

Abstract
The present study examined the effect of chronic activation of 5′-AMP-activated protein kinase (AMPK) on the metabolic profile, including uncoupling protein-3 (UCP-3) and myosin heavy chain (MHC)-based fibre phenotype of rodent fast-twitch tibialis anterior muscle. Sprague-Dawley rats were given daily injections of 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR), a known activator of AMPK, or vehicle (control) for 28 days. After AICAR treatment, UCP-3 expression at the mRNA level was elevated 1.6 ± 0.1-fold (P < 0.006) and corresponded to a 3.3 ± 0.2-fold increase in UCP-3 protein content (P < 0.0001). In addition, the activities of the mitochondrial reference enzymes citrate synthase (EC 4.1.3.7) and 3-hydroxyacyl-CoA-dehydrogenase (EC 1.1.1.35), which are known to increase in proportion to mitochondrial volume density, were elevated 1.6-fold (P < 0.006), while the activity of lactate dehydrogenase (EC 1.1.1.27) was reduced to 80 % of control (P < 0.02). No differences were detected after AICAR treatment in the activities of the glycolytic reference enzymes glyceraldehydephosphate dehydrogenase (EC 1.2.1.12) or phosphofructokinase (EC 2.7.1.11), nor were MHC-based fibre-type transitions observed, using immunohistochemical or electrophoretic analytical methods. These changes could not be attributed to variations in inter-organ signalling by metabolic substrates or insulin. We conclude that an AMPK-dependent pathway of signal transduction does mimic some of the metabolic changes associated with chronic exercise training, but does not affect expression of the MHC-based structural phenotype. Thus, the metabolic and MHC-based fibre types do not appear to be regulated in a co-ordinated way, but may be independently modified by different signalling pathways.
Increased contractile activity, such as chronic exercise training or chronic low-frequency stimulation (CLFS), induces in fast-twitch skeletal muscle a set of adaptive responses in an intensity- and time-dependent manner. Collectively, these changes affect the performance of the contractile machinery with regard to both energy supply and energy expenditure. Increases in mitochondrial content serve to enhance the aerobic-oxidative capacity of energy supply for sustained performance, while fast-to-slower fibre-type transitions form the basis for more economical energy expenditure (Pette & Staron, 2000). The CLFS protocol has been proven to evoke maximum responses of these two systems. In rat muscle, CLFS induces pronounced increases in mitochondrial enzyme activities of terminal substrate oxidation concomitant with decreases in glycolytic and glycogenolytic enzyme activities. In addition, we have recently shown that it leads to an upregulation of the mitochondrial uncoupling protein-3 (UCP-3) (Putman et al. 2002). As also shown in previous studies, CLFS of adult rat fast-twitch muscle leads to fibre-type transitions, mainly within the fast-fibre population and in the direction from type IIB to type IIA (Jaschinski et al. 1998; Putman et al. 2000).
Increasing evidence indicates that metabolic and myofibrillar adaptive responses to increased contractile activity are under the control of different pathways of signal transduction (for review see Pette, 2002). A calcineurin-dependent pathway seems to be involved in the upregulation of slow myosin (Naya et al. 2000; Bigard et al. 2000; Meissner et al. 2001). The Ca2+-calmodulin protein kinase-related pathway (CaMKIV) induces mitochondrial biogenesis (Wu et al. 2002) and also activates the myosin heavy chain (MHC) IIA promoter (Allen & Leinwand, 2002). An independent pathway of signal transduction involving AMP-activated protein kinase (AMPK) has been identified as being related to the metabolic adaptation to increased contractile activity (Hutber et al. 1997; Holmes et al. 1999; Winder et al. 2000; Winder, 2001). With regard to enhanced mitochondrial biogenesis, therefore, two independent pathways, namely a CaMK- and the AMPK-related chains of signal transduction appear to be involved in metabolic adaptation. Whereas the CaMK pathway seems to regulate both metabolic and myofibrillar adaptations, it is not clear whether the AMPK pathway is solely involved in mitochondrial biogenesis or additionally contributes to myofibrillar adaptations. To our knowledge, this question has not been addressed.
In order to study this possibility, a model of chronically activating AMPK in vivo was used. Chronic AMPK activation was induced by chronic administration of 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR), a known activator of the enzyme (Corton et al. 1995; Salt et al. 1998). For this purpose, adult male rats were injected daily with the activator for a period of 28 days. To investigate metabolic effects, the activities of mitochondrial references enzymes of aerobic-oxidative metabolism, which are known to increase in direct proportion to increases in mitochondrial volume density (Holloszy & Coyle, 1984; Reichmann et al. 1985; Harlan & Williams, 1988; Hoppeler, 1990; Pette & Staron, 1997), were measured. Because uncoupling protein-3 has recently been reported to also increase in proportion to mitochondrial biogenesis (Jones et al. 2003) and to be independently regulated by AMPK (Zhou et al. 2000; Stoppani et al. 2002), we also examined mRNA and protein expression levels of this mitochondrial carrier. Cytosolic enzymes of anaerobic energy metabolism were also studied and immunohistochemical and electrophoretic analyses were performed to examine the distribution profiles of MHC-based fibre types.
METHODS
Animals
Ten adult male Sprague-Dawley rats were obtained from the local animal facility of the University of Alberta and used in the present study. All experiments were completed in accordance with the guidelines of the Canadian Council for Animal Care and received ethical approval from the University of Alberta. The animals were maintained under controlled environmental conditions (22 °C and 12 h alternating light and dark cycles), and received standard chow and water ad libitum.
AICAR treatment
Rats received either daily subcutaneous injections of 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR, Sigma, St Louis, MO, USA; 0.1 mg (kg body weight)−1 for 2 weeks and 0.5 mg (kg body weight)−1 for the remaining 2 weeks of the study; n = 5), or an equal volume of the vehicle (sterile buffered saline, pH 7.4; n = 5) for the same duration. At the end of the treatment period rats were anaesthetized (40 mg (kg body weight)−1 sodium pentobarbital) and gastrocnemius muscles were freeze-clamped, excised and immediately frozen in liquid nitrogen (-196 °C); blood samples were simultaneously collected in heparinized syringes by cardiac puncture. Animals were subsequently killed by exsanguination and the tibialis anterior (TA) muscles were excised and frozen in melting isopentane (-156 °C).
Plasma metabolites and insulin
Plasma free fatty acids (FFA) and whole blood lactate (Lac−) concentrations were measured according to Putman et al. (1993). Plasma glucose was determined using a Beckman Glucose Analyser II (Model 16517; Mississauga, Canada). Plasma insulin was quantified by radioimmunoassay (RIA; Linco, Rat Insulin RIA Kit, St Charles, MO, USA).
AMPK activity
AMPK activity was assayed in extracts of freeze-clamped gastrocnemius by measuring the rate of incorporation of terminal phosphate of 32P-ATP into the synthetic AMARA peptide (AMARAASAAALARRR; Alberta Peptide Institute, University of Alberta, Canada) at 30 °C (Kudo et al. 1995).
