

Abstract
Chronic hypoxia is associated with elevated sympathetic activity and hypertension in patients with chronic pulmonary obstructive disease. However, the effect of chronic hypoxia on systemic and regional sympathetic activity in healthy humans remains unknown. To determine if chronic hypoxia in healthy humans is associated with hyperactivity of the sympathetic system, we measured intra-arterial blood pressure, arterial blood gases, systemic and skeletal muscle noradrenaline (norepinephrine) spillover and vascular conductances in nine Danish lowlanders at sea level and after 9 weeks of exposure at 5260 m. Mean blood pressure was 28% higher at altitude (P < 0.01) due to increases in both systolic (18% higher, P < 0.05) and diastolic (41% higher, P < 0.001) blood pressures. Cardiac output and leg blood flow were not altered by chronic hypoxia, but systemic vascular conductance was reduced by 30 % (P < 0.05). Plasma arterial noradrenaline (NA) and adrenaline concentrations were 3.7- and 2.4-fold higher at altitude, respectively (P < 0.05). The elevation of plasma arterial NA concentration was caused by a 3.8-fold higher whole-body NA release (P < 0.001) since whole-body noradrenaline clearance was similar in both conditions. Leg NA spillover was increased similarly (× 3.2, P < 0.05). These changes occurred despite the fact that systemic O2 delivery was greater after altitude acclimatisation than at sea level, due to 37 % higher blood haemoglobin concentration. In summary, this study shows that chronic hypoxia causes marked activation of the sympathetic nervous system in healthy humans and increased systemic arterial pressure, despite normalisation of the arterial O2 content with acclimatisation.
Acute hypoxia, sensed by the peripheral and medullary chemoreceptors (Marshall, 1994; Reis et al. 1994), is a powerful activator of the sympathetic nervous system as reflected by the increase of systemic and regional sympathetic tone during acute hypoxia. Hypoxia-mediated sympathetic activation is currently envisaged as a defence mechanism to assure O2 supply to critical organs by means of raising cardiac output and regulating regional conductances (Rowell et al. 1989; Leuenberger et al. 1991; Duplain et al. 1999; Calbet, 2000). In contrast, hypoxia either directly, or through its metabolic effects, causes vasodilatation in most vascular beds, such that sympathetic tone should be increased to avoid exaggerated vasodilatation and hypotension (Hilton & Eichholtz, 1925; Rowell et al. 1989; Laughlin et al. 1996). Elevated sympathetic activity has also been reported in patients with chronic hypoxaemia (defined as a low arterial pressure of O2 (Pa,O2)) (Heindl et al. 2001), as well as in patients with sleep apnoea who are intermittently hypoxaemic (Imadojemu et al. 2002). However, the effect that prolonged exposure to hypoxia has on global and regional sympathetic activity in the healthy human remains unknown. It is also unknown to what extent the sympathetic discharge to the skeletal muscle vascular bed contributes to the overall sympathetic activity.
Increased sympathetic activation has been reported in classical studies in which the effect of chronic hypoxia on sympathetic function in man has been generally assessed through the determination of catecholamine concentrations in urine, venous plasma and, to a lesser extent, in arterial plasma (Rostrup, 1998). However, measurement of urinary excretion of either catecholamines or their metabolites is rather non-specific and is currently considered largely obsolete as a test of human sympathetic nervous system activity (Esler, 2000). As a test of sympathetic activity, plasma noradrenaline (NA) concentration has two major limitations. The first is that no indication is provided on regional sympathetic activation when it is well known that sympathetic nervous system responses show regional differentiation (Esler et al. 1984; Esler, 2000). A second drawback is that plasma noradrenaline is a poor index of sympathetic tone because the noradrenaline plasma concentration is determined not only by the rate of its release but also by the rate at which it is removed from the circulation (Esler, 2000). A better alternative to assess systemic and regional sympathetic nervous function is the determination of noradrenaline spillover rate based on the isotope dilution method (Esler et al. 1984).
Several adaptations develop in healthy humans whereby, within a few weeks at altitude, the arterial O2 content (Ca,O2) values return to levels similar or higher than those observed at sea level (Calbet et al. 2003). If the hyperactivity of the sympathetic system observed in acute hypoxia is caused by reduced Ca,O2, sympathetic activity should be attenuated or normalised as the acclimatisation to altitude progresses. In contrast, Hansen & Sander (2003) have recently shown enhanced sympathetic nerve activity in healthy humans as demonstrated by microneurographic recordings of the vascular sympathetic fibres of the peroneal nerve after 4 weeks of residence at 5260 m of altitude. Several aspects were not clarified in the study of Hansen & Sander (2003). Firstly, the contribution of skeletal muscle sympathetic overactivity to systemic sympathetic activation. Secondly, the influence of sympathetic overactivity on systemic and peripheral haemodynamics. Finally, it remains to be ascertained if, after a more prolonged sojourn at altitude and hence better acclimatisation, enhanced blood haemoglobin concentration and higher Pa,O2 (Calbet et al. 2003), sympathetic activity still remains elevated.
To test the hypothesis that systemic and skeletal muscle sympathetic activity are normalised after prolonged acclimatisation at high altitude, the systemic and skeletal muscle noradrenaline spillover was measured in nine healthy Danish lowlanders at sea level and after 9 weeks of exposure at 5260 m. In addition, the arterial blood gases and acid-base balance were also assessed to determine their potential influence on sympathetic activity. Finally, blood pressure, systemic and leg vascular conductances, as well as O2 transport were also measured to determine the functional repercussions of any possible alteration of sympathetic tone caused by chronic hypoxia.
