonate-dependent chloride secretion or chloride-dependent bicarbonate secretion, the authors favouring the former. Subsequent flux studies, however, showed it was the latter mechanism that was operative (Lee et al. 1998). An important difference appeared to exist between the nature of the basal current and that obtained after stimulation, as the stimulated current was sensitive to blockers of the Na+-K+-2Cl− cotransporter (Shen et al. 1994; Singh et al. 1997). Thus it was argued that the stimulated SCC was due to electrogenic chloride secretion, while the basal current was due to bicarbonate secretion. Devor et al. (1999) showed that the nature of the stimulus apparently determined the nature of the transported ion. Forskolin, acting via cAMP, produced a bicarbonate secretion, whereas EBIO (1-ethyl-2-benzimidazolone) produced chloride secretion. In this study we have used 7,8-benzoquinoline, an agent with similar actions to EBIO (Duszyk et al. 2001; Cuthbert, 2003), to stimulate Calu-3 monolayers. The chance observation that the effect of 7,8-benzoquinoline was inhibited by acetazolamide prompted us to re-examine the question of the bicarbonate dependence of stimulated SCC responses in Calu-3 monolayers.
METHODS
Calu-3 cell culture
Calu-3 cells (from the American Type Culture Collection) were grown on 75 cm2 culture flasks containing Eagle's minimal essential medium (Vitacell, ATCC, Virginia, USA) with 10 % fetal calf serum (Gibco BRL), 100 μM ml−1 kanamycin and 1.25 mg ml−1 fungizone, and incubated at 37 °C in humidified air containing 5 % CO2. Cells were collected by trypsinisation and subcultured either on Snapwell polycarbonate membrane inserts (1 cm2, 0.4 μM pore size) (Costar UK Ltd, Buckinghamshire, UK) or untreated glass coverslips (1 cm2). Cultures were re-fed every 3-4 days; the inserts were used between 17 and 24 days after subculture and the cells on coverslips were used after 4 days. All experimental procedures used cells from passages 3-10.
SSC recording and modifications of the standard SCC procedure
The Snapwell inserts, bearing the cultured monolayers, were inserted into CHM5 Ussing chambers with associated electrodes (WPI, Hertfordshire, UK) and voltage-clamped at zero potential using a WPI Dual Voltage Clamp-1000 (WPI). Both sides of the epithelium were bathed in 5 or 6 ml of Krebs Henseleit solution (KHS) that was continually circulated through the half-chambers, maintained at 37 °C and continuously bubbled with 95 %O2-5 %CO2. Basal characteristics of Calu-3 monolayers (transepithelial potential, basal SCC and resistance) are given elsewhere (Cuthbert & MacVinish, 2003). Bicarbonate-free bathing solution was buffered with Hepes to pH 7.4 and bubbled with 100 % O2. SCCs were recorded continuously using an ADInstruments PowerLab/8SP (NSW 2154, Australia) and displayed on a computer screen.
Nystatin treatment (180-360 mg ml−1) of the apical membranes was used to examine the effects of 7,8-benzoquinoline on the basolateral membranes of Calu-3 epithelia. In these experiments the apical bathing solution was changed to potassium gluconate Krebs solution (PGK) and the basolateral solution to sodium gluconate Krebs solution (SGK), thus imposing a K+ gradient in the apical-to-basolateral direction. For examining the effects of agents on the apical membranes the method described by Cuthbert (2001) was used. A high K+-containing solution (PGK) was used to depolarise the basolateral membrane while the apical membrane remained bathed in KHS, thus imposing an apical-to-basolateral Cl− gradient.
Measurement of 36Cl− fluxes in Calu-3 monolayers
Calu-3 monolayers were used in pairs, each bathed in 6 ml KHS on each side. After the SCC had stabilised, a few microcuries of 36Cl− was added to the basolateral side of one monolayer and to the apical side of the other. A period of 20 min was allowed for the radioactive fluxes to achieve equilibrium, after which samples were taken from the side to which the radioisotope had not been added. The sample size was 2 ml and the bathing solution was immediately topped up with 2 ml of fresh KHS. A second set of samples were taken 20 min later using the same protocol, and 7,8-benzoquinoline (210 μM) was then added to both sides of the epithelia. A third set of samples was taken after a further 20 min. The SCC current was recorded throughout the experiment and small samples (100 μl) were taken from the ‘hot’ sides periodically. Unidirectional Cl− fluxes were calculated from the specific activity, making allowance for the dilution caused by removing samples. The areas under the SCC traces were obtained using the integrator facility on the ADI PowerLab /8SP. The paired experiment was repeated 12 times.
Measurement of intracellular pH in Calu-3 cells
Calu-3 cells were grown on coverslips as described above and exposed to the pH-sensitive fluorescent dye BCECF (2′,7′-bis(2-carboxyethyl)-5-(6)-carboxyfluoroscein; Molecular Probes, The Netherlands) in its AM (acetoxymethyl ester) form (BCECF-AM, 0.5 μM in 0.1 %DMSO) for 7-8 min. After loading, individual coverslips were washed in KHS and mounted in a cuvette so that both the incident and reflected light beams were at 45 deg to the cell layer. The cuvette contained 2.5 ml KHS and the contents were maintained at 37 °C and stirred with a magnetic flea. Fluorescence was recorded every 5 s with an F-2000 Hitachi fluorescence spectrophotometer at 526 nm with excitation alternately at 440 nm and 502 nm with 5 nm excitation and emission slits. The pHi was calculated from the fluorescence ratio for BCECF, which varies linearly with pH over the range 6.9-7.9.
Solutions
KHS had the following composition (mM): 117 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3 and 11.1 glucose (pH 7.4). The modified solutions PGK (SGK) contained (mM): 120 potassium gluconate (sodium gluconate), 25 NaHCO3, 3.3 KH2PO4, 0.8 K2HPO4, 1.2 MgCl2, 4 CaCl2 and 10 glucose. Hepes-buffered solution had the following composition (mM): 142 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.2 KH2PO4, 10 Hepes and 11.1 glucose. This solution was adjusted to pH 7.4 with NaOH and bubbled with 100 % O2.
Materials
The following were obtained from Sigma-Aldrich Co. Ltd, Poole, Dorset, UK: amiloride, bumetanide, acetazolamide, nystatin, charybdotoxin (ChTX) and 7,8-benzoquinoline. BCECF-AM was from Molecular Probes Europe BV, Leiden, The Netherlands. EBIO (1-ethyl-2-benzimidazolone) was from Tocris, Bristol, UK and the di-sodium salt of 4,4′-dinitrostilbene-2,2′-disulphonic acid (DNDS) was from the Tokyo Kasei Kogyo Co. Ltd, Tokyo, Japan. The non-penetrating carbonic anhydrase inhibitors were synthesised in the Dipartimento di Chimica, University of Florence (Scozzafava et al. 2000; Supuran et al. 2003). Addition of drugs to the Calu-3 monolayers will be shown throughout as (ap) for apical addition, (bl) for basolateral addition and (bs) when added to both sides of the epithelium.
Statistical evaluation
Where appropriate, Students t test was used to evaluate differences, with P < 0.05 being considered significant.