SDS-PAGE, Western blot analyses
Frozen muscle powder was homogenized in 10 volumes of the following solution: 10 % glycerol, 0.5 % SDS, 0.5 % β-mercaptoethanol, 0.25 % (w/v) of protease inhibitor cocktail (Roche Biochemicals), 25 mM Tris-Cl (pH 6.8). The homogenate was heated for 15 min at 96 °C and cleared by centrifugation (15 min, 13 000 g, 4 °C). Supernatant fractions were collected, and protein contents determined using the Bradford assay (BioRad). To ensure equal loading, samples were diluted to the same protein concentrations with homogenization buffer containing 0.1 % bromophenol blue. Electrophoresis was performed for 105 min at 150 V on a 12 % polyarcylamide mini-gel (BioRad Protean II). Proteins were electrophoretically transferred to a PVDF membrane (40 min, 200 mA), using a semi-dry technique (Hoeffer, Mulitemp III). Membranes were blocked overnight at 4 °C in a buffer containing 2.5 % skim milk powder, 0.5 % protease free and fat-free bovine serum albumin. Primary anti-UCP-3 antibody (Affinity Bioreagents Inc.) was applied overnight at +4 °C in blocking solution (1:1000). After washing, the membranes were incubated for 45 min with horseradish peroxidase-conjugated anti-rabbit IgG (Vector Laboratories Inc., 1:2000 in blocking solution). Immunoreactivity was subsequently detected with the ECL Plus chemiluminescence substrate (BioRad) and documented. To quantify acetylCoA carboxylase (ACC) phosphorylation, gels were run as before; primary monoclonal anti-phospho-ACC antibody (Cell Signalling Technology, Beverly, MA, USA) was applied overnight at 4 °C. Membranes were subsequently washed and developed as described above. Under the Western blot conditions used, UCP-3 was not detected in extracts of tissues known to not express significant amounts of UCP-3 at the protein level (i.e. neonatal rat liver and white adipose tissue), thus confirming the specificity of the anti-UCP-3 antibody used. All samples were evaluated in triplicate. Immunoblots were evaluated by integrating densitometry using GeneSnap and GeneTools (Chemigenius Gel Documentation System, Syngene, UK).
UCP-3 and α-actin mRNA determination
TA muscle samples were powdered under liquid nitrogen and total RNA was isolated (Putman et al. 2000). The concentrations of RNA extracts were subsequently quantified by measuring the absorbance at 260 nm using a microplate reader (Spectra Max 190, Molecular Devices, Sunnyvale, CA, USA) and 96-well flat bottom UV-transparent plates (Corning Inc. Costar). Samples were also examined at 280 nm to judge purity. Samples were subsequently diluted to 1 μg (μl)−1 and reverse transcription (RT) was performed for 1 h at 37 °C using 1 μg of total RNA with random oligo (dT15) primers (Invitrogen, Life Technologies, Burlington, Ontario, Canada), and the Moloney Murine Leukemia Virus (M-MLV) DNA polymerase (Invitrogen, Life Technologies, Burlington, Ontario, Canada). PCR was performed using the following UCP-3 primers: 5′-AAG AGT GCA GAG CGT GCA GTA-3′ (forward) and 5′-ACA GAA ACC AGC TCC AAA G-3′ (reverse) (Sigma Genosys), which yielded a 537 bp amplicon corresponding to a unique non-homologous transintronic region near the 5′-end of the UCP-3 mRNA. Primers for rat UCP-3 were designed within non-homologous regions with other UCP homologues using Genejockey software (Genbank accession number AB006614; Matsuda et al. 1997). To confirm that no cross-reactivity existed with other UCP homologues, the PCR product was screened using Amplify (Sigma). For determination of α-actin mRNA, PCR was performed using the primers, 5′-GCG GTG CTG TCT CTC TAT GC-3′ (forward) and 5′-CGG TGA GGA TTT TCA TCA GG-3′ (reverse) (Sigma Genosys). A single PCR product of 127 bp was produced, which corresponded to nt417-nt589 of the translated region. Primers were also designed using Genejockey software against rat α-actin (Genbank accession number J00692). The resulting UCP-3 and α-actin amplicons were validated for linearity and sequenced (Molecular Biology Services Unit, University of Alberta).
Reactions were carried out in an ICycler (BioRad Laboratories, Mississauga, Ontario, Canada) using Taq DNA polymerase (Sigma, Canada). UCP-3 mRNA was amplified under the following conditions: 94 °C for 5 min, followed by 28 cycles consisting of 94 °C for 30 s, 52 °C for 1 min 15 s, and 72 ° C for 1 min 15 s, and finally a 7 min extension at 72 °C. α-Actin was amplified in separate tubes under identical conditions, with the exception of a 55 °C primer annealing temperature. Both UCP-3 and α-actin PCR were performed using aliquots from the same RT reaction. All samples were analysed in triplicate. PCR products were resolved on a 2 % agarose gel, stained with ethidium bromide, and evaluated by integrating densitometry using GeneSnap and GeneTools software (ChemiGenius, Syngene, UK.), and expressed as the ratio of UCP-3 mRNA to α-actin mRNA. α-Actin mRNA was not altered by AICAR treatment.
Enzyme measurements
Measurements of citrate synthase (CS, EC 4.1.3.7), 3-hydroxyacyl-CoA dehydrogenase (HADH, EC 1.1.1.35), and lactate dehydrogenase (LDH, EC 1.1.1.27) were performed on 10 % whole muscle extracts prepared according to (Suarez et al. 1986). CS activity was measured at 30 °C according to Srere (1969). HADH activity was also determined at 30 °C according to Bass et al. (1969), with the exception that the final concentration of acetoacetyl-CoA was reduced to 0.06 mM.
In order to assure complete extraction of soluble and structure-bound activities, glyceraldehyde phosphate dehydrogenase (GAPDH, EC 1.2.1.12) and phosphofructokinase (PFK, EC 2.7.1.11) were extracted in a high-salt medium containing 5 mM EDTA, 0.1 M sodium potassium phosphate buffer (pH 7.2), according to (Reichmann et al. 1983), with the addition of 0.1 % Triton X-100. To stabilize PFK, fructose 1,6-bisphosphate and dithithreitol (DTT) were added to an aliquot of the supernatant fraction yielding 1 mM and 2 mM final concentrations, respectively. PFK and GAPDH activities were immediately measured at 30 °C according to Bass et al. (1969). Soluble protein was subsequently determined using the Bradford assay (BioRad).
Immunohistochemistry for MHC isoforms
To classify individual fibre types, the following monoclonal antibodies directed against adult MHC isoforms were used: MHCIβ (NOQ7.5.4D) (Putman et al. 2000); MHCIα (IgG, F88 12F8, 1) (Putman et al. 1999); MHCIIA (SC-71), MHC-embryonic (BF-45) and MHCIIB (BF-F3) (Schiaffino et al. 1989). Staining of 12-μm-thick, frozen sections was completed according to Putman et al. (2000). Types Iβ, IIA, and IIB fibres were identified by their reactions with the corresponding anti-MHC monoclonal antibodies. Type IID/X fibres were identified as those fibres that remained unstained by this panel of antibodies. This method allowed accurate determination of all four adult fibre types. Quantitative evaluation of MHC-based fibre types was performed using a computer program (Putman et al. 2000). Up to three separate areas were examined for each muscle, which corresponded to an average of 537 ± 66 and 532 ± 37 fibres for each of the control and AICAR-treated muscles, respectively. A total of 5345 fibres were analysed.
