

Abstract
We investigated the effect of baroreflex-induced sympathetic activation, produced by lower body negative pressure (LBNP) at −40 mmHg, on cerebrovascular responsiveness to hyper- and hypocapnia in healthy humans. Transcranial Doppler ultrasound was used to measure blood flow velocity (CFV) in the middle cerebral artery during variations in end-tidal carbon dioxide pressure (PET,CO2) of +10, +5, 0, −5, and −10 mmHg relative to eupnoea. The slopes of the linear relationships between PET,CO2 and CFV were computed separately for hyper- and hypocapnia during the LBNP and no-LBNP conditions. LBNP decreased pulse pressure, but did not change mean arterial pressure. LBNP evoked an increase in ventilation that resulted in a 9 ± 2 mmHg decrease in PET,CO2, which was corrected by CO2 supplementation of the inspired air. LBNP did not affect cerebrovascular CO2 response slopes during steady-state hypercapnia (3.14 ± 0.24 vs. 2.96 ± 0.26 cm s−1 mmHg−1) or hypocapnia (1.31 ± 0.18 vs. 1.32 ± 0.19 cm s−1 mmHg−1), or the CFV responses to voluntary apnoea (+51 ± 19 vs.+50 ± 18 %). Thus, cerebrovascular CO2 responsiveness was not altered by baroreflex-induced sympathetic activation. Our data challenge the concept that sympathetic activation restrains cerebrovascular responses to alterations in CO2 pressure.
The extraparenchymal cerebral arteries, down to and including the pial arteries, are richly innervated by sympathetic nerve fibres (Nielsen & Owman, 1967; Falck, et al. 1968; Nelson & Rennels, 1970; Edvinsson, 1975). Previous investigators have shown that electrical stimulation of the superior cervical ganglion, the stellate ganglion, or the cervical sympathetic trunk can elicit substantial reductions in cerebral blood flow in cortical and brainstem regions (Meyer et al. 1967; James et al. 1969; Harper et al. 1972; Aubineau et al. 1975). This experimental evidence notwithstanding, the traditional thinking is that, under physiological conditions, the role of the sympathetic nervous system in control of cerebral blood flow is limited mainly to modulation of the cerebrovascular responses to alterations in arterial CO2 pressure Pa,CO2 (James et al. 1969; Kobayashi et al. 1971; Harper et al. 1972; Wei et al. 1980).
Previous investigators have hypothesized that activation of the sympathetic nervous system produces cerebral vasoconstriction during orthostatic stress in humans (Giller et al. 1992; Levine et al. 1994). Augmented sympathetic outflow to the cerebral circulation is a putative cause of decreased cerebral blood flow and lightheadedness in individuals with idiopathic orthostatic intolerance (Jordan et al. 1998); however, this sign and symptom have also been linked to the hyperventilation-induced hypocapnia that often occurs when such patients assume the upright posture (Lagi et al. 2001). In healthy humans, cerebrovascular responses to experimental manipulations of CO2 tension were observed to be diminished during head-up tilt and augmented during ganglionic blockade, effects the authors attributed to sympathetic activation and deactivation, respectively (Jordan et al. 2000). In a previous study from our laboratory, we used a comparable ganglionic blockade protocol but did not replicate the finding that removal of basal sympathetic tone augmented cerebrovascular responses to hyper- and hypocapnia (Przybylowski et al. 2003). The purpose of the present study, therefore, was to test the hypothesis that augmentation of sympathetic vasoconstrictor outflow diminishes cerebrovascular CO2 responsiveness. Accordingly, in healthy subjects, we measured cerebral blood flow velocity (CFV) during manipulations of CO2 pressure from +10 mmHg above to −10 mmHg below eupnoea under control conditions and also during simulated orthostatic stress.
METHODS
Subjects
Ten healthy, non-smoking subjects (5 women, 5 men), aged 22 ± 3 (mean ± S.D.) years, participated in this study. The study requirements were explained in detail to all subjects, and all gave informed, written consent prior to participation. The experimental protocol was approved by the University of Wisconsin-Madison Health Sciences Human Subjects Committee. Participants did not have pulmonary, neurological, or cardiovascular disease, as assessed by history and physical examination.
General procedures
All experiments were conducted with subjects lying supine with their lower extremities, up to the level of the iliac crests, inside an airtight lower body negative pressure (LBNP) chamber. The room temperature was maintained at 24 ± 1 °C. Ventilation was measured through a mouthpiece connected to a pneumotachograph (Model 3700; Hans Rudolph, Kansas City, MO, USA). Expired air was sampled from the mouthpiece and the end-tidal CO2 tension (PET,CO2) was measured with an infrared gas analyser (Model CD3A; Ametek, Pittsburgh, PA, USA). Heart rate was measured from the electrocardiogram. Blood pressure was measured indirectly on a beat-by-beat basis by photoelectric plethysmography (Finapres 2300; Ohmeda, Louisville, CO, USA) as well as at one minute intervals by an automated arm cuff sphygmomanometer (Dinamap 1846SX/P; Critikon, Tampa, FL, USA).
Measurement of cerebral flow velocity
A 2 MHz pulsed Doppler ultrasound system (Neurovision 500 M; Multigon Industries, Younkers, NY, USA) was used to measure peak cerebral blood flow velocity (CFV) in the proximal (M1) segment of the middle cerebral artery. The middle cerebral artery was insonated through the right temporal window using search techniques that have been described previously (Otis & Ringelstein, 1996). After obtaining the best-quality signal, the probe was secured using a headband device to provide a fixed angle of insonation.
