

Abstract
Congestive heart failure (CHF) induces alterations in energy metabolism and mitochondrial function that span cardiac as well as skeletal muscles. Whether these defects originate from altered mitochondrial DNA copy number and/or mitochondrial gene transcription is not known at present, nor are the factors that control mitochondrial capacity in different muscle types completely understood. We used an experimental model of CHF induced by aortic banding in the rat and investigated mitochondrial respiration and enzyme activity of biochemical mitochondrial markers in cardiac, slow and fast skeletal muscles. We quantified mitochondrial DNA (mtDNA), expression of nuclear (COX IV) and mitochondrial (COX I) encoded cytochrome c oxidase subunits as well as nuclear factors involved in mitochondrial biogenesis and in the necessary coordinated interplay between nuclear and mitochondrial genomes in health and CHF. CHF induced a decrease in oxidative capacity and mitochondrial enzyme activities with a parallel decrease in the mRNA level of COX I and IV, but no change in mtDNA content. The expression of the peroxisome proliferator activated receptor gamma co-activator 1α (PGC-1α) gene was downregulated in CHF, as well as nuclear respiratory factor 2 and mitochondrial transcription factor A, which act downstream from PGC-1α. Most interestingly, only the level of PGC-1α expression was strongly correlated with muscle oxidative capacity in cardiac and skeletal muscles, both in healthy and CHF rats. Mitochondrial gene transcription is reduced in CHF, and PGC-1α appears as a potential modulator of muscle oxidative capacity under these experimental conditions.
Heart failure originates from the impairment of the ability of the cardiac pump to meet requirements of the body, and progresses towards alterations of other essential functions of the organism. Patients suffering from congestive heart failure (CHF) always complain of early muscular fatigue and exercise intolerance. Alterations in skeletal muscle vascular function, and intrinsic skeletal muscle abnormalities have been reported in this disease (Lunde et al. 2001). Alterations in energy metabolism play a substantial role in the functional defects such as decreased cardiac contractility, and decreased contraction and resistance to fatigue of the skeletal muscle. It was shown that energetic deficiencies in CHF affect the mitochondrial compartment and include decreased respiration rate, activity and expression of mitochondrial enzymes (De Sousa et al. 1999, 2000, 2001; Ide et al. 2001; Marin-Garcia et al. 2001). These alterations are encountered in cardiac muscle as well as in skeletal muscles and diaphragm, and thus led to the proposal of a metabolic myopathy in heart failure. This raises the possibility that some circulating factor(s), activated in CHF, may impair mitochondrial gene replication and thus the mitochondrial DNA copy number and/or mitochondrial gene transcription, but this remains to be determined.
Cardiac and skeletal muscles differ in their contractile, metabolic properties and mitochondrial content. These properties can be regulated by a variety of physiological and pathophysiological processes, allowing the muscle cells to adapt to various environmental constraints. Mitochondria occupy 20–30 % of cell volume in cardiac myocytes, ≈6 % in oxidative skeletal muscle like soleus (SOL), and only 2–3 % in superficial glycolytic parts of gastrocnemius muscle (GAS). The cellular mechanisms that determine the oxidative capacity of a particular muscle type are not completely understood.
Mitochondria have their own genomic system, the mitochondrial DNA (mtDNA), a covalently closed-circular double-stranded DNA molecule. Mammalian mtDNA contains only two promoters, the light-strand and heavy-strand promoters (LSP and HSP, respectively), from which transcripts are produced and then processed to yield the individual mRNAs encoding 13 subunits of the oxidative phosphorylation system (OXPHOS), ribosomal and transfer RNAs (Attardi & Schatz, 1988; Shadel & Clayton, 1997). Transcription from the LSP also produces an RNA primer that is necessary for initiating mtDNA replication. The rest of the OXPHOS subunits as well as other mitochondrial proteins and the factors involved in mtDNA maintenance are encoded by the nucleus. Thus, mitochondrial biogenesis and function depend on the coordinated expression of nuclear and mitochondrial genomes. Recent advances have started to elucidate the factors that regulate mtDNA replication and transcription as well as the expression of nuclear-encoded mitochondrial genes in physiological and pathological conditions. Based on these studies, two transcription factors play a key role in nucleo-mitochondrial communication: nuclear respiratory factor 1 or 2 (NRF-1, NRF-2) and mitochondrial transcription factor A (mtTFA).
Transcription factors NRF-1 and NRF-2 bind and activate the promoters of various nuclear genes that encode for components of OXPHOS as well as factors required for mtDNA transcription and replication, such as mtTFA (Scarpulla, 2002). Electrical stimulation of cardiomyocytes leads to NRF-1 mRNA induction and increased expression of mitochondrial proteins (Xia et al. 1997). Induction or increased binding activity of NRF-1 have been observed in skeletal muscle during training or following active bouts of exercise (Murakami et al. 1998), following AMP kinase activation or administration of a creatine analogue (Bergeron et al. 2001), all situations associated with an increased mitochondrial content. Targeted disruption of NRF-1 in mice induces a dramatic decrease in the amount of mtDNA and is lethal during embryonic development, suggesting that NRF-1 is required for mtDNA maintenance and respiratory chain function during early embryogenesis (Huo & Scarpulla, 2001). MtTFA is a nucleus-encoded protein, which binds upstream of the LSP and HSP of mtDNA and promotes transcription of mtDNA in an in vitro system. It also has an important role in regulating mtDNA replication, as initiation of replication of the leading strand of mtDNA is dependent on an RNA primer produced by transcription from the LSP (Parisi & Clayton, 1991; Dairaghi et al. 1995). Disruption of the mtTFA gene causes depletion of mtDNA, loss of mitochondrial transcripts, loss of mtDNA-encoded polypeptides and severe respiratory chain deficiency (Larsson et al. 1998). Moreover, targeted disruption of mtTFA in cardiomyocytes induces a dilated cardiomyopathy with atrioventricular heart conduction blocks and severe respiratory chain deficiency (Wang et al. 1999; Li et al. 2000). Thus, these results establish a critical role for mtTFA in the regulation of mtDNA copy number in vivo and in mitochondrial biogenesis.