Analysis of MHC isoforms
MHC isoforms were electrophoretically resolved, in duplicate, under denaturing conditions on 7 % PAA separating gels containing glycerol according to Hämäläinen & Pette (1996), detected by silver staining (Oakley et al. 1980) and evaluated by densitometry (ChemiGenius, Syngene, UK). High-loading conditions (i.e. 2 μg of total extract per lane) were used to accurately quantify the proportion of MHCI present, while low-loading conditions (i.e. 0.75 μg of total extract per lane) were employed to resolve the three fast MHC isoforms, (i.e. MHCIIA, MHCIID/X and MHCIIB).
Statistical analyses
Data are presented as means ± S.E.M. Differences between the control and AICAR groups were assessed by a two-tailed independent-samples Student's t test. Differences were considered significant at P < 0.05.
RESULTS
Animal body weights
The body weights of the control and AICAR groups were similar at the beginning (mean ± S.E.M., AICAR vs. control, 471 ± 5 and 466 ± 6 g, P ≥ 0.50) and the end of the study (AICAR vs. control: 560 ± 17 and 579 ± 16 g, P ≥ 0.44). The absolute weight gained by the two groups over the course of the experiment was also similar (AICAR vs. control: 94 ± 22 and 107 ± 19 g, P ≥ 0.66).
Plasma metabolites and insulin
The concentrations of plasma glucose (P ≥ 0.74), and whole blood Lac− (P ≥ 0.97) were not altered by AICAR treatment (Table 1). In contrast, the concentrations of plasma free fatty acids (P < 0.05) and insulin (P < 0.02) were 43 % and 86 % lower, respectively, in the AICAR group (Table 1).
Table 1.
Concentations of plasma free fatty acids, glucose and insulin, and whole-blodd lactate in control and AICAR-treated rats
| Measure | ||||
|---|---|---|---|---|
| Condition | FFA (mm) | Glucose (mm) | Lactate (mm) | Insulin (pm) |
| Control | 0.23 ± 0.03 | 13.2 ± 0.5 | 4.01 ± 1.30 | 908 ± 254 |
| AICAR | 0.13 ± 0.03* | 12.9 ± 0.7 | 3.97 ± 0.47 | 1.30 ± 13* |
AICAR different from control.
AMPK activity and ACC phosphorylation
Chronic AICAR treatment was not associated with increased AMPK activities, as determined in vitro (AMPK, control vs. AICAR: 616 ± 71 vs. 606 ± 121 pmol min−1 (mg protein)−1, P ≥ 0.95). However, because the response of AMPK to chronic AICAR exposure is known to be blunted when measured in vitro (Winder et al. 2000; Buhl et al. 2002), in vivo AMPK activation was confirmed by examining the phosphorylation state of a known AMPK target, ACC. This is an established method of confirming in vivo AMPK activity (Nielsen et al. 2003). In the present study chronic AICAR administration resulted in a 9.5 ± 1.9-fold increase (n = 4 each condition, P < 0.005) in phosphorylation of ACC (Fig. 1).
Figure 1. AcetylCoA carboxylase phosphorylation.
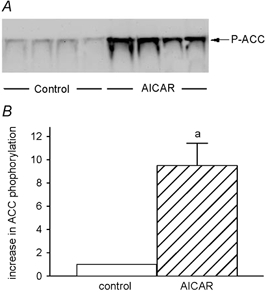
Representative immunoblot of control and AICAR-treated muscles for phosphorylated ACC (Ser79) (A) and the densitometric evaluation (B). a, AICAR different from control.
UCP-3 protein levels
A representative immunoblot for UCP-3 protein is shown in Fig. 2A and reveals marked increases in the muscles of the AICAR-treated animals. According to densitometric evaluation of triplicate measures of extracts from each muscle, the increase in UCP-3 protein content of the AICAR-treated muscles exceeded that of the controls by a factor of 3.3 ± 0.2 (P < 0.0001) (Fig. 2B).
Figure 2. UCP-3 protein content.
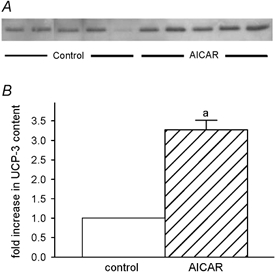
Representative immunoblot of control and AICAR-treated TA muscles for UCP-3 protein (A) and the densitometric evaluation of UCP-3 protein content (B). a, AICAR different from control.
UCP-3 mRNA
The signal intensities for UCP-3 mRNA also revealed a strong upregulation in the muscles of AICAR-treated rats (Fig. 3A). Relative to α-actin mRNA, densitometric evaluation showed a 1.6 ± 0.1-fold increase (from 0.29 ± 0.03 to 0.45 ± 0.03, P < 0.006) of the signal for UCP-3 mRNA (Fig. 3B).
Figure 3. UCP-3 mRNA expression.
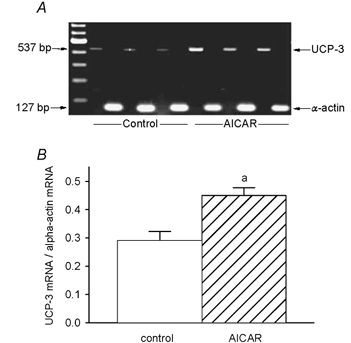
A, relative mRNA expression of UCP-3 and α-actin, as determined by RT-PCR in control and AICAR-treated TA muscles. B, sequences were amplified separately, visualized by ethidium bromide staining and quantified by integrating densitometry. a, AICAR different from control.
Enzyme activities
Mitochondrial citrate synthase (CS) and 3-hydroxyacyl-CoA dehydrogenase (HADH) activities were both 1.6-fold elevated (P < 0.006) over control in the muscles of AICAR-treated rats. This increase was true for both absolute (Fig. 4) and specific activities, as the soluble protein content was largely unaffected by the treatment (control vs. AICAR: 128.0 ± 9.3 vs. 107.1 ± 6.0 mg protein (g muscle)−1, P ≥ 0.10). Thus, CS and HADH activities amounted to 0.36 ± 0.02 and 0.19 ± 0.02 U mg−1 of soluble protein in the control muscles, and increased to 0.57 ± 0.03 (P < 0.0001) and 0.32 ± 0.02 U mg−1 (P < 0.003) of soluble protein in the AICAR muscles, respectively. Conversely, the absolute activity of the cytosolic lactate dehydrogenase was reduced to ≈80 % of that observed in the control muscles (P < 0.02) (Fig. 5). As referred to soluble protein, LDH activity was 12.4 ± 0.8 U (mg protein)−1 in the control and 10.0 ± 0.5 U (mg protein)−1 in the AICAR group (P < 0.04). Whilst the glycolytic enzymes phosphofructokinase (PFK) and glyceraldehyde phosphate dehydrogenase (GAPDH) were slightly elevated in AICAR-treated muscles (Fig. 5), the differences were not statistically significant for either enzyme (i.e. PFK, P ≥ 0.24; GAPDH, P ≥ 0.37). Similarly the specific activities of the PFK and GAPDH were slightly elevated in AICAR-treated muscles but did not reach statistical significance; PFK and GAPDH-specific activities were 0.33 ± 0.05 and 7.1 ± 0.9 U (mg soluble protein)−1 in the control muscles, and 0.44 ± 0.03 (P ≥ 0.08) and 8.6 ± 0.5 U (mg soluble protein)−1 (P ≥ 0.17) in the AICAR muscles, respectively. The increase in UCP-3 was 2.1 ± 0.3- and 2.0 ± 0.4-fold greater than the increases in CS and HADH (P < 0.005), respectively.