RESULTS
Inhibition of anion secretion by bumetanide and acetazolamide in Calu-3 monolayers
The nature of the SCC increase caused by 7,8-benzoquinoline (bs) was investigated using the inhibitors bumetanide (bl) and acetazolamide (bs), on the presumption that they would block Cl− and HCO3− secretion, respectively. Assuming each removes a specific component of the current, then the relative contributions of the two inhibitors should not vary with the order in which they are added. In Fig. 1A, the ratio of the current removed by bumetanide to that removed by acetazolamide was 0.75 when acetazolamide was given prior to bumetanide. Put another way, the current removed by acetazolamide was 1.3 times larger than that removed by bumetanide. However, when the order of addition was reversed, that is bumetanide was added before acetazolamide, the ratio was 6.2 (Fig. 1B), or, in other words, the current removed by acetazolamide was less than one-sixth of that removed by bumetanide. The two halves of the experiment shown in Fig. 1 were carried out on the same Calu-3 monolayer with washing between. The near-identity of the two 7,8-benzoquinoline responses indicates that the effect of this agent and the responses to bumetanide and acetazolamide are fully reversible. The experiment was repeated a total of five times and the mean response to 7,8-benzoquinoline was 68.5 ± 5.8 μA cm−2. The current removed when bumetanide (20 μM, bl) was given first was 45.4 ± 7.9 μA cm−2 compared with 10.0 ± 2.4 μA cm−2 when it was applied second, these two values being significantly different (P < 0.03, n = 5). Similarly the responses to acetazolamide (100 μM, bs) were 30.9 ± 4.3 μA cm−2 when it was given first and 7.3 ± 1.1 μA cm−2 when it was given second, the two values again being significantly different (P < 0.001, n = 5). The bumetanide/acetazolamide ratios were 0.3 ± 0.1 and 6.7 ± 1.9 (P < 0.01, n = 5), depending on the order of application. The result indicates that either the secretions of Cl− and HCO3− are related in a complex way or that the inhibitors are not accurately reporting the secretion of the ions whose transport they supposedly specifically inhibit.
Figure 1. Inhibition of short circuit current (SCC) responses to 7,8-benzoquinoline in a Calu-3 epithelium.
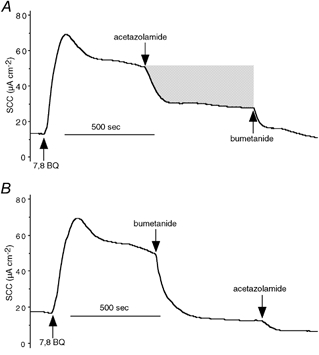
SCC traces from a single Calu-3 monolayer stimulated with 7,8-benzoquinoline (250 μM, applied to both sides (bs)). Inhibitors of the current, namely acetazolamide (100 μM, bs) and bumetanide (20 μM, basolaterally (bl)), were applied sequentially (A), after which the monolayer was washed and the responses repeated, but with the order of addition of the inhibitors reversed (B). The shaded area in A represents the current removed by acetazolamide and is equal to 0.21 μequiv. The abbreviation 7,8 BQ refers to 7,8-benzoquinoline in this and other figures.
To investigate whether this phenomenon was specific for 7,8-benzoquinoline or whether similar results could be obtained with other anion secretagogues, a comparison of the actions of EBIO and 7,8-benzoquinoline was made, as shown in Fig. 2. Here each Calu-3 monolayer was exposed to both 7,8-benzoquinoline (bs) and EBIO (bs) with washing between, both agents producing responses with very similar profiles in each monolayer. With either agonist the bumetanide/acetazolamide ratio was greater when bumetanide was given first compared with when it was given after acetazolamide. The bumetanide/acetazolamide ratio when acetazloamide was given first appeared to be greater after EBIO (1.8; Fig. 2C) than when 7,8-benzoquinoline was used as agonist (0.3; Fig. 2D) Indeed this latter value is the same as that from the earlier set of experiments (0.3 ± 0.1, n = 5). To be sure the ratio given in Fig. 2C with EBIO was different from that given in Fig. 2D with 7,8-benzoquinoline, more experiments with EBIO were performed. In six experiments the SCC increase caused by EBIO (600 μM, bs) was 134.2 ± 16.3 μA cm−2 and was reduced by 43.9 ± 7.1 to 90.3 ± 16.2 μA cm−2 by acetazolamide (100 μM, bs), a significant reduction (P < 0.002, paired t test). Subsequently, bumetanide (20 μM bl) reduced the current by a further 93.3 ± 10.5 μA cm−2. The bumetanide/acetazolamide ratio was 2.3 ± 0.3 (n = 6) and was significantly different from the ratio obtained when 7,8-benzoquinoline (250 μM) was used as the agonist, namely 0.3 ± 0.1 (n = 5, P < 0.0005). Thus while EBIO and 7,8-benzoquinoline show the same phenomenon with respect to the potency of the inhibitors depending on the order of application, there appear to be quantitative differences between the two secretagogues.
Figure 2. Inhibition of SCC reponses to 7,8-benzoquinoline and EBIO.
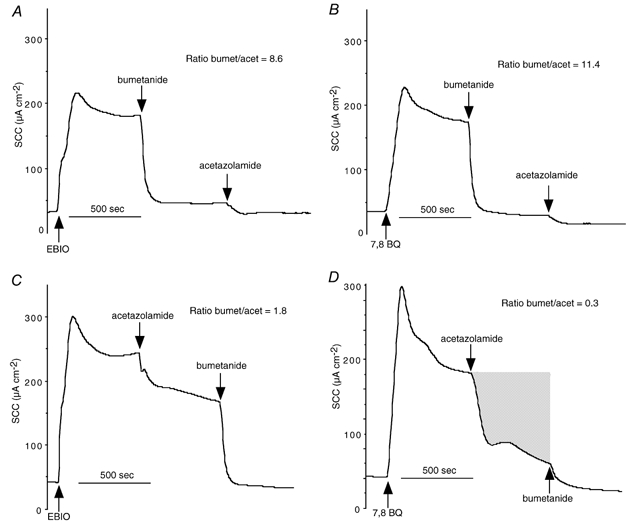
SCC traces from two Calu-3 monolayers stimulated with EBIO (1-ethyl-2-benzimidazolone; 600 μM, bs) Inhibitors, namely acetazolamide (100 μM, bs) and bumetanide (20 μM, bl), were applied sequentially (A and C), after which the monolayers were thoroughly washed. The monolayers were then re-stimulated with 7,8-benzoquinoline (250 μM, bs) and the inhibitory responses to acetazolamide and bumetanide were repeated (B and D). The ratios of the current removed by bumetanide to that removed by acetazolamide were measured (ratio bumet/acet) and are given in each panel. The shaded area in D represents the current removed by acetazolamide and is equal to 0.52 μequiv.
Can removal of HCO3−/CO2 mimic the effects of acetazolamide?
In Hepes-buffered solution, without HCO3−/CO2, responses to 7,8-benzoquinoline were attenuated. Figure 3 shows results from 12 monolayers, all from the same batch, half of which were suspended in KHS while the others were in the modified solution without HCO3−/CO2. Peak responses to 7,8-benzoquinoline (250 μM, bs) were 109 ± 9.1 μA cm−2 (n = 6) in KHS but only 83 ± 4.7 μA cm−2 (n = 6, P < 0.03) in the modified solution. Similarly if the charge transfer in 10 min was measured the response in KHS was greater than in the absence of HCO3−/CO2 (0.551 ± 0.041 μequiv versus 0.394 ± 0.021 μequiv, n = 6, P < 0.007). Acetazolamide (100 μM, bs) reduced the current remaining after 10 min by 43.9 ± 4.4 μA cm−2 in KHS, but had no effect on the current in the modified solution. Subsequently, bumetanide (20 μM, bl) reduced the current by 49.0 ± 3.8 μA cm−2 in KHS and by 39.2 ± 2.9 μA cm−2 in the Hepes solution.