Upstream of these transcription factors, attention has been focused recently on a transcriptional co-activator of peroxisome proliferator activated receptor gamma (PPARγ), known as PGC-1α. PGC-1α has been described as a versatile co-activator that can co-activate PPARα to promote fatty acid oxidation (FAO) enzyme expression, or PPARγ/thyroid hormone receptor β to promote uncoupling proteins expression for adaptive thermogenesis, but also can activate mitochondrial biogenesis through its interaction with NRFs (Knutti & Kralli, 2001; Lehman & Kelly, 2002; Scarpulla, 2002). PGC-1α gene expression parallels increases in energy-producing capacity during cardiac perinatal development, and overexpression of PGC-1α in cardiac myocytes in culture or in cardiac muscle of transgenic mice induces increased mitochondrial content, fatty acid cycle enzymes and respiration (Lehman et al. 2000). In addition, recent findings showed that a cardiac-specific repression of PGC-1α expression by a mutant histone deacetylase 5 (HDAC5) results in loss of and morphological changes to mitochondria and in downregulation of mitochondrial enzymes (Czubryt et al. 2003). These data establish a direct cause and effect relationship between PGC-1α gene expression and oxidative capacity. As such, PGC-1α behaves as a key regulator of mitochondrial function capacity and participates in the transduction of physiological stimuli to energy production in the heart. PGC-1α mRNA is higher in oxidative and cardiac muscles than in glycolytic muscles (Larrouy et al. 1999; Lin et al. 2002), and is increased by low intensity prolonged exercise and by activation of AMP kinase (Terada et al. 2002). It can activate the expression and activity of the NRFs, which in turn upregulate the expression of nuclear genes of mitochondria and of the nuclear-encoded mtTFA (Wu et al. 1999; Scarpulla, 2002).
We hypothesized that heart failure can affect oxidative capacity via an impairment of mitochondrial gene replication and/or transcription. Using a model of CHF in rats induced by aortic banding, we measured mtDNA content and mRNA expression of one nuclear- and one mitochondrial-encoded subunit of respiratory chain complex IV (COX IV and COX I) in control and failing cardiac and skeletal muscles. In addition, we also examined whether the known factors involved in the nucleo-mitochondrial communication (NRFs, mtTFA and PGC-1α) quantitatively correlate with the level of oxidative capacity in different healthy muscles types and whether their mRNA expression is altered and could account for the decreased oxidative capacity in CHF by correlating each mRNA level to oxygen consumption rates and activity of oxidative enzymes.
The results demonstrate that mitochondrial gene transcription rather than replication is decreased in CHF and that PGC-1α correlates with the oxidative capacity in failing cardiac and skeletal muscles as well as in healthy muscles.
METHODS
Rat model of CHF
This study conformed to the guidelines of the animal welfare committee of INSERM. Animals were operated on by Charles River Laboratories (France) and sham-operated and CHF rats were purchased 3 weeks after surgery. Animals were randomly assigned to one of two groups (aortic stenosis (CHF) or sham-operated (Sham)) and were anaesthetized with 60 mg kg−1 pentobarbital, I.P. Aortic stenosis was induced by placing a stainless steel haemoclip of 0.6-mm width on the ascending aorta via a thoracic incision as previously described (De Sousa et al. 1999; Feldman et al. 1993). The sham-operated rats underwent the same procedure without placement of the clip. Six months after surgery, the mortality rate was about 70 % in the CHF group, and 11 CHF and 11 Sham rats were studied. Animals were anesthetized with an intraperitoneal injection of urethane (0.2g (100 g body weight)−1) and the right and left ventricles (RV and LV, respectively), oxidative soleus (SOL) and superficial glycolytic parts of gastrocnemius (GAS) muscles were isolated and weighed. Parts of these muscles were rapidly frozen for biochemical, DNA and RNA analysis and other parts were immediately used for respiration experiments.
Enzyme assay
After homogenization of frozen tissue samples in an ice-cold buffer ((50 mg wet weight) ml−1) containing (mm): Hepes 5 (pH 8.7), EGTA 1, dithiothreitol 1, MgCl2 5 and Triton X-100 (0.1 %), an incubation was performed for 60 min at 4 °C to ensure complete enzyme extraction. The total activities of citrate synthase (CS) and creatine kinase (CK) were assayed (30 °C, pH 7.5) with coupled enzyme systems as previously described (De Sousa et al. 1999). CK isoenzymes were separated using agarose (1 %) gel electrophoresis performed at 250 V for 90 min and individual isoenzymes were resolved by incubating the gels with a coupled enzyme system. Cytochrome c oxidase (COX) activity was determined as previously described (Wharton & Tzagoloff, 1967).
Functional properties of mitochondria
Respiratory parameters of the total mitochondrial population were studied in situ in freshly saponin-skinned fibres as previously described (Veksler et al. 1987). Briefly, thin fibre bundles were excised from LV, SOL and GAS muscles and incubated for 30 min at 4 °C in solution S (see below) containing 50 μg ml−1 saponin to permeabilize the sarcolemma. Respiratory rates were determined with a Clark electrode (Strathkelvin Instruments, UK) in an oxygraphic cell containing 3 ml respiration solution (solution R, see below) at 22 °C with continuous stirring and were expressed as (μmol O2) min−1 (g dry weight)−1. Solutions S and R contained (mm): EGTA-CaEGTA buffer 10 (free Ca2+ concentration 100 nm), MgCl2 1, taurine 20, dithiothreitol 0.5, imidazole 20 and ionic strenght 160 mm (potassium methane sulfonate). Solution S (pH 7.1) also contained 5 mm MgATP and 15 mm phophocreatine, whereas solution R contained 5 mm glutamate, 2 mm malate, 3 mm phosphate and 2 mg ml−1 fatty acid free bovine serum albumin. The ADP-stimulated respiration (ADP) above basal oxygen consumption (O2) was plotted as a function of [ADP] with or without creatine (20 mm). ADP was calculated using a non-linear fitting of the Michaelis-Menten equation. Maximal respiration rate (max) was (ADP+O2). From one to three measurements were made for each muscle.
Southern blot analysis of total DNA
Total cellular DNA was extracted from LV, SOL and GAS muscles by standard methods including successive steps of proteinase K digestion, organic extraction and ethanol precipitation. To measure mtDNA levels, a Southern blot analysis was performed using concomitant hybridization with a cDNA probe for mtDNA corresponding to nt231–695 of the rat mitochondrial 12S rRNA gene (GenBank accession number 854269) and a cDNA probe for nDNA corresponding to nt872–1525 of the rat nuclear-encoded 18S rRNA gene (GenBank accession number M11188) as a control for the amount of nuclear DNA. For each muscle type, total DNA (2 μg) was digested with BamHI restriction enzyme, separated by electrophoresis through 0.7 % agarose gel and transferred onto nylon membrane (Hybond-N+, Amersham Biosciences, Orsay, France). A small amount of a plasmid (25 ng) carrying a sequence complementary to the mtDNA probe was added with the test samples and was used as a positive control. All membranes were prehybridized at 57 °C for 60 min with hybridization buffer (AlkPhos Direct Hybridization buffer, Amersham), hybridized to the labelled probes at 57 °C overnight, and then washed twice at 57 °C for 20 min each in primary wash buffer (2 M urea, 0.1 % SDS, 50 mm sodium phosphate, 150 mm NaCl, 1 mm MgCl2, 2 g blocking reagent (Amersham Biosciences)) and twice at room temperature for 20 min in secondary wash buffer (50 mm Tris, 100 mm NaCl, 2 mm MgCl2). Each probe was labelled with a labelling kit using alkaline phosphatase (AlkPhos Direct, Amersham). Signals were detected by chemoluminescent reagents (CDP-Star, Amersham) and quantified using an image analyser (Bio-Rad, Marnes-la-Coquette, France) to determine the ratio of mtDNA to nDNA.