METHODS
Subjects
Nine Danish lowlanders (four females and five males) volunteered to participate in these studies. Their mean (± S.E.M.) age, height and weight were 24.3 ± 0.5 years, 176 ± 3 cm and 74 ± 4 kg, respectively. Six of these subjects also participated in the experiments carried out by Hansen & Sander (2003) after 4 weeks of residence at 5260 m of altitude. All subjects were physically active and had a normal resting ECG, as well as normal liver, kidney and thyroid function. Fasting plasma glucose and electrolyte concentrations, in addition to resting blood pressure and blood pressure response to an incremental exercise test to exhaustion, were also normal. Their blood haemoglobin concentration ([Hb]) was also into the normal range in males and females (145 ± 4 and 124 ± 3 g l−1). Plasma concentrations of ferritin at the beginning of the experiment were 69 ± 19 and 22 ± 8 μg l−1, while plasma transferrin concentrations were 31.3 ± 0.6 and 33.0 ± 1.4 μmol l−1 in the males and females, respectively. The subjects were informed about the procedures and risks of the study before giving written informed consent to participate as approved by the Copenhagen-Fredriksberg Ethical Committee.
General procedures and experimental protocol
The present study was conducted at altitude after 9 weeks residence at 5260 m on Mount Chacaltaya (Bolivia). Approximately 7-10 days later, the subjects returned to Copenhagen and after 6-9 months of residence at sea level the measurements were repeated under normoxic conditions. An 18 gauge catheter (Hydrocath, Ohmeda, Swindon, UK) was inserted, under local anaesthesia (2 % lidocaine (lignocaine)), into either the right or left femoral vein, 2 cm below the inguinal ligament and advanced 7 cm distally for venous sampling and injection of cold saline. A thin polyethylene-coated thermistor (model 94-030-2.5F, T. D. Probe, Edwards Edslab, Baxter, Irvine, CA, USA) was then inserted 3 cm below the inguinal ligament and advanced proximally 10 cm into the same femoral vein. An 18 gauge catheter was also placed into the femoral artery 2 cm below the inguinal ligament for arterial sampling and blood pressure measurement. An additional catheter was placed in a vein in the left forearm for the injection of the cardio-green dye.
After a rest period of 15 min in the supine position a priming bolus of 30 μCi of tritiated NA (I-[7,8-3H]NA, specific activity 36.0 Ci mmol−1 at 1.0 mCi ml−1, TRK584, Nycomed Amersham, Buckinghamshire, UK) was administered over 5 min followed by a constant infusion at 0.7 μCi min−1, delivered with a Harvard infusion pump (Harvard pump, Harvard Apparatus, Millis, MA, USA). Femoral arterial and venous samples were obtained 10, 15 and 20 min after the start of the constant infusion. Femoral venous blood flow (i.e. leg blood flow) was measured in the femoral vein by constant-infusion thermodilution (Andersen & Saltin, 1985), before and immediately after the withdrawal of each blood sample. Cardiac output was measured by indocyanine green (ICG, Akorn Inc., IL, USA) dye dilution (Dow, 1956) at the end of the constant infusion period. Intra-arterial blood pressure was monitored continuously by a disposable transducer (T100209A, Baxter, Unterschleissheim, Germany) placed at the level of the left atrium. A 3-lead electrocardiogram was measured and displayed on a monitor during the catheterisation phase and the experimental period. The blood pressure transducer and the ECG electrodes were interfaced with a monitor (Dialogue 2000, Danica, Copenhagen, Denmark), which was, in turn, connected to the data acquisition system (MacLab 16/s ADInstruments, Sydney, Australia). Systolic, diastolic and mean arterial pressures were computed from the recorded pressure wave.
Blood haemoglobin concentration ([Hb]) and O2 saturation (SO2) were measured with a co-oximeter (OSM 3 Hemoximeter, Radiometer, Copenhagen, Denmark). PO2, PCO2, pH, HCO3−, and actual base excess (BE) were determined with a blood gas analyser (ABL 5, Radiometer). Plasma blood lactate levels were measured with an electrolyte metabolite analyser (EML 105, Radiometer). Plasma noradrenaline (NA) and adrenaline concentrations were determined by HPLC with electrochemical detection (Hallman et al. 1978). Total body NA clearance and spillover to plasma were measured with the technique of Esler et al. (1979) as follows:
where the infusion rate of [3H]NA is measured as d.p.m. per minute and the arterial plasma [3H]NA concentration is measured as d.p.m. per litre (d.p.m., disintegrations per minute of tritiated NA).
where NA clearance is measured as litres per minute and arterial NA concentration is measured as nanomoles per litre. Leg noradrenaline spillover rates were calculated according to the Fick principle, with adjustment for NA uptake across the forearm, using fractional extraction of [3H]NA:
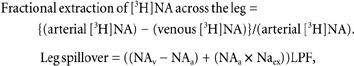 |
where NAa and NAv are the NA concentrations in the arterial and venous effluent plasma, NAex is the fractional extraction of [3H]NA in single passage through the leg, and LPF is the leg plasma flow in litres per minute.
Leg clearance was calculated as: LPF × NAex. Since NA clearance and spillover at 10, 15 and 20 min of the infusion period were similar, the mean of these three measurements was taken as the representative value.
Statistical analysis
Differences in the measured variables among conditions were assessed with ANOVA for repeated measures, with sex as a between-subjects factor with two levels. Student's paired t test was applied when appropriate to determine if the observed differences between the means were significant or not. Significance was accepted at P < 0.05. Data are reported as means ± S.E.M.
RESULTS
Effects of chronic hypoxia on arterial blood gases and acid-base balance
After 9 weeks at 5260 m Pa,O2 and Sa,O2 were lower than at sea level (Table 1). However, the effect of hypoxia on O2 transport was efficiently counterbalanced by a 37 % elevation of blood [Hb] compared with that at sea level (Table 1). As a result, despite the decreases in Pa,O2 and Sa,O2, Ca,O2 at rest after acclimatisation at altitude was 21 % higher than it was during normoxia at sea level. As depicted in Table 1, chronic hypoxia caused a mild respiratory alkalosis (arterial pH = 7.48) that was, in part, metabolically compensated.
Table 1.