Figure 4. Mitochondrial reference enzyme activities.
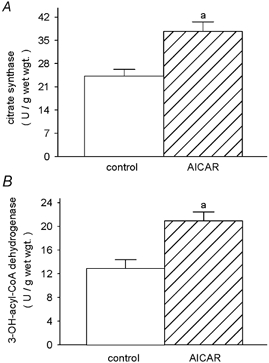
Activities of the mitochondrial enzymes citrate synthase (EC 4.1.3.7) (A) and 3-hydroxyacyl-CoA dehydrogenase (EC 1.1.1.35) (B) in TA muscles of control and AICAR-treated rats. a, AICAR different from control.
Figure 5. Glycolytic reference enzyme activities.
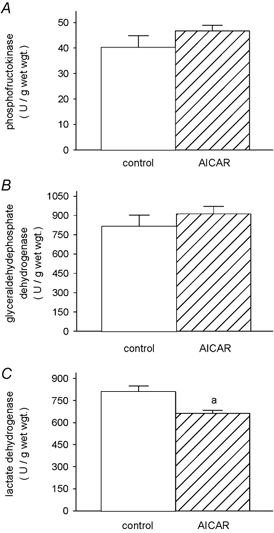
Activities of the glycolytic enzymes phosphofructokinase (EC 2.7.1.11) (A) glyceraldehyde phosphate dehydrogenase (EC 1.2.1.12) (B) and lactate dehydrogenase (EC 1.1.1.27) (C) in TA muscles of control and AICAR-treated rats. a, AICAR different from control.
MHC-based fibre types
The method used to evaluate fibre-type distribution in the muscles of control and AICAR-treated rats is illustrated in Fig. 6. As shown in Table 2, no differences were detected in the proportions of the four major immunohistochemically delineated fibre types, i.e. Iβ (P ≥ 0.28), IIA (P ≥ 0.72), IID/X (P ≥ 0.58) and IIB (P ≥ 0.96). The proportions of types Iβ, IIA, IID/X and IIB fibres were approximately, 3 %, 15 %, 27 % and 55 %, respectively. There were no extrafusal fibres present in the control or AICAR-treated muscles, which expressed MHC-embryonic or MHCIα. Similarly, quantitative evaluation of MHC isoform contents by SDS-PAGE did not reveal differences in the proportions of the various MHC isoforms between the control and AICAR-treated groups (MHCI, P ≥ 0.14; MHCIIA, P ≥ 0.37; MHCIID/X, P ≥ 0.62; MHCIIB, P ≥ 0.14) (Table 3).
Figure 6. Examples of the immunohistochemical staining used.
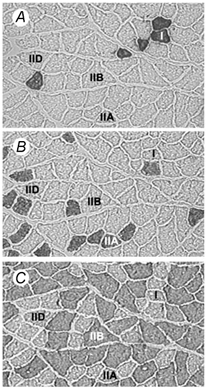
Staining was used to identify: A, type I (anti-MHCI, clone NOQ7.5.4D); B, type IIA (anti-MHCIIA, clone SC-71); C, type IIB (anti-MHCIIB, clone BF-F3) fibres. There were no fibres present which expressed MHC-embryonic. Note that the fibre types marked I, IIA and IIB were identified by their reaction with the corresponding MHC antibody, while IID/X fibres (denoted IID), were identified by the absence of staining.
Table 2.
Percentage fibre typr distribution in control and AICAR-treated rat TA muscle as determined by immunohistochemical analysis
| Fibre type (%) | ||||
|---|---|---|---|---|
| Condition | I | IIA | IID/X | IIB |
| Control | 3.3 ± 0.6 | 17.8 ± 3.3 | 24.5 ± 6.3 | 54.6 ± 9.4 |
| AICAR | 2.1 ± 0.9 | 15.4 ± 5.2 | 28.7 ± 3.6 | 53.9 ± 8.0 |
Table 3.
MHC isoform contents in control and AICAR-treated rat TA muscles as determined by SDS-PAGE analysis
| MHC content (%) | ||||
|---|---|---|---|---|
| Condition | MHCI | MHCIIA | MHCIID/X | MHCIIB |
| Control | 4.0 ± 0.6 | 11.0 ± 1.2 | 23.3 ± 0.9 | 61.8 ± 0.8 |
| AICAR | 2.3 ± 0.8 | 9.3 ± 1.3 | 22.3 ± 1.5 | 66.1 ± 2.4 |
DISCUSSION
The purpose of the present study was to assess the effects of chronic AICAR administration on the metabolic and MHC-based fibre-type profiles of skeletal muscle, which are known targets for adaptive change in response to altered functional demands. The present study provides new empirical evidence that enhances our understanding of the regulatory aspects of muscle gene expression mediated by reductions in the intracellular phosphorylation potential that accompany exercise training or chronic low-frequency stimulation (Green et al. 1992). We demonstrate that a known activator of AMPK mimics some but not all of the changes in skeletal muscle that are typically associated with exercise training. The importance of our findings relates to the widely held belief that the metabolic and structural phenotypes are co-ordinately regulated (Pette, 2002). We provide the first evidence that these two aspects of the adaptive potential of skeletal muscle fibres are discordantly controlled.
Administration of AICAR to rodents in vivo has been shown to activate skeletal muscle AMPK (Winder, 2001) and has been interpreted as mimicking, at least to some extent, the activation of AMPK in response to exercise (Winder & Hardie, 1996; Fujii et al. 2000; Wojtaszewski et al. 2000) and acute electrical stimulation (Vavvas et al. 1997; Hutber et al. 1997). Furthermore, chronic administration of AICAR has been shown to induce increases in the activities of some mitochondrial enzymes and glucose regulatory enzymes in skeletal muscle (Winder, 2001), and thus AMPK activation is believed to underlie some of the adaptive changes associated with prolonged exercise training. As shown from the large increase in ACC phosphorylation, AICAR treatment was successful in activating AMPK in vivo. This assay has recently been shown to accurately reflect the in vivo AMPK activity (Park et al. 2002; Nielsen et al. 2003), and is most appropriately applied to studies employing a model of long-term AMPK activation by AICAR.
Several recent reports have noted that, when measured in vitro using the traditional assay method, AMPK activity appears blunted after long-term AICAR exposure (Winder et al. 2000; Buhl et al. 2002; Nielsen et al. 2003). It is important to note, however, that direct assay of AMPK in vitro only reflects the phosphorylation state at the time of extraction, while allosteric regulatory influences cannot be measured. This limitation is especially relevant to interpreting the results of the present study, as allosteric regulation of AMPK appears to play a prominent role in determining AMPK activity in trained muscle and in response to chronic AICAR treatment. The results of the present study are in agreement with previous studies that have examined the effects of chronic AICAR administration on skeletal muscle metabolism, but further extend the findings of those studies to include the examination of additional mitochondrial enzymes, of glycolytic reference enzyme activities, of uncoupling protein-3 mRNA and protein expression, and of the pattern of MHC protein isoform expression and fibre-type distribution.