Real-time quantitative RT-PCR analysis
Total muscle RNA was isolated from frozen tissue samples (50–100 mg) using the Trizol reagent technique according to the manufacturer's instructions (Invitrogen, Cergy Pontoise, France). Oligo-dT first-strand cDNA was synthesized from 5 μg total RNA using Superscript II reverse transcriptase (Invitrogen).
Real-time PCR was performed using SYBR Green technology on a LightCycler rapid thermal cycler (Roche Diagnostics, Meylan, France). Forward and reverse primers (Table 1) were each designed in a different exon of the target gene sequence, eliminating the possibility of amplifying genomic DNA. For the mitochondrial COX I gene, which was devoid of intronic sequences, we checked using the negative reverse transcription product that contamination of genomic DNA did not interfere with quantification. For each set of primers, a basic local alignment search tool (BLAST) search revealed that sequence homology was obtained only for the target gene. Prior optimization was conducted for each set of primers, which consisted of determining optimal primer and MgCl2 concentrations, the template concentration and verifying the efficiency of the amplification. To confirm the specificity of the amplification, the PCR product was subjected to a melting curve analysis and agarose gel electrophoresis. PCR amplification was performed in duplicate in a total reaction volume of 15 μl. The reaction mixture consisted of 1 μl diluted template, 1.5 μl FastStart DNA Master SYBR Green I kit (× 10), 3 mm (COX I, mtTFA, NRF-1, NRF-2α, PGC-1α) or 4 mm (COX IV, β-actin) MgCl2 and 0.5 μm forward and reverse primers (except for NRF-2α and PGC-1α, when 0.3 μm were used). After an 8-min activation of Taq polymerase, amplification was allowed to proceed for 30–40 cycles, each consisting of denaturation at 95 °C for 10 s, annealing at 55 (COX I, mtTFA), 56 (NRF-1), 58 (COX IV) or 65 °C (NRF-2α, PGC-1α) for 5–10 s and extension at 72 °C for 5–24 s, depending on the target gene. Fivefold serial dilutions from soleus total RNA were analysed for each target gene and allowed us to construct linear standard curves from which the concentrations of the test sample were calculated. Results were normalized to β-actin transcription to compensate for variation in input RNA amounts and efficiency of reverse transcription, and then multiplied by total RNA (mg wet weight)−1 to correct for variation in the amount of total RNA per amount of tissue to compare expression level of target genes between the three types of muscles.
Table 1.
Primers used for real-time PCR amplification
| Gene | GenBank accession number | Forward Primer (5′–3′) Reverse Primer (5′–3′) | PCR product size (bp) |
|---|---|---|---|
| PGC-1α | AF049330 | AGTGTGCTGCTCTGGTTGGTG | 613 |
| GGAGGGTCATCGTTTGTGGTC | |||
| NRF-1 | NM–010938 | TTACTCTGCTGTGGCTGATGG | 92 |
| CCTCTGATGCTTGCGTCGTCT | |||
| NRF-2α | M74515 | AGGTGAGGAGATGGGCTGC | 604 |
| CGTTGTCCCCATTTTTGCG | |||
| MtTFA | AJ312746 | GAAAGCACAAATCAAGAGGAG | 175 |
| CTGCTTTTCATCATGAGACAG | |||
| COX I | X14848 | AGCAGGAATAGTAGGGACAGC | 520 |
| TGAGAGAAGTAGTAGGACGGC | |||
| COX IV | X15029 | TGGGAGTGTTGTGAAGAGTGA | 273 |
| GCAGTGAAGCCGATGAAGAAC | |||
| β-actin | X00351 | GGGTCAGAAGGATTCCTATG | 238 |
| GGTCTCAAACATGATCTGGG |
PGC-1α, peroxisome proliferator activated receptor gamma co-activator 1α; NRF-1, nuclear respiratory factor 1; NRF-2α, nuclear respiratory factor 2 DNA-binding alpha subunit; mtTFA, mitochondrial transcription factor A; COX I and COX IV, cytochrome c oxidase subunits I and IV.
Statistics
Data are expressed as means ± s.e.m. One way ANOVA, followed by Newman-Keuls post hoc test was used to assess the differences between control muscles. Effects of CHF were assessed with Student's t test within each muscle. Values of P < 0.05 were considered significant.
RESULTS
Anatomical data
As CHF rats showed markedly decreased body weight (−34 %), the weight of organs was expressed per tibial length, an index of growth independent of body fat or fluid homeostasis (Table 2). CHF induced significant absolute and relative cardiac hypertrophy (+83 %) due to increase in relative weights of both ventricles and also induced increase in lung weight. Furthermore, obvious clinical evidence of cardiac decompensation (brown and ‘nutmeg’ liver, pleural and peritoneal effusions and enlarged atria) was observed in CHF rats.
Table 2.
Anatomicl data
| Sham (n = 11) | CHF (n = 11) | P vs. sham | |
|---|---|---|---|
| Body weight (g) | 732 ± 22 | 484 ± 33 | < 0.001 |
| Heart weight (g) | 1.62 ± 0.07 | 2.93 ± 0.15 | < 0.001 |
| Heart weight/TL (g cm−1) | 0.35 ± 0.01 | 0.64 ± 0.03 | < 0.001 |
| Right ventricle weight/TL (g cm−1) | 0.06 ± 0.01 | 0.11 ± 0.01 | < 0.001 |
| Left ventricle weight/TL (g cm−1) | 0.24 ± 0.01 | 0.38 ± 0.02 | < 0.001 |
| Soleus weight/TL (g cm−1) | 0.062 ± 0.002 | 0.051 ± 0.003 | < 0.05 |
| Lung weight/TL (g cm−1) | 0.37 ± 0.01 | 0.61 ± 0.06 | < 0.001 |
TL indicates tibia length. Values are means ± s.e.m.