Cerebrovascular responses to hyper- and hypocapnia
Baseline data were collected during at least 5 min of eupnoeic breathing with self-selected frequency and tidal volume. After a stable baseline was established, CO2 was added to the inspirate until a PET,CO2 of +10 mmHg above baseline was attained. After 5 min of this level of hypercapnia, the subject's tidal volume and frequency were noted, and then s/he was instructed to maintain that same level of tidal volume and frequency, using audio and visual feedback, while the inspired CO2 was reduced in steps to achieve PET,CO2 values of +5, 0, −5 and −10 mmHg relative to baseline eupnoeic breathing. At least 5 min of steady-state data were collected at each level of PET,CO2.
We calculated mean values for CFV (velocity-time integral, see Data analysis, below) during steady-state eupcapnia, hypercapnia and hypocapnia. Linear regression analyses were performed to determine the slopes of the relationships between PET,CO2and CFV. Separate slopes were computed for hypercapnia and hypocapnia. The day-to-day reproducibility of this measure of cerebrovascular responsiveness to CO2 was assessed in a separate group of nine subjects (6 males, 3 females; aged 34 ± 11 years (mean ± S.D.). The group mean values for CO2 response slopes measured on day 1 vs. day 2 were virtually identical (2.87 ± 0.18 vs. 2.91 ± 0.18 cm s−1 mmHg−1 for hypercapnia and 1.29 ± 0.10 vs. 1.14 ± 0.14 cm s−1 mmHg−1 for hypocapnia, both P > 0.05). The coefficients of variation (standard deviation of the difference scores/grand mean × 100) for hypercapnia and hypocapnia were 12 and 20 %, respectively.
Experimental protocols
Each subject underwent a familiarization trial of LBNP prior to data collection. Then, the cerebrovascular response of the subject to hyper- and hypocapnia was assessed twice: once with LBNP maintained at −40 mmHg and again without LBNP. The order in which the two trials were performed was randomized (in four subjects the LBNP trial was performed first, and in six subjects the no-LBNP trial was performed first). At least 5 min of baseline data were acquired before initiation of the CO2 response tests. After the CO2 response tests were completed, six of the subjects performed 20 s-breath holds starting from functional residual capacity in the control condition and during LBNP at −40 mmHg (3–4 trials in each condition). We previously determined the day-to-day reproducibility of CFV increases during 20 s-breath holds (coefficient of variation, 20 %).
All variables were recorded continuously on paper (Astro-Med K2G; Grass Instruments, West Warwick, RI, USA) and videotape (Model 400A PCM; Vetter, Rebersburg, PA, USA). The signals were also routed to a computer (sampling rate, 120 Hz) for off-line analysis using custom-written software.
Data analysis
Measurements were performed using custom-made software. Mean CFV for each cardiac cycle was determined from the integral of the maximal frequency shift over one cardiac cycle divided by the length of the corresponding cardiac cycle (i.e. velocity-time integral). The beat-by-beat values for CFV, breath-by-breath values for PET,CO2 and minute-by-minute values for mean arterial pressure (MAP) obtained with the arm cuff sphygmomanometer, calculated as 1/3 pulse pressure + diastolic pressure, were averaged over the 5 min at each level of CO2. The slopes of the linear relationships between CFV and PET,CO2 during hyper- and hypocapnia in the 0 and −40 mmHg conditions were compared by Student's paired t tests. Five minute averages of MAP were compared using a 2-way analysis of variance with repeated measures on the CO2 and LBNP factors. To assess the CFV and MAP responses to breath holds, the peak values at the termination of each breath hold were computed as three-beat averages of the actual highest cardiac cycle and the two adjacent cardiac cycles. The change in CFV and MAP caused by each breath hold was computed as the difference between the peak value and the mean of the baseline values. For each subject, haemodynamic responses to the 3–4 breath hold trials in each condition were averaged and these average values were used in computation of the group means. P values < 0.05 were considered statistically significant. Except where otherwise noted, data are expressed as means ± s.e.m.
RESULTS
Haemodynamic and ventilatory responses to LBNP alone
Application of LBNP at −40 mmHg produced an increase in heart rate and a decrease in pulse pressure with no change in MAP (Table 1; n = 10). Application of LBNP produced an increase in minute ventilation that was caused mainly by an increase in tidal volume. This increase in minute ventilation resulted in a decrease in PET,CO2. CFV was lower than baseline during LBNP at −40 mmHg when subjects breathed spontaneously (i.e. they hyperventilated); however, when PET,CO2 was returned to the control level via supplementation of the inspired air, CFV was the same as in the no-LBNP condition.
Table 1.