Femoral arterial and venous blood gases and acid-base balance at sea level and after 9 weeks of residence at 5260 m
| Sea level | 5260 m chronic hypoxia | |
|---|---|---|
| Hba(g l−1) | 132 ± 5 | 181 ± 4* |
| Hcta | 41 ± 1 | 50 ± 1* |
| Pa,O2 (mmHg) | 95 ± 2 | 49 ± 1* |
| Pfv,O2 (mmHg) | 29 ± 2 | 26 ± 2 |
| SaO2(%) | 96.7 ± 0.1 | 85 ± 1.1* |
| Sfv,O2 (%) | 58 ± 3.7 | 42.4 ± 3.9 |
| Ca,O2 (ml l−1) | 182 ± 6 | 220 ± 6* |
| Cfv,O2(ml l−1) | 107 ± 7 | 117 ± 10 |
| Pa,CO2(mmHg) | 38 ± 1 | 21 ± 1* |
| Pfv,CO2 (mmHg) | 47 ± 2 | 30 ± 2* |
| PHa | 7.42 ± 0.01 | 7.48 ± 0.01 |
| PHfv | 7.38 ± 0.01 | 7.44 ± 0.01 |
| [HCO3−]a (mmol l−1) | 24.1 ± 0.5 | 15.6 ± 0.5* |
| [HCO3−]fv (mmol l−1) | 26.9 ± 0.8 | 20 ± 0.8* |
| BEa (mmol l−1) | −0.1 ± 0.3 | −3.4 ± 0.4 |
| BEfv (mmol l−1) | −0.4 ± 0.4 | −3.8 ± 0.2* |
| Laca (mmol l−1) | 0.7 ± 0.1 | 1.5 ± 0.2* |
| Lacfv (mmol l−1) | 0.8 ± 0.1 | 1.4 ± 0.1* |
a, arterial; fv, femoral venous; Hb, haemoglobin; Hct, haematocrit; PaO2, arterial partial pressure of O2; Pfv,O2; femoral venous partial pressure of O2; SaO2, arterial O2 saturation; Sfv, O2, femoral venous O2 saturation; CaO2, arterial O2 content; Cfv,O2, femoral venous O2 content; Pa,CO2, arterial partial pressure of CO2; Cfv, CO2, femoral venous pressure of CO2; BE, base excess; Lac, lactate.
Significantly different from sea level (P < 0.05).
Heart rate and blood pressure and vascular conductance
Resting heart rate was increased from 59 ± 1 beats min−1 at sea level to 75 ± 3 beats min−1 (P < 0.001) after 9 weeks of residence at altitude. In contrast, resting cardiac output was not altered by chronic hypoxia (5.48 ± 0.34 and 4.93 ± 0.42 l min−1, at sea level and after 9 weeks at 5260 m, respectively). As depicted in Fig. 1A, chronic hypoxia resulted in higher blood pressure. Mean blood pressure was 28 % greater at altitude (P < 0.01) due to an enhancement of both systolic (18 % higher, P < 0.05) and diastolic (41 % higher, P < 0.001) blood pressures. The increment of diastolic blood pressure contributed significantly more than systolic blood pressure to the elevation of mean blood pressure. Consequently, systemic vascular conductance was reduced by 30 % at altitude (P < 0.05) (Fig. 1B). Since leg blood flow was also similar in both conditions, leg vascular conductance tended to be reduced to the same proportion, being 27 % lower at altitude (10.3 ± 1.3 and 7.6 ± 1.4 ml mmHg−1 min−1, P = 0.10) (Fig. 1B).
Figure 1. Blood pressure and vascular conductances.
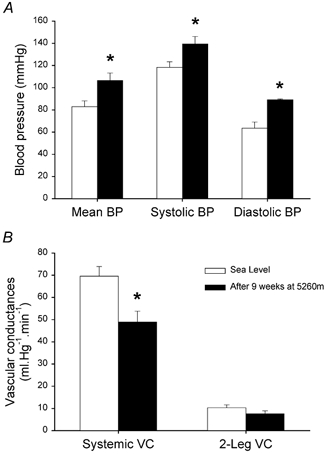
Arterial blood pressure (BP) and vascular conductances (VC) after 9 weeks at 5260 m (▪) and 6-9 months after the subjects had returned from the expedition to sea level conditions (□). * P < 0.05.
Plasma catecholamine kinetics
Both catecholamines were increased at altitude (Fig. 2A). Plasma arterial noradrenaline concentration was 3.7-fold higher at altitude (P < 0.001), while arterial adrenaline was 2.4 times higher (P < 0.05). The elevation of plasma arterial noradrenaline concentration was associated with a 3.8-fold increase in whole-body noradrenaline release (P < 0.001) since whole-body noradrenaline clearance was similar in both conditions (Fig. 2C). Leg NA spillover was also increased in about the same proportion, with an increase in chronic hypoxia which was 3.2 times the value at sea level (P < 0.05) (Fig. 2B). Consequently, the contribution of skeletal muscle to whole-body NA spillover was similar at sea level and at altitude.
Figure 2. Catecholamine kinetics.
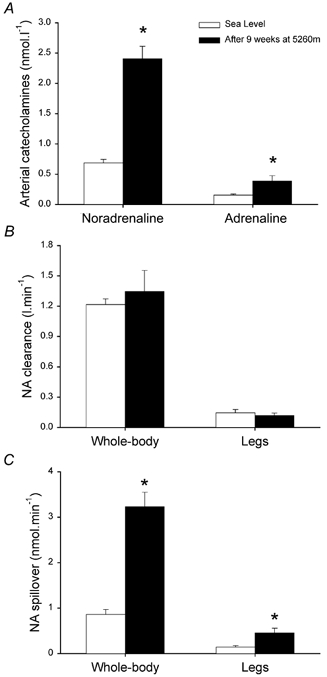
Arterial plasma catecholamine concentrations and catecholamine kinetics after 9 weeks at 5260 m (▪) and 6-9 months after the subjects had returned from the expedition to sea level conditions (□). NA, noradrenaline; * P < 0.05.