Reference enzyme activities
In the present study we demonstrate increases in the activities of the mitochondrial enzymes citrate synthase (CS) and 3-hydroxyacyl-CoA dehydrogenase (HADH), after chronic administration of AICAR in vivo. The 1.6-fold increase in CS observed in our study was similar to the increase (≈1.5-fold) previously reported for the white portion of rat gastrocnemius muscle after AICAR administration for 28 days (Winder et al. 2000). HADH activity, however, was reportedly not altered in that same study, which is in contrast to our novel observation of a 1.6-fold elevation in fast-twitch muscle. This discrepancy may be due to the different muscles studied or to the lower concentration of acetoacetyl-CoA used to assay HADH in the present study, which was necessary to eliminate the inhibitory effects on enzyme activity in vitro. The parallel increase in CS and HADH activities demonstrated in AICAR-treated rats examined in the present study resembles quite strikingly similar increases induced by chronic contractile activity, such as exercise training by treadmill running (Green et al. 1983) or CLFS (Simoneau & Pette, 1988; Pette & Vrbová, 1992). In this regard, our findings are in agreement with the proposed hypothesis that chronic AICAR administration mimics the metabolic effects of chronically increased contractile activity (Winder, 2001). We also show that LDH activity is decreased, while the activities of PFK and GAPDH remained unaltered by AICAR treatment. These findings are also similar to those observed after prolonged endurance exercise training (Green et al. 1983) and to some extent CLFS (Pette & Vrbová, 1992). Collectively our data concerning changes in the reference enzymes of energy metabolism are consistent with the hypothesis that an AMPK-dependent pathway is responsible for regulating the metabolic phenotype of skeletal muscle fibres, in a manner similar to exercise training.
The increases in CS and HADH activities can be explained by enhanced mitochondrial biogenesis, as the increase in the activities of these marker enzymes of terminal substrate oxidation (i.e. the citric acid and cycle and β-oxidation of fatty acids) are widely accepted as reliable and valid indicators of changes in mitochondrial volume density (Holloszy & Coyle, 1984; Reichmann et al. 1985; Harlan & Williams, 1988; Pette & Staron, 1990; Hoppeler, 1990). Interestingly it has recently been reported that chronic activation of AMPK, secondary to a decrease in the intracellular phosphorylation potential in rat fast-twitch muscles by chronic feeding of β-guanidinopropionic acid, results in several-fold increases in nuclear respiratory factor-1 (NRF-1). This transcription factor for mitochondrial transcription and replication was shown to be elevated 10-fold at the mRNA level and accompanied by a two-fold increase in mitochondrial content (Bergeron et al. 2001). Similarly, expression of PGC-1α, another putative regulator of mitochondrial biogenesis, is increased in proportion to AMPK activity and is associated with mitochondrial biogenesis (Zong et al. 2003).
UCP-3 expression and content
To our knowledge modest up-regulation of UCP-3, at the protein level, by AICAR-induced AMPK activation has only been demonstrated acutely in vitro by Zhou et al. (2000), who showed 15-30 % increases in UCP-3 protein in vitro after 30-200 min of a single exposure to a high concentration of AICAR (Zhou et al. 2000). By comparison, we observed a 330 % increase in UCP-3 protein content in whole muscle extracts (i.e. 11- to 22-fold greater), which can only have occurred from the cumulative effect of the repeated daily AICAR injections. More recently, Stoppani et al. (2002) reported transcriptional activation of UCP-3, in vivo, after acute exposure to AICAR but no change in UCP-3 protein content. The results of the present study extend the findings of those studies to include 1.6- and 3.3-fold increases in UCP-3 mRNA and protein content, respectively, after chronic administration of AICAR in vivo.
The greater UCP-3 content observed in the present study is probably related, in part, to the increase in mitochondrial content. However, as judged from the 3.3-fold increase in content, UCP-3 accumulation was proportionally greater than the increase in mitochondrial reference enzyme activities or mitochondrial volume density, indicating an increase in the concentration of UCP-3 within the mitochondrial compartment. This probably resulted in part from increased transcriptional activation mediated by AMPK (Zhou et al. 2000; Stoppani et al. 2002). While it is also possible that UCP-3 expression was influenced by insulin signalling or fatty acid signalling (Samec et al. 1998a,b; Gong et al. 1999; Himms-Hagen & Harper, 2001; Schrauwen et al. 2001; Young et al. 2001; Son et al. 2001; Buhl et al. 2002), reductions in the plasma concentrations of these factors observed in the AICAR group would seem to indicate reduced inter-organ signalling and thus preclude such possibilities. Greater UCP-3 protein content was probably further enhanced as the result of post-transcriptional regulatory events, as has been reported in vivo (Rehnmark et al. 1992; Sivitz et al. 1999; Pecqueur et al. 2001). Interestingly UCP-3 expression and content are reduced with long-term training (Boss et al. 1998), while mitochondrial biogenesis is known to continually increase under these conditions (Pette & Vrbová, 1992; Pette & Staron, 2000). Collectively these findings suggest that UCP-3 expression is not strictly related to mitochondrial biogenesis as recently suggested (Jones et al. 2003), but may also be influenced by changes in the capacity of terminal substrate oxidation that accompanies training (Schrauwen et al. 2001).
Our findings appear most relevant when considered in the context of the established relationship between UCP-3 content and basal metabolic rate (Schrauwen et al. 1999; Harper et al. 2001; Lanouette et al. 2002), as well as its putative role in a futile cycle involving mitochondrial fatty acid import and export (Himms-Hagen & Harper, 2001). It is thus interesting to speculate that upregulation of UCP-3 by AICAR treatment may accelerate the basal oxidation of fatty acids by skeletal muscle and prove to be beneficial in the treatment of obesity and type II diabetes (Buhl et al. 2001). It does not, however, seem to mediate such changes by altering the plasma concentrations of fatty acids or insulin during 28 days of AICAR treatment, but is limited to events within muscle fibres.
Co-ordinated regulation of metabolic and MHC phenotypes
The AMPK-induced changes in UCP-3 expression and metabolic properties of fast-twitch muscle observed in our study resemble some of those induced by endurance training, and to some extent CLFS. Unlike exercise training or CLFS, however, they do not include changes in MHC content or patterns of expression, as determined by extensive immunohistochemical fibre-type analyses or by SDS-PAGE analyses of purified myosin. In this regard, AMPK-induced phenotypic changes seem to differ from those induced by exercise or CLFS. Thus our data indicate that an AMPK-related pathway is primarily involved in the upregulation of mitochondrial enzymes of terminal substrate oxidation. Moreover, our data demonstrate that adaptive metabolic changes can occur within rodent fast-twitch muscles independently of fibre-type transitions.