Haemodynamic and spontaneous ventilatory responses to LBNP at −40 mmHg
| LBNP (mmHg) | HR (beats min−1) | MAP (mmHg) | PP (mmHg) | VE (1 min−1) | VT (1) | Rf (breaths min−1) | PET,CO2 (mmHg) | CFV (cm s−1) |
|---|---|---|---|---|---|---|---|---|
| 0 | 77 ± 8 | 81 ± 3 | 63 ± 6 | 7.6 ± 0.7 | 0.5 ± 0.1 | 15 ± 1 | 40.5 ± 1.1 | 53.5 ± 9.2 |
| −40 | 93 ± 9* | 85 ± 5 | 54 ± 5* | 11.5 ± 1.5* | 0.9 ± 0.2 | 16 ± 1 | 31.9 ± 1.7* | 40.7 ± 6.9* |
Note that during these measurements no attempt was made to control VT, breathing frequency, or PET,CO2. HR, heart rate; PP, pulse pressure; VE, ventilation; VT, tidal volume and Rf, breathing frequency, n = 10;
P < 0.05, −40 vs. 0 mmHg LBNP.
Responses to hyper- and hypocapnia without LBNP
In all subjects, supplementation of the inspired CO2 produced graded increases in CFV, whereas voluntary hyperventilation caused graded reductions in CFV (Fig. 1 and Fig. 2, open circles). Mean values for the slopes of the linear relationships between CFV and PET,CO2 were 2.96 ± 0.3 cm s−1 mmHg−1 for hypercapnia and 1.32 ± 0.19 cm s−1 mmHg−1 for hypocapnia.
Figure 1. Individual cerebral flow velocity responses to hyper- and hypocapnia with (•) and without (○) lower body negative pressure (LBNP).
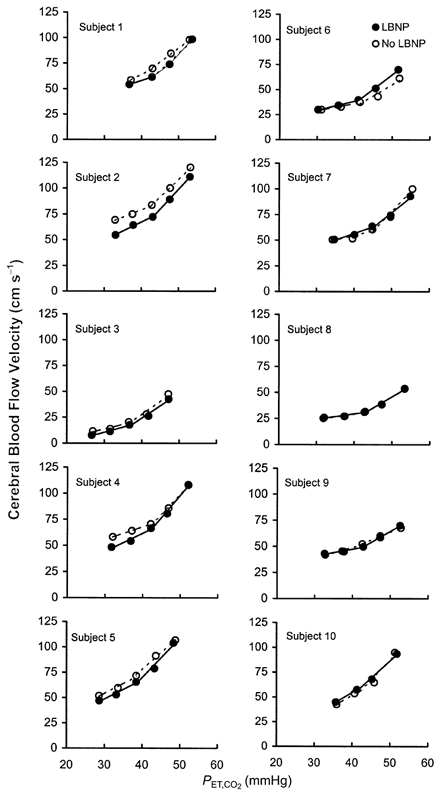
Continuous and dashed lines represent linear regressions of velocity on PET,CO2, calculated separately for hyper- and hypocapnia.
Figure 2. Relationships between PET,CO2 and CFV and mean arterial pressure (MAP) with (•) and without (○) LBNP.
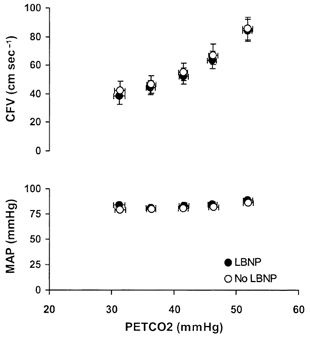
Data shown are means ± s.e.m., n = 10.
The higher level of hypercapnia (PET,CO2, +10 mmHg above baseline) produced small, but consistent elevations in MAP (P < 0.05). MAPs at all other levels of PET,CO2were not different from baseline (P > 0.05; Fig. 2).
Responses to hyper- and hypocapnia during LBNP
Since our goal was to compare CFV responses over the same range of PCO2 values with and without LBNP, we supplemented the inspired CO2 as necessary to restore baseline PET,CO2 prior to the initiation of the hyper- and hypocapnia trials. Baseline CFV, measured under isocapnic conditions at equal tidal volumes and breathing frequencies, was unaffected by application of LBNP (55 ± 6 and 52 ± 6 cm s−1, P > 0.05; Table 2).
Table 2.
Haemodynamic and ventilatory responses to LBNP at −40 mmHg when ventilation was maintained at the level elicited during hypercapnia (PPET,CO2+ 10 mmHg above eupnoea) and PPET,CO2 was held constant at control levels
| LBNP (mmHg) | HR (beats min−1) | MAP (mmHg) | PP (mmHg) | VE (1 min−1) | VT (1) | Rf (breaths min−1) | PPET,CO2 (mmHg) | CFV (cm s−1) |
|---|---|---|---|---|---|---|---|---|
| 0 | 70 ± 6 | 80 ± 2 | 61 ± 4 | 29.6 ± 5.6 | 1.5 ± 0.3 | 20 ± 1 | 41.6 ± 0.8 | 55.0 ± 6.4 |
| -40 | 88 ± 6* | 83 ± 2 | 54 ± 3* | 29.8 ± 5.5 | 1.5 ± 0.3 | 20 ± 1 | 41.6 ± 0.8 | 52.4 ± 5.6 |
n = 10;
P < 0.05, −40 vs. 0 mmHg LBNP.
Application of LBNP did not alter the slope of the CFV responses to hypercapnia (3.14 ± 0.24 vs. 2.96 ± 0.26 cm s−1 mmHg−1, P > 0.05) or to hypocapnia (1.31 ± 0.18 vs. 1.32 ± 0.19 cm s−1 mmHg−1, P > 0.05; Fig. 2). The order of trials had no effect on CFV responsiveness. The between-trial difference in slopes was the same in subjects who performed the LBNP trial first vs. those who performed the LBNP trial second, both for hypercapnia (0.03 ± 0.31 vs. 0.27 ± 0.18 cm s−1 mmHg−1) and for hypocapnia (0.19 ± 0.08 vs.– 0.14 ± 0.17 cm s−1 mmHg−1) (both P > 0.05).