In the same rat model of CHF (De Sousa et al. 1999; Momken et al. 2003) we showed, using the Langendorff-perfused heart model, systolic and diastolic abnormalities in CHF hearts (e.g. increased left ventricular end-diastolic pressure from 10 ± 4 to 62 ± 18 mmHg), and using in vivo echocardiography an impairment of systolic function (30 % decrease in LV shortening, and 21 % decrease in ejection fraction). Collectively, these results confirmed the development of severe CHF in these animals. Decreased soleus weight showed muscle atrophy.
Enzyme assay and functional properties of mitochondria
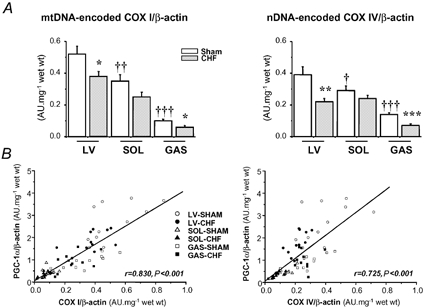
Healthy cardiac muscle exhibited the highest levels of CS, COX, and mitochondrial CK (mi-CK) activities and of oxidative capacities, followed by SOL and then GAS muscles (Fig. 1). Both in LV and GAS, CHF induced a significant decrease in CS (LV, −37 %; GAS, −53 %), COX (LV, −37 %; GAS, −68 %) and mi-CK activities (LV, −63 %; GAS, −72 %). Oxidative capacity was reduced by 21 and 33 % during CHF in LV and GAS, respectively. The response of SOL muscle to CHF was less pronounced: CS and mi-CK were significantly reduced by 22 and 33 %, respectively, whereas decreases in COX activity and maximal respiration rate (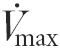 ) were not significant.
) were not significant.
Figure 1. Enzymatic activities and mitochondrial function in left ventricle (LV), soleus (SOL) and gastrocnemius (GAS) of sham-operated and CHF rats.
CS, citrate synthase; COX, cytochrome c oxidase; mi-CK, mitochondrial creatine kinase and , maximal respiration rate. Values are means ± s.e.m. (n = 11). *P < 0.05; **P < 0.01; ***,†††P < 0.001 versus respective Sham group (*) or versus Sham LV (†).
Mitochondrial DNA content
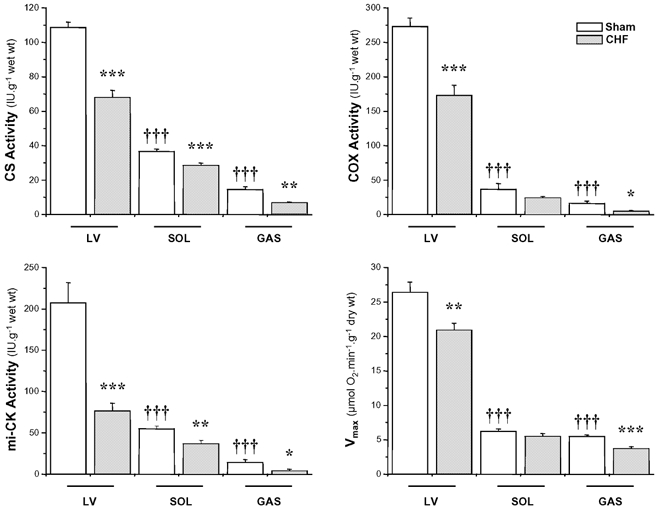
The mtDNA content, expressed as the ratio of mtDNA to nDNA was measured by a Southern blot analysis. Results presented in Fig. 2 did not reveal a significant difference in the mtDNA content between healthy and CHF cardiac or skeletal muscles.
Figure 2. Southern blot analysis of total DNA from left ventricle (LV), soleus (SOL) and gastrocnemius (GAS) of sham-operated (S) and CHF rats.
A, representative Southern blots for each muscle type with signals resulting from mitochondrial DNA (mtDNA) and nuclear DNA (nDNA). A positive control corresponding to a plasmid carrying sequence complementary to the mitochondrial probe was included and provided a signal at 4365 bp. B, mtDNA-to-nDNA ratio was determined after quantification of each signal. Values are means ± s.e.m. (n = 11).
Levels of mtDNA-encoded COX subunit I and nDNA-encoded COX subunit IV mRNA
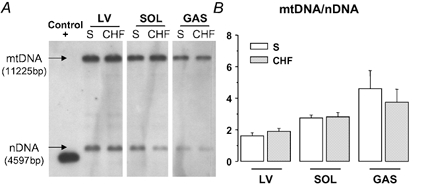
As previously observed (Habets et al. 1999), total RNA content was twofold lower in Sham GAS than in Sham LV or SOL (LV: 0.98 ± 0.03, SOL: 0.92 ± 0.05 and GAS: 0.52 ± 0.02 μg (mg wet wt)−1, P < 0.001). Moreover, LV in CHF showed an increase in total RNA content (Sham LV: 0.98 ± 0.03, CHF LV: 1.08 ± 0.03 μg (mg wet wt)−1, P < 0.01). The levels of expression of the two COX subunits (Fig. 3A) and of the key factors involved in mitochondrial biogenesis (Fig. 4) were normalized to β-actin values, as the expression of this gene was the most stable compared to other tested reference genes (data not shown). They were multiplied by total RNA (mg wet weight)−1, to compare the three muscles (Habets et al. 1999).
Figure 3. Real-time quantitative RT-PCR analysis of mRNA expression of mitochondrial DNA (mtDNA)-encoded cytochrome c oxidase subunit I (COX I) and nuclear DNA (nDNA)-encoded cytochrome c oxidase subunit IV (COX IV) in left ventricle (LV), soleus (SOL) and gastrocnemius (GAS) of sham-operated and CHF rats.
A, results are given as means ± s.e.m. of values normalized to β-actin transcription and multiplied by total RNA (mg wet weight)−1. *,†P < 0.05; **,††P < 0.01; ***,†††P < 0.001 versus respective Sham group (*) or versus Sham LV (†). B, correlations between the mRNA levels of COX I or COX IV and PGC-1α (peroxisome proliferator activated receptor gamma co-activator 1α). r, the correlation coefficient; P, the statistical significance.
Figure 4. Real-time quantitative RT-PCR analysis of mRNA expression of peroxisome proliferator activated receptor gamma co-activator 1α (PGC-1α), nuclear respiratory factor 1 (NRF-1), nuclear respiratory factor 2 DNA-binding subunit α (NRF-2α) and mitochondrial transcription factor A (mtTFA) in left ventricle (LV), soleus (SOL) and gastrocnemius (GAS) of sham-operated and CHF rats.