Figure 3. Responses to 7,8-benzoquinoline in Calu-3 monolayers in HCO3−/CO2-free conditions.
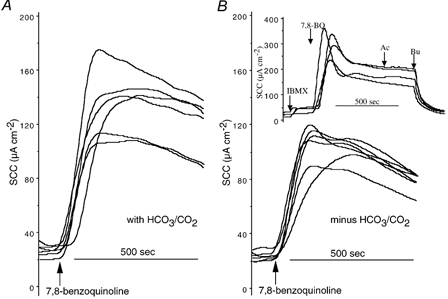
Responses to 7,8-benzoquinoline (250 μM, bs) in Krebs Henseleit solution (KHS) (A) and in Hepes-buffered solution (B) are shown. The inset shows responses in four further monolayers when IBMX (100 μM, bs) was added prior to 7,8-benzoquinoline. Ac, acetazolamide (100 μM, bs); Bu, bumetanide (20 μM, bl). All monolayers were from the same batch.
It will have been noted from earlier figures (compare Fig. 1 and Fig. 2) that the responses to a given concentration of 7,8-benzoquinoline are variable. While we have not studied this systematically, others have reported that human airway cells vary in their responses to 7,8-benzoquinoline-like agents (Caci et al. 2003) and have attributed this to variations in endogenous cAMP generation. For this reason all individual sets of experiments reported here are carried out with monolayers from the same batch. To gain further insight into the role of HCO3−/CO2 in the responses to 7,8-benzoquinoline, we repeated the experiment above with Calu-3 monolayers that responded poorly in as much as the responses were smaller than expected compared to those illustrated in Fig. 3. The peak responses to 7,8-benzoquinoline (250 μM, bs) and the charge transfer in 10 min were not different whether the bathing solution was KHS or Hepes buffer (Fig. 4) (52.1 ± 5.3 μA cm−2 and 0.254 ± 0.027 μequiv, resepctively, in KHS and 48.6 ± 4.5 μA cm−2 and 0.214 ± 0.033 μequiv, respectively, in Hepes (all values n = 6, P = n.s.). Based on the suggestion that the endogenous cAMP production might be low, a further 12 monolayers from the same batch were treated with IBMX (3-isobutyl-1-methylxanthine; 100 μM, bs) and the experiment repeated. Peak responses in KHS and Hepes were not different (171.7 ± 24.4 μA cm−2 in KHS and 222.7 ± 41.4 μA cm−2 in Hepes, n = 6, n.s.), whereas the charge transfer in 10 min due to the combined effects of IBMX and 7,8-benzoquinoline was significantly greater in the KHS-buffered monolayers (1.29 ± 0.075 versus 0.87 ± 0.06 μequiv, n = 6, P < 0.002). More intriguingly the form of the responses was different in the two solutions, being well maintained in KHS and declining rapidly from the peak in Hepes. The plateau responses after 10 min in KHS were sensitive to acetazolamide (100 μM, bs) while those in Hepes buffer were not (89.5 ± 17.9 μA cm−2versus 0.0 ± 6.1 μA cm−2, n = 3, P < 0.01). The change in form was not peculiar to poorly responding monolayers as it was also shown with more sensitive epithelia (see inset to Fig. 3).
Figure 4. Effect of IBMX on 7,8-benzoquinoline responses in HCO3−/CO2-free conditions.
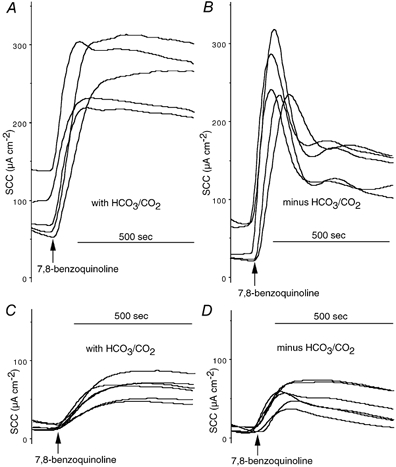
Shown are the responses to 7,8-benzoquinoline (250 μM, bs) in KHS (A and C) or in HCO3−/CO2-free buffer (B and D). IBMX (3-isobutyl-1-methylxanthine; 100 μM, bs) was added before 7,8-benzoquinoline in A and B. All monolayers were from the same batch.
Effects of DNDS on the responses to 7,8-benzoquinoline
According to the model of transport in Calu-3 monolayers (Devor et al. 1999) HCO3− secretion depends, in part, on the presence of an electrogenic Na+-HCO3− cotransporter in the basolateral surface of the cells, which is inhibited by high concentrations of DNDS. Consequently experiments were made in which both DNDS (3 mM, bl), to inhibit the cotransporter, and acetazolamide (100 μM, bs) were given together. Figure 5A gives one example in which the both agents were added after 7,8-benzoquinoline (250 μM, bs), producing substantial inhibition, with some recovery, the remaining current being removed by bumetanide. After washing away the inhibitors and 7,8-benzoquinoline, the same monolayer was allowed to recover. When the epithelium was subsequently exposed to DNDS-acetazolamide, which caused an immediate reduction in the basal SCC, the response to 7,8-benzoquinoline was attenuated (Fig. 5B). Two other identical experiments were carried out. The ratio of the peak response to 7,8-benzoquinoline given before DNDS-acetazolamide to that when 7,8-benzoquinoline was given after the inhibitors was 3.4 ± 1.0 (n = 3). However, the responses to DNDS-acetazolamide did not appear to be much greater than when acetazolamide was added alone. Consequently, further experiments were made where the two agents were added sequentially (Fig. 5C). In this series acetazolamide caused a reduction of 53.1 % in the response to 7,8-benzoquinoline that was increased to 66.5 % by addition of DNDS (Fig. 5D). DNDS (3 mM bl) given alone caused inhibition of the 7,8-benzoquinoline response by 50 % (see next section), confirming that the responses to DNDS and acetazolamide were not additive.
Figure 5. Inhibition of SCC responses to 7,8-benzoquinoline by DNDS.
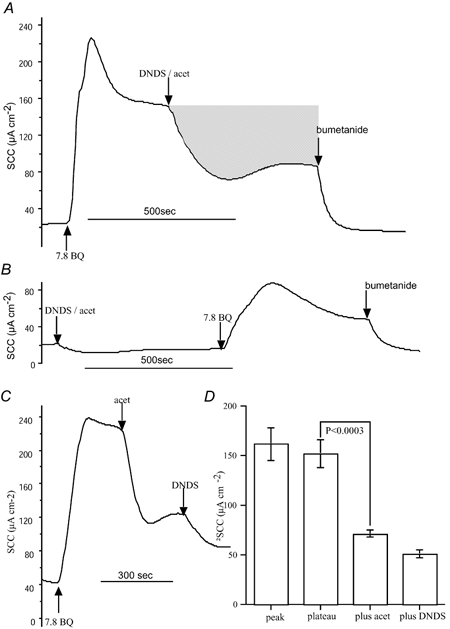
The SCC produced in response to 7,8-benzoquinoline (250 μM, bs) in a Calu-3 monolayer was inhibited by subsequent addition of the di-sodium salt of 4,4′-dinitrostilbene-2,2′-disulphonic acid (DNDS) plus acetazolamide given together. DNDS and acetazolamide were used at concentrations of 3 mM (bl) and 100 μM (bs), respectively. Finally bumetanide (20 μM, bl) was added. After the experiment shown in A the tissue was washed and a further experiment carried out as in B. Here DNDS and acetazolamide were added first, followed by 7,8-benzoquinoline. Note that the response is severely curtailed but reaches a maximal value of 88 μA, not different from the steady-state value after DNDS and acetazolamide in A (89 μA). The shaded area in A represents the current removed by DNDS plus acetazolamide and is equivalent to 0.43 μequiv. C, the effects of sequential addition of acetazolamide (100 μM, bs) and DNDS (3 mM, bl) on the response to 7,8-benzoquinoline (250 μM, bs) are shown. D gives the composite data from 6 experiments.