Application of LBNP did not affect the influence of alterations in PET,CO2 on MAP (Fig. 2). Similar to the no-LBNP condition, MAP was higher during the hypercapnia at +10 mmHg than at other levels of PCO2 (P < 0.05).
Responses to breath holds with and without LBNP
LBNP had no effect on the increases in CFV evoked by 20 s breath holds (Fig. 3; n = 6; +50 ± 18 vs.+51 ± 19 %, P > 0.05). Likewise, the MAP responses to breath hold were unaffected by LBNP (+6 ± 2 vs.+8 ± 2 mmHg, P > 0.05).
Figure 3. Cerebrovascular responses to 20 s breath holds with (•) and without (○) LBNP.
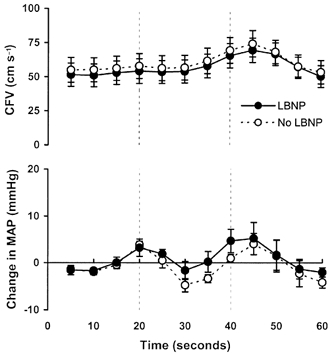
The dashed vertical lines indicate the duration of the apnoea. Data shown are means ± s.e.m., n = 6.
DISCUSSION
Summary of findings
In the present study we found that baroreflex-mediated increases in sympathetic vasoconstrictor outflow did not alter the cerebrovascular responses to hyper- and hypocapnia. Moreover, such sympathetic activation had no effect on the increase in CFV produced by apnoea. These findings, taken together with recent evidence from our laboratory that removal of basal sympathetic tone also had no effect on these responses (Przybylowski et al. 2003), calls into question the concept that sympathetic activation constrains, and sympathetic ablation enhances, cerebrovascular CO2 responsiveness (Jordan et al. 1998, 2000). The following discussion details the assumptions and evidence that underlie these conclusions.
Critique of methods
Doppler ultrasonography measures flow velocity rather than blood flow. Nevertheless, we believe that velocity is a reasonable estimate of flow in our experiments because: (1) the diameter of the middle cerebral artery varies by less than ±4 % during changes in arterial pressure (Giller et al. 1993), CO2 tension (Giller et al. 1993; Valdueza et al. 1997; Serrador et al. 2000) or gravitational stress (Serrador et al. 2000), and (2) velocity and flow through the middle cerebral artery are highly correlated (Bishop et al. 1986; Kirkham et al. 1986). Our estimates of cerebrovascular CO2 reactivity (6 % mmHg−1 for hypercapnia and 3 % mmHg−1 for hypocapnia) are consistent with previous reports based on measurements of tissue nitrous oxide uptake (Kety & Schmidt, 1948), functional magnetic resonance imaging (Rostrup et al. 1994; Kastrup et al. 2001), positron emission tomography (Ramsay et al. 1993; Nishimura et al. 1999), and 133Xe washout (Tominaga et al. 1976).
We consider it unlikely that our negative findings were secondary to Type II hypothesis testing errors. The differences in cerebrovascular CO2 response slopes during LBNP vs. no-LBNP were small (+0.18 ± 0.16 cm s−1 mmHg−1 for hypercapnia and −0.01 ± 0.12 cm s−1 mmHg−1 for hypocapnia) and the 95 % confidence intervals for these differences (−0.18 to −0.54 cm s−1 mmHg−1 for hypercapnia and −0.27 to 0.26 cm s−1 mmHg−1 for hypocapnia) are both narrowly centred around zero difference, indicating that the true value of this parameter is very close to the null (Hoenig & Heisey, 2001). Furthermore, the coefficients of variation for the difference in CO2 responses slopes of individual subjects (17 and 28 % for hyper- and hypocapnia, respectively) were similar to those observed in tests of the day-to-day reproducibility of these measurements. The order in which subjects were exposed to the LBNP vs. no-LBNP conditions did not affect the difference in slopes.
Baroreflex effects on the cerebral circulation
Our conclusions are predicated on the assumption that LBNP, a stimulus that is known to increase sympathetic vasoconstrictor outflow to forearm, calf and splanchnic vascular beds via baroreflex unloading (Johnson et al. 1974; Abboud et al. 1979; Jacobsen et al. 1993), also increases sympathetic outflow to the cerebral circulation. From a teleological perspective, it seems inappropriate that baroreflex-induced sympathetic vasoconstriction, a mechanism for maintaining cerebral perfusion and avoiding syncope, would occur in the brain. Moreover, such vasoconstriction presumably could not assist in defending blood pressure in the face of orthostatic stress because the cerebral circulation is located above the level of the heart. Nevertheless, the neural substrate for such a reflex pathway is known to exist. Baroreflex deactivation increases efferent activity in the cervical sympathetic trunk (Tafil-Klawe et al. 1989) and electrical stimulation of the cervical sympathetic trunk causes vasoconstriction in multiple regions of the brain (Meyer et al. 1967). Although previous investigators demonstrated that carotid sinus baroreceptor activation had no effect on cerebral blood flow (Rapela et al. 1967; Heistad & Marcus, 1976), haemorrhage-induced baroreceptor deactivation (reductions in MAP to 60 and 45 % of baseline) did cause mild cerebral vasoconstriction in anaesthetized cats (Gross et al. 1979). The smaller degree of baroreflex unloading produced by LBNP in our study (8 mmHg reduction in pulse pressure, no change in MAP), although equivalent to that produced by assumption of the upright posture, had no effect on baseline CFV or on the cerebrovascular responses to hyper- and hypocapnia.