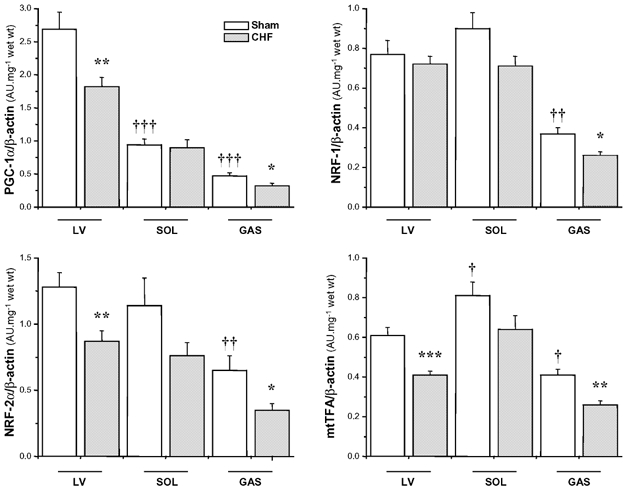
Results are given as means ± s.e.m. of values normalized to β-actin transcription and multiplied by total RNA (mg wet weight)−1. *,†P < 0.05; **,††P < 0.01; ***,†††P < 0.001 versus respective Sham group (*) or versus Sham LV (†).
In healthy animals, mtDNA-encoded COX I and nDNA-encoded COX IV subunits exhibited a similar pattern of mRNA expression, being higher in LV, 1.5 times lower in SOL and 3–5 times lower in GAS for COX IV and COX I, respectively. In CHF, we observed a parallel decrease in the expression of mRNA for these two subunits of 27 and 43 % in LV, and 67 and 74 % in GAS muscles for COX I and COX IV, respectively. A depressed expression of COX I and COX IV was also present in failing SOL but did not reach significance level.
mRNA levels of nuclear factors associated with mitochondrial biogenesis and function
In healthy animals, PGC-1α mRNA expression was higher in LV, being 3 times lower in SOL (P < 0.001) and 6 times lower in GAS (P < 0.001). The NRF-1, NRF-2α and mtTFA transcripts did not present a similar tissue distribution: their expression was comparable in Sham LV and SOL muscles and was significantly lower in GAS (Fig. 4).
In LV and GAS, CHF led to significant 30–40 % reductions in PGC-1α, NRF-2α and mtTFA gene expression. SOL again presented a less severe response to CHF. Generalized metabolic defects were associated with a global decrease in the expression of key factors involved in the regulation of mitochondrial biogenesis and function.
In healthy tissues, strong positive correlations (P < 0.001) were observed between PGC-1α mRNA expression and CS (r = 0.856), COX (r = 0.837), mi-CK (r = 0.649) activities or oxidative capacities (r = 0.868). No significant correlation was found between NRFs or mtTFA mRNA, and mitochondrial parameters (for example for COX activity: r = 0.204 with NRF-1, r = 0.324 with NRF-2α, r = −0.022 with mtTFA). When both control and CHF groups were pooled, we again found a significant positive correlation between PGC-1α mRNA and mitochondrial parameters (Fig. 5). Interestingly, if we correlated expression of COX I or COX IV subunit mRNA with expression of transcription factors or PGC-1α mRNA, significant correlations were only obtained with PGC-1α mRNA (Fig. 3B).
Figure 5. Correlations between the mRNA expression level of PGC-1α and enzymatic activities of citrate synthase (CS), cytochrome c oxidase (COX), mitochondrial creatine kinase (mi-CK) or maximal respiration rate () in left ventricle (LV), soleus (SOL) and gastrocnemius (GAS) of sham-operated and CHF rats.
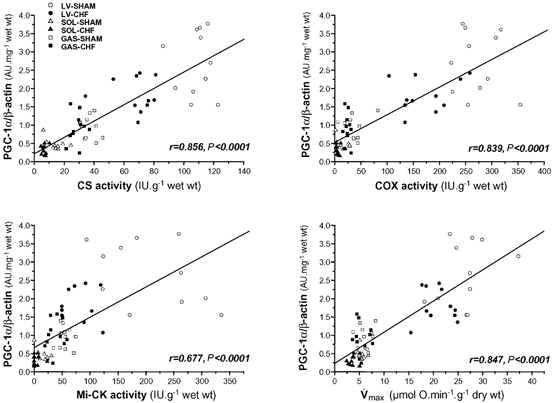
r, the correlation coefficient; P, the statistical significance.
DISCUSSION
The present results show that (1) CHF induces a decrease in oxidative capacity and mitochondrial enzyme activities in both cardiac and skeletal muscles. (2) This is accompanied by a concurrent decrease in the expression of nuclear- and mitochondrial-encoded subunits of complex IV of the respiratory chain with no impairment of the mitochondrial DNA copy number, suggesting dysfunction in mitochondrial gene transcription rather than in replication, associated with a decrease in nuclear-encoded mitochondrial gene expression. (3) The downregulation of PGC-1α, NRF-2α and mtTFA gene expression in cardiac and skeletal muscles observed in CHF could explain the alterations in mitochondrial gene transcription and thereby in respiratory chain function. (4) PGC-1α mRNA levels strongly correlated with expression of COX I or COX IV subunit mRNA as well as with mitochondrial markers, suggesting that this factor, in healthy and diseased muscles, may modulate the tissue-specific oxidative capacity.
Oxidative capacity in failing cardiac and skeletal muscles
Aortic banding is a well-characterized model of heart failure (Feldman et al. 1993) that leads to haemodynamic and peripheral alterations. Despite the diversity in the pathogenesis of heart failure, defects in energy metabolism have been implicated in the disease progression (Ingwall 1993; Dzeja et al. 2000; Lehman & Kelly, 2002; Ventura-Clapier et al. 2002). Proteomic analysis has shown that many of the cardiac proteins altered in CHF are involved in energy metabolism (Heinke et al. 1999). Marked reductions in the complexes of the respiratory chain have been described in both cardiac and skeletal muscles in canine and rat heart failure models (Ide et al. 2001; Marin-Garcia et al. 2001). A dysfunction of respiratory complex I was also shown in human failing myocardium (Scheubel et al. 2002).