It will be noted that in Figs 1, 2 and 5 sections of the records are filled in to show the reduction in charge transfer caused by the addition of acetazolamide. If these shaded areas represent HCO3− secretion, rather than Cl− secretion, there should be a major discrepancy between the net flux of Cl− and the charge transfer indicated by the SCC.
Inhibition of anion secretion by DNDS and by amiloride in Calu-3 monolayers
While DNDS had apparently little extra effect when added together with acetazolamide it was necessary to examine its effects when given alone. DNDS (3 mM, bl) reduced the response to 7,8-benzoquinoline from 109.1 ± 14.8 μA cm−2 (n = 6) to 54.4 ± 5.5 μA cm−2 (n = 6, P < 0.006), that is a reduction of 50.1 %. Further addition of amiloride (1 mM), again to the basolateral side, produced a further small reduction in current to 45.6 ± 5.5 μA cm−2 (n = 6), giving a total inhibition of 58.2 %, comparable to the inhibition produced by acetazolamide given alone after 7,8-benzoquinoline. Reversing the order of application produced a slightly different result. Addition of amiloride (1 mM, bl) reduced the response to 7,8-benzoquinoline from 93.3 ± 11.9 μA cm−2 (n = 5) to 53.6 ± 8.1 μA cm−2 (n = 5, P < 0.003, paired t test), that is a reduction of 42.6 % that was not further increased by DNDS. Examples are given in Fig. 6A and B, together with the composite data from all experiments. In the presence of amiloride and DNDS, acetazolamide was able to cause some further inhibition of SCC (data not shown). Thus a combination of amiloride, at concentrations known to inhibit sodium-proton exchange, and DNDS, at concentrations that inhibit the chloride-bicarbonate exchanger and the electrogenic sodium-bicarbonate cotransporter, produces inhibition of the responses to 7,8-benzoquinoline of a magnitude comparable to those produced by acetazolamide.
Figure 6. Inhibition by amiloride of the response to 7,8-benzoquinoline in Calu-3 epithelia.
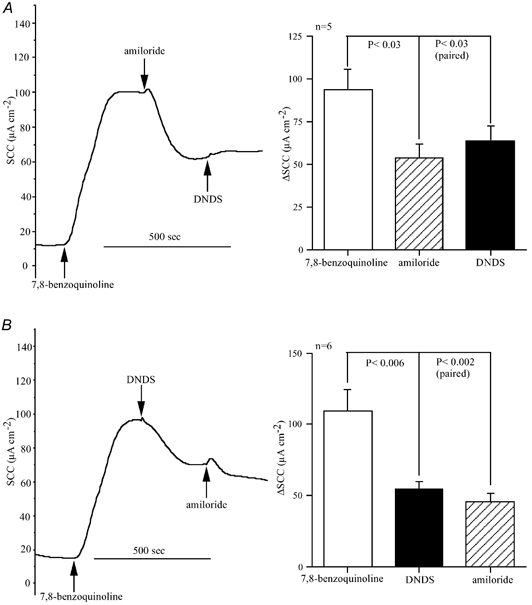
The traces show the effects of amiloride (1 mM, bl) and DNDS (3 mM, bl) on the responses to 7,8-benzoquinoline (500 μM) on two Calu-3 monolayers. The order of addition of the two inhibitors was reversed in A and B. Also shown are cumulative data from identical experiments to those shown in A and B.
Measurement of net chloride fluxes in Calu-3 monolayers using 36Cl−
Measurement of Cl− fluxes using 36Cl− as a tracer were made in 24 Calu-3 monolayers, 12 each for flux measurements in the basolateral-to-apical (bl-ap) and apical-to-basolateral (ap-bl) directions. SCC was measured simultaneously throughout and the charge transfer during the control period and during the action of 7,8-benzoquinoline was measured. The results are given in Table 1, all measurements being given as microequivalents per square centimetre per hour. It is shown that there is a small increase in the apical-to-basolateral flux after 7,8-benzoquinoline, but a larger increase in flux in the opposite direction, giving an overall increase in net transport in the basolateral-to-apical direction. Overall, it is shown that the net flux of Cl− ions is not significantly different from the net flux obtained by integration of the SCC response to 7,8-benzoquinoline. This means that under SCC conditions the whole of the response to 7,8-benzoquinoline can be explained as an increase in net flux of Cl− ions in the basolateral-to-apical direction.
Table 1.
Simulataneous measurement of 36Cl− fluxes and SCC in calu-3 monolayers
| Control period | Plus 7,8-BQ | Difference | P | n | |
|---|---|---|---|---|---|
| 36Cl− flux (ap–bl) (μequiv cm−2h−1) | 1.51 ± 0.24 | 2.25 ± 0.32 | 0.73 ± 0.22 | < 0.0001 | 12 |
| 36Cl− flux (bl–ap) (μequiv cm−2h−1) | 2.75 ± 0.74 | 5.34 ± 0.76 | 2.59 ± 0.27 | 12 | |
| 36Cl− net flux (bl–ap) (μequiv cm−2h−1) | 1.24 ± 0.77 | 3.09 ± 0.82 | 1.86 ± 0.34 | n.s. | 12 |
| SCC (μequiv cm−2h−1) | 1.11 ± 0.35 | 2.79 ± 0.18 | 1.68 ± 0.12 | 24 |
The resistance of the monolayers used in these experiments was 256 ± 24 Ω cm−2 (n = 4) and the time from seeding to use was 20.2 ± 0.2 days (n = 24). Mean values and s.e.m. are given throught. The values for bl–ap fluxes are significantly different from the values for ap–bl fluxes (P < 0.001), while the value for the net flux is not significantly different from the value derived from the SCC responses. The concentration of 7,8-benzoquinoline (7,8-BQ) used was 210 μM. SCC, short circuit current. n.s., not significant.
From this study there is little to suggest that that bicarbonate secretion contributes substantially to the responses. It is possible that Calu-3 monolayers also secrete K+ along with Cl−, which would reduce the SCC response in comparison to the net flux of Cl−. To test this possibility the effects of 7,8-benzoquinoline (250 μM, bs) were measured both before and after the addition of the non-specific K+ channel blocker Ba2+ to the apical bathing solution. The response to 7,8-benzoquinoline given after Ba2+ (5 mM) was 89.5 ± 6.7 μA cm−2 (n = 5), which was not different from that obtained in the absence of Ba2+ (107.2 ± 9.8 μA cm−2, n = 5, P = n.s.). Clearly there was no evidence that addition of Ba2+ to the apical bathing solution increased the SCC response to 7,8-benzoquinoline.