It is important to note that other investigators have observed a decrease in CFV during immersion of the hand in ice water (cold pressor test) that was blocked by clonidine, an inhibitor of central sympathetic outflow (Micieli et al. 1994). In our study, activation of the sympathetic nervous system via LBNP-induced unloading of arterial and cardiopulmonary baroreceptors, alone and in combination with stimulation of chemoreceptors during breath hold, failed to alter cerebrovascular responsiveness to CO2. We cannot, on the basis of our data, speculate whether other methods employed to increase sympathetic outflow in our study would be sufficient to influence CFV. In addition, because we measured cerebrovascular responses during sustained application of LBNP, it is possible that we may have overlooked the transient effects of abrupt increases in sympathetic outflow on CFV. In experimental animals, previous investigators have observed lack of sustained cerebral vasoconstriction (i.e. an ‘escape’ phenomenon) during electrical stimulation of the cervical sympathetic trunk (Baumbach & Heistad, 1983).
Effect of LBNP on cerebral perfusion pressure
Cerebral perfusion pressure, the difference between MAP and intracranial pressure when the latter exceeds central venous pressure (Weyland et al. 2000; Munis & Lozada, 2000), is a major determinant of CFV. Although we did not measure intracranial pressure or central venous pressure in our study, it is likely that both were reduced during LBNP (Jacobsen et al. 1993; Tankisi et al. 2002). If so, cerebral perfusion pressure would have been higher during LBNP vs. baseline conditions. The finding that CFV was unchanged in spite of increased perfusion pressure suggests that LBNP elicited an autoregulatory response. This autoregulation apparently had no affect on cerebrovascular CO2 responsiveness, because the changes in CFV produced by alterations in PCO2 were the same with and without LBNP.
Comparison with previous studies of cerebrovascular regulation during baroreceptor unloading
Our findings are consistent with those of previous investigators who observed comparable cerebrovascular CO2 reactivity in the supine and sitting positions (Mayberg et al. 1996), but they conflict with those of a recent study that found reduced cerebrovascular CO2 responsiveness during head-up tilt (Jordan et al. 2000). The reasons for this discrepancy are not obvious; however, there may be differences in the amount of sympathetic activation caused by actual vs. simulated orthostatic stress. In the previous study, sympathetic stimulation was produced by head-up tilt, a manoeuvre that not only unloads baroreceptors but also activates the vestibulosympathetic reflex (Ray & Monahan, 2002). In addition, in the previous study, CO2 response slopes were calculated by linear regression of CFV on Pa,CO2 over the entire range of observed CO2 tensions. It is possible that an apparent reduction in CO2 responsiveness during head-up tilt occurred secondarily to the somewhat lower PCO2 values observed during hyper- and hypocapnia during tilt vs. supine and the alinear nature of the cerebral blood flow:PCO2 relationship (the response slopes are known to be steeper during hyper- than hypocapnia (Reivich, 1964)). In our experiments we ensured that PET,CO2 was the same in the sympathetic activation vs. no sympathetic activation conditions and we constructed separate slopes for responses to hyper- and hypocapnia.
Effects of sympathetic activation on baseline CFV: role of hypocapnia
Our finding that application of LBNP did not alter baseline CFV is also inconsistent with several other recent studies (Giller et al. 1992; Levine et al. 1994; Serrador et al. 2000). We suspect that this difference may be attributable to strict maintenance of eucapnia during LBNP in our subjects. In two of the previous studies, the PET,CO2 responses to LBNP were not reported (Giller et al. 1992; Levine et al. 1994). In the other study, LBNP-induced decreases in PET,CO2 occurred, but were not statistically significant (Serrador et al. 2000). In several other previous reports, hyperventilation-induced hypocapnia was a consistent, statistically significant finding during simulated orthostatic stress with LBNP and also during upright tilt (Kobayashi et al. 1980; Ahn et al. 1989; Cencetti et al. 1997; Cooke et al. 1999). In our subjects, we observed a drop in CFV at the onset of LBNP when PET,CO2 levels were allowed to fall; however, when we restored PET,CO2 to control by supplementing the inspired CO2, we also restored the control level of CFV (Tables 1 and 2). The cause of hyperventilation during orthostatic stress, although not understood, may result from baroreflex-chemoreflex interactions (Somers et al. 1991) or perhaps the vestibulorespiratory reflex (Monahan et al. 2002). Previous investigators speculated that this increase in ventilation reinforces the cardiovascular adjustments to central hypovolaemia by facilitating venous return via the respiratory pump and enhancing vasoconstriction via reduced Pa,CO2 (Hildebrandt et al. 2000).