Muscle oxidative capacities have been assessed by measuring respiration of mitochondria in permeabilized fibres with no limitation of substrates, ADP or oxygen (Saks et al. 1998). The main mitochondrial pathways were investigated by measuring activity of key enzymes: CS, an enzyme of the Krebs cycle, COX, the complex IV of the respiratory chain, and mi-CK, an intermembrane space enzyme involved in energy transfer between mitochondria and cytosol in oxidative muscles (Ventura-Clapier et al. 1998). We describe here a significant depression in mitochondrial function and biochemical markers in both heart and gastrocnemius. In soleus, only CS and mi-CK were significantly reduced, showing a lower sensitivity of this postural muscle to heart failure. Whether this results from mitochondrial gene expression and/or mitochondrial replication defects is not known at present. It appears that the defects in energy metabolism in CHF were not due to a decrease in the mitochondrial DNA content. Similar results were obtained in pacing-induced CHF (Marin-Garcia et al. 2001), and humans (Sack et al. 1996) although a decrease in this content was described in a murine heart failure model early after myocardial infarction (Ide et al. 2001). This is in line with the increased number of smaller mitochondria without alteration in total mitochondrial volume reported in failing myocardium (Sabbah et al. 1992).
On the other hand, metabolic defects in heart failure are more likely to result from disturbed transcription of OXPHOS genes, as shown by the coordinated decrease in the nuclear- (COX IV) and the mitochondrial-encoded (COX I) COX subunit gene expression. Expression of the main nuclear factors involved in the regulation of mitochondrial genes showed marked alterations in CHF. MtTFA gene expression was clearly diminished in muscles of failing rats, although this decrease did not reach significance for soleus. MtTFA promotes mitochondrial gene transcription and replication. It was recently established that replication of mtDNA was stimulated by small amounts of mtTFA and remained highly active over a large range of mtTFA concentrations whereas transcription was activated only at high levels of mtTFA (Falkenberg et al. 2002). This may explain the depression of mitochondrial gene transcription without alteration in mitochondrial gene replication. Expression of NRF-2α, for which recognition sites have been described on genes encoding mtTFA and mitochondrial proteins in rodents (Scarpulla, 2002), was dramatically downregulated in failing rat cardiac and fast muscles, while NRF-1 was significantly downregulated only in gastrocnemius.
During hypertrophy, myocardial substrate utilization shifts from fatty acid to glucose oxidation with a concomitant decrease in FAO enzyme expression (Sack et al. 1996). This metabolic shift is linked to a downregulation of PGC-1α expression and deactivation of PPAR α nuclear transcription factor (Vega et al. 2000; Lehman & Kelly, 2002). Thus, as PGC-1α is also involved in mitochondrial biogenesis and function through its interaction with NRFs, it was interesting to investigate its expression in cardiac and skeletal muscles in advanced heart failure (six months after aortic banding). PGC-1α mRNA was significantly decreased in CHF gastrocnemius and heart, the two muscles that were the most affected by CHF, while no significant decrease was observed in soleus, which was less affected. Moreover, PGC-1α gene expression positively correlated with expression of COX subunit genes, oxidative capacity and mitochondrial markers in healthy and diseased muscles. Taken together, these results suggest that the decreased oxidative capacity in cardiac and skeletal muscles in CHF may eventually result from a decrease in mitochondrial gene expression linked to PGC-1α, NRFs and mtTFA downregulation.
Nothing is known at present concerning the signalling pathways leading to a downregulation of PGC-1α and decreased energy metabolism in CHF. In the progression from compensated hypertrophy to failure, there is a generalized hyperactivation of hormones (the renin- angiotensin-aldosterone system, endothelin-1, and adrenaline (epinephrine)), neuromediators (noradrenaline (norepinephrine)) and cytokines (interleukins, and tumor necrosis factors (TNFs)). Different signalling pathways that are not activated during the compensated phase of hypertrophy are stimulated later when the compensatory mechanisms are overwhelmed (Haq et al. 2001). Mitogen-actived protein kinases as well as the protein kinase Akt or protein kinase B (PKB) pathways, which are not activated in hypertrophied heart, are highly activated in heart failure irrespective of the cause of the disease (Haq et al. 2001). TNFα, angiotensin II and endothelin-1, which are dramatically increased in heart failure, may potentially activate the Akt pathway (Molkentin & Dorn, 2001). Cardiac-specific expression of a constitutively active mutant of Akt mediates a nearly threefold downregulation of PGC-1α mRNA expression (Cook et al. 2002). Overexpression of TNFα induces heart failure, accompanied by mitochondrial abnormalities and impaired DNA repair activity (Li et al. 2001) and TNFα has been proposed to play a contributory role in mitochondrial defects in CHF (Marin-Garcia et al. 2001). This provides a possible link between neurohumoral activation, decreased PGC-1α expression, and altered cardiac and skeletal muscle bioenergetics in CHF and needs further investigation.
Oxidative capacity in healthy cardiac and skeletal muscles
Muscle and cardiac mitochondrial content can be modulated by neuronal, hormonal or mechanical parameters. The four markers of oxidative capacity (maximal respiration rate, CS, COX and mi-CK) fitted to the known mitochondrial content of the different tissues with the rank order: left ventricle >> soleus > gastrocnemius, but factor(s) that determine the oxidative capacity in different muscles are not known. PGC-1α has been proposed as a very likely candidate (Lehman et al. 2000; Lin et al. 2002). PGC-1α is expressed very highly in soleus and to a lower extent in fast muscles, and amongst transgenic lines containing different levels of PGC-1α its expression is proportional to the expression of mitochondrial markers (Lin et al. 2002). In heart muscle it has been identified as a critical regulator of cardiac mitochondrial number and function in response to energy demands (Lehman et al. 2000). The present results show that oxygen consumption, CS and COX activities strongly correlated with PGC-1α mRNA in cardiac and skeletal muscles. Mi-CK, which is expressed in very low amounts in glycolytic fibres but is increased in oxidative fibres (Ventura-Clapier et al. 1998; Qin et al. 1999), correlated as well with PGC-1α mRNA. These results extend previous data and add support to the contention that PGC-1α may orchestrate a set of genes implicated in the control of muscle oxidative capacity.
Opposite to what was found for PGC-1α, NRF-1 or NRF-2α expression did not linearly correlate with muscle oxidative capacity or biochemical markers. NRF1 and NRF-2α mRNA levels were similar in heart and soleus whilst half the amount was found in gastrocnemius. However, as not only the expression but also the transcriptional activity of the NRFs may be directly controlled by PGC-1α (Wu et al. 1999; Scarpulla, 2002), a PGC-1α-mediated change in the transcriptional activity of NRFs could also take part in the oxidative capacity differences between cardiac and soleus muscles.