Effects of acetazolamide on the apical and basolateral membrane responses to 7,8-benzoquinoline
A number of other investigations were carried out to examine if acetazolamide had other actions on Calu-3 monolayers other than those due to the inhibition of carbonic anhydrase. Benzoquinolines and phenanthrolines are known to activate basolateral K+ channels, as well as activating an apical Cl− conductance to increase overall transepithelial anion transport (Duszyk et al. 2001; Cuthbert, 2003). We have investigated whether acetazolamide affects the actions of 7,8-benzoquinoline at either of these membrane locations. Briefly, by treating the apical membrane with nystatin to create membrane pores permeable to small ions and in the presence of an apical-to-basolateral K+ gradient, effects on basolateral K+ channels can be investigated. Similarly if the basolateral membrane is depolarised with a high K+ solution, in the presence of an apical-to-basolateral Cl− gradient, effects on the apical membrane can be recorded as changes in negative SCC. Using these methods the effects of acetazolamide on the responses to 7,8-benzoquinoline were investigated. Figure 7A shows an example of the response to 7,8-benzoquinoline (250 μM, bs) in a nystatin-treated Calu-3 monolayer, where it caused an increase in SCC of 350 μA cm−2. Addition of acetazolamide caused a minor reduction in the plateau response of approximately 5 %. This experiment, typical of three others, cannot signify a major closure of K+ channels in the basolateral membrane that were affected by the subsequent addition of ChTX (50 nM, bl). Figure 7B shows an example of the effects of 7,8-benzoquinoline on anion conductance in an ‘apical membrane only’ preparation, where the change in slope indicates the increase in anion permeability. Acetazolamide had no effect on the trajectory of the response. In a total of four experiments the slope of the -SCC versus time trace was 0.152 ± 0.063 μA s−1 and increased to 0.325 ± 0.096 μA s−1 after 7,8-benzoquinoline, 1 mM (bs) (P < 0.017, paired t test). In none of the experiments did acetazolamide affect the responses.
Figure 7. Effects of 7,8-benzoquinoline on apical and basolateral membranes of Calu-3 cells.
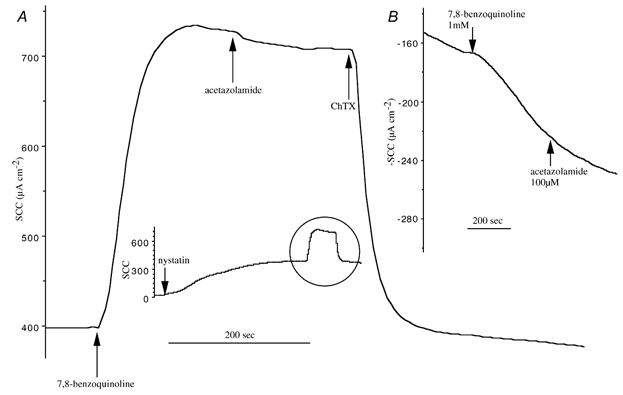
A, the effect of 7,8-benzoquinoline (250 μM, bs) on a Calu-3 monolayer previously exposed to nystatin on the apical side (360 μg ml−1 for 30 min) in the presence of an apical-to-basolateral K+ gradient. Afterwards acetazolamide (100 μM, bs) and ChTx (50 nM, bl) were added as indicated. The inset illustrates a complete experiment that is typical of 3 others. B, the response to 7,8-benzoquinoline (1 mM, bs) on a Calu-3 monolayer depolarised by high K+ on the basolateral side and in the presence of an apical-to-basolateral Cl− gradient. Acetazolamide (100 μM, bs) was added as indicated and the result is typical of 3 other experiments.
Effect of 7,8-benzoquinoline on pHi
Calu-3 cells grown on glass coverslips were used to study intracellular pH changes using BCECF as a reporter molecule, as described in the Methods section. We are aware this condition does not exactly mimic short-circuited epithelia, but is an indication of pH changes that are likely to occur following 7,8-benzoquinoline. Addition of 7,8-benzoquinoline (250 μM) in the absence of acetazolamide caused pHi to decrease by 0.19 ± 0.04 pH units from a value of 7.49 ± 0.09 (both values n = 8). In the presence of acetazolamide (100 μM) the pH was reduced by 0.34 pH units (mean of 2) by 7,8-benzoquinoline (250 μM), the former agent causing no discernable change in pH on its own. Examples are given in Fig. 8.
Figure 8. Measurement of pHi using BCECF fluorescence.
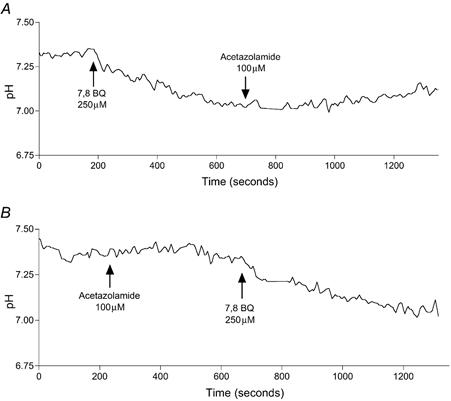
Fluorescence in Calu-3 monolayers was measured at 526 nm with excitation alternately at 440 nm and 502 nm. The fluorescence ratio was used to calculate pHi values, as described in the Methods. Addition of 7,8-benzoquinoline (A) caused a fall in pH of 0.3 units. Acetazolamide (100 μM) alone did not affect pHi (B) or prevent pH lowering by 7,8-benzoquinoline.
Effects of non-penetrating carbonic anhydrase inhibitors on reponses to 7,8-benzoquinoline in Calu-3 epithelial monolayers
Acetazolamide does not differentiate between membrane bound and cytosolic forms of carbonic anhydrase (CA), yet the membrane-bound form CAIV can display channel type characteristics (Fanjul et al. 2002). Therefore we examined whether any part of the current generated by 7,8-benzoquinoline could be attributed to CAIV. Three non-penetrating carbonic anhydrase inhibitors were used to examine for effects on 7,8-benzoquinoline responses. Each of the inhibitors CAI 1-3 was investigated on four occasions. After 7,8-benzoquinoline causes its peak response, the SCC declines slowly. The inhibitors were added at this time and on no occasion was there any change in current to indicate an inhibitory effect. Subsequent addition of acetazolamide produced a rapid inhibition of more than 50 % of the remaining current. The inhibitors, CAI 1-3 were used at concentrations that were 8, 13 and 25 times their Ki values for CAIV, respectively. Consequently we can conclude that, as the inhibitors did not penetrate cells, the effects of acetazolamide must be exerted at intracellular sites. An example of the lack of response to CAI 1-3 is given in Fig. 9.
Figure 9. The lack of effect of the charged carbonic anhydrase inhibitors CAI 1-3 on responses to 7,8-benzoquinoline.
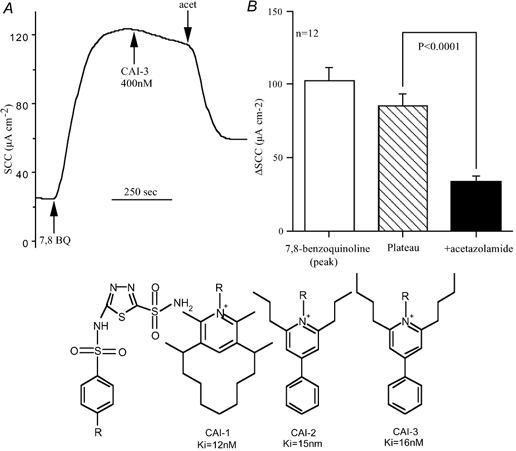
A, typical response to 7,8-benzoquinoline (250 μM, bs) in which CAI-3 (400 nM, ap) was added during the plateau phase. This experiment was repeated four times each with all three CAI inhibitors. In no experiment did any discernable change in response occur. The concentrations of the inhibitors used were 100 nM (8 × Ki) for CAI-1, 200 nM (13 × Ki) for CAI-2, and 400 nM (25 × Ki) for CAI-3. Finally acetazolamide (100 μM, bs) was added in each experiment. B, the mean response to 7,8-benzoquinoline, the mean plateau response at the time acetazolamide was added and the remaining current after acetazolamide. Acetazolamide caused a significant reduction in SCC (P < 0.0001); acet, acetazolamide. The structure of the CAI inhibitors is given below. Each has a common sulphonamide grouping with different R groups.