That metabolic and contractile properties may be co-ordinately regulated follows from recent work on the calcineurin-induced upregulation of slow-fibre type-specific myofibrillar protein isoforms and metabolic properties (Chin et al. 1998; Olson & Williams, 2000; Meissner et al. 2001). Similarly, a Ca2+-calmodulin kinase-dependent pathway has been identified in stimulating mitochondrial biogenesis and shifting contractile properties towards the slow-fibre type (Wu et al. 2002). The latter two pathways are activated by increases in free cytosolic Ca2+, whereas increases in 5′-AMP concomitant with a reduction in the ATP phosphorylation potential trigger the AMPK-related pathway. CLFS or exercise can induce almost immediate and persistent decreases in the intracellular [ATP]/[ADPfree] ratio concomitant with increases in 5′-AMP and a decline in the phosphocreatine content (Green et al. 1992; Conjard et al. 1998; Conjard & Pette, 1999). The latter change may additionally activate AMPK by alleviating the inhibitory influence of high phosphocreatine concentration (Ponticos et al. 1998). In addition, repeated muscle fibre contractions are known to induce a rapid and persistent increase in free cytosolic Ca2+ (Carroll et al. 1999). Thus enhanced contractile activity creates the intracellular conditions of increased 5′-AMP and Ca2+ concentrations that are both necessary and sufficient to simultaneously activate all three pathways, and probably accounts for the apparent co-ordinated regulation of metabolic changes with MHC-based fibre type transitions typically observed after exercise training and CLFS (Pette & Vrbová, 1992; Pette & Staron, 2000).
Acknowledgments
The authors thank Dr G. D. Lopaschuk for assistance with the anti-phospho-ACC blots and the AMPK activity measures. This study was funded by research grants from the Natural Sciences and Engineering Council of Canada (to C. T. Putman and W. T. Dixon) and the Alberta Heritage Foundation for Medical Research (AHFMR) (to C. T. Putman). C. T. Putman is a Heritage Medical Scholar of AHFMR.
REFERENCES
- Allen DL, Leinwand LA. Intracellular calcium and myosin isoform transitions. Calcineurin and calcium-calmodulin kinase pathways regulated preferential activation of the IIa myosin heavy chain promoter. J Biol Chem. 2002;277:45323–45330. doi: 10.1074/jbc.M208302200. [DOI] [PubMed] [Google Scholar]
- Bass A, Brdiczka D, Eyer P, Hofer S, Pette D. Metabolic differentiation of distinct muscle types at the level of enzymatic organization. Eur J Biochem. 1969;10:198–206. doi: 10.1111/j.1432-1033.1969.tb00674.x. [DOI] [PubMed] [Google Scholar]
- Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, Young LH, Semenkovich CF, Shulman GI. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab. 2001;281:E1340–1346. doi: 10.1152/ajpendo.2001.281.6.E1340. [DOI] [PubMed] [Google Scholar]
- Bigard X, Sanchez H, Zoll J, Mateo P, Rousseau V, Veksler V, Ventura-Clapier R. Calcineurin co-regulates contractile and metabolic components of slow muscle phenotype. J Biol Chem. 2000;275:19653–19660. doi: 10.1074/jbc.M000430200. [DOI] [PubMed] [Google Scholar]
- Boss O, Samec S, Desplanches D, Mayet MH, Seydoux J, Muzzin P, Giacobino JP. Effect of endurance training on mRNA expression of uncoupling proteins 1, 2, and 3 in the rat. FASEB J. 1998;12:335–339. doi: 10.1096/fasebj.12.3.335. [DOI] [PubMed] [Google Scholar]
- Buhl ES, Jessen N, Pold R, Ledet T, Flyvbjerg A, Pedersen SB, Pedersen O, Schmitz O, Lund S. Long-term AICAR administration reduces metabolic disturbances and lowers blood pressure in rats displaying features of the insulin resistance syndrome. Diabetes. 2002;51:2199–2206. doi: 10.2337/diabetes.51.7.2199. [DOI] [PubMed] [Google Scholar]
- Buhl ES, Jessen N, Schmitz O, Pedersen SB, Pedersen O, Holman GD, Lund S. Chronic treatment with 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside increases insulin-stimulated glucose uptake and GLUT4 translocation in rat skeletal muscles in a fiber type-specific manner. Diabetes. 2001;50:12–17. doi: 10.2337/diabetes.50.1.12. [DOI] [PubMed] [Google Scholar]
- Carroll S, Nicotera P, Pette D. Calcium transients in single fibers of low-frequency stimulated fast-twitch muscle of rat. Am J Physiol Cell Physiol. 1999;277:C1122–1129. doi: 10.1152/ajpcell.1999.277.6.C1122. [DOI] [PubMed] [Google Scholar]
- Chin ER, Olson EN, Richardson JA, Yang Q, Humphries C, Shelton JM, Wu H, Zhu W, Bassel-duby R, Williams RS. A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev. 1998;12:2499–2509. doi: 10.1101/gad.12.16.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conjard A, Pette D. Phosphocreatine as a marker of contractile activity in single muscle fibres. Pflugers Arch. 1999;438:278–282. doi: 10.1007/s004240050910. [DOI] [PubMed] [Google Scholar]
- Conjard A, Peuker H, Pette D. Energy state and myosin heavy chain isoforms in single fibres of normal and transforming rabbit muscles. Pflugers Arch. 1998;436:962–969. doi: 10.1007/s004240050730. [DOI] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-Aminoimidazole-4-carboxamide ribonucleoside: a specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- Gong DW, He Y, Reitman ML. Genomic organization and regulation by dietary fat of the uncoupling protein 3 and 2 genes. Biochem Biophys Res Commun. 1999;256:27–32. doi: 10.1006/bbrc.1999.0239. [DOI] [PubMed] [Google Scholar]
- Green HJ, Düsterhöft S, Dux L, Pette D. Metabolite patterns related to exhaustion, recovery and transformation of chronically stimulated rabbit fast-twitch muscle. Pflugers Arch. 1992;420:359–366. doi: 10.1007/BF00374471. [DOI] [PubMed] [Google Scholar]
- Green HJ, Reichmann H, Pette D. Fiber type specific transformations in the enzyme activity pattern of rat vastus lateralis muscle by prolonged endurance training. Pflugers Arch. 1983;399:216–222. doi: 10.1007/BF00656718. [DOI] [PubMed] [Google Scholar]
- Harlan WR, Williams RS. Activity-induced adaptations in skeletal muscles of iron-deficient rabbits. J Appl Physiol. 1988;65:782–787. doi: 10.1152/jappl.1988.65.2.782. [DOI] [PubMed] [Google Scholar]
- Harper ME, Dent RM, Bezaire V, Antoniou A, Gauthier A, Monemdjou S, McPherson R. UCP3 and its putative function: consistencies and controversies. Biochem Soc Trans. 2001;29:768–773. doi: 10.1042/bst0290768. [DOI] [PubMed] [Google Scholar]