DISCUSSION
This study is the first to report that whole-body and skeletal muscle NA spillover are markedly elevated in healthy humans after 9 weeks of residence at an altitude of 5260 m, despite dramatically improved O2 content induced by acclimatisation due to the enhancement of blood [Hb]. This is in contrast to the hypothesis that systemic and skeletal muscle sympathetic activity are normalised after prolonged altitude acclimatisation. The observed sympathetic overactivity was accompanied by enhanced systemic blood pressure and reduced systemic vascular conductance. Thus, it appears that the sympathetic nervous system plays an important role in the pathogenesis of hypoxia-induced hypertension.
In agreement with these results, previous studies have shown increased plasma NA concentration after exposures to altitudes greater than 4000 m (Mazzeo et al. 1991, 1995; Antezana et al. 1994; Kanstrup et al. 1999). Noradrenaline plasma concentration doubled and tripled after 24 h and 5 days of residence at 4559 m, respectively (Kanstrup et al. 1999). More moderate elevations of noradrenaline and adrenaline were reported by Mazzeo et al. (1991, 1995) after 3 weeks at 4300 m. However, a similar period of sojourn at a higher altitude (6542 m, in the Bolivian Andes) resulted in a 3.4 times higher resting plasma noradrenaline concentration than at sea level (Antezana et al. 1994). In contrast, despite absolute noradrenaline values being three times higher at the barometric pressure equivalent to the summit of Mount Everest, non-significant differences were reported in five subjects who were progressively decompressed during 40 days in a hypobaric chamber (Young et al. 1992). This was most likely due to great inter-individual variability. The present study shows that even after a longer period of time at altitude, which resulted in a ≈20 % greater arterial O2 content than at sea level, plasma catecholamine concentrations still remain elevated.
A novel finding of the present investigation is that the increase of arterial plasma NA concentration is caused by enhanced spillover of the neurotransmitter into the bloodstream since NA clearance was unaffected during chronic hypoxia. These findings suggest that skeletal muscle and systemic sympathetic tone are elevated in the healthy human after prolonged exposure to hypoxic environments (Esler et al. 1984; Esler 2000). The evidence, however, is indirect. In fact, NA spillover may be increased without a change of sympathetic discharge if neuronal reuptake before entering the bloodstream, and/or the rate of enzymatic breakdown of naturally released NA, are reduced during chronic hypoxia (Eisenhofer et al. 1996). However, during the same expedition, 5 weeks earlier, local sympathetic nerve activity was measured directly by peroneal microneurography in six of the subjects who participated in the present investigation (Hansen & Sander, 2003). Expressed in bursts per minute, sympathetic nerve activity was three times greater after 4 weeks at 5260 m (Hansen & Sander, 2003). After 9 weeks permanently at altitude, the magnitude of leg sympathetic overactivity (× 3.2), as assessed by the NA spillover method, agrees amazingly well with that observed 5 weeks earlier by Hansen & Sander (2003). The later finding suggests that sympathetic overactivity persists at altitude. Even though, as a result of the acclimatisation process, subjects reached Ca,O2 and Pa,O2 values similar to those observed in the natives living in at the same altitude (Wagner et al. 2002).
A less accentuated elevation in NA spillover has been reported in acute hypoxia (Leuenberger et al. 1991). Following 20-30 min acute exposure to a level of hypoxia of similar magnitude to that used in the present study, NA spillover was increased by 46 % while NA clearance was reduced by 20 % (Leuenberger et al. 1991). The present study indicates that altitude acclimatisation results in a restoration of sea level NA clearance while spillover is remarkably elevated, reaching values similar to those reported, for example, in patients with compensated chronic heart failure (Azevedo et al. 2000). Interestingly, this investigation also demonstrates that skeletal muscle NA spillover is increased during chronic hypoxia in the same proportion as whole-body spillover. Assuming that NA spillover was similar in all skeletal muscles, it can be estimated that the contribution of skeletal muscle NA spillover to whole-body spillover was around 25-30 % at sea level as well at altitude. A similar contribution of skeletal muscle to whole-body NA spillover has been reported in sea level conditions (Esler et al. 1984).
Previous researchers have shown elevations in resting blood pressure at altitudes greater than 4000 m (Mazzeo et al. 1991; Antezana et al. 1994). The increment of mean blood pressure observed here is identical to that reported by Mazzeo et al. (1991). Even though the degree of hypoxaemia was greater in the investigation of Antezana et al. (1994), resting systolic blood pressure was increased a little less (10 %) than in the present study (18 %), while diastolic blood pressure was only increased by 17 % compared with the 40 % enhancement observed in the current study. Apart from the fact that Antezana et al. (1994) measured blood pressure with a sphygmomanometer, the two main differences between their study and the current investigation are the higher altitude, i.e. the greater level of hypoxia in the subjects studied by Antezana et al. (1994), and the longer time of exposure to chronic hypoxia in the present study. This indicates that not only may the magnitude of the hypoxic stimulus be an important factor in determining the extent of the hypertensive response (Richalet et al. 1988; Antezana et al. 1994), the duration of the exposure also appears to be relevant. In fact, during the course of other experiments carried out after 4 weeks of residence in the same facilities, mean blood pressure was increased by 10 mmHg (determined by automated oscillometry; Hansen & Sander, 2003), while 5 weeks later it was 26 mmHg (determined intra-arterially) greater than at sea level.