In conclusion, we found that baroreflex-induced sympathetic activation had no influence on cerebrovascular responses to CO2, one of the most powerful regulators of vascular tone in this region. Our findings do not exclude the possibility that sympathetic activation that is more intense or that is evoked via different mechanisms might influence cerebral blood flow, although chemoreceptor activation during breath holds failed to uncover a difference. Nevertheless, they are consistent with the traditional view that the sympathetic nervous system plays a minor role, if any, in regulation of cerebral blood flow under physiological conditions. The major importance of sympathetically mediated vasoconstriction in the cerebral circulation may be to protect the blood-brain barrier when the limits of autoregulation are exceeded (e.g. during acute hypertensive episode; Mayhan et al. 1987).
Acknowledgments
The authors would like to thank Ms Patricia Mecum for assistance with manuscript preparation. This research was supported by the Medical Research Service of the US Department of Veterans Affairs and by a Wisconsin/Hilldale Undergraduate/Faculty Research Fellowship (G. L. and J. B. S.).
REFERENCES
- Abboud FM, Eckberg DL, Johannsen UJ, Mark AL. Carotid and cardiopulmonary baroreceptor control of splanchnic and forearm vascular resistance during venous pooling in man. J Physiol. 1979;286:173–184. doi: 10.1113/jphysiol.1979.sp012612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn B, Sakakibara Y, Paulev PE, Masuda A, Nishibayashi Y, Nakamura W, Honda Y. Circulatory and respiratory responses to lower body negative pressure in man. Jpn J Physiol. 1989;39:919–929. doi: 10.2170/jjphysiol.39.919. [DOI] [PubMed] [Google Scholar]
- Aubineau PF, Sercombe R, Seylaz J. Continuous recordings of local cerebral blood flow during cervical sympathetic nerve blockade or stimulation. J Physiol. 1975;246:104–106P. [PubMed] [Google Scholar]
- Baumbach GL, Heistad DD. Effects of sympathetic stimulation and changes in arterial pressure on segmental resistance of cerebral vessels in rabbits and cats. Circ Res. 1983;52:527–533. doi: 10.1161/01.res.52.5.527. [DOI] [PubMed] [Google Scholar]
- Bishop CC, Powell S, Rutt D, Browse NL. Transcranial Doppler measurement of middle cerebral artery blood flow velocity: a validation study. Stroke. 1986;17:913–915. doi: 10.1161/01.str.17.5.913. [DOI] [PubMed] [Google Scholar]
- Cencetti S, Bandinelli G, Lagi A. Effect of PCO2changes induced by head-upright tilt on transcranial Doppler recordings. Stroke. 1997;28:1195–1197. doi: 10.1161/01.str.28.6.1195. [DOI] [PubMed] [Google Scholar]
- Cooke WH, Hoag JB, Crossman AA, Kuusela TA, Tahvanainen KU, Eckberg DL. Human responses to upright tilt: a window on central autonomic integration. J Physiol. 1999;517:617–628. doi: 10.1111/j.1469-7793.1999.0617t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvinsson L. Neurogenic mechanisms in the cerebrovascular bed. Autonomic nerves, amine receptors and their effects on cerebral blood flow. Acta Physiol Scand. 1975;427(Suppl.):1–35. [PubMed] [Google Scholar]
- Falck B, Nielsen KC, Owman C. Adrenergic innervation of the pial circulation. Scand J Clin Lab Invest. 1968;(Suppl. 102):VI. doi: 10.3109/00365516809168994. [DOI] [PubMed] [Google Scholar]
- Giller CA, Bowman G, Dyer H, Mootz L, Krippner W. Cerebral arterial diameters during changes in blood pressure and carbon dioxide during craniotomy. Neurosurgery. 1993;32:737–741. [PubMed] [Google Scholar]
- Giller CA, Levine BD, Meyer Y, Buckey JC, Lane LD, Borchers DJ. The cerebral hemodynamics of normotensive hypovolemia during lower-body negative pressure. J Neurosurg. 1992;76:961–966. doi: 10.3171/jns.1992.76.6.0961. [DOI] [PubMed] [Google Scholar]
- Gross PM, Heistad DD, Strait MR, Marcus ML, Brody MJ. Cerebral vascular responses to physiological stimulation of sympathetic pathways in cats. Circ Res. 1979;44:288–294. doi: 10.1161/01.res.44.2.288. [DOI] [PubMed] [Google Scholar]
- Harper AM, Deshmukh VD, Rowan JO, Jennett WB. The influence of sympathetic nervous activity on cerebral blood flow. Arch Neurol. 1972;27:1–6. doi: 10.1001/archneur.1972.00490130003001. [DOI] [PubMed] [Google Scholar]
- Heistad DD, Marcus ML. Total and regional cerebral blood flow during stimulation of carotid baroreceptors. Stroke. 1976;7:239–243. doi: 10.1161/01.str.7.3.239. [DOI] [PubMed] [Google Scholar]
- Hildebrandt W, Ottenbacher A, Schuster M, Baisch F, Bartsch P. Increased hypoxic ventilatory response during hypovolemic stress imposed through head-up-tilt and lower-body negative pressure. Eur J Appl Physiol. 2000;81:470–478. doi: 10.1007/s004210050070. [DOI] [PubMed] [Google Scholar]