DISCUSSION
The major transporting activity in Calu-3 monolayers stimulated with 7,8-benzoquinoline has been shown to be the electrogenic transport of chloride ions, based upon the equivalence of SCC responses to the net transport measured with 36Cl−. Others have shown that under basal conditions approximately one-quarter of the current is due to sodium-dependent glucose absorption while the rest is eliminated by Cl− or HCO3− removal (Lee et al. 1998). Devor et al. (1999) concluded that after forskolin the stimulated current was due to HCO3− secretion, while after EBIO the dominant transported species was Cl−, with a minor contribution from HCO3−. However, our results differ in that we show that the carbonic anhydrase inhibitor acetazolamide is able to cause significant inhibition of the 7,8-benzoquinoline-stimulated current. This finding is not unique to 7,8-benzoquinoline since acetazolamide also significantly reduced the Cl− secretory response to EBIO. Furthermore we show that DNDS significantly reduces the response to 7,8-benzoquinoline, while Devor et al. (1999) report that both acetazolamide and DNDS have only small (< 10 %) inhibitory effects on the responses to EBIO. Not only did acetazolamide and DNDS significantly reduce the response to 7,8-benzoquinoline in our experiments, but so did amiloride when applied at a high concentration to the basolateral surface to inhibit the Na+-H+ exchanger. The responses to amiloride and DNDS were not additive; when applied after DNDS, amiloride caused further minor inhibition, while when DNDS was added after amiloride, no further inhibition resulted. This suggests that the transporting mechanisms inhibited by DNDS and amiloride work in concert. Furthermore the inhibitory responses to acetazolamide and DNDS were not additive, again suggesting that both interact, at least partially, via a common mechanism. We consider that our data can be used to provide a case for the existence of HCO3−-dependent Cl− secretion in Calu-3 monolayers stimulated with 7,8-benzoquinoline. In elaborating this proposition frequent reference will be made to the diagram shown in Fig. 10. With two exceptions evidence exists for the presence and location of all the transporting components indicated.
Figure 10. Diagram of a Calu-3 human epithelial transporting cell.
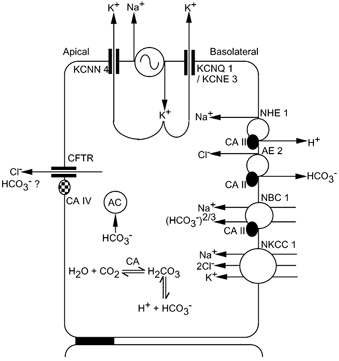
Evidence for all the transporting components shown, except two, is available and cited in the text. The exceptions are transmembrane carbonic anhydrase (CAIV) and cytosolic adenylyl cyclase (AC). Abbreviations are: sodium-proton exchanger, NHE1; Cl−-HCO3− exchanger, AE2; electrogenic sodium-bicarbonate transporter, NBC1; Na+-K+-2Cl− cotransporter, NKCC1; cytosolic carbonic anhydrase, CAII; transmembrane carbonic anhydrase, CAIV; cytosolic adenylyl cyclase, AC; intermediate conductance calcium-sensitive potassium channel, KCNN4; cAMP-sensitive potassium channel, KCNQ1/KCNE3; and cystic fibrosis transmembrane conductance regulator, CFTR. Sodium-potassium-ATPase is indicated by the alternating sign.
As acetazolamide has no known pharmacology except the inhibition of carbonic anhydrase, it is instructive to ask where it acts in the context of the diagram in Fig. 10. Clearly the cytoplasmic form of carbonic anhydrase, CAII, is inhibited, reducing the formation of cytosolic bicarbonate ions. CAII has also been shown to bind to and form a complex with NHE1, AE2 and NBC1 (Vince & Reithmeier, 2000; Gross et al. 2002; Li et al. 2002). Evidence that these three transporters are present in Calu-3 cells has also been obtained. RT-PCR detected the expression of the electrogenic Na+-HCO3− transporter, NBC1, and the Cl−-HCO3− exchanger, AE2, and functional evidence was provided for NHE1, the sodium-proton exchanger sensitive to high concentrations of amiloride (Loffing et al. 2000; Inglis et al. 2002). Human bronchi have also been shown to contain mRNA for NHE1 (Dudeja et al. 1999). The three anion exchangers AE1, AE2 and AE3 all bind cytosolic CAII (Vince & Reithmeier, 2000), as does the C-terminal region of NHE1, the latter being enhanced by low pH and by phosphorylation (Li et al. 2002). Cytoplasmic carbonic anhydrase CAII also forms a complex with NBC1. Phosphorylation of Ser982 alters the stoichiometry of NBC1 from 2:1 to 3:1 and thermodynamic arguments indicate that the phosphorylated form of NBC1 is exporting while the non-phosphorylated form is importing. Acetazolamide only inhibits NBC1 when the CAII-NBC1 complex is in the 3:1 form (Gross et al. 2002). It is also known that cells expressing NHE1 and CAII have twice the transport rate of cells expressing NHE1 alone (Li et al. 2002). The term ‘metabolon’ has been coined for complexes of membrane transporters with carbonic anhydrase as a device for co-ordinating metabolism with enhanced transporting activity. The hypothesis is that increasing substrate availability adjacent to the transport mechanism reduces the diffusion time and is responsible for enhancing transport rate. Thus NHE1, AE2 and NBC1 are all targets for acetazolamide, although inhibition of CAII will not completely inhibit transport. It is noted that 7,8-benzoquinoline causes cytosolic acidification which would facilitate the binding of CAII to NHE1. Therefore it is not surprising that DNDS and amiloride produce similar effects on 7,8-benzoquinoline responses to acetazolamide. The consequence is that the reduction in NaCl entering the cell results in a reduced source of Cl− available for transepithelial transport. This mechanism of Cl− secretion was proposed for the equine trachea to explain the source of basal electrogenic Cl− transport (Tessier et al. 1990). DNDS inhibits both AE2 and NBC1; however, it is unlikely that NBC1 functions as an importer of HCO3− after stimulation with 7,8-benzoquinoline because of membrane hyperpolarisation, so the major effect of DNDS is likely to be on AE2.
Recently, from a study of the corneal endothelium, it has been shown that cytosolic soluble, non-forskolin-sensitive adenylyl cyclase contributes significantly to baseline cAMP production, sufficiently so to influence CFTR activity and basal transport (Sun et al. 2003). It was found that phosphorylation of adenylyl cyclase was increased by 150 % in the presence of HCO3− and increased cAMP levels by 42 %. While the presence of soluble adenylyl cyclase has not been investigated in Calu-3 cells, the mechanism described for the cornea provides a further way in which a reduction in intracellular HCO3− can influence anion transport. Inhibition of carbonic anhydrase will reduce the availability of HCO3− in cells, especially if hyperpolarisation due to 7,8-benzoquinoline has inhibited or reversed the effect of NBC1, and could influence apical membrane Cl− conductance via an effect on cAMP generation.