However, the question remains: why are sympathetic activity and blood pressure elevated in well-acclimatised healthy humans sojourning at high-altitude? Although the chemoreceptors, in particular the aortic bodies, are also sensitive to low Ca,O2 (Lahiri, 1980), sympathetic activity appears to rise with acclimatisation notwithstanding improved Ca,O2 resulting from enhanced [Hb] and Sa,O2, combined with the decrease in the affinity of Hb for O2 (Calbet et al. 2003). Despite the overcompensation of Ca,O2, the partial pressure of oxygen in arterial blood barely improves during acclimatisation (Calbet et al. 2003). Thus, the persistently lowered Pa,O2 is likely to be the stimulus triggering and maintaining high efferent sympathetic outflow in chronic hypoxia via its action on the peripheral and central chemoreceptors (Gonzalez et al. 1994; Marshall, 1994; Reis et al. 1994; Sun & Reis, 1995). In fact, investigations conducted with patients suffering from obstructive sleep apnoea have shown increased blood pressure and sympathetic activation dependent on the reduction of Pa,O2 (Narkiewicz et al. 1999; Imadojemu et al. 2002), thereby supporting a role for intermittent hypoxaemia in the pathogenesis of sympathetic overactivity. The present investigation suggests that a similar mechanism may also operate in healthy humans living at altitude.
In addition to this chemoreflex, several processes may modulate sympathetic activity in chronic hypoxia, such as hyperventilation and feedback from high-pressure and low-pressure baroreceptors. Acclimatisation to hypoxia involves a slow and progressive rise in ventilation, which in turn produces a slow increase in end-tidal PO2, a slow decrease in end-tidal PCO2, and a progressive respiratory alkalosis (Clar et al. 1999). These changes were shown to start within the first few hours of the acclimatisation process (Clar et al. 1999) and were still present after 9 weeks at 5260 m in the current investigation. This increase in ventilation appears to be mediated by the peripheral chemoreceptors, which become more sensitive to hypoxia and CO2 after a few hours in hypoxia (Dwinell et al. 1997; Fatemian & Robbins 2001). So it seems reasonable to suggest that the sensitisation of the peripheral chemoreceptors could account for the sympathetic overactivity observed in chronic hypoxia, a mechanism similar to that previously suggested by Trzebski et al. (1982) in mildly hypertensive men.
During the Chacaltaya expedition, however, Hansen & Sander (2003) revealed that breathing 100 % oxygen for 15 min, to eliminate chemoreflex activation, did not attenuate skeletal muscle sympathetic activity. This implies that additional mechanisms must be involved. It may be argued, however, that hyperoxia could fail to decrease sympathetic outflow after altitude acclimatisation, since hyperoxic breathing at altitude is accompanied by an elevation of arterial Pa,CO2 (from 20 to 23 mmHg (van Hall et al. 2001)). This, in turn, could maintain sympathetic overactivity via stimulation of central chemoreceptors despite hyperoxic inhibition of peripheral chemoreceptors (Xie et al. 2001). This response could be facilitated in the presence of hypocapnia and high sensitivity to CO2 as it occurs during altitude acclimatisation (Dwinell et al. 1997). However, the elevation of Pa,CO2 with hyperoxic breathing appears too small to account for the lack of suppression of sympathetic outflow by hyperoxia at altitude reported by Hansen & Sander (2003).
In turn, muscle sympathetic nerve activity may be modulated depending on respiratory activity. In the anaesthetised, vagotomised and barodenervated animal, rostral ventrolateral medulla pre-sympathetic neurons and peripheral sympathetic nerves discharge in synchrony with phrenic nerve activity during inspiration and reach their lowest activity during expiration (Boczek-Funcke et al. 1992). The magnitude of respiratory related sympathetic activity is accentuated with hypercapnia or hypoxia (Dempsey et al. 2002). The influence of the respiratory pattern on muscle sympathetic nerve activity has been thoroughly studied by Dempsey et al. (2002). In the intact human, muscle sympathetic nerve activity displays its nadir at peak inspiration (when lung volume is at its peak) and its maximum at peak expiration (when lung volume is at its minimum). When humans breathe at increased tidal volume (with Pa,CO2 clamped at physiological level and without respiratory muscle fatigue) both muscle sympathetic nerve activity and vasoconstrictor tone are attenuated. In contrast to the observations made on experimental animals, ingenious human experiments performed by Dempsey et al. (2002) provided evidence that central respiratory motor output of any magnitude is unlikely to be a significant determinant of sympathetic vasoconstrictor activity. Moreover, Seals et al. (1990) have shown that even ‘exercise-like’ hyperpnoea does not modify the amount of sympathetic activity per minute. Thus, it is unlikely that chronic hypoxia-induced hyperventilation (data not shown) had an influence on sympathetic activation in our experimental conditions.
The elevation of NA spillover at altitude occurred in spite of a marked increase in arterial blood pressure, which should have blunted sympathetic activation through stimulation of arterial baroreceptors (Somers et al. 1991), unless the baroreceptors have been reset to a higher set point, as described in sleep apnoea patients (Carlson et al. 1996). In addition, a 15-20 % reduction of circulating blood volume is common at altitude. This has been associated with decreased cardiac filling pressures (Alexander & Grover, 1983) and therefore reduced stimulation of low-pressure baroreceptors. The latter would attenuate negative feedback, allowing for a greater level of sympathoexcitation. However, such a mechanism is unlikely since it has been reported that basal muscle sympathetic nerve activity is not increased after 18 days of 6 deg head-down bed rest, despite 8 % lower plasma volume and 27 % lower pulmonary capillary wedge pressure after bed rest (Pawelczyk et al. 2001).
Several other factors may have played a role in the elevation of blood pressure with chronic hypoxia; for example, increased blood viscosity, decreased basal release or production of nitric oxide (NO) (Fletcher, 2001), and increased NO scavenging due to enhanced blood [Hb] (Pawloski et al. 2001). Blood viscosity increases with haematocrit, therefore the increment of haematocrit observed in the current investigation (9 units) should have caused the relative viscosity to go up by approximately 1 unit. Given the initial haematocrit, an enhancement of blood viscosity close to 20 % would have been expected (Cinar et al. 1999). According to the Hagen-Poiseuille equation, to maintain resting flow at the same level as before acclimatisation, blood pressure would have had to increase by 20 % or the cross-sectional radius of the vascular system by 4.7 %. The fact that systemic vascular conductance was actually reduced suggests that no vasodilatation occurred as a consequence of the elevation in haematocrit, but rather, chronic hypoxia was associated with increased vasoconstrictor tone.