- Hoenig JM, Heisey DM. The abuse of power: the pervasive fallacy of power calculations for data analysis. Am Stat. 2001;55:1–6. [Google Scholar]
- Jacobsen TN, Morgan BJ, Scherrer U, Vissing SF, Lange RA, Johnson N, Ring WS, Rahko PS, Hanson P, Victor RG. Relative contributions of cardiopulmonary and sinoaortic baroreflexes in causing sympathetic activation in the human skeletal muscle circulation during orthostatic stress. Circ Res. 1993;73:367–378. doi: 10.1161/01.res.73.2.367. [DOI] [PubMed] [Google Scholar]
- James IM, Millar RA, Purves MJ. Observations on the extrinsic neural control of cerebral blood flow in the baboon. Circ Res. 1969;25:77–93. doi: 10.1161/01.res.25.1.77. [DOI] [PubMed] [Google Scholar]
- Johnson JM, Rowell LB, Niederberger M, Eisman MM. Human splanchnic and forearm vasoconstrictor responses to reductions of right atrial and aortic pressures. Circ Res. 1974;34:515–524. doi: 10.1161/01.res.34.4.515. [DOI] [PubMed] [Google Scholar]
- Jordan J, Shannon JR, Black BK, Paranjape SY, Barwise J, Robertson D. Raised cerebrovascular resistance in idiopathic orthostatic intolerance: evidence for sympathetic vasoconstriction. Hypertension. 1998;32:699–704. doi: 10.1161/01.hyp.32.4.699. [DOI] [PubMed] [Google Scholar]
- Jordan J, Shannon JR, Diedrich A, Black B, Costa F, Robertson D, Biaggioni I. Interaction of carbon dioxide and sympathetic nervous system activity in the regulation of cerebral perfusion in humans. Hypertension. 2000;36:383–388. doi: 10.1161/01.hyp.36.3.383. [DOI] [PubMed] [Google Scholar]
- Kastrup A, Kruger G, Neumann-Haefelin T, Moseley ME. Assessment of cerebrovascular reactivity with functional magnetic resonance imaging: comparison of CO2 and breath holding. Magn Reson Imaging. 2001;19:13–20. doi: 10.1016/s0730-725x(01)00227-2. [DOI] [PubMed] [Google Scholar]
- Kety SS, Schmidt CF. The effects of altered arterial tensions of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal young men. J Clin Invest. 1948;27:484–492. doi: 10.1172/JCI101995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkham FJ, Padayachee TS, Parsons S, Seargeant LS, House FR, Gosling RG. Transcranial measurement of blood velocities in the basal cerebral arteries using pulsed Doppler ultrasound: velocity as an index of flow. Ultrasound Med Biol. 1986;12:15–21. doi: 10.1016/0301-5629(86)90139-0. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Waltz AG, Rhoton AL., Jr Effects of stimulation of cervical sympathetic nerves on cortical blood flow and vascular reactivity. Neurology. 1971;21:297–302. doi: 10.1212/wnl.21.3.297. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Loeppky JA, Venters MD, Luft UC. Circulation and respiration response to arm exercise and lower body negative pressure. Med Sci Sports Exerc. 1980;12:244–249. [PubMed] [Google Scholar]
- Lagi A, Cencetti S, Corsoni V, Georgiadis D, Bacalli S. Cerebral vasoconstriction in vasovagal syncope: any link with symptoms? A transcranial Doppler study (comment) Circulation. 2001;104:2694–2698. doi: 10.1161/hc6172.099397. [DOI] [PubMed] [Google Scholar]
- Levine BD, Giller CA, Lane LD, Buckey JC, Blomqvist CG. Cerebral versus systemic hemodynamics during graded orthostatic stress in humans. Circulation. 1994;90:298–306. doi: 10.1161/01.cir.90.1.298. [DOI] [PubMed] [Google Scholar]
- Mayberg TS, Lam AM, Matta BF, Visco E. The variability of cerebrovascular reactivity with posture and time. J Neurosurg Anesthesiol. 1996;8:268–272. doi: 10.1097/00008506-199610000-00002. [DOI] [PubMed] [Google Scholar]
- Mayhan WG, Werber AH, Heistad DD. Protection of cerebral vessels by sympathetic nerves and vascular hypertrophy. Circulation. 1987;75(suppl I):I107–I112. [PubMed] [Google Scholar]
- Meyer JS, Yoshida K, Sakamoto K. Autonomic control of cerebral blood flow measured by electromagnetic flowmeters. Neurology. 1967;17:638–648. doi: 10.1212/wnl.17.7.638. [DOI] [PubMed] [Google Scholar]
- Micieli G, Tassorelli C, Bosone D, Cavallini A, Viotti E, Nappi G. Intracerebral vascular changes induced by cold pressor test: a model of sympathetic activation. Neurol Res. 1994;16:163–167. doi: 10.1080/01616412.1994.11740219. [DOI] [PubMed] [Google Scholar]
- Monahan KD, Sharpe MK, Drury D, Ertl AC, Ray CA. Influence of vestibular activation on respiration in humans. Am J Physiol Regul Integr Comp Physiol. 2002;282:R689–694. doi: 10.1152/ajpregu.00568.2001. [DOI] [PubMed] [Google Scholar]
- Munis JR, Lozada LJ. Giraffes, siphons and starling resistors. Cerebral perfusion pressure revisited. J Neurosurg Anesthesiol. 2000;12:290–296. doi: 10.1097/00008506-200007000-00029. [DOI] [PubMed] [Google Scholar]