It is shown that removal of HCO3−/CO2 from the bathing fluid reduces the responses to 7,8-benzoquinoline, supporting the contention that acetazolamide is acting via inhibition of carbonic anhydrase. However, when the sensitivity to 7,8-benzoquinoline was poor, in that the SCC response was smaller than expected, the effect disappeared. Although no systematic study has been made, our impression is that poor responses are generally obtained in old monolayers from high passage numbers, especially when used 4-5 days post feeding. Caci et al. (2003) have also encountered this phenomenon with human airway epithelial cells and contend that it is due to variation in endogenous cAMP concentrations. Our investigations in poorly responding monolayers support this view, as addition of IBMX restored the responses to 7,8-benzoquinoline and the effects of HCO3−/CO2 removal. An important conclusion from this result is that the bicarbonate dependence is most important when the responses are large.
Thus there are a variety of ways in which inhibition of carbonic anhydrase with acetazolamide or removal of HCO3−/CO2 can reduce the transport of Cl− ions across Calu-3 epithelia. However this creates some problems of interpretation. For example, when bumetanide is given after 7,8-benzoquinoline the inhibition appears to be too large (greater than 50 %, while acetazolamide also inhibits by 50 % or more). The bumetanide-sensitive Na+-K+-2Cl− cotransporter is certainly present in Calu-3 cells, as demonstrated by the presence of mRNA for, and protein expression of, this transporter by Liedtke et al. (2001). Its basolateral location is evidenced by the sidedness of the effects of bumetanide. However, as has been shown, the alternate NHE1 plus AE2 mechanism for Cl− secretion is demonstrable only at higher transport rates. Thus one possibility is that, as bumetanide blocks Cl− secretion via inhibition of the Na+-K+-2Cl− cotransporter, the NHE1-AE2 mechanism downregulates since the transport rate is lowered. This possibility will require investigation. A further point is that the effects of acetazolamide after 7,8-benzoquinoline may be exaggerated if the basal HCO3− secretion present in unstimulated Calu-3 monolayers continues after stimulation with 7,8-benzoquinoline. This may be unlikely if NBC1 is compromised by the addition of hyperpolarising 7,8-benzoquinoline. Yet a further complication is the sensitivity of the Na+-K+-2Cl− cotransporter to Cl−i (Haas & McBrayer, 1994; Putney et al. 1999). If blocking NHE1 and AE2 or alternatively inhibiting carbonic anhydrase reduces Cl−i, then the activity of the cotransporter will be upregulated. We note that when inhibitors are used there is some rebound after maximal inhibition is achieved (Fig. 5 and Fig. 6), which could represent upregulation of the Na+-K+-2Cl− cotransporter, although we have not specifically investigated this.
We have searched for other possible actions of acetazolamide on the channels known to be present in both apical and basolateral membranes. Turning first to the apical membrane, both functional and expression studies show the presence of CFTR in Calu-3 cells (Haws et al. 1994; Liedtke et al. 2001) and it is known that lowering pHi reduces the chloride conductance (GCl) of CFTR in sweat gland epithelia (Reddy et al. 1998). Addition of 7,8-benzoquinoline caused a modest lowering of pHi but provided no clue that pH changes were relevant to the inhibitory effects of acetazolamide on SCC. We also investigated if acetazolamide acted upon the basolateral K+ channels so crucial for the stimulation by 7,8-benzoquinoline. That two types of basolateral epithelial K+ channels exist in Calu-3 cells follows from the demonstration of appropriate mRNAs (Cowley & Linsdell, 2002). However, in nysatin-treated monolayers acetazolamide had only a miniscule inhibitory effect on the current, possibly as a result of cells that had not been permeabilised with nystatin.
It is hardly necessary to comment on the presence of the ubiquitous Na+-K+-ATPase in the diagram, since it is found in all cells, but its basolateral location was proven from the sidedness of the effects of ouabain on SCC in Calu-3 monolayers (Singh et al. 1997). The inclusion of a membrane-bound form of carbonic anhydrase, CAIV in the apical membrane does require comment since it has not been looked for in Calu-3 cells. CAIV is found in HCO3−-secreting human pancreatic duct cells, where trafficking to the cell membrane is dependent upon CFTR and where it may show channel characteristics (Fanjul et al. 2002). Thus some fraction of the current inhibited by acetazolamide may have been due to HCO3− movement through CAIV. There is a continuing debate as to whether, in HCO3−-secreting epithelia, HCO3− effluxes through CFTR or whether there is a parallel arrangement of CFTR with a Cl−-HCO3− exchanger. However no evidence for the presence of AE2 in the apical membrane of Calu-3 cells has been found (Lee et al. 1998; Loffing et al. 2000). Acetazolamide does not differentiate between membrane bound and cytosolic CAs, hence we used non-permeable CA inhibitors to investigate the possibility that membrane-bound carbonic anhydrase in the apical membrane was involved in anion transport. The inhibitors CAI 1-3 had no discernable effect on the responses to 7,8-benzoquinoline. Thus CAIV does not appear to be involved in the actions of 7,8-benzoquinoline on Calu-3 cells.
Finally, we must conclude that activities of AE2 and/or NHE1 are increased by 7,8-benzoquinoline, as no Cl− transport is present in the basal state. We do not know if this activity is direct or a consequence of other changes. Presumably this activity is also demonstrated by EBIO, which we showed behaved similarly to 7,8-benzoquinoline. The quantitative differences between EBIO and 7,8-benzoquinoline could be explained by differences in the relative activities on different cellular targets in both the apical and basolateral membranes.
In summary, 7,8-benzoquinoline activates Cl− secretion by co-ordinate actions at basolateral K+ channels and apical Cl− channels, presumably CFTR. Cl− enters the cell through the basolateral membrane using the Na+-K+-2Cl− cotransporter and a parallel arrangement of NHE1 with AE2, these latter two being sensitive to acetazolamide because of their association with CAII. Amiloride and DNDS can achieve the same effect as acetazolamide, again through inhibition of one of the processes for Cl− entry. Transport mediated by NHE1 together with AE2 is important when the transport rate is high. The electrogenic sodium-bicarbonate cotransporter probably has little effect on transepithelial transport in the presence of 7,8-benzoquinoline, since it is inhibited or reversed by membrane hyperpolarisation. Thus a fraction of the current stimulated by 7,8-benzoquinoline and inhibited by acetazolamide can be said to represent HCO3−-dependent Cl− secretion.
Acknowledgments
Support from the Cystic Fibrosis Trust and the Leverhulme Trust is gratefully acknowledged. We thank Caroline Taylor for expert help with the fluorescence measurements.