Despite substantial enhancement of NA release in chronic hypoxia, vascular conductance was only reduced by approximately 30 %, i.e. less than expected for this level of NA spillover (Jie et al. 1987). Thus, some vasodilating mechanisms, perhaps hypoxia-mediated, must have opposed the action of NA (Weisbrod et al. 2001), and/or the responsiveness of the vascular smooth muscle fibres to the action of vasoconstrictor agents is partly blunted in chronic hypoxia (Heistad et al. 1972; Doyle &Walker, 1991).
The dramatic elevation of sympathetic activity and resting blood pressure with chronic hypoxia in healthy humans has some important clinical implications. Human populations living at moderate altitude and many lowlanders travelling to the mountains are potentially exposed to the risk of developing hypertension and sympathetic overactivity. The time needed to restore normal arterial blood pressure and sympathetic activity after altitude exposure remains to be determined. The influence that altitude-induced sympathetic overactivity may have on concomitant pathologies is unknown. Duplain et al. (1999) have shown that mountaineers prone to high-altitude pulmonary oedema display exaggerated sympathetic vasoconstrictor activation, which may contribute to the enhanced pulmonary hypertensive response to hypoxia in these subjects. The fact that life expectancy is decreased in heart (Kaye et al. 1995) and kidney (Zoccali et al. 2002) failure patients with high NA spillover further emphasises the importance of these findings.
In summary, this study shows that chronic hypoxia causes increased systemic arterial pressure and massive activation of the sympathetic nervous system in healthy humans, despite improved arterial O2 content with acclimatisation. Strikingly, sympathetic activation reaches levels similar to those observed in chronic heart failure patients. It is particularly important to establish the functional and pathophysiological consequences that may be derived from the chronic hypoxia-induced sympathetic overactivity.
Acknowledgments
Special thanks are owed to Harriet Wagner, Carsten Nielsen, Karin Hansen and Birgitte Jessen for their excellent technical assistance. The Academia de Ciencias de Bolivia and especially Dr Carlos Aguirre are also acknowledged for all their help and support in setting up the expeditionary laboratory on the heights of Mount Chacaltaya. The work of Nicola Leadbeater in reviewing the English style and grammar of the paper is greatly appreciated. This study was supported by a grant from The Danish National Research Foundation (504-14). José A. L. Calbet was on leave from the Department of Physical Education at the University of Las Palmas de Gran Canaria.
REFERENCES
- Alexander JK, Grover RF. Mechanism of reduced cardiac stroke volume at high altitude. Clin Cardiol. 1983;6:301–303. doi: 10.1002/clc.4960060612. [DOI] [PubMed] [Google Scholar]
- Andersen P, Saltin B. Maximal perfusion of skeletal muscle in man. J Physiol. 1985;366:233–249. doi: 10.1113/jphysiol.1985.sp015794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antezana AM, Kacimi R, Le Trong JL, Marchal M, Abousahl I, Dubray C, Richalet JP. Adrenergic status of humans during prolonged exposure to the altitude of 6,542 m. J Appl Physiol. 1994;76:1055–1059. doi: 10.1152/jappl.1994.76.3.1055. [DOI] [PubMed] [Google Scholar]
- Azevedo ER, Newton GE, Floras JS, Parker JD. Reducing cardiac filling pressure lowers norepinephrine spillover in patients with chronic heart failure. Circulation. 2000;101:2053–2059. doi: 10.1161/01.cir.101.17.2053. [DOI] [PubMed] [Google Scholar]
- Boczek-Funcke A, Habler HJ, Janig W, Michaelis M. Respiratory modulation of the activity in sympathetic neurones supplying muscle, skin and pelvic organs in the cat. J Physiol. 1992;449:333–361. doi: 10.1113/jphysiol.1992.sp019089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calbet JA. Oxygen tension and content in the regulation of limb blood flow. Acta Physiol Scand. 2000;168:465–472. doi: 10.1046/j.1365-201x.2000.00698.x. [DOI] [PubMed] [Google Scholar]
- Calbet JA, Boushel R, Radegran G, Sondergaard H, Wagner PD, Saltin B. Why is VO2max after altitude acclimatization still reduced despite normalization of arterial O2 content? Am J Physiol Regul Integr Comp Physiol. 2003;284:R304–316. doi: 10.1152/ajpregu.00156.2002. [DOI] [PubMed] [Google Scholar]
- Cinar Y, Demir G, Pac M, Cinar AB. Effect of hematocrit on blood pressure via hyperviscosity. Am J Hypertens. 1999;12:739–743. doi: 10.1016/s0895-7061(99)00011-4. [DOI] [PubMed] [Google Scholar]
- Clar C, Dorrington KL, Robbins PA. Ventilatory effects of 8 h of isocapnic hypoxia with and without beta-blockade in humans. J Appl Physiol. 1999;86:1897–1904. doi: 10.1152/jappl.1999.86.6.1897. [DOI] [PubMed] [Google Scholar]
- Dempsey JA, Sheel AW, St Croix CM, Morgan BJ. Respiratory influences on sympathetic vasomotor outflow in humans. Respir Physiol Neurobiol. 2002;130:3–20. doi: 10.1016/s0034-5687(01)00327-9. [DOI] [PubMed] [Google Scholar]