- Nelson E, Rennels M. Innervation of intracranial arteries. Brain. 1970;93:475–490. doi: 10.1093/brain/93.3.475. [DOI] [PubMed] [Google Scholar]
- Nielsen KC, Owman C. Adrenergic innervation of pial arteries related to the circle of Willis in the cat. Brain Res. 1967;6:773–776. doi: 10.1016/0006-8993(67)90134-5. [DOI] [PubMed] [Google Scholar]
- Nishimura S, Suzuki A, Hatazawa J, Nishimura H, Shirane R, Yasui N, Yoshimoto T. Cerebral blood-flow responses to induced hypotension and to CO2 inhalation in patients with major cerebral artery occlusive disease: a positron-emission tomography study. Neuroradiology. 1999;41:73–79. doi: 10.1007/s002340050709. [DOI] [PubMed] [Google Scholar]
- Otis SM, Ringelstein EB. The transcranial Doppler examination: principles and applications of transcranial Doppler sonography. In: Tegeler CH, Babikian VL, Gomez CR, editors. Neurosonology. St Louis, MO USA: Mosby; 1996. pp. 113–128. [Google Scholar]
- Przybylowski T, Bangash M-F, Reichmuth K, Morgan BJ, Skatrud JB, Dempsey JA. Mechanisms of the cerebrovascular response to apnoea. J Physiol. 2003;548:323–332. doi: 10.1113/jphysiol.2002.029678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay SC, Murphy K, Shea SA, Friston KJ, Lammertsma AA, Clark JC, Adams L, Guz A, Frackowiak RS. Changes in global cerebral blood flow in humans: effect on regional cerebral blood flow during a neural activation task. J Physiol. 1993;471:521–534. doi: 10.1113/jphysiol.1993.sp019913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapela CE, Green HD, Denison AB., Jr Baroreceptor reflexes and autoregulation of cerebral blood flow in the dog. Circ Res. 1967;21:559–568. doi: 10.1161/01.res.21.4.559. [DOI] [PubMed] [Google Scholar]
- Ray CA, Monahan KD. The vestibulosympathetic reflex in humans: neural interactions between cardiovascular reflexes. Clin Exp Pharm Physiol. 2002;29:98–102. doi: 10.1046/j.1440-1681.2002.03614.x. [DOI] [PubMed] [Google Scholar]
- Reivich M. Arterial PCO2 and cerebral hemodynamics. Am J Physiol. 1964;206:25–35. doi: 10.1152/ajplegacy.1964.206.1.25. [DOI] [PubMed] [Google Scholar]
- Rostrup E, Larsson HB, Toft PB, Garde K, Thomsen C, Ring P, Sondergaard L, Henriksen O. Functional MRI of CO2 induced increase in cerebral perfusion. NMR Biomed. 1994;7:29–34. doi: 10.1002/nbm.1940070106. [DOI] [PubMed] [Google Scholar]
- Serrador JM, Picot PA, Rutt BK, Shoemaker JK, Bondar RL. MRI measures of middle cerebral artery diameter in conscious humans during simulated orthostasis. Stroke. 2000;31:1672–1678. doi: 10.1161/01.str.31.7.1672. [DOI] [PubMed] [Google Scholar]
- Somers VK, Mark AL, Abboud FM. Interaction of baroreceptor and chemoreceptor reflex control of sympathetic nerve activity in normal humans. J Clin Invest. 1991;87:1953–1957. doi: 10.1172/JCI115221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafil-Klawe M, Klawe J, Majcherczyk S, Trzebski A. Sympatho-inhibitory baroreflex in conscious rabbits: simultaneous recordings of sympathetic and aortic nerve activity. J Auton Nerv Syst. 1989;28:227–232. doi: 10.1016/0165-1838(89)90150-1. [DOI] [PubMed] [Google Scholar]
- Tankisi A, Rolighed LJ, Rasmussen M, Dahl B, Cold GE. The effects of 10 degrees reverse trendelenburg position on ICP and CPP in prone positioned patients subjected to craniotomy for occipital or cerebellar tumours. Acta Neurochir. 2002;144:665–670. doi: 10.1007/s00701-002-0957-y. [DOI] [PubMed] [Google Scholar]
- Tominaga S, Strandgaard S, Uemura K, Ito K, Kutsuzawa T. Cerebrovascular CO2 reactivity in normotensive and hypertensive man. Stroke. 1976;7:507–510. doi: 10.1161/01.str.7.5.507. [DOI] [PubMed] [Google Scholar]
- Valdueza JM, Balzer JO, Villringer A, Vogl TJ, Kutter R, Einhaupl KM. Changes in blood flow velocity and diameter of the middle cerebral artery during hyperventilation: assessment with MR and transcranial Doppler sonography. Am J Neuroradiol. 1997;18:1929–1934. [PMC free article] [PubMed] [Google Scholar]
- Wei EP, Kontos HA, Patterson JL., Jr Dependence of pial arteriolar response to hypercapnia on vessel size. Am J Physiol. 1980;238:697–703. doi: 10.1152/ajpheart.1980.238.5.H697. [DOI] [PubMed] [Google Scholar]
- Weyland A, Buhre W, Grund S, Ludwig H, Kazmaier S, Weyland W, Sonntag H. Cerebrovascular tone rather than intracranial pressure determines the effective downstream pressure of the cerebral circulation in the absence of intracranial hypertension. J Neurosurg Anesthesiol. 2000;12:210–216. doi: 10.1097/00008506-200007000-00002. [DOI] [PubMed] [Google Scholar]