REFERENCES
- Basbaum CB, Jany B, Finkbeiner WE. The serous cell. Ann Rev Physiol. 1990;52:97–113. doi: 10.1146/annurev.ph.52.030190.000525. [DOI] [PubMed] [Google Scholar]
- Caci E, Folli C, Zegarra-Moran O, Ma T, Springsteel MF, Sammelson RE, Nantz MH, Kurth MJ, Verkman AS, Galietta M. CFTR activation in human bronchial epithelial cells by novel benzoflavone and benzimidazolone compounds. Am J Physiol Lung Cell Mol Physiol. 2003;285:180–188. doi: 10.1152/ajplung.00351.2002. [DOI] [PubMed] [Google Scholar]
- Cowley EA, Linsdell P. Characteristaion of basolateral K+ channels underlying anion secretion in the human airway cell line Calu-3. J Physiol. 2002;538:747–757. doi: 10.1113/jphysiol.2001.013300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert AW. Assessment of CFTR chloride channel openers in intact normal and cystic fibrosis murine epithelia. Br J Pharmacol. 2001;132:659–668. doi: 10.1038/sj.bjp.0703859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert AW. Benzoquinolines and chloride secretion in murine colonic epithelia. Br J Pharmacol. 2003;138:1528–1534. doi: 10.1038/sj.bjp.0705203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert AW, MacVinish LJ. Mechanisms of anion secretion in Calu-3 human airway epithelial cells by 7,8-benzoquinoline. Br J Pharmacol. 2003 doi: 10.1038/sj.bjp.0705403. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor DC, Singh AK, Lambert LC, Deluca A, Frizzell RA, Bridges RJ. Bicarbonate and chloride secretion in Calu-3 airway epithelial cells. J Gen Physiol. 1999;113:743–760. doi: 10.1085/jgp.113.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudeja PK, Hafez N, Tyagi S, Gailey CA, Toofanfard M, Alrefai WA, Nazir TM, Ramaswamy K, Al-Bazzaz FJ. Expressionof the Na+/H+ and the Cl−/HCO3− exchanger isoforms in proximal and distal human airways. Am J Physiol. 1999;276:L971–978. doi: 10.1152/ajplung.1999.276.6.L971. [DOI] [PubMed] [Google Scholar]
- Duszyk M, MacVinish LJ, Cuthbert AW. Phenanthrolines-a new class of CFTR chloride channel openers. Br J Pharmacol. 2001;134:853–864. doi: 10.1038/sj.bjp.0704328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt JF, Yankaskas JR, Ernst SA, Yang Y, Marino CR, Boucher RC, Cohn JA, Wilson JM. Submucosal glands are the predominant sites of CFTR expression in the human bronchus. Nat Genet. 1992;2:240–248. doi: 10.1038/ng1192-240. [DOI] [PubMed] [Google Scholar]
- Fanjul M, Salvador C, Alvarez L, Cantet S, Hollande E. Targeting of carbonic anhydrase IV to plasma membranes is altered in cultured human pancreatic duct cells expressing a mutated (deltaF508) CFTR. Eur J Cell Biol. 2002;81:437–447. doi: 10.1078/0171-9335-00264. [DOI] [PubMed] [Google Scholar]
- Gross E, Pushkin A, Abuladze N, Fedotoff O, Kurtz I. Regulation of the sodium bicarbonate cotransporter kNBC1 function: role of the Asp986, Asp988 and kNBC1-carbonic anhydrase II binding. J Physiol. 2002;544:679–685. doi: 10.1113/jphysiol.2002.029777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas M, McBrayer DG. Na-K-Cl cotransport in nystatin-treated tracheal cells:regulation by isoproterenol, apical UTP and [Cl]i. Am J Physiol. 1994;266:C1440–1452. doi: 10.1152/ajpcell.1994.266.5.C1440. [DOI] [PubMed] [Google Scholar]
- Haws C, Finkbeiner WE, Widdicombe JH, Wine JJ. CFTR in Calu-3 human airway cells :channel properties and role in cAMP-activated Cl− conductance. Am J Physiol. 1994;266:L502–512. doi: 10.1152/ajplung.1994.266.5.L502. [DOI] [PubMed] [Google Scholar]
- Inglis SK, Finlay L, Raminger SJ, Richard K, Ward NR, Wilson SM, Olver RE. Regulation of intracellular pH in Calu-3 human airway cells. J Physiol. 2002;538:527–539. doi: 10.1113/jphysiol.2001.012806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Penland CM, Widdicombe JH, Wine JJ. Evidence that Calu-3 human airway cells secrete bicarbonate. Am J Physiol. 1998;274:L450–453. doi: 10.1152/ajplung.1998.274.3.L450. [DOI] [PubMed] [Google Scholar]
- Li X, Alvarez B, Casey JR, Reithmeier RA, Fliegel L. Carbonic anhydrase II binds to and enhances activity of the Na+/H+ exchanger. J Biol Chem. 2002;277:36085–36091. doi: 10.1074/jbc.M111952200. [DOI] [PubMed] [Google Scholar]
- Liedtke CM, Cody D, Cole TS. Differential regulation of Cl− transport proteins by PKC in Calu-3 cells. Am J Physiol Lung Cell Mol Physiol. 2001;280:L739–7447. doi: 10.1152/ajplung.2001.280.4.L739. [DOI] [PubMed] [Google Scholar]
- Loffing J, Moyer BD, Reynolds D, Shmukler BE, Alper SL, Stanton BA. Functional and molecular characteristaion of an anion exchanger in airway serous cells. Am J Physiol Cell Physiol. 2000;279:C1016–1023. doi: 10.1152/ajpcell.2000.279.4.C1016. [DOI] [PubMed] [Google Scholar]
- Moon S, Singh M, Krouse ME, Wine JJ. Calcium-stimulated Cl− secretion in Calu-3 human airway cells requires CFTR. Am J Physiol. 1997;273:L1208–1219. doi: 10.1152/ajplung.1997.273.6.L1208. [DOI] [PubMed] [Google Scholar]
- Pilewski JM, Frizzell RA. Role of CFTR in airway disease. Physiol Rev. 1999;79:S215–S255. doi: 10.1152/physrev.1999.79.1.S215. [DOI] [PubMed] [Google Scholar]
- Putney LK, Vibat CRT, O'Donnell ME. Intracellular chloride regulates Na-K-Cl cotransport activity in human trabecular meshwork cells. Am J Physiol. 1999;277:C373–383. doi: 10.1152/ajpcell.1999.277.3.C373. [DOI] [PubMed] [Google Scholar]
- Reddy MM, Kopito R, Quinton PM. Cytosolic pH regulates GCl through control of phosphorylation states of CFTR. Am J Physiol. 1998;275:C1040–1047. doi: 10.1152/ajpcell.1998.275.4.C1040. [DOI] [PubMed] [Google Scholar]
- Scozzafava A, Briganti F, Ilies MA, Supuran CT. Carbonic anhydrase inhibitors: synthesis of membrane-impermeant low molecular weight sulfonamides posessing in vivo selectivity for the membrane-bound vs. cytosolic isozymes. J Med Chem. 2000;43:292–300. doi: 10.1021/jm990479+. [DOI] [PubMed] [Google Scholar]
- Shen B-Q, Finkbeiner WE, Wine JJ, Mrsny RJ, Widdicombe JH. Calu-3: a human airway epithelial cell line that shows cAMP-dependent Cl− secretion. Am J Physiol. 1994;266:L493–501. doi: 10.1152/ajplung.1994.266.5.L493. [DOI] [PubMed] [Google Scholar]
- Singh M, Krouse M, Moon S, Wine JJ. Most basal ISC in Calu-3 human airway cells is bicarbonate-dependent Cl− secretion. Am J Physiol. 1997;272:L690–698. doi: 10.1152/ajplung.1997.272.4.L690. [DOI] [PubMed] [Google Scholar]
- Sun XC, Zhai CB, Cui M, Chen Y, Levin LR, Buck J, Bonanno JA. HCO3−-dependent soluble adenylyl cyclase activates the cystic fibrosis transmembrane conductance regulator in corneal endothelium. Am J Physiol. 2003;284:C1114–1122. doi: 10.1152/ajpcell.00400.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev. 2003;23:146–189. doi: 10.1002/med.10025. [DOI] [PubMed] [Google Scholar]
- Tessier GJ, Traynor TR, Kannan MS, O'Grady SM. Mechanisms of sodium and chloride transport across equine tracheal epithelium. Am J Physiol. 1990;259:L459–467. doi: 10.1152/ajplung.1990.259.6.L459. [DOI] [PubMed] [Google Scholar]
- Vince JW, Reithmeier RAF. Identification of the carbonic anhydrase II binding site in the Cl−/HCO3− anion exchanger. Biochemistry. 2000;39:5527–5533. doi: 10.1021/bi992564p. [DOI] [PubMed] [Google Scholar]