- Hämäläinen N, Pette D. Slow-to-fast transitions in myosin expression of rat soleus muscle by phasic high-frequency stimulation. FEBS Lett. 1996;399:220–222. doi: 10.1016/s0014-5793(96)01325-7. [DOI] [PubMed] [Google Scholar]
- Himms-Hagen J, Harper ME. Physiological role of UCP3 may be export of fatty acids from mitochondria when fatty acid oxidation predominates: an hypothesis. Exp Biol Med. 2001;226:78–84. doi: 10.1177/153537020122600204. [DOI] [PubMed] [Google Scholar]
- Holloszy JO, Coyle EF. Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J Appl Physiol. 1984;56:831–838. doi: 10.1152/jappl.1984.56.4.831. [DOI] [PubMed] [Google Scholar]
- Holmes BF, Kurth-Kraczek EJ, Winder WW. Chronic activation of 5′-AMP-activated protein kinase increases GLUT-4, hexokinase, and glycogen in muscle. J Appl Physiol. 1999;87:1990–1995. doi: 10.1152/jappl.1999.87.5.1990. [DOI] [PubMed] [Google Scholar]
- Hoppeler H. The range of mitochondrial adaptation in muscle fibers. In: Pette D, editor. The Dynamic State of Muscle Fibres. Berlin New York: De Gruyter; 1990. pp. 567–586. [Google Scholar]
- Hutber CA, Hardie DG, Winder WW. Electrical stimulation inactivates muscle acetylCoA carboxylase and increases AMP-activated protein kinase. Am J Physiol Endocrinol Metab. 1997;272:E262–266. doi: 10.1152/ajpendo.1997.272.2.E262. [DOI] [PubMed] [Google Scholar]
- Jaschinski F, Schuler MJ, Peuker H, Pette D. Changes in myosin heavy chain mRNA and protein isoforms of rat muscle during forced contractile activity. Am J Physiol Cell Physiol. 1998;274:C365–370. doi: 10.1152/ajpcell.1998.274.2.C365. [DOI] [PubMed] [Google Scholar]
- Jones TE, Baar K, Ojuka E, Chen M, Holloszy JO. Exercise induces an increase in muscle UCP3 as a component of the increase in mitochondrial biogenesis. Am J Physiol Endocrinol Metab. 2003;284:E96–101. doi: 10.1152/ajpendo.00316.2002. [DOI] [PubMed] [Google Scholar]
- Kudo N, Barr AJ, Barr RL, Desai S, Lopaschuk GD. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5′-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J Biol Chem. 1995;270:17513–17520. doi: 10.1074/jbc.270.29.17513. [DOI] [PubMed] [Google Scholar]
- Lanouette CM, Chagnon YC, Rice T, Perusse L, Muzzin P, Giacobino JP, Gagnon J, Wilmore JH, Leon AS, Skinner JS, Rao DC, Bouchard C. Uncoupling protein 3 gene is associated with body composition changes with training in HERITAGE study. J Appl Physiol. 2002;92:1111–1118. doi: 10.1152/japplphysiol.00726.2001. [DOI] [PubMed] [Google Scholar]
- Matsuda J, Hosoda K, Itoh H, Son C, Doi K, Tanaka T, Fukunaga Y, Inoue G, Nishimura H, Yoshimasa Y, Yamori Y, Nakao K. Cloning of rat uncoupling protein-3 and uncoupling protein-2 cDNAs: their gene expression in rats fed high-fat diet. FEBS Lett. 1997;418:200–204. doi: 10.1016/s0014-5793(97)01381-1. [DOI] [PubMed] [Google Scholar]
- Meissner JD, Gros G, Scheibe RJ, Scholz M, Kubis HP. Calcineurin regulates slow myosin, but not fast myosin or metabolic enzymes, during fast-to-slow transformation in rabbit skeletal muscle cell culture. J Physiol. 2001;533:215–226. doi: 10.1111/j.1469-7793.2001.0215b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naya FJ, Mercer B, Shelton J, Richardson JA, Williams RS, Olson EN. Stimulation of slow skeletal muscle fiber gene expression by calcineurin in vivo. J Biol Chem. 2000;275:4545–4548. doi: 10.1074/jbc.275.7.4545. [DOI] [PubMed] [Google Scholar]
- Nielsen JN, Mustard KJ, Graham DA, Yu H, MacDonald CS, Pilegaard H, Goodyear LJ, Hardie DG, Richter EA, Wojtaszewski JF. 5′-AMP-activated protein kinase activity and subunit expression in exercise-trained human skeletal muscle. J Appl Physiol. 2003;94:631–641. doi: 10.1152/japplphysiol.00642.2002. [DOI] [PubMed] [Google Scholar]
- Oakley BR, Kirsch DR, Morris NR. A simplified ultrasensitive silver stain for detecting proteins in polyacrylamide gels. Anal Biochem. 1980;105:361–363. doi: 10.1016/0003-2697(80)90470-4. [DOI] [PubMed] [Google Scholar]
- Olson EN, Williams RS. Remodeling muscles with calcineurin. Bioessays. 2000;22:510–519. doi: 10.1002/1521-1878(200011)22:11<1049::AID-BIES14>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Park SH, Gammon SR, Knippers JD, Paulsen SR, Rubink DS, Winder WW. Phosphorylation-activity relationships of AMPK and acetyl-CoA carboxylase in muscle. J Appl Physiol. 2002;92:2575–2482. doi: 10.1152/japplphysiol.00071.2002. [DOI] [PubMed] [Google Scholar]
- Pecqueur C, Alves-Guerra MC, Gelly C, Levi-Meyrueis C, Couplan E, Collins S, Ricquier D, Bouillaud F, Miroux B. Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J Biol Chem. 2001;276:8705–8712. doi: 10.1074/jbc.M006938200. [DOI] [PubMed] [Google Scholar]
- Pette D. The adaptive potential of skeletal muscle fibers. Can J Appl Physiol. 2002;27:423–448. doi: 10.1139/h02-023. [DOI] [PubMed] [Google Scholar]
- Pette D, Staron RS. Cellular and molecular diversities of mammalian skeletal muscle fibers. Rev Physiol Biochem Pharmacol. 1990;116:1–76. doi: 10.1007/3540528806_3. [DOI] [PubMed] [Google Scholar]
- Pette D, Staron RS. Mammalian skeletal muscle fiber type transitions. Int Rev Cytol. 1997;170:143–223. doi: 10.1016/s0074-7696(08)61622-8. [DOI] [PubMed] [Google Scholar]
- Pette D, Staron RS. Myosin isoforms, muscle fiber types, and transitions. Microsc Res Tech. 2000;50:500–509. doi: 10.1002/1097-0029(20000915)50:6<500::AID-JEMT7>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Pette D, Vrbová G. Adaptation of mammalian skeletal muscle fibers to chronic electrical stimulation. Rev Physiol Biochem Pharmacol. 1992;120:115–202. doi: 10.1007/BFb0036123. [DOI] [PubMed] [Google Scholar]
- Ponticos M, Lu QL, Morgan JE, Hardie DG, Partridge TA, Carling D. Dual regulation of the AMP-activated protein kinase provides a novel mechanism for the control of creatine kinase in skeletal muscle. EMBO J. 1998;17:1688–1699. doi: 10.1093/emboj/17.6.1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putman CT, Conjard A, Peuker H, Pette D. Alpha-cardiac like myosin heavy chain MHCI-alpha is not upregulated in transforming rat muscle. J Muscle Res Cell Motil. 1999;20:155–162. doi: 10.1023/a:1005430115402. [DOI] [PubMed] [Google Scholar]
- Putman CT, Düsterhöft S, Pette D. Satellite cell proliferation in low-frequency stimulated fast muscle of hypothyroid rat. Am J Physiol Cell Physiol. 2000;279:C682–690. doi: 10.1152/ajpcell.2000.279.3.C682. [DOI] [PubMed] [Google Scholar]