- Dow P. Estimations of cardiac output and central blood volume by dye dilution. Physiol Rev. 1956;36:77–102. doi: 10.1152/physrev.1956.36.1.77. [DOI] [PubMed] [Google Scholar]
- Doyle MP, Walker BR. Attentuation of systemic vasoreactivity in chronically hypoxic rats. Am J Physiol. 1991;260:R1114–1122. doi: 10.1152/ajpregu.1991.260.6.R1114. [DOI] [PubMed] [Google Scholar]
- Duplain H, Vollenweider L, Delabays A, Nicod P, Bartsch P, Scherrer U. Augmented sympathetic activation during short-term hypoxia and high-altitude exposure in subjects susceptible to high-altitude pulmonary edema. Circulation. 1999;99:1713–1718. doi: 10.1161/01.cir.99.13.1713. [DOI] [PubMed] [Google Scholar]
- Dwinell MR, Janssen PL, Pizarro J, Bisgard GE. Effects of carotid body hypocapnia during ventilatory acclimatization to hypoxia. J Appl Physiol. 1997;82:118–124. doi: 10.1152/jappl.1997.82.1.118. [DOI] [PubMed] [Google Scholar]
- Eisenhofer G, Friberg P, Rundqvist B, Quyyumi AA, Lambert G, Kaye DM, Kopin IJ, Goldstein DS, Esler MD. Cardiac sympathetic nerve function in congestive heart failure. Circulation. 1996;93:1667–1676. doi: 10.1161/01.cir.93.9.1667. [DOI] [PubMed] [Google Scholar]
- Esler M. The sympathetic system and hypertension. Am J Hypertens. 2000;13:99S–105S. doi: 10.1016/s0895-7061(00)00225-9. [DOI] [PubMed] [Google Scholar]
- Esler M, Jackman G, Bobik A, Kelleher D, Jennings G, Leonard P, Skews H, Korner P. Determination of norepinephrine apparent release rate and clearance in humans. Life Sci. 1979;25:1461–1470. doi: 10.1016/0024-3205(79)90371-0. [DOI] [PubMed] [Google Scholar]
- Esler M, Jennings G, Leonard P, Sacharias N, Burke F, Johns J, Blombery P. Contribution of individual organs to total noradrenaline release in humans. Acta Physiol Scand Suppl. 1984;527:11–16. [PubMed] [Google Scholar]
- Fatemian M, Robbins PA. Selected contribution: chemoreflex responses to CO2 before and after an 8-h exposure to hypoxia in humans. J Appl Physiol. 2001;90:1607–1614. doi: 10.1152/jappl.2001.90.4.1607. [DOI] [PubMed] [Google Scholar]
- Fletcher EC. Invited review: Physiological consequences of intermittent hypoxia: systemic blood pressure. J Appl Physiol. 2001;90:1600–1605. doi: 10.1152/jappl.2001.90.4.1600. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A, Rigual R. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiol Rev. 1994;74:829–898. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- Hallman H, Farnebo LO, Hamberger B, Johnsson G. A sensitive method for the determination of plasma catecholamines using liquid chromatography with electrochemical detection. Life Sci. 1978;23:1049–1052. doi: 10.1016/0024-3205(78)90665-3. [DOI] [PubMed] [Google Scholar]
- Hansen J, Sander M. Sympathetic neural overactivity in healthy humans after prolonged exposure to hypobaric hypoxia. J Physiol. 2003;546:921–929. doi: 10.1113/jphysiol.2002.031765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindl S, Lehnert M, Criee CP, Hasenfuss G, Andreas S. Marked sympathetic activation in patients with chronic respiratory failure. Am J Respir Crit Care Med. 2001;164:597–601. doi: 10.1164/ajrccm.164.4.2007085. [DOI] [PubMed] [Google Scholar]
- Heistad DD, Abboud FM, Mark AL, Schmid PG. Impaired reflex vasoconstriction in chronically hypoxemic patients. J Clin Invest. 1972;51:331–337. doi: 10.1172/JCI106818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton R, Eichholtz F. The influence of chemical factors on the coronary circulation. J Physiol. 1925;59:413–425. doi: 10.1113/jphysiol.1925.sp002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imadojemu VA, Gleeson K, Gray KS, Sinoway LI, Leuenberger UA. Obstructive apnea during sleep is associated with peripheral vasoconstriction. Am J Respir Crit Care Med. 2002;165:61–66. doi: 10.1164/ajrccm.165.1.2009062. [DOI] [PubMed] [Google Scholar]
- Jie K, van Brummelen P, Vermey P, Timmermans PB, Van Zwieten PA. Modulation of noradrenaline release by peripheral presynaptic alpha 2-adrenoceptors in humans. J Cardiovasc Pharmacol. 1987;9:407–413. doi: 10.1097/00005344-198704000-00005. [DOI] [PubMed] [Google Scholar]
- Kanstrup IL, Poulsen TD, Hansen JM, Andersen LJ, Bestle MH, Christensen NJ, Olsen NV. Blood pressure and plasma catecholamines in acute and prolonged hypoxia: effects of local hypothermia. J Appl Physiol. 1999;87:2053–2058. doi: 10.1152/jappl.1999.87.6.2053. [DOI] [PubMed] [Google Scholar]
- Kaye DM, Lefkovits J, Jennings GL, Bergin P, Broughton A, Esler MD. Adverse consequences of high sympathetic nervous activity in the failing human heart. J Am Coll Cardiol. 1995;26:1257–1263. doi: 10.1016/0735-1097(95)00332-0. [DOI] [PubMed] [Google Scholar]
- Lahiri S. Role of arterial O2 flow in peripheral chemoreceptor excitation. Fed Proc. 1980;39:2648–2652. [PubMed] [Google Scholar]
- Laughlin MH, Korthuis RJ, Duncker DJ, Bache RJ. Control of blood flow to cardiac and skeletal muscle during exercise. In: Rowell LB, Shepherd JT, editors. Handbook of Physiology. Exercise: Regulation and Integration of Multiple Systems. Vol. 12. Bethesda, MD, USA: American Physiological Society; 1996. pp. 705–769. [Google Scholar]
- Leuenberger U, Gleeson K, Wroblewski K, Prophet S, Zelis R, Zwillich C, Sinoway L. Norepinephrine clearance is increased during acute hypoxemia in humans. Am J Physiol. 1991;261:H1659–1664. doi: 10.1152/ajpheart.1991.261.5.H1659. [DOI] [PubMed] [Google Scholar]
- Marshall JM. Peripheral chemoreceptors and cardiovascular regulation. Physiol Rev. 1994;74:543–594. doi: 10.1152/physrev.1994.74.3.543. [DOI] [PubMed] [Google Scholar]
- Mazzeo RS, Bender PR, Brooks GA, Butterfield GE, Groves BM, Sutton JR, Wolfel EE, Reeves JT. Arterial catecholamine responses during exercise with acute and chronic high-altitude exposure. Am J Physiol. 1991;261:E419–424. doi: 10.1152/ajpendo.1991.261.4.E419. [DOI] [PubMed] [Google Scholar]
- Mazzeo RS, Brooks GA, Butterfield GE, Podolin DA, Wolfel EE, Reeves JT. Acclimatization to high altitude increases muscle sympathetic activity both at rest and during exercise. Am J Physiol. 1995;269:R201–207. doi: 10.1152/ajpregu.1995.269.1.R201. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, van de Borne PJ, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation. 1999;99:1183–1189. doi: 10.1161/01.cir.99.9.1183. [DOI] [PubMed] [Google Scholar]
- Pawelczyk JA, Zuckerman JH, Blomqvist CG, Levine BD. Regulation of muscle sympathetic nerve activity after bed rest deconditioning. Am J Physiol Heart Circ Physiol. 2001;280:H2230–2239. doi: 10.1152/ajpheart.2001.280.5.H2230. [DOI] [PubMed] [Google Scholar]
- Pawloski JR, Hess DT, Stamler JS. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409:622–626. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- Reis DJ, Golanov EV, Ruggiero DA, Sun MK. Sympatho-excitatory neurons of the rostral ventrolateral medulla are oxygen sensors and essential elements in the tonic and reflex control of the systemic and cerebral circulations. J Hypertens Suppl. 1994;12:S159–180. [PubMed] [Google Scholar]
- Richalet JP, Larmignat P, Rathat C, Keromes A, Baud P, Lhoste F. Decreased cardiac response to isoproterenol infusion in acute and chronic hypoxia. J Appl Physiol. 1988;65:1957–1961. doi: 10.1152/jappl.1988.65.5.1957. [DOI] [PubMed] [Google Scholar]
- Rostrup M. Catecholamines, hypoxia and high altitude. Acta Physiol Scand. 1998;162:389–399. doi: 10.1046/j.1365-201X.1998.00335.x. [DOI] [PubMed] [Google Scholar]
- Rowell LB, Johnson DG, Chase PB, Comess KA, Seals DR. Hypoxemia raises muscle sympathetic activity but not norepinephrine in resting humans. J Appl Physiol. 1989;66:1736–1743. doi: 10.1152/jappl.1989.66.4.1736. [DOI] [PubMed] [Google Scholar]
- Seals DR, Suwarno NO, Dempsey JA. Influence of lung volume on sympathetic nerve discharge in normal humans. Circ Res. 1990;67:130–141. doi: 10.1161/01.res.67.1.130. [DOI] [PubMed] [Google Scholar]
- Somers VK, Mark AL, Abboud FM. Interaction of baroreceptor and chemoreceptor reflex control of sympathetic nerve activity in normal humans. J Clin Invest. 1991;87:1953–1957. doi: 10.1172/JCI115221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun MK, Reis DJ. Decerebration does not alter hypoxic sympathoexcitatory responses in rats. J Auton Nerv Syst. 1995;53:77–81. doi: 10.1016/0165-1838(94)00175-j. [DOI] [PubMed] [Google Scholar]
- Trzebski A, Tafil M, Zoltowski M, Przybylski J. Increased sensitivity of the arterial chemoreceptor drive in young men with mild hypertension. Cardiovasc Res. 1982;16:163–172. doi: 10.1093/cvr/16.3.163. [DOI] [PubMed] [Google Scholar]
- van Hall G, Calbet JA, Sondergaard H, Saltin B. The re-establishment of the normal blood lactate response to exercise in humans after prolonged acclimatization to altitude. J Physiol. 2001;536:963–975. doi: 10.1111/j.1469-7793.2001.00963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner PD, Araoz M, Boushel R, Calbet JA, Jessen B, Radegran G, Spielvogel H, Sondegaard H, Wagner H, Saltin B. Pulmonary gas exchange and acid-base state at 5,260 m in high-altitude Bolivians and acclimatized lowlanders. J Appl Physiol. 2002;92:1393–1400. doi: 10.1152/japplphysiol.00093.2001. [DOI] [PubMed] [Google Scholar]
- Weisbrod CJ, Minson CT, Joyner MJ, Halliwill JR. Effects of regional phentolamine on hypoxic vasodilatation in healthy humans. J Physiol. 2001;537:613–621. doi: 10.1111/j.1469-7793.2001.00613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie A, Skatrud JB, Puleo DS, Morgan BJ. Exposure to hypoxia produces long-lasting sympathetic activation in humans. J Appl Physiol. 2001;91:1555–1562. doi: 10.1152/jappl.2001.91.4.1555. [DOI] [PubMed] [Google Scholar]
- Young PM, Sutton JR, Green HJ, Reeves JT, Rock PB, Houston CS, Cymerman A. Operation Everest II: metabolic and hormonal responses to incremental exercise to exhaustion. J Appl Physiol. 1992;73:2574–2579. doi: 10.1152/jappl.1992.73.6.2574. [DOI] [PubMed] [Google Scholar]
- Zoccali C, Mallamaci F, Parlongo S, Cutrupi S, Benedetto FA, Tripepi G, Bonanno G, Rapisarda F, Fatuzzo P, Seminara G, Cataliotti A, Stancanelli B, Malatino LS, Cateliotti A. Plasma norepinephrine predicts survival and incident cardiovascular events in patients with end-stage renal disease. Circulation. 2002;105:1354–1359. doi: 10.1161/hc1102.105261. [DOI] [PubMed] [Google Scholar]