MtTFA gene expression did not strictly follow the muscle oxidative capacity either. In fact there was no difference in mRNA expression between heart and soleus, despite a fivefold higher oxidative capacity in heart. However, very recently two mitochondrial transcription factors named TFB1M and TFB2M were discovered that co-associate with mitochondrial RNA polymerase and mtTFA, and are necessary for the transcriptional activity of both mitochondrial promoters (Falkenberg et al. 2002). It needs to be determined whether PGC-1α can regulate their expression. Further studies are needed to establish the mechanisms linking PGC-1α expression and mitochondrial content.
Conclusions
This study shows that depressed mitochondrial function in cardiac and skeletal muscles during CHF is linked to altered expression of mitochondrial protein genes. The results identify several transcription factors whose gene expression is downregulated in CHF, establishing a correlation between the expression of PGC-1α, NRFs, mtTFA and mitochondrial function. They also support the possible importance of PGC-1α in modulating the tissue-specific mitochondrial capacity. Altered mitochondrial function and energy transfer in CHF may limit calcium homeostasis and myofibrillar function and participate in cardiac pump failure and skeletal muscle weakening. Implication of PGC-1α as a possible determinant of oxidative capacity during heart failure may unveil a new potential therapeutic target to escape from the vicious circle of an energy-depleted heart.
Acknowledgments
We thank R. Fischmeister for continuous support. R.V.-C. is supported by the ‘Centre National de la Recherche Scientifique’.
REFERENCES
- Attardi G, Schatz G. Biogenesis of mitochondria. Annu Rev Cell Biol. 1988;4:289–333. doi: 10.1146/annurev.cb.04.110188.001445. [DOI] [PubMed] [Google Scholar]
- Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, Young LH, Semenkovich CF, Shulman GI. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Met. 2001;281:E1340–1346. doi: 10.1152/ajpendo.2001.281.6.E1340. [DOI] [PubMed] [Google Scholar]
- Cook SA, Matsui T, Li L, Rosenzweig A. Transcriptional effects of chronic Akt activation in the heart. J Biol Chem. 2002;277:22528–22533. doi: 10.1074/jbc.M201462200. [DOI] [PubMed] [Google Scholar]
- Czubryt MP, Mcanally J, Fishman GI, Olson EN. Regulation of peroxisome proliferator-activated receptor gamma coactivator 1alpha (PGC-1alpha) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci U S A. 2003;100:1711–6. doi: 10.1073/pnas.0337639100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dairaghi DJ, Shadel GS, Clayton DA. Addition of a 29 residue carboxyl-terminal tail converts a simple HMG box-containing protein into a transcriptional activator. J Mol Biol. 1995;249:11–28. doi: 10.1006/jmbi.1995.9889. [DOI] [PubMed] [Google Scholar]
- De Sousa E, Veksler V, Bigard X, Mateo P, Serrurier B, Ventura-Clapier R. Dual influence of disease and increased load on diaphragm muscle in heart failure. J Mol Cell Cardiol. 2001;33:699–710. doi: 10.1006/jmcc.2000.1336. [DOI] [PubMed] [Google Scholar]
- De Sousa E, Veksler V, Bigard X, Mateo P, Ventura-Clapier R. Heart failure affects mitochondrial but not myofibrillar intrinsic properties of skeletal muscle. Circulation. 2000;102:1847–1853. doi: 10.1161/01.cir.102.15.1847. [DOI] [PubMed] [Google Scholar]
- De Sousa E, Veksler V, Minajeva A, Kaasik A, Mateo P, Mayoux E, Hoerter J, Bigard X, Serrurier B, Ventura-Clapier R. Subcellular creatine kinase alterations-implications in heart failure. Circ Res. 1999;85:68–76. doi: 10.1161/01.res.85.1.68. [DOI] [PubMed] [Google Scholar]
- Dzeja PP, Redfield MM, Burnett JC, Terzic A. Failing energetics in failing hearts. Curr Cardiol Rep. 2000;2:212–217. doi: 10.1007/s11886-000-0071-9. [DOI] [PubMed] [Google Scholar]
- Falkenberg M, Gaspari M, Rantanen A, Trifunovic A, Larsson NG, Gustafsson CM. Mitochondrial transcription factors B1 and B2 activate transcription of human mtDNA. Nat Genet. 2002;31:289–294. doi: 10.1038/ng909. [DOI] [PubMed] [Google Scholar]
- Feldman AM, Weinberg EO, Ray PE, Lorell BH. Selective changes in cardiac gene expression during compensated hypertrophy and the transition to cardiac decompensation in rats with chronic aortic banding. Circ Res. 1993;73:184–192. doi: 10.1161/01.res.73.1.184. [DOI] [PubMed] [Google Scholar]
- Habets PE, Franco D, Ruijter JM, Sargeant AJ, Pereira JA, Moorman AF. RNA content differs in slow and fast muscle fibers: implications for interpretation of changes in muscle gene expression. J Histochem Cytochem. 1999;47:995–1004. doi: 10.1177/002215549904700803. [DOI] [PubMed] [Google Scholar]
- Haq S, Choukroun G, Lim H, Tymitz KM, Del Monte F, Gwathmey J, Grazette L, Michael A, Hajjar R, Force T, Molkentin JD. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation. 2001;103:670–677. doi: 10.1161/01.cir.103.5.670. [DOI] [PubMed] [Google Scholar]
- Heinke MY, Wheeler CH, Yan JX, Amin V, Chang D, Einstein R, Dunn MJ, Dos Remedios CG. Changes in myocardial protein expression in pacing-induced canine heart failure. Electrophoresis. 1999;20:2086–2093. doi: 10.1002/(SICI)1522-2683(19990701)20:10<2086::AID-ELPS2086>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Huo L, Scarpulla RC. Mitochondrial DNA instability and peri-implantation lethality associated with targeted disruption of nuclear respiratory factor 1 in mice. Mol Cell Biol. 2001;21:644–654. doi: 10.1128/MCB.21.2.644-654.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–535. doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- Ingwall JS. Is cardiac failure a consequence of decreased energy reserve. Circulation. 1993;87(suppl. VII):58–62. [Google Scholar]
- Knutti D, Kralli A. PGC-1, a versatile coactivator. Trends Endocrinol Metab. 2001;12:360–365. doi: 10.1016/s1043-2760(01)00457-x. [DOI] [PubMed] [Google Scholar]
- Larrouy D, Vidal H, Andreelli F, Laville M, Langin D. Cloning and mRNA tissue distribution of human PPARgamma coactivator-1. Int J Obes Relat Metab Disord. 1999;23:1327–1332. doi: 10.1038/sj.ijo.0801106. [DOI] [PubMed] [Google Scholar]
- Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman JJ, Kelly DP. Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin Exp Pharmacol Physiol. 2002;29:339–345. doi: 10.1046/j.1440-1681.2002.03655.x. [DOI] [PubMed] [Google Scholar]
- Li H, Wang J, Wilhelmsson H, Hansson A, Thoren P, Duffy J, Rustin P, Larsson NG. Genetic modification of survival in tissue-specific knockout mice with mitochondrial cardiomyopathy. Proc Natl Acad Sci U S A. 2000;97:3467–3472. doi: 10.1073/pnas.97.7.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YY, Chen D, Watkins SC, Feldman AM. Mitochondrial abnormalities in tumor necrosis factor-alpha-induced heart failure are associated with impaired DNA repair activity. Circulation. 2001;104:2492–2497. doi: 10.1161/hc4501.098944. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- Lunde PK, Sjaastad I, Schiotz Thorud HM, Sejersted OM. Skeletal muscle disorders in heart failure. Acta Physiol Scand. 2001;171:277–294. doi: 10.1046/j.1365-201x.2001.00830.x. [DOI] [PubMed] [Google Scholar]
- Marin-Garcia J, Goldenthal MJ, Moe GW. Abnormal cardiac and skeletal muscle mitochondrial function in pacing- induced cardiac failure. Cardiovasc Res. 2001;52:103–110. doi: 10.1016/s0008-6363(01)00368-6. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Dorn GW., II Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu Rev Physiol. 2001;63:391–426. doi: 10.1146/annurev.physiol.63.1.391. [DOI] [PubMed] [Google Scholar]
- Momken I, Kahapip J, Bahi L, Badoual T, Hittinger L, Ventura-Clapier R, Veksler V. Does angiotensin-converting enzyme inhibition improve the energetic status of cardiac and skeletal muscles in heart failure induced by aortic stenosis in rats. J Mol Cell Cardiol. 2003;35:399–407. doi: 10.1016/s0022-2828(03)00044-0. [DOI] [PubMed] [Google Scholar]
- Murakami T, Shimomura Y, Yoshimura A, Sokabe M, Fujitsuka N. Induction of nuclear respiratory factor-1 expression by an acute bout of exercise in rat muscle. Biochim Biophys Acta. 1998;1381:113–122. doi: 10.1016/s0304-4165(98)00018-x. [DOI] [PubMed] [Google Scholar]
- Parisi MA, Clayton DA. Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science. 1991;252:965–969. doi: 10.1126/science.2035027. [DOI] [PubMed] [Google Scholar]
- Qin WN, Khuchua Z, Boero J, Payne RM, Strauss AW. Oxidative myocytes of heart and skeletal muscle express abundant sarcomeric mitochondrial creatine kinase. Histochem J. 1999;31:357–365. doi: 10.1023/a:1003748108062. [DOI] [PubMed] [Google Scholar]
- Sabbah HN, Sharov V, Riddle JM, Kono T, Lesch M, Goldstein S. Mitochondrial abnormalities in myocardium of dogs with chronic heart failure. J Mol Cell Cardiol. 1992;24:1333–1347. doi: 10.1016/0022-2828(92)93098-5. [DOI] [PubMed] [Google Scholar]
- Sack MN, Rader TA, Park SH, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 1996;94:2837–2842. doi: 10.1161/01.cir.94.11.2837. [DOI] [PubMed] [Google Scholar]
- Saks VA, Veksler VI, Kuznetsov AV, Kay L, Sikk P, Tiivel T, Tranqui L, Olivares J, Winkler K, Wiedemann F, Kunz WS. Permeabilized cell and skinned fiber techniques in studies of mitochondrial function in vivo. Mol Cell Biochem. 1998;184:81–100. [PubMed] [Google Scholar]
- Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim Biophys Acta. 2002;1576:1–14. doi: 10.1016/s0167-4781(02)00343-3. [DOI] [PubMed] [Google Scholar]
- Scheubel RJ, Tostlebe M, Simm A, Rohrbach S, Prondzinsky R, Gellerich FN, Silber RE, Holtz J. Dysfunction of mitochondrial respiratory chain complex I in human failing myocardium is not due to disturbed mitochondrial gene expression. J Am Coll Cardiol. 2002;40:2174–2181. doi: 10.1016/s0735-1097(02)02600-1. [DOI] [PubMed] [Google Scholar]
- Shadel GS, Clayton DA. Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem. 1997;66:409–435. doi: 10.1146/annurev.biochem.66.1.409. [DOI] [PubMed] [Google Scholar]
- Terada S, Goto M, Kato M, Kawanaka K, Shimokawa T, Tabata I. Effects of low-intensity prolonged exercise on PGC-1 mRNA expression in rat epitrochlearis muscle. Biochem Biophys Res Comm. 2002;296:350–354. doi: 10.1016/s0006-291x(02)00881-1. [DOI] [PubMed] [Google Scholar]
- Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veksler VI, Kuznetsov AV, Sharov VG, Kapelko VI, Saks VA. Mitochondrial respiratory parameters in cardiac tissue: a novel method of assessment by using saponin-skinned fibers. Biochim Biophys Acta. 1987;892:191–196. doi: 10.1016/0005-2728(87)90174-5. [DOI] [PubMed] [Google Scholar]
- Ventura-Clapier R, De Sousa E, Veksler V. Metabolic myopathy in heart failure. News Physiol Sci. 2002;17:191–196. doi: 10.1152/nips.01392.2002. [DOI] [PubMed] [Google Scholar]
- Ventura-Clapier R, Kuznetsov A, Veksler V, Boehm E, Anflous K. Functional coupling of creatine kinases in muscles: species and tissue specificity. Mol Cell Biochem. 1998;184:231–247. [PubMed] [Google Scholar]
- Wang JM, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P, Bruning JC, Kahn CR, Clayton DA, Barsh GS, Thoren P, Larsson NG. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nature Genetics. 1999;21:133–137. doi: 10.1038/5089. [DOI] [PubMed] [Google Scholar]
- Wharton DC, Tzagoloff A. Cytochrome oxidase from beef heart mitochondria. Methods Enzymol. 1967;10:245–250. [Google Scholar]
- Wu ZD, Puigserver P, Andersson U, Zhang CY, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Xia Y, Buja LM, Scarpulla RC, McMillin JB. Electrical stimulation of neonatal cardiomyocytes results in the sequential activation of nuclear genes governing mitochondrial proliferation and differentiation. Proc Natl Acad Sci U S A. 1997;94:11399–11404. doi: 10.1073/pnas.94.21.11399. [DOI] [PMC free article] [PubMed] [Google Scholar]