- Putman CT, O'Brien CL, Kirisci M, Leisner E, Pette D. Chronic low-frequency stimulation up-regulates mitcohondrial uncoupling protein-3 in rat tibialis anterior. FASEB J. 2002;16:A445. [Google Scholar]
- Putman CT, Spriet LL, Hultman E, Lindinger MI, Lands LC, McKelvie RS, Cederblad G, Jones NL, Heigenhauser GjF. Pyruvate dehydrogenase activity and acetyl-group accumulation during exercise after different diets. Am J Physiol Endocrinol Metab. 1993;265:E752–760. doi: 10.1152/ajpendo.1993.265.5.E752. [DOI] [PubMed] [Google Scholar]
- Rehnmark S, Bianco AC, Kieffer JD, Silva JE. Transcriptional and posttranscriptional mechanisms in uncoupling protein mRNA response to cold. Am J Physiol Endocrinol Metab. 1992;262:E58–67. doi: 10.1152/ajpendo.1992.262.1.E58. [DOI] [PubMed] [Google Scholar]
- Reichmann H, Hoppeler H, Mathieu-Costello O, Von Bergen F, Pette D. Biochemical and ultrastructural changes of skeletal muscle mitochondria after chronic electrical stimulation in rabbits. Pflugers Arch. 1985;404:1–9. doi: 10.1007/BF00581484. [DOI] [PubMed] [Google Scholar]
- Reichmann H, Srihari T, Pette D. Ipsi- and contralateral fibre transformations by cross-reinnervation. A principle of symmetry. Pflugers Arch. 1983;397:202–208. doi: 10.1007/BF00584358. [DOI] [PubMed] [Google Scholar]
- Salt IP, Celler JW, Hawley SA, Prescott A, Woods A, Carling D, Hardie DG. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. Biochem J. 1998;334:177–187. doi: 10.1042/bj3340177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samec S, Seydoux J, Dulloo AG. Interorgan signaling between adipose tissue metabolism and skeletal muscle uncoupling protein homologs: is there a role for circulating free fatty acids? Diabetes. 1998a;47:1693–1698. doi: 10.2337/diabetes.47.11.1693. [DOI] [PubMed] [Google Scholar]
- Samec S, Seydoux J, Dulloo AG. Role of UCP homologues in skeletal muscles and brown adipose tissue: mediators of thermogenesis or regulators of lipids as fuel substrate. FASEB J. 1998b;12:715–724. doi: 10.1096/fasebj.12.9.715. [DOI] [PubMed] [Google Scholar]
- Schrauwen P, Saris WH, Hesselink MK. An alternative function for human uncoupling protein 3: protection of mitochondria against accumulation of nonesterified fatty acids inside the mitochondrial matrix. FASEB J. 2001;15:2497–2502. doi: 10.1096/fj.01-0400hyp. [DOI] [PubMed] [Google Scholar]
- Schrauwen P, Xia J, Bogardus C, Pratley RE, Ravussin E. Skeletal muscle uncoupling protein 3 expression is a determinant of energy expenditure in Pima Indians. Diabetes. 1999;48:146–149. doi: 10.2337/diabetes.48.1.146. [DOI] [PubMed] [Google Scholar]
- Simoneau J-A, Pette D. Species-specific effects of chronic nerve stimulation upon tibialis anterior muscle in mouse, rat, guinea pig and rabbit. Pflugers Arch. 1988;412:86–92. doi: 10.1007/BF00583735. [DOI] [PubMed] [Google Scholar]
- Sivitz WI, Fink BD, Donohoue PA. Fasting and leptin modulate adipose and muscle uncoupling protein: divergent effects between messenger ribonucleic acid and protein expression. Endocrinology. 1999;140:1511–1519. doi: 10.1210/endo.140.4.6668. [DOI] [PubMed] [Google Scholar]
- Son C, Hosoda K, Matsuda J, Fujikura J, Yonemitsu S, Iwakura H, Masuzaki H, Ogawa Y, Hayashi T, Itoh H, Nishimura H, Inoue G, Yoshimasa Y, Yamori Y, Nakao K. Up-regulation of uncoupling protein 3 gene expression by fatty acids and agonists for PPARs in L6 myotubes. Endocrinology. 2001;142:4189–4194. doi: 10.1210/endo.142.10.8446. [DOI] [PubMed] [Google Scholar]
- Srere PA. Citrate synthase. In: Lowenstein JM, editor. Citric Acid Cycle. New York London: Academic Press; 1969. pp. 3–11. [Google Scholar]
- Stoppani J, Hildebrandt AL, Sakamoto K, Cameron-smith D, Goodyear LJ, Neufer PD. AMP-activated protein kinase activates transcription of the UCP3 and HKII genes in rat skeletal muscle. Am J Physiol Endocrinol Metab. 2002;283:E1239–1248. doi: 10.1152/ajpendo.00278.2002. [DOI] [PubMed] [Google Scholar]
- Suarez RK, Brown GS, Hochachka PW. Metabolic sources of energy for hummingbird flight. Am J Physiol Regul Integr Comp Physiol. 1986;251:R537–542. doi: 10.1152/ajpregu.1986.251.3.R537. [DOI] [PubMed] [Google Scholar]
- Vavvas D, Apazidis A, Saha AK, Gamble J, Patel A, Kemp BE, Witters LA, Ruderman NB. Contraction-induced changes in acetyl-CoA carboxylase and 5′-AMP-activated kinase in skeletal muscle. J Biol Chem. 1997;272:13255–13261. doi: 10.1074/jbc.272.20.13255. [DOI] [PubMed] [Google Scholar]
- Winder WW. Energy-sensing and signaling by AMP-activated protein kinase in skeletal muscle. J Appl Physiol. 2001;91:1017–1028. doi: 10.1152/jappl.2001.91.3.1017. [DOI] [PubMed] [Google Scholar]
- Winder WW, Hardie DG. Inactivation of acetylCoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol Endocrinol Metab. 1996;270:E299–304. doi: 10.1152/ajpendo.1996.270.2.E299. [DOI] [PubMed] [Google Scholar]
- Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol. 2000;88:2219–2226. doi: 10.1152/jappl.2000.88.6.2219. [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JF, Nielsen P, Hansen BF, Richter EA, Kiens B. Isoform-specific and exercise intensity-dependent activation of 5′-AMP-activated protein kinase in human skeletal muscle. J Physiol. 2000;528:221–226. doi: 10.1111/j.1469-7793.2000.t01-1-00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Kanatous SB, Thurmond FA, Gallardo T, Isotani E, Bassel-Duby R, Williams SW. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science. 2002;296:349–352. doi: 10.1126/science.1071163. [DOI] [PubMed] [Google Scholar]
- Young ME, Patil S, Ying J, Depre C, Ahuja HS, Shipley GL, Stepkowski SM, Davies PJ, Taegtmeyer H. Uncoupling protein 3 transcription is regulated by peroxisome proliferatoractivated receptor (alpha) in the adult rodent heart. FASEB J. 2001;15:833–845. doi: 10.1096/fj.00-0351com. [DOI] [PubMed] [Google Scholar]
- Zhou M, Lin BZ, Coughlin S, Vallega G, Pilch PF. UCP-3 expression in skeletal muscle: effects of exercise, hypoxia, and AMP-activated protein kinase. Am J Physiol Endocrinol Metab. 2000;279:E622–629. doi: 10.1152/ajpendo.2000.279.3.E622. [DOI] [PubMed] [Google Scholar]
- Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2003;99